

Abstract
The Escherichia coli RNA degradosome is a multienzyme assembly that functions in transcript turnover and maturation of structured RNA precursors. We have developed a procedure to reconstitute the RNA degradosome from recombinant components using modular coexpression vectors. The reconstituted assembly can be purified on a scale that has enabled biochemical and biophysical analyses, and we compare the properties of recombinant and cell-extracted RNA degradosomes. We present evidence that auxiliary protein components can be recruited to the ‘superprotomer’ core of the assembly through a dynamic equilibrium involving RNA intermediaries. We discuss the implications for the regulation of RNA degradosome function in vivo.
Keywords: RNA degradosome, RNase E, DEAD box helicase, polynucleotide phosphorylase, Hfq
Introduction
The multienzyme RNA degradosome of Escherichia coli contributes to the steady-state profile of transcripts.1 Each mRNA species has an intrinsic sensitivity to ribonuclease activity that affects response to transcription rate changes.2 In addition to its degradative role, the degradosome also helps to mature precursors of structured RNAs into their active forms. The substrates processed by the degradosome include the precursors of transfer RNA, 5S ribosomal RNA, 6S RNA (which modulates sigma factor function), and transfer-messenger RNA (which rescues stalled ribosomes).1
The RNA degradosome is composed of four main components: the hydrolytic endoribonuclease E (RNase E), the phosphorolytic exoribonuclease polynucleotide phosphorylase (PNPase; EC 2.7.7.8), the ATP-dependent RNA helicase B (RhlB; EC 3.6.1), and the glycolytic enzyme enolase (EC 4.2.1.11).3–5 The organizing scaffold for the degradosome is provided by RNase E, which is a large protein of 1061 amino acids (118 kDa). The N-terminal half of RNase E encompasses an endoribonuclease domain,6 and this has extensive sequence similarity with its paralogue, RNase G.7 The N-terminal half of RNase E forms a homotetramer,8,9 and the quaternary organization is likely to be functionally important, since disruption of the oligomeric state compromises organism fitness.10 While the N-terminal half has a globular structure, the C-terminal half of RNase E is natively unfolded in isolation11 but provides the recruitment sites for other degradosome components through small recognition motifs,12 as summarized schematically in Fig. 1. The C-terminal half of RNase E also contains two RNA-binding regions that can interact with substrates, such as the 9S precursor for 5S ribosomal RNA, to which it binds avidly with dissociation constants in the nanomolar range.11,13
Fig. 1. Organization of the E. coli RNA degradosome assembly.3–13 .

The endonuclease RNase E has a structured catalytic domain (residues 1–529) and an unstructured scaffold region (residues 530-1061). The latter possesses ‘microdomains’ that contain the binding sites for the DEAD box RNA helicase RhlB, the glycolytic enzyme enolase, and the 3’-exoribonuclease PNPase. Two RNA-binding regions flanking the RhlB binding site are also located within the scaffold region.
RNase E has cutting preferences for RNA with a 5’-terminal monophosphate;14–16 in E. coli, some of these substrates are generated from nascent transcripts by the activity of a specialized phosphatase that removes pyrophosphate from the triphosphate terminus.17,18 Turnover of the mRNA follows from cleavage at internal sites by RNase E, which produces a new 5′ terminus with a monophosphate that can be subsequently cleaved. The fragments can also be digested in a 3′-to-5′ direction by exoribonucleases such as PNPase (77 kDa). Although RNase E and PNPase can function independently, their association within the RNA degradosome may enable the degradation process to be coordinated.19
The RhlB helicase (50 kDa) of the degradosome is a family member of the DEAD box motif ATPases, which include enzymes with demonstrated unwinding, translocase, or remodeling activities.20–24 In the context of RNA turnover, RNA helicases can remove secondary structure in RNAs that would otherwise hinder ribonuclease activity. Hence, the association of RhlB with the ribonucleases of the degradosome facilitates degradation of structured substrates.12,13 Other DEAD box helicases (SrmB, RhlE, and CsdA) can associate with RNase E,25 and these may bind to sites outside the RhlB recognition region.26
The remaining canonical degradosome component, enolase (45 kDa), is a glycolytic enzyme. Although its role in the degradosome is unknown presently, degradosome-bound enolase has been implicated in controlling the stability of mRNA that encodes a transmembrane component of the glucose transporter.27 The rate of degradation of the transcript by RNase E is increased when the glycolytic pathway is blocked by the accumulation of glucose-6-phosphate or its analogue.27
Other proteins are present in substoichiometric amounts from isolated degradosomes, including polyphosphate kinase, poly(A) polymerase, ribosomal proteins, and the molecular chaperones DnaK and GroEL.4,28,29 In addition to these minor variations, the composition of the degradosome can also undergo larger changes in response to environmental conditions. At 15 °C, E. coli cells produce a set of cold shock proteins, including the DEAD box helicase CsdA, that can associate with the degradosome.30 PNPase expression is also increased in response to cold shock.31 The compositional changes of the degradosome following cold exposure may account, in part, for changes in mRNA stability associated with cold shock response.32 Degradosome composition and function may also be modulated through its interactions with the proteins RraA (regulator of ribonuclease activity A) and RraB (regulator of ribonuclease activity B), which inhibit the nucleolytic activity of RNase E.25 Another potential interaction may occur between the degradosome and the cytoskeleton protein MinD, which may account for the apparent association of the degradosome with the cytoskeleton.33
The ribonucleolytic activity of the degradosome can be directed by the RNA chaperone Hfq (host factor I for phage Qβ), an abundant 11.2-kDa RNA-binding protein of the highly conserved Sm-like family.34 In E. coli, Hfq recruits certain small regulatory RNAs (sRNAs) to RNase E for directed destruction of both target mRNAs and sRNAs, enabling rapid responses to environmental changes.29,35,36 Hfq can also function with other sRNAs to stabilize transcripts and to promote translation,37 but these functions probably are not degradosome-mediated.
The emerging view is that the degradosome is a dynamic complex with variable components that modulate its activity under different environmental conditions. An evaluation of these and other properties of the degradosome could be made possible if recombinant materials were available on a large scale. Protocols have been established to reconstitute a minimal RNA degradosome that lacks the catalytic domain of RNase E and the enolase component.38,39 Here, we describe the preparation of the complete recombinant degradosome and the evaluation of its physical and functional properties. Our observations suggest that RNA itself may be a key modulator of degradosome composition and function by influencing a dynamic equilibrium with auxiliary protein components.
Results
Preparation of recombinant degradosome assemblies using modular coexpression vectors
We designed coexpression vectors that are schematically outlined in Fig. 2a. RNase E and RhlB were coexpressed in one culture, and PNPase and enolase were coexpressed in a separate culture (see Supplementary Information for further details). The combined lysate was applied to a Ni2+-affinity column, and an apparent complex was eluted with a gradient of imidazole. This complex remained intact by size-exclusion chromatography using a preparative S500 column (Fig. 2b). The complex eluted broadly from the S500 column without a clear maximum, and the strong absorbance at 254 nm compared to 280 nm indicated that the material coeluted with nucleic acid (Fig. 2b). Much of this copurifying nucleic acid could be removed at the Ni2+-affinity matrix step by including 1.0 M NaCl and 0.5 M urea in the washing buffer. The material eluted from the sizing column with the high-salt/urea treatment gave a maximum with a broad Gaussian-shaped profile (Fig. 2b). The broad elution profiles indicated that the complex is likely to be heterogeneous in size or nucleic acid content. Evaluation of these specimens by electron microscopy revealed discrete particles (data not shown). The estimated mass of the high-salt/urea purified recombinant degradosome from S500 size-exclusion chromatography is in excess of 50 MDa, but this may indicate an ‘open’ structure1 rather than a self-closing assembly.40 The average size estimated by dynamic light scattering varied between 400 and 1000 Å. However, each fraction from size-exclusion chromatography exhibited a low polydispersity index, indicating that the samples are monodisperse within each fraction. The size variation between fractions was likely to be due, in part, to copurification of large RNA species (see below).
