

Cellular senescence, a stable cell division arrest caused by severe damage and stress, is a hallmark of aging in humans. Here, we review mechanisms of senescence that are related to mitochondria and their interorganelle communication, and the involvement of proteostasis networks and metabolic control in the senescent phenotype. We also explore possibilities to target these mechanisms as new opportunities for pharmacological interventions with the aim of tissue rejuvenation.
Keywords: calcium signaling homeostasis, caloric restriction mimetic, interorganellar connectivity, lysosome, mitochondria, mitophagy, proteostasis, RNA modification, senescence, translational control
Abstract
Cellular senescence, a stable cell division arrest caused by severe damage and stress, is a hallmark of aging in vertebrates including humans. With progressing age, senescent cells accumulate in a variety of mammalian tissues, where they contribute to tissue aging, identifying cellular senescence as a major target to delay or prevent aging. There is an increasing demand for the discovery of new classes of small molecules that would either avoid or postpone cellular senescence by selectively eliminating senescent cells from the body (i.e., ‘senolytics’) or inactivating/switching damage‐inducing properties of senescent cells (i.e., ‘senostatics/senomorphics’), such as the senescence‐associated secretory phenotype. Whereas compounds with senolytic or senostatic activity have already been described, their efficacy and specificity has not been fully established for clinical use yet. Here, we review mechanisms of senescence that are related to mitochondria and their interorganelle communication, and the involvement of proteostasis networks and metabolic control in the senescent phenotype. These cellular functions are associated with cellular senescence in in vitro and in vivo models but have not been fully exploited for the search of new compounds to counteract senescence yet. Therefore, we explore possibilities to target these mechanisms as new opportunities to selectively eliminate and/or disable senescent cells with the aim of tissue rejuvenation. We assume that this research will provide new compounds from the chemical space which act as mimetics of caloric restriction, modulators of calcium signaling and mitochondrial physiology, or as proteostasis optimizers, bearing the potential to counteract cellular senescence, thereby allowing healthy aging.
Abbreviations
- AMPK
adenosine monophosphate‐activated protein kinase
- ASC
adipose‐derived stem cells
- BNIP3L/Nix
BCL2/adenovirus E1B 19 kDa protein‐ interacting protein 3‐like
- CR
caloric restriction
- DIRAS3
DIRAS family GTPase 3
- Dnm1l
dynamin 1 like (also known as Drp1)
- DR
dietary restriction
- eIF4G
eukaryotic translation initiation factor 4G
- ER
endoplasmic reticulum
- ETC
electron transport chain
- FAHD1
FAH domain containing protein 1
- FUNDC1
FUN14 Domain Containing 1
- IGF‐1
insulin‐like growth factor‐1
- MAM
mitochondria‐associated endoplasmic reticulum membranes
- MAPK
mitogen‐activated protein kinase
- MCU
mitochondrial calcium uniporter
- MDV
mitochondria‐derived vesicles
- METTL5
methyltransferase like 5
- MICU
mitochondrial calcium uptake protein
- mTOR
mammalian target of rapamycin
- NAD+
nicotinamide adenosine dinucleotide (oxidized form)
- NSUN5
NOP2/Sun RNA methyltransferase 5
- PI3K
phosphoinositide 3‐kinase
- PINK1
PTEN‐induced Kinase 1
- PRMT1
Protein methyl transferase 1
- ROS
reactive oxygen species
- SASP
senescence‐associated secretory phenotype
- TCA cycle
tricarboxylic acid cycle
Introduction
Aging is a complex process driving progressive decline of functionality and regenerative potential of tissues. One hallmark of aging is cellular senescence, a state of stable cell division arrest caused by severe damage and stress, leading to cellular dysfunctions among others in metabolic signaling, intra‐organelle signaling, proteostasis, and mitochondria. Senescence is involved in tissue homeostasis, embryonic development as well as inhibition of tumor progression [1]. During aging, senescent cells accumulate in multiple organs and compromise tissue function, essentially caused by the unique property of senescent cells to secrete a bunch of pro‐inflammatory and damage‐inducing molecules, commonly referred to as the senescence‐associated secretory phenotype (SASP). SASP components are causally involved in molecular and cellular changes giving rise to pathological manifestations and frailty. Hence, senescence is most likely a defining feature of human age‐related diseases, including obesity, diabetes mellitus type 2, cardiovascular disease, skin aging, and cancer [2, 3] (Fig. 1).
Fig. 1.