Fig. 2. Schematic of the cloning strategy and purification results of recombinant degradosome assemblies.

(a) DNAs encoding for RNase E and RhlB were inserted into the vector pRSFDuet-1; C-terminal histidine-tagged PNPase, together with enolase, were inserted into a pETDuet-1 vector. (b) The elution profile from a preparative S500 size-exclusion column, comparing samples from low-salt or high-salt/urea washes at the Ni2+-affinity matrix step. SDS-PAGE shows an example of a concentrated pre-S500 fraction and of fractions collected at certain elution volumes for the different S500 runs: (a and b) low-salt wash, 4–12% polyacrylamide bis-Tris gel (lanes 1–5, 50–54 ml; lanes 6–7, 75 and 80 ml); (c) high-salt/urea wash, 10% polyacrylamide bis-Tris (lanes 1–5,65–77 ml). In the portions of the chromatogram labelled “a” and “b” excess PNPase is clearly visible, and the portion “d” contains mainly free PNPase. The faint band migrating below enolase is the outer membrane protein OmpF. Molecular weight markers are indicated in kilodaltons. (c) The S500 elution profile of the C-terminally truncated RNase E 1–762/RhlB subassembly previously subjected to a wash step on a Ni2+-affinity column with high-salt/urea buffer. The sharp peak eluting at ~45 ml contains protein aggregates. The broad peak labelled “e” with a low A254/280 nm ratio was analyzed by SDS-PAGE (lanes 1–4 fractions eluting at 68–71 ml) and was found to contain the degradosome subassembly (cartoon insert).
Using pRSF_rnerhlB, we prepared truncated hexahistidine-tagged RNase E constructs that encompass the RhlB recognition site and its flanking RNA-binding regions [RNase E(1–762), a segment of RNase E corresponding to residues 1–762, encompassing the catalytic N-terminal domain and the helicase binding site; RNase E(1–825), a segment of RNase E containing residues 1–825, encompassing the catalytic N-terminal domain and the helicase binding site] but lacked the C-terminal portion that engages enolase and PNPase. The truncated RNase E coexpression constructs gave good yields, and the complex could be purified readily. SDS-PAGE analysis of the post-S500 fractions showed that RNase E (1–762 or 1–825) and RhlB were present in an approximate protomer stoichiometry of 1:1 (Fig. 2c, lanes 1–4).
Comparison of recombinant and endogenously expressed degradosomes
To examine whether the recombinant complex was similar to the in vivo degradosome, we isolated the full-length RNase E from an E. coli strain in which the genomically encoded enzyme has a C-terminally fused FLAG, an octapeptide epitope tag with the amino acid sequence DYKDDDDK.29 As the rne-FLAG gene was under the control of the endogenous promoter, complexes containing RNase E were likely to have the same subunit composition as in wild-type cells. Comparison of the recombinant material and anti-FLAG immunoprecipitate by denaturing gel electrophoresis indicated a similar composition of subunits, which run at the same apparent molecular masses as the recombinant proteins and were confirmed as RNase E, PNPase, enolase, and RhlB by matrix-assisted laser deso-rption/ionization time-of-flight mass spectrometry of protease-generated fragments (Fig. 3a). The immunoprecipitate also contained components of the pyruvate dehydrogenase (PDH) complex, as reported earlier.29 Precipitation of PDH is caused by cross-reactivity of the anti-FLAG antibody with a FLAG-like sequence in the E1 subunit. We found that recombinant degradosome also contains sub-stoichiometric amounts of the outer membrane protein OmpF (Fig. 2b). The protein was not seen in the cell-extracted degradosome, suggesting that its association with the recombinant degradosome is an artifact. Nonetheless, the similarity of the immunoprecipitate and recombinant-based assemblies indicated that the reconstitution procedure successfully generates a degradosome assembly.
Fig. 3. Comparison of recombinant and cell-extracted degradosome.

(a) SDS-PAGE analysis. Lane 1, the cell-extracted FLAG-tagged pull-down degradosome assembly grown at 37 °C; lane 2, recombinant assembly purified using high-salt/urea; lane 3, recombinant low-salt preparation. The change in the electrophoretic mobility of PNPase in lanes 2 and 3 is due to the presence of a hexahistidine tag. Molecular weight markers (in kDa) are indicated. (b) Native gel electrophoresis of degradosome samples. Lane 1, recombinant assembly; lane 2, FLAG-tagged degradosome. Bands were detected by immunoblotting with anti-RNase-E antibodies. After treatment with RNase A, the recombinant and cell-extracted degradosomes migrate at similar positions (right).
The cell-extracted and recombinant degradosome samples were compared by native gel electrophoresis. The cell-extracted samples ran with faster mobility than the recombinant material (Fig. 3b), and the UV absorbance spectra indicated that they contained different amounts of nucleic acid. The nucleic acid was most likely to be RNA, since the electrophoretic mobility of the degradosome was substantially shifted on treatment with RNase A. Recombinant and cell-extracted degradosome migrated more closely after ribonuclease treatment (Fig. 3b). Electrophoretic analysis of nucleic acid extracted from the low-salt purified recombinant degradosome revealed multiple RNA species, with the most abundant resembling ribosomal RNAs in size (e.g., Fig. 4b). These findings are consistent with earlier reports that cell-extracted degradosome contains a number of RNA species, including ribosomal RNA.42
Fig. 4. Processing of 9S RNA.

(a) Sequence and predicted secondary structural elements of 9S RNA. The in-vitro-transcribed 9S RNA used in this work has a G at positions +1 and + 2. The locations of the two major cleavage sites (indicated by the letters a and b in the schematic) and the minor cleavage site (c in the schematic) are indicated with downward arrows. (b and c) Denaturing PAGE gels stained with SYBR Gold reveal the processed products of 9S RNA by the recombinant degradosome and RNase E(1–762)/RhlB subassembly. Lane 1 in (b) represents RNA size markers, and lane 2 represents in-vitro-transcribed 9S RNA. 9S RNA processing was tested in the absence (lanes 3–4) and in the presence (lanes 5–10) of additives. The band at 126 nt corresponds to p5S, a precursor of the mature 120-nt 5S rRNA. In (c), the addition of EDTA is seen to inhibit 9S RNA processing by wild-type (wt) RNase E in the recombinant degradosome assembly (lane 1) and in the RNase E(1–762) RhlB subassembly (lane 4). In the absence of EDTA, processing occurs (lanes 2, 3, 5, and 6). The 9S secondary structure schematic was adopted from Cormack and Mackie.41
Processing of ribosomal RNA precursor 9S RNA by the degradosome
One well-established activity assay for RNase E in vitro is the processing of 9S RNA, which is a precursor of 5S ribosomal RNA.43 Recombinant degradosome and truncated RNase E/RhlB were incubated with purified 9S RNA transcribed in vitro. This substrate was processed into the products expected from cuts at the three RNase E cleavage sites (Fig. 4a), and the pattern of digest products was similar to those reported earlier for preparations of cell-isolated degradosomes.3 The presence of ethylenediaminetetraacetic acid (EDTA) prevents RNA degradation, consistent with the expectation that the activity of RNase E is magnesium-dependent (Fig. 4c, lane 1).
The degradosome used in initial assays was the RNase E N305D mutant, which has been shown to decrease catalytic activity in the context of the isolated catalytic domain for 13-nt single-stranded RNA substrates.9 We observed that this RNase E mutant still retains processing capabilities for a large structured RNA in the context of the full-length protein and RNase E(1–762) (lanes 3–10 and 5–6 in Fig. 4b and c, respectively), but not as great as for the degradosome reconstituted with wild-type RNase E (not shown). The activity could be favored by interaction with the arginine-rich domain of RNase E;44 in the assays used here, an excess of enzyme was used so that even weakened activity would still result in processing. 9S RNA processing was also observed for the degradosome subassembly comprising RNase E N305D(1–762) and RhlB (Fig. 4c, lanes 5 and 6).
In the presence of phosphate, which is required for the nucleolytic activity of PNPase, the 9S processing reaction was slightly more effective, probably because the competing degradosome-bound RNA was partially degraded by the phosphorolytic activity of PNPase (Fig. 4b, lanes 5 and 6). Addition of ATP and Mg2+ was expected to stimulate the helicase activity of RhlB, but this resulted in the generation of a distinct ladder of polynucleotides (apparent as smear in lanes 7 and 8 in Fig. 4b). This ladder was likely due to the reverse (polymerization) activity of PNPase from ADP. However, in the presence of both phosphate and ATP, the equilibrium was expected to be driven in the direction of degradation by PNPase; indeed, the processing of 9S RNA and degradation of the background RNA were most efficient when these additives were present (Fig. 4b, lanes 9 and 10). Helicase ATPase activity and PNPase-catalyzed ribonucleotide polymerization may be linked in a cycle that enhances the overall rates of exoribonucleolytic degradation.
Composition of the degradosome varies with physiological conditions and RNA content
The subunit stoichiometry of the degradosome preparations was determined by SDS-PAGE and densitometry (Fig. 5 and Supplementary Fig. S1). The ratio of components varied according to extraction conditions of the recombinant material or physiological conditions of the cell-extracted FLAG-tagged degradosome. However, the simplest stoichiometry observed was a 1:1 complex of RNase E/PNPase protomers in the high-salt/urea-extracted degradosome (Fig. 5a). A similar ratio was observed for the FLAG-tagged degradosome under regular growth conditions (Fig. 5c).
Fig. 5. Stoichiometry of the RNA degradosome.
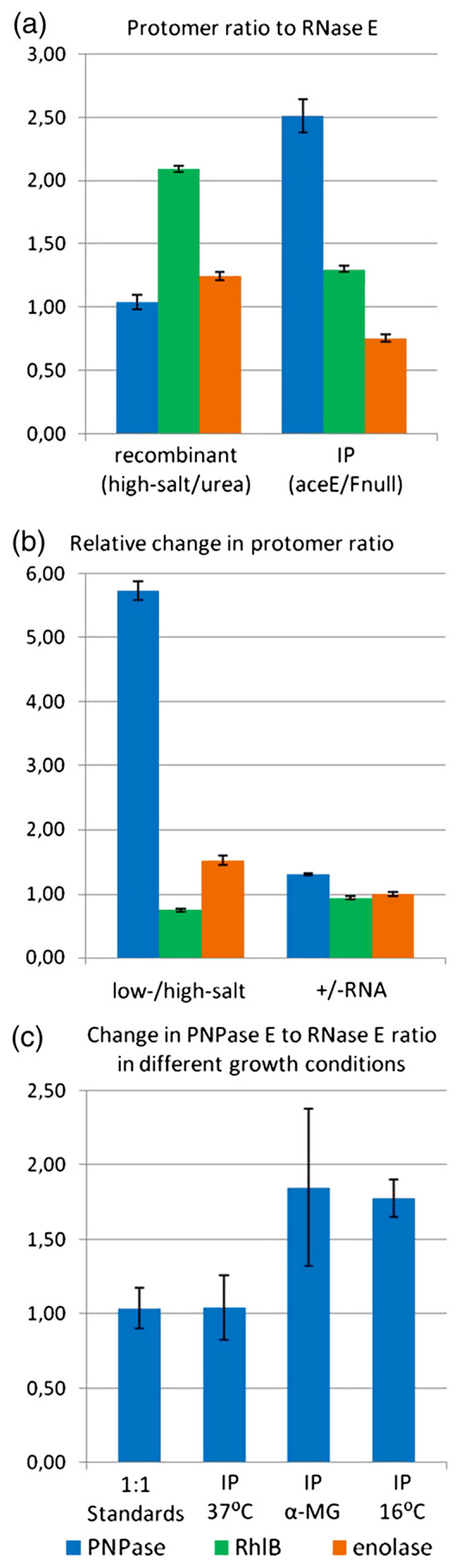
Band intensities obtained from gel densitometry were used to calculate internal ratios of PNPase, RhlB, and enolase to RNase E. (a) Internal ratios for high-salt/urea-treated recombinant degradosome and for FLAG-tagged degradosome purified from aceE/aceF-null cells to avoid crossreaction of the PDH with the FLAG antibody. The ratios were normalized using 1:1:1:1 standards prepared from purified recombinant components (see Supplementary Fig. S1). The RNase E level is set to unity. (b) An example of the change in internal ratios for components of the recombinant degradosome. Ratios for low-salt-extracted degradosome were divided by ratios of high-salt/urea-extracted degradosome. High-salt/urea degradosome was also purified with the addition of extra RNA, and the internal ratios were divided by ratios for the unmodified sample. Samples were taken from fractions at the same elution volume from size-exclusion chromatography. (c) PNPase/RNase E ratio determined for FLAG-tagged degradosomes purified from cells subjected to different growth conditions. αMG cells are cells grown in the presence of α-D-methylglucoside. Error bars are standard deviations that account for loading errors and background subtraction, and were estimated by running the samples in triplicate on SDS-PAGE.
The degradosome composition was found to be very sensitive to nucleic acid content. The low-salt procedure generated recombinant degradosome that was enriched in both RNA and PNPase components, in comparison with the complex derived from the high-salt/urea procedure. Correspondingly, the addition of exogenous RNA during the purification procedure resulted in the enrichment of PNPase in the recombinant degradosome eluted from the sizing column (Fig. 5b). This indicates that exogenous RNA may have aided in the recruitment of additional PNPase to the degradosome.
Stress conditions may influence the degradosome composition in vivo. E. coli carrying the rne-FLAG gene was subjected to either cold shock (16 °C) or phosphosugar stress (Supplementary Information). The glucose analogue α-D-methylglucoside accumulates in the cells as α-methylglucoside 6-phosphate, which induces phosphosugar stress.27 Densitometry analysis of immunoprecipitates indicates that, under these stress conditions, the amount of PNPase associated with the degradosome increases. The value for the ratio was roughly two PNPase monomers per RNase E monomer in our preparations (Fig. 5c).
The stoichiometry was also estimated for immunoprecipitates prepared from an aceE/aceF (E1 subunit of PDH/E2 subunit of PDH)-null strain of E. coli that lacks the PDH E1 and E3 subunits that were the contaminants in rne-FLAG preparations. Immunoprecipitates from aceE/aceF-null cells gave a purer degradosome; however, the PNPase component is nearly three times that estimated from the tagged degradosome from wild-type control cells (Fig. 5a). PNPase enrichment in the aceE/aceF-null cells was perhaps in response to stress arising from metabolic perturbation in the absence of PDH.
Interactions of RNase E with Hfq
Hfq is an RNA chaperone required for RNase-E-mediated degradation of ptsG mRNA.36 When E. coli is subjected to phosphosugar stress, an sRNA (SgrS) which binds Hfq is induced, which in turn associates with RNase E in the degradosome.29,45 In agreement with earlier findings, Hfq was observed by immunoblotting to be preferentially enriched in FLAG-tagged RNase E immunoprecipitates under phosphosugar stress conditions compared with cells grown in normal media (data not shown).
The coimmunoprecipitation with Hfq was dependent on the C-terminal 360 residues of RNase E,29 suggesting that the interaction involves this scaffold region, which includes the binding sites for RhlB, enolase, and PNPase (Fig. 6a). The Hfq/RNase E interaction was probed using purified recombinant Hfq and two RNase E constructs spanning this region (Fig. 6a). RNase E(628–843) (a segment of RNase E containing residues 628–843 of the C-terminal domain, encompassing the two RNA-binding regions and the helicase recognition site) included the RhlB recognition motif and an RNA-binding region. The CTD (a hexa-histidine-tagged C-terminal domain of RNase E comprising residues 1–26 fused to residues 498–1061) construct contained the binding sites for all degradosome components and the two RNA-binding regions. Protein-protein interactions were tested with the cross-linker dimethyl suberimidate (DMS) and analyzed by SDS-PAGE and immunoblotting. Highly purified Hfq failed to interact with either RNase E(628–843) or CTD; however, in positive controls, RNase E(628–843) formed a cross-linked species with RhlB (Supplementary Fig. S2). An interaction between CTD and Hfq was tested by isothermal titration calorimetry, but no heat change was observed (results not shown).
Fig. 6. The interaction between Hfq and RNase E is RNA-mediated.

(a) Schematic cartoon depicting the RNase E constructs used to investigate RNase E/Hfq interactions. Hfq (green) is reported to bind RNase E in the 701–1061 region.29 (b) SDS-PAGE analysis of Hfq and RNase E samples treated with the cross-linking reagent DMS. With increasing amounts of RNA, a new band (indicated by a box) attributed to a Hfq/RNase E(628–843)/RNA interaction appeared. Distribution of higher-molecular-weight species, which can be seen in the smeared pattern was also formed; this diminished with higher concentrations of RNA. Molecular weight markers (in kDa) are indicated. (c) Immunoblot against His-tagged RNase E CTD. Mixtures of RNase E CTD, Hfq, and bulk yeast RNA were treated with DMS and resolved by SDS-PAGE, followed by immunoblotting. A band is detected (*) when RNase E CTD is incubated with Hfq and RNA, but is not present if either Hfq or RNA is omitted, suggesting the formation of a ternary complex. (d) Immunoblot against Hfq, following native electrophoresis of mixtures of RNase E(628–843), Hfq, and bulk yeast RNA. In the presence of RNase E (628–843), the mobility of the Hfq/RNA complex is retarded (lane 4), suggesting an interaction between these components. The Hfq antibody cross-reacts weakly with the RNase E(628–843)/RNA complex, causing background labeling. Hfq does not enter the gel in the absence of RNA.
Hfq samples that were not purified with a butyl Sepharose column were found to contain avidly bound RNA that originated from the E. coli expression host. This E. coli RNA/Hfq complex readily formed cross-links in the presence of glutaraldehyde with RNase E(628–843) and CTD (Supplementary Fig. S3). This interaction was further explored using RNA-free Hfq in the presence of bulk yeast RNA. Denaturing PAGE revealed a supershifted species, which becomes more intense with addition of more RNA (Fig. 6b). A supershifted species was also observed in an immunoblot using an anti-His antibody to detect the His-tagged RNase E CTD (Fig. 6c). Similarly, a supershifted species of Hfq and RNase E (628–843) was visualized by anti-Hfq immunoblotting in the presence of bulk RNA (Fig. 6d). Hfq has also been reported to coimmunoprecipitate with PNPase.46 We found that this interaction was also RNA-mediated (Supplementary Fig. S4). These results indicate that Hfq may be recruited to the degradosome through a mediating RNA.
Discussion
We have described here a method for preparing RNA degradosome from recombinant components. Using this material, we have explored the subunit composition of the fundamental unit of the degradosome, which we refer to as the “superprotomer” in view of its complexity and its likelihood to associate into a higher-order quaternary structure. Our analyses indicate that RNase E and PNPase are present in equimolar ratio in ‘resting-state’ preparations with minimal copurifying RNA. An equimolar ratio is consistent with earlier findings for the stoichiometry of PNPase with the isolated recognition site from RNase E,11 and with recent crystallographic analysis of the E. coli PNPase/RNase E complex (Salima Nurmohamed, personal communication). It agrees with the observed stoichiometry of RNase E/PNPase from isolated cellular degradosome, but we must emphasize that this ratio is for normal growth conditions. We will return to this point below in discussions of the changes occurring during stress conditions. For enolase and RhlB, crystallographic and biophysical measurements indicate that one enolase dimer and one helicase protomer interact with their respective isolated recognition sites within RNase E.13,47,48 However, for the recombinant and cell-extracted degradosomes, the stoichiometries vary with RNA content.
The RNase E/PNPase/enolase/RhlB “superprotomer” is likely to associate into a higher-order assembly, since the scaffolding component (namely, RNase E) is itself a tetramer. Additionally, PNPase is a stable trimer49 and could bridge RNase E tetramers in a higher-order organization. In principle, three RNase E tetramers and four PNPase trimers could form a self-closing assembly composed of 12 protomers, which would satisfy all possible binding sites. The ideal composition of such an assembly is 12:12:24:12 RNase E/PNPase/enolase/RhlB.40 Our experimentally determined stoichiometries are consistent with this model for RNase E and PNPase in an unstressed physiological state, but the RhlB and enolase components can vary and may be more dynamic elements of the degradosome. The idealized assembly would have a mass in excess of 4 MDa, but our S500 results indicate that the complex is much larger still. We cannot rule out the possibility of an extended nonglobular shape and some aggregation that might result in the nonideal behavior on the size-exclusion column.
A recent study indicates that the degradosome can be found in superhelical assemblies that are associated with the cytoskeleton,33 and an earlier microscopy study revealed it to be localized near the cytoplasmic membrane.50 It is not clear whether the degradosome spontaneously forms a helical network in these cellular bodies, but our electron microscopy images suggest that purified degradosome is not a filamentous oligomer. The helical bodies observed in vivo may arise from interactions of the degradosome with components of the cytoskeleton.
Our findings show that the PNPase content of the degradosome can change with physiological conditions, and this is most likely due to ancillary RNA. The PNPase content in immunoprecipitated degradosome changes in response to phosphosugar stress, temperature shock, and perhaps metabolic adjustment associated with the loss of the PDH complex. Cellular PNPase levels may be environment-sensitive; for example, cold shock can boost PNPase expression,32 and the cellular ratio of total PNPase to RNase E can vary with growth stage.50 In the recombinant degradosome preparations, we observe that the PNPase stoichiometry increases with the content of copurifying RNA. PNPase itself could be the source of the copurifying RNA: recombinant PNPase avidly binds RNA, and we observe that it copurifies with ribosomal RNA fragments (Górna et al., unpublished results). It is possible that accessory protein/RNA complexes could be recruited to the degradosome in a dynamic equilibrium that changes the composition and directs the function of the degradosome. One possible function of this additional recruitment might be to assist PNPase in its capacity to perform quality control checks for ribosomal RNA.51,52
We also explored interactions of RNase E with the Hfq protein, which mediates the function of some sRNAs in E. coli. Under conditions of sRNA expression, Hfq coimmunoprecipitates with FLAG-tagged RNase E,29 potentially explaining how the sRNA targets its complementary mRNA for degradation. In our study, no evidence for direct interaction between RNase E and Hfq could be identified, but the addition of RNA was sufficient to induce an interaction, suggesting that an sRNA (or sRNA/mRNA complex) may be responsible for the coimmunoprecipitation of these proteins under stress conditions. Morita et al. observed that Hfq remained bound to RNase E even after incubation with micrococcal nuclease, suggesting that the proteins interact directly.29 However, none of the techniques used here (e.g., cross-linking, nondenaturing PAGE, and isothermal titration calorimetry) was able to detect a direct interaction between the purified proteins. This prompts an alternative explanation that micrococcal nuclease was unable to digest a tightly bound RNA that maintained an indirect RNase E/Hfq interaction. There are, of course, other explanations for the absence of an Hfq/RNase E interaction in vitro. Posttranslational modification of one or either protein may be required, perhaps in an environmental-stimulusdependent manner. However, our finding of avidly bound RNA in both FLAG-tagged RNase E immunoprecipitate and recombinant RNase E preparations, as well as during Hfq purification, supports the hypothesis that the interaction of Hfq and RNase E is RNA-mediated. Our hypothesis may serve to explain how Hfq is recruited to RNase E under conditions of sRNA expression.29
It has been observed that the composition of the cellular degradosome can be altered by the regulators RraA and RraB.25 These observations lead to a picture of a dynamic nature of the degradosome, whose composition and function are modulated by regulators. Our data suggest that RNA may affect the protein composition of the degradosome. In the case of PNPase, the RNA is likely to be a structured molecule and, for Hfq, its ternary complex with transcript/sRNA is likely to form a transient interaction with the degradosome prior to RNase-E-mediated cleavage (Fig. 7). We thus envisage that defined RNA species, perhaps recognized by their specific folds, may be modulators of the degradosome, through corecruitment of accessory factors in dynamic equilibria that modify its function.
Fig. 7. Schematic summary of a hypothetical model of canonical degradosome superprotomer core and its compositional variation by binding protein/RNA complexes.

The recruitment of additional proteins is proposed to be mediated through RNA species that form binding interactions with two different proteins. The arrows indicate the recruitment of the auxiliary RNA/protein complexes that may be engaged at the RNA-binding sites of the RNase E C-terminal domain.
Materials and Methods
Construction, expression, and purification of recombinant degradosome
Coexpression vectors for the expression of recombinant components of the degradosome assembly in E. coli were constructed using pRSFDuet-1 (Kanr) and pETDuet-1 (Ampr) plasmids (Novagen) (see Fig. 2a). The genes encoding the individual degradosome components were amplified by polymerase chain reaction (PCR) from pET11a vectors (provided by A. J. Carpousis). PCR primers included restriction enzyme sites for facile cloning of the products into the multiple cloning sites of the coexpression vectors. The N305D mutation in the catalytic domain was generated by introducing a single nucleotide change in the RNase E gene (AAC-to-GAC) using a procedure based on the Stratagene Quikchange mutagenesis kit. The clones were sequenced to corroborate the vector construction and insertion of the intended mutation.
E. coli BL21(DE3) cells were transformed with pRSF_ rnerhlB or pETD_pnpeno, and a number of transformants were transferred to 5 ml of 2xYT medium supplemented with 50 μg/ml kanamycin or 100 μg/ml carbenicillin, respectively, and incubated at 37 °C. Five milliliters of the liquid precultures was used to separately inoculate 2-l Erlenmeyer flasks containing 500 ml of 2xYT and either 50 μg/ml kanamycin or 100 μg/ml carbenicillin, with incubation continued at 37 °C until an OD600 nm of 0.5 had been reached, at which point expression of the recombinant genes in both cultures was induced with 1 mM IPTG. The temperature of the pETD_pnpeno cultures was decreased to 25 °C. After 3 h, the pETD_pnpeno and pRSF_rnerhlB cultures were mixed, and the cells were harvested and resuspended in buffer A [50 mM Tris–HCl pH 7.8,100 mM NaCl, 50 mM KCl, 5 mM imidazole, 5 mM MgSO4, and 5% (vol/vol) glycerol]. Cells were lysed by passing several times through an EmulsiFlex-05 cell disruptor (Avestin) until the lysate was free flowing. The lysate was clarified by centrifugation (37,500g, 30 min, 4 °C), and the soluble fraction was loaded onto a Ni-NTA HiTrap column (GE Healthcare). Extensive washing with either low-salt or high-salt/urea was followed by an isocratic elution with buffer A supplemented with 0.5 M imidazole. Fractions containing RNase E, RhlB, enolase, and PNPase (assessed by SDS-PAGE) were pooled and concentrated with 100-kDa cutoff Centricon units (Vivascience) and loaded onto an S500 size-exclusion column (GE Healthcare) equilibrated with buffer C [50 mM Tris–HCl pH 7.8, 150 mM NaCl, 100 mM KCl, 5 mM MgSO4, and 5% (vol/vol) glycerol]. Samples were evaluated by SDS-PAGE and subjected to trypsin digestion, followed by matrix-assisted laser desorption/ioniza-tion time-of-flight mass spectrometry analysis of the peptide fragments. Analysis of the change in degrado-some stoichiometry upon addition of extra RNA was performed for high-salt/urea purified degradosome with hexa-histidine tags on both RNase E and PNPase. For that purpose, a sample containing roughly 40 mg of concentrated degradosome purified by metal-affinity chromatography was divided into two, and one of the aliquots was supplemented with 2 mg of yeast ribonucleic acid (Sigma) and incubated for 30 min at 25 °C, then both samples were purified by size-exclusion chromatography under identical conditions.
Preparation of individual degradosome components and RNase E constructs
RhlB, PNPase, and enolase were overexpressed as previously described by Callaghan et al. 11 The full-length N-terminal histidine-tagged RNase E N305D was expressed in BL21(DE3) cells at 37 °C, with cells harvested 3 h after induction with IPTG. The cell lysate was extracted and purified in buffers containing 6 M urea. The C-terminal RNase E constructs consisting of either residues 628–843 or the entire CTD were purified as previously described.11
Preparation of Hfq
E. coli BL21(DE3) cells transformed with pEH-10-(hfq) were a kind gift of Dr. Isabella Moll (Max Perutz Laboratory, Vienna). Isolation and purification were performed as described by Vassillieva et al.53 Purified Hfq was dialyzed against 50 mM sodium phosphate (pH 8.0) and 150 mM NaCl.
Isolation of degradosome assemblies from cells
E. coli K12 strain TM522 has the genotype W3110mlc rne-FLAG, where the C-terminal FLAG-tagged RNase E replaces the endogenous RNase E.29 Details of experimental procedures are provided in Supplementary Information.
Preparation of 9S RNA
A 263-bp template encoding E. coli 9S RNA was produced from the pKK233-2 plasmid54 (kindly provided by A. J. Carpousis and L. Poljak, Centre National de la Recherche Scientifique, Toulouse) using two rounds of PCR. The first, using primers 5’-GAGCGTTCACCGACAAAC-3’ and 5’-GGAGCTGTTTTGGCGGATGAGAGAAG-3’, introduced an A-to-G substitution (underlined). The second, using primers 5′-CGAAAGGCCCAGTCTTTCGACTG-3′ and 5′-GTAATACGACTCACTATAGGAGCTGTTTTGGCGGAT-GAGAG-3’, introduced a T7 RNA polymerase consensus (underlined). The second PCR product was used as a template in a standard in vitro transcription reaction.
Degradosome activity assays
To assay the RNA processing activity of the recombinant degradosome, reactions were set up with 1 or 2 pmol of 9S RNA and 25–28 pmol of RNase E monomer (assuming 1:1:1:1 stoichiometry for the four degradosome components and using the Christian-Warburg method to correct for nucleic acid contamination55). For low-salt purified recombinant degradosome containing RNase E N305D mutant, 30-μl reactions were set up in buffer C supplemented with 0.1 U/μl RNaseOUT™ (Invitrogen), 1 mM DTT, and, where indicated, 1 mM ATP and 1 mM MgCl2 or 10 mM potassium phosphate. Samples were assembled on ice without RNA and preincubated (37 °C, 20 min), then 1 μl of 1 μM 9S RNAwas added, and the reaction mixture was incubated for 5 min at 25 °C. Reactions were quenched with 5 μl of 10 mg/ml Tritirachium album Proteinase K (Sigma-Aldrich) and 50 μl of 2× Proteinase K buffer [200 mM Tris–HCl pH 7.5, 25 mM EDTA, 300 mM NaCl, and 2% (wt/vol) SDS]. Samples were incubated (37 °C, 30 min) then RNA-purified by phenol extraction and ethanol precipitation. For wild-type RNase E recombinant degradosome and N305D RNase E(1–762)/RhlB, 4 μl of 6.3 μM RNase E construct in buffer C was incubated with 2 μl of 1 μM 9S RNA in deionized water and either 1 μl of 50 mM Mops (pH 7.4) and 50 mM NaCl or 1 μl of 70 mM EDTA. The reaction was assembled on ice without RNA, then RNAwas added and the reaction was incubated for 30 or 60 min at 37 °C. Reactions were quenched by adding 1 vol of 2× Proteinase K buffer with 0.2 mg/ml Proteinase K and incubating for 30 min at 50 °C. After the addition of formamide/urea loading dye,56 the samples were analyzed on an 8% polyacrylamide gel containing 7 M urea. Nucleic acids were detected using SYBR® Gold stain (Invitrogen). TotalLab (Nonlinear Dynamics) was used to estimate the molecular weight of the cleavage products.
Electrophoretic mobility shift assays
Samples of FLAG-tagged degradosome (5 μl) in immunoprecipitation buffer (20 mM Tris-Cl pH 8.0, 0.1M KCl, 5 mM Mg sulfate, 10% v/v glycerol, 0.1% v/v Tween 20,1 tablet/50 ml of EDTA-free protease inhibitor (Roche)) buffer or recombinant high-salt/urea purified degrado-some in buffer C were treated with 2 μl of 10 mg/ml RNase A, where indicated, analyzed on 0.5% (wt/vol) agarose gel, and transferred to PVDF P-immobilon membrane (Millipore). The membrane was probed for RNase E using rabbit polyclonal anti-RNase E primary antibody (a gift of A. J. Carpousis). For the RNase E(628–843)/Hfq binding assay, 5 μl of 150 μM protein solutions was mixed, where indicated, with yeast ribonucleic acid (Sigma), incubated for 30 min on ice, and analyzed on 0.5% (wt/vol) agarose gel in 0.5× Tris-borate-EDTA buffer. Proteins were electrotransferred onto PVDF and incubated with rabbit anti-Hfq antibody (kindly provided by Isabella Moll, Max Perutz Laboratory). PVDF membranes were next incubated with horseradish-peroxidase-conjugated goat antirabbit secondary antibody (Sigma) and visualized using enhanced chemiluminescence (GE Healthcare).
Densitometry of protein gels
Samples of degradosome were analyzed on a 4–12% bisTris gel (Invitrogen) and stained with SimplyBlue SafeStain (Invitrogen), and the intensities of protein bands were quantified using GeneTools software (Syngene). To avoid the problem of nonlinearity in the relationship between integrated intensities and protein quantity, the internal ratios of band intensities within each lane were used for comparison between the lanes. A 1:1:1:1 mixture of purified recombinant His6-RNase E, PNPase, RhlB, and enolase was prepared as a standard sample and loaded in a concentration range onto a gel to calculate a theoretical internal ratio of each component to RNase E. Errors were estimated by running each degradosome sample on the gel in triplicate.
Chemical cross-linking
RNase E(628–843) was exchanged into cross-linking buffer (50 mM Mops pH 7.4 and 50 mM NaCl) using a PD-10 column (GE Healthcare), and Hfq and RNase E CTD were prepared as described above. Chemical cross-linking was performed using a 70-mM stock of DMS (Pierce) freshly prepared in 0.5 M sodium borate (pH 9.2). Cross-linking reactions (volume, 10 μl) contained 115 pmol of RNase E(628–843), 708 pmol of Hfq, and yeast RNA (Sigma) at an indicated concentration in 10 mM DMS, 40 mM Mops, and 100 mM sodium borate (pH 8.7). For immunoblotting, cross-linking reactions were prepared with 12 pmol of RNase E CTD, 70 pmol of Hfq, 2 mg/ml yeast RNA, 10 mM DMS, 70 mM sodium borate, 10 mM potassium phosphate, 20 mM NaCl, and 20 mM Mops (pH 8.5). Reactions were incubated for 45 min at 25 °C. For results described in Supplementary Information, proteins were cross-linked at a concentration of 0.4–0.7 mg/ml in a buffer of 50 mM sodium phosphate (pH 8.0), 150 mM NaCl, 2 mM DTT, and 1 mg/ml DMS for 30 min at 25 °C. All reactions were quenched by addition of 50 mM Tris–HCl (pH 8.0) and analyzed by SDS-PAGE. For immunoblotting, the proteins were transferred to PVDF P-immobilon membrane, followed by incubation with alkaline-phosphatase-conjugated mouse anti-His6 antibody (Sigma) and detection by colorimetric reaction with FAST BCIP/NBP (Sigma).
Supplementary Material
Acknowledgements
We thank Leonora Poljak and Agamemnon Carpousis (Centre National de la Recherche Scien-tifique) for generously providing plasmids encoding the individual degradosome components and 9S precursor of 5S ribosomal RNA, and for the antibody against RNase E. We thank Kiyoshi Nagai and Daniel Krummel (Laboratory of Molecular Biology, Cambridge) for helpful advice on preparing RNA, and Isabella Moll (Max Perutz Laboratory) for providing the Hfq expression vector, advice on protein purification, and antibody against Hfq. We acknowledge gratefully Hiroji Aiba (Nara University) for the generous gift of E. coli strain TM522 and its aceE/aceF-null derivative, and Len Packman (Department of Biochemistry, Cambridge) for mass spectrometry analysis. We thank Xue Yuan Pei and Richard Henderson for help and advice with electron microscopy. We thank Salima Nurmo-hamed for assistance and advice with the isothermal titration calorimetry experiments. We thank Hir-oyoshi Matsumura for help in designing some of the earlier expression constructs for RNase E, and A. J. Carpousis, Gadi Schuster, Joel Belasco, and Kenny McDowall for helpful discussions. M.G. was supported by a European Union training fellowship of the European Union Early-Stage Training Site ChemBioCam. This work is supported by the Wellcome Trust.
Abbreviations used
- RNase E
ribonuclease E
- PNPase
polynucleotide phosphorylase
- RhlB
RNA helicase B
- sRNA
small regulatory RNA
- PDH
pyruvate dehydrogenase
- EDTA
ethylenediaminetetraacetic acid
- DMS
dimethyl suberimidate
References
- 1.Carpousis AJ. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol. 2007;61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- 2.Carpousis AJ, Vanzo NF, Raynal LC. mRNA degradation. A tale of poly(A) and multiprotein machines. Trends Genet. 1999;15:24–28. doi: 10.1016/s0168-9525(98)01627-8. [DOI] [PubMed] [Google Scholar]
- 3.Carpousis AJ, Van Houwe G, Ehretsmann C, Krisch HM. Copurification of E. coli RNAase E and PNPase: evidence for a specific association between two enzymes important in RNA processing and degradation. Cell. 1994;76:889–900. doi: 10.1016/0092-8674(94)90363-8. [DOI] [PubMed] [Google Scholar]
- 4.Miczak A, Kaberdin VR, Wei CL, Lin-Chao S. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc Natl Acad Sci USA. 1996;93:3865–3869. doi: 10.1073/pnas.93.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Py B, Higgins CF, Krisch HM, Carpousis AJ. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature. 1996;381:169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- 6.McDowall KJ, Cohen SN. The N-terminal domain of the rne gene product has RNase E activity and is non-overlapping with the arginine-rich RNA-binding site. J Mol Biol. 1996;255:349–355. doi: 10.1006/jmbi.1996.0027. [DOI] [PubMed] [Google Scholar]
- 7.McDowall KJ, Hernandez RG, Lin-Chao S, Cohen SN. The ams-1 and rne-3071 temperature-sensitive mutations in the ams gene are in close proximity to each other and cause substitutions within a domain that resembles a product of the Escherichia coli mre locus. J Bacteriol. 1993;175:4245–4249. doi: 10.1128/jb.175.13.4245-4249.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Callaghan AJ, Grossmann JG, Redko YU, Ilag LL, Moncrieffe MC, Symmons MF, et al. Quaternary structure and catalytic activity of the Escherichia coli ribonuclease E amino-terminal catalytic domain. Biochemistry. 2003;42:13848–13855. doi: 10.1021/bi0351099. [DOI] [PubMed] [Google Scholar]
- 9.Callaghan AJ, Marcaida MJ, Stead JA, McDowall KJ, Scott WG, Luisi BF. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature. 2005;437:1187–1191. doi: 10.1038/nature04084. [DOI] [PubMed] [Google Scholar]
- 10.Caruthers JM, Feng Y, McKay DB, Cohen SN. Retention of core catalytic functions by a conserved minimal ribonuclease E peptide that lacks the domain required for tetramer formation. J Biol Chem. 2006;281:27046–27051. doi: 10.1074/jbc.M602467200. [DOI] [PubMed] [Google Scholar]
- 11.Callaghan AJ, Aurikko JP, Ilag LL, Gunter Grossmann J, Chandran V, Kuhnel K, et al. Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J Mol Biol. 2004;340:965–979. doi: 10.1016/j.jmb.2004.05.046. [DOI] [PubMed] [Google Scholar]
- 12.Vanzo NF, Li YS, Py B, Blum E, Higgins CF, Raynal LC, et al. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev. 1998;12:2770–2781. doi: 10.1101/gad.12.17.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandran V, Poljak L, Vanzo NF, Leroy A, Miguel RN, Fernandez-Recio J, et al. Recognition and cooperation between the ATP-dependent RNA helicase RhlB and ribonuclease RNase E. J Mol Biol. 2007;367:113–132. doi: 10.1016/j.jmb.2006.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang X, Diwa A, Belasco JG. Regions of RNase E important for 5’-end-dependent RNA cleavage and autoregulated synthesis. J Bacteriol. 2000;182:2468–2475. doi: 10.1128/jb.182.9.2468-2475.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackie GA. Ribonuclease E is a 5’-end-dependent endonuclease. Nature. 1998;395:720–723. doi: 10.1038/27246. [DOI] [PubMed] [Google Scholar]
- 16.Tock MR, Walsh AP, Carroll G, McDowall KJ. The CafA protein required for the 5’-matura-tion of 16S rRNA is a 5’-end-dependent ribonuclease that has context-dependent broad sequence specificity. J Biol Chem. 2000;275:8726–8732. doi: 10.1074/jbc.275.12.8726. [DOI] [PubMed] [Google Scholar]
- 17.Celesnik H, Deana A, Belasco JG. Initiation of RNA decay in Escherichia coli by 5’ pyrophosphate removal. Mol Cell. 2007;27:79–90. doi: 10.1016/j.molcel.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deana A, Celesnik H, Belasco JG. The bacterial enzyme RppH triggers messenger RNA degradation by 5’ pyrophosphate removal. Nature. 2008;451:355–358. doi: 10.1038/nature06475. [DOI] [PubMed] [Google Scholar]
- 19.Xu F, Cohen SN. RNA degradation in Escherichia coli regulated by 3’ adenylation and 5’ phosphorylation. Nature. 1995;374:180–183. doi: 10.1038/374180a0. [DOI] [PubMed] [Google Scholar]
- 20.Bleichert F, Baserga SJ. The long unwinding road of RNA helicases. Mol Cell. 2007;27:339–352. doi: 10.1016/j.molcel.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Cordin O, Banroques J, Tanner NK, Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Iost I, Dreyfus M. DEAD-box RNA helicases in Escherichia coli . Nucleic Acids Res. 2006;34:4189–4197. doi: 10.1093/nar/gkl500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jankowsky E, Fairman ME. RNA heli-cases—one fold for many functions. Curr Opin Struct Biol. 2007;17:316–324. doi: 10.1016/j.sbi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 24.Bhaskaran H, Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature. 2007;449:1014–1018. doi: 10.1038/nature06235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao J, Lee K, Zhao M, Qiu J, Zhan X, Saxena A, et al. Differential modulation of E. coli mRNA abundance by inhibitory proteins that alter the composition of the degradosome. Mol Microbiol. 2006;61:394–406. doi: 10.1111/j.1365-2958.2006.05246.x. [DOI] [PubMed] [Google Scholar]
- 26.Khemici V, Toesca I, Poljak L, Vanzo NF, Carpousis AJ. The RNase E of Escherichia coli has at least two binding sites for DEAD-box RNA helicases: functional replacement of RhlB by RhlE. Mol Microbiol. 2004;54:1422–1430. doi: 10.1111/j.1365-2958.2004.04361.x. [DOI] [PubMed] [Google Scholar]
- 27.Morita T, Kawamoto H, Mizota T, Inada T, Aiba H. Enolase in the RNA degradosome plays a crucial role in the rapid decay of glucose transporter mRNA in the response to phosphosugar stress in Escherichia coli . Mol Microbiol. 2004;54:1063–1075. doi: 10.1111/j.1365-2958.2004.04329.x. [DOI] [PubMed] [Google Scholar]
- 28.Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli . Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 29.Morita T, Maki K, Aiba H. RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev. 2005;19:2176–2186. doi: 10.1101/gad.1330405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prud’homme-Genereux A, Beran RK, Iost I, Ramey CS, Mackie GA, Simons RW. Physical and functional interactions among RNase E, polynucleotide phosphorylase and the cold-shock protein, CsdA: evidence for a ‘cold shock degradosome’. Mol Microbiol. 2004;54:1409–1421. doi: 10.1111/j.1365-2958.2004.04360.x. [DOI] [PubMed] [Google Scholar]
- 31.Beran RK, Simons RW. Cold-temperature induction of Escherichia coli polynucleotide phosphorylase occurs by reversal of its autoregulation. Mol Microbiol. 2001;39:112–125. doi: 10.1046/j.1365-2958.2001.02216.x. [DOI] [PubMed] [Google Scholar]
- 32.Yamanaka K, Inouye M. Selective mRNA degradation by polynucleotide phosphorylase in cold shock adaptation in Escherichia coli . J Bacteriol. 2001;183:2808–2816. doi: 10.1128/JB.183.9.2808-2816.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taghbalout A, Rothfield L. RNase E and the other constituents of the RNA degradosome are components of the bacterial cytoskeleton. Proc Natl Acad Sci USA. 2007;104:1667–1672. doi: 10.1073/pnas.0610491104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brennan RG, Link TM. Hfq structure, function and ligand binding. Curr Opin Microbiol. 2007;10:125–133. doi: 10.1016/j.mib.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 35.Baker CS, Eory LA, Yakhnin H, Mercante J, Romeo T, Babitzke P. CsrA inhibits translation initiation of Escherichia coli hfq by binding to a single site overlapping the Shine-Dalgarno sequence. J Bacteriol. 2007;189:5472–5481. doi: 10.1128/JB.00529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aiba H. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr Opin Microbiol. 2007;10:134–139. doi: 10.1016/j.mib.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 37.Gottesman S. The small RNA regulators of Escherichia coli roles and mechanisms. Annu Rev Microbiol. 2004;58:303–328. doi: 10.1146/annurev.micro.58.030603.123841. [DOI] [PubMed] [Google Scholar]
- 38.Coburn GA, Miao X, Briant DJ, Mackie GA. Reconstitution of a minimal RNA degrado-some demonstrates functional coordination between a 3’ exonuclease and a DEAD-box RNA helicase. Genes Dev. 1999;13:2594–2603. doi: 10.1101/gad.13.19.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackie GA, Coburn GA, Miao X, Briant DJ, Prud’homme-Genereux A. Preparation of Escherichia coli Rne protein and reconstitution of RNA degradosome. Methods Enzymol. 2001;342:346–356. doi: 10.1016/s0076-6879(01)42557-2. [DOI] [PubMed] [Google Scholar]
- 40.Marcaida MJ, DePristo MA, Chandran V, Carpousis AJ, Luisi BF. The RNA degradosome: life in the fast lane of adaptive molecular evolution. Trends Biochem Sci. 2006;31:359–365. doi: 10.1016/j.tibs.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Cormack RS, Mackie GA. Structural requirements for the processing of Escherichia coli 5S ribosomal RNA by RNase E in vitro . J Mol Biol. 1992;228:1078–1090. doi: 10.1016/0022-2836(92)90316-c. [DOI] [PubMed] [Google Scholar]
- 42.Bessarab DA, Kaberdin VR, Wei CL, Liou GG, Lin-Chao S. RNA components of Escherichia coli degradosome: evidence for rRNA decay. Proc Natl Acad Sci USA. 1998;95:3157–3161. doi: 10.1073/pnas.95.6.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carpousis AJ, Leroy A, Vanzo N, Khemici V. Escherichia coli RNA degradosome. Methods Enzymol. 2001;342:333–345. doi: 10.1016/s0076-6879(01)42556-0. [DOI] [PubMed] [Google Scholar]
- 44.Kaberdin VR, Walsh AP, Jakobsen T, McDowall KJ, von Gabain A. Enhanced cleavage of RNA mediated by an interaction between substrates and the arginine-rich domain of E. coli ribonuclease E. J Mol Biol. 2000;301:257–264. doi: 10.1006/jmbi.2000.3962. [DOI] [PubMed] [Google Scholar]
- 45.Morita T, El-Kazzaz W, Tanaka Y, Inada T, Aiba H. Accumulation of glucose 6-phosphate or fructose 6-phosphate is responsible for destabilization of glucose transporter mRNA in Escherichia coli . J Biol Chem. 2003;278:15608–15614. doi: 10.1074/jbc.M300177200. [DOI] [PubMed] [Google Scholar]
- 46.Mohanty BK, Maples VF, Kushner SR. The Sm-like protein Hfq regulates polyadenylation dependent mRNA decay in Escherichia coli . Mol Microbiol. 2004;54:905–920. doi: 10.1111/j.1365-2958.2004.04337.x. [DOI] [PubMed] [Google Scholar]
- 47.Chandran V, Luisi BF. Recognition of enolase in the Escherichia coli RNA degradosome. J Mol Biol. 2006;358:8–15. doi: 10.1016/j.jmb.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 48.Worrall JA, Howe FS, McKay AR, Robinson CV, Luisi BF. Allosteric activation of the ATPase activity of the Escherichia coli RhlB RNA helicase. J Biol Chem. 2008;283:5567–5576. doi: 10.1074/jbc.M708620200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Symmons MF, Jones GH, Luisi BF. A duplicated fold is the structural basis for polynucleotide phosphorylase catalytic activity, processivity, and regulation. Structure. 2000;8:1215–1226. doi: 10.1016/s0969-2126(00)00521-9. [DOI] [PubMed] [Google Scholar]
- 50.Liou GG, Jane WN, Cohen SN, Lin NS, Lin-Chao S. RNA degradosomes exist in vivo in Escherichia coli as multicomponent complexes associated with the cytoplasmic membrane via the N-terminal region of ribonuclease E. Proc Natl Acad Sci USA. 2001;98:63–68. doi: 10.1073/pnas.011535498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng Z-F, Deutscher MP. Quality control of ribosomal RNA mediated by polynucleotide phosphorylase and RNase R. Proc Natl Acad Sci USA. 2003;100:6388–6393. doi: 10.1073/pnas.1231041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deutscher MP. Degradation of RNA in bacteria: comparison of mRNA and stable RNA. Nucleic Acids Res. 2006;34:659–666. doi: 10.1093/nar/gkj472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vassillieva I, Nikulin A, Blasi U, Moll I, Garber M. Crystallization of Hfq protein: a bacterial gene-expression regulator. Acta Crystallogr Sec D. 2003;59:1061–1063. doi: 10.1107/s0907444903006929. [DOI] [PubMed] [Google Scholar]
- 54.Amann E, Brosius J. ATG vectors for regulated high-level expression of cloned genes in Escherichia coli . Gene. 1985;40:183–190. doi: 10.1016/0378-1119(85)90041-1. [DOI] [PubMed] [Google Scholar]
- 55.Warburg O, Christian W. Isolierung und Kristallisation des Gärungsferments Enolase. Biochem Z. 1942;310:384–421. [Google Scholar]
- 56.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd. Cold Spring Harbor Laboratory; Cold Spring Harbor, New York: 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.