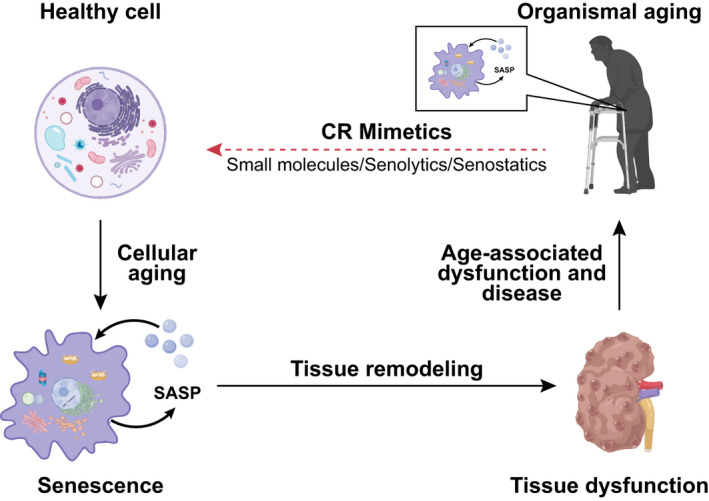
The role of senescence and its reversion in healthy aging and age‐related dysfunction and disease. With increasing age, senescent cells accumulate in several human tissues, due to a process known as cellular aging. Senescent cells secrete a plethora of extracellular proteins, lipids, and other bioactive material, collectively referred to as the senescence‐associated secretory phenotype (SASP). SASP components trigger changes in adjacent cells resulting in tissue remodeling and subsequently age‐associated tissue dysfunction of human organs, such as the kidney. Age‐associated dysfunction in several tissues contributes to organismal aging. Currently, small molecules with the potential to trigger the elimination of senescent cells (referred to as senolytics) or to dampen their detrimental influence on the organism (referred to as senostatics) are under development. Such compounds, with a potential for tissue rejuvenation, may provide new therapeutic opportunities for age‐associated dysfunctions and diseases. Based on the beneficial effects of caloric restriction (CR) on human healthspan, compounds which mimic beneficial effects of caloric restriction may be particularly suitable candidates for the development of senolytics and/or senostatics.
Although considerable progress has been achieved in the field of cellular senescence, it is still not clear whether the appearance of senescent cells is causative to or a mere consequence of the aging process. Elimination of senescent cells has been shown to extend both lifespan and health span of different organisms [2]. Still, there is an increasing demand for the discovery of new classes of small molecules, which would either avoid or postpone cellular senescence by selectively eliminating senescent cells from the body (i.e., ‘senolytics’) or by inactivating/modulating damage‐inducing properties of senescent cells (i.e., ‘senostatics/senomorphics’) [4, 5], such as the SASP.
Recent literature suggests that age‐related dysfunctions can be counteracted by senolysis in animal models; accordingly, there are attempts to eliminate senescent cells in order to delay aging. While there is evidence in favor of this hypothesis, alternative scenarios can be envisaged as well. An example might be the activation of the p16/Rb and/or p21/p53 stress response pathways, which boost key conserved molecular mechanisms driving cellular senescence. Still, one cannot exclude that in some instances, the activation of the p16/Rb and p21/p53 pathways primarily serves as marker of dysfunctional cells ready to be eliminated.
Blocking the mechanisms that cause senescence may not resolve the dysfunction but just one of its consequences. Thus, elimination of senescent cells can cause detrimental rather than beneficial effects to the organism [6, 7], which is not too surprising given the participation of cellular senescence in mammalian development [8] and during wound healing [9], a process driven by SASP components.
Several molecular and signaling pathways related to cellular dysfunction (cell cycle arrest, DNA and protein damage response, etc.) that contribute to induction and maintenance of the senescent state are conserved in evolution. Thus, simple and easy‐to‐manipulate model organisms such as the yeast Saccharomyces cerevisiae (S. cerevisiae) or the nematode Caenorhabditis elegans (C. elegans) are frequently used to elucidate fundamental aspects of cell damage and disruption of homeostasis, which promote senescence in vertebrates [10, 11, 12, 13, 14, 15]. However, there is little evidence for senescence in these simple models, which are evolutionarily quite distant from humans. Consequently, mammalian aging models, including mice, and human organoids, such as human skin equivalents and adipose spheroids, have to be used to gain insights into the translatability from simple to more complex organisms [16].
In order to overcome negative effects of cellular senescence on human health [17], it is essential to identify aging‐related pathways and mediators to elaborate senescence‐associated mechanisms, including maintenance of mitochondrial functions, interorganelle communication, proteostasis, and metabolic signaling in selected cellular and organismal aging models. The identification of pathways and cellular targets along with lead compounds may pave the way for the development of treatment strategies to counteract age‐associated cellular dysfunction and, ultimately, pathophysiological processes, to promote healthy aging [18].
Mitochondria‐related mechanisms of senescence
Aside from being the cell's power plants, mitochondria serve as metabolic hubs, contributors to signal transduction, autophagy, and programmed cell death, and are in a unique position to mediate or modify aging‐associated processes, including cellular senescence. In fact, it has been shown that impairment of mitochondrial function is sufficient to induce senescence in a variety of cell types [19, 20, 21]. On the other hand, cells lacking mitochondria do not respond to most senescence‐inducing stress factors [22], highlighting the key role for mitochondria in senescence induction and execution.
Reactive oxygen species (ROS) are generated continuously by mitochondria as the result of oxidative metabolism [23] and are known to cause damage to DNA, proteins, and lipid complexes, including mitochondria themselves [21], leading to impaired cellular function and eventually cell death [24, 25, 26, 27]. Besides mitochondria, peroxisomes contribute to cellular ROS levels (Fig. 2). A more detailed description of the interactions between mitochondria and peroxisomes is provided in chapter 3.
Fig. 2.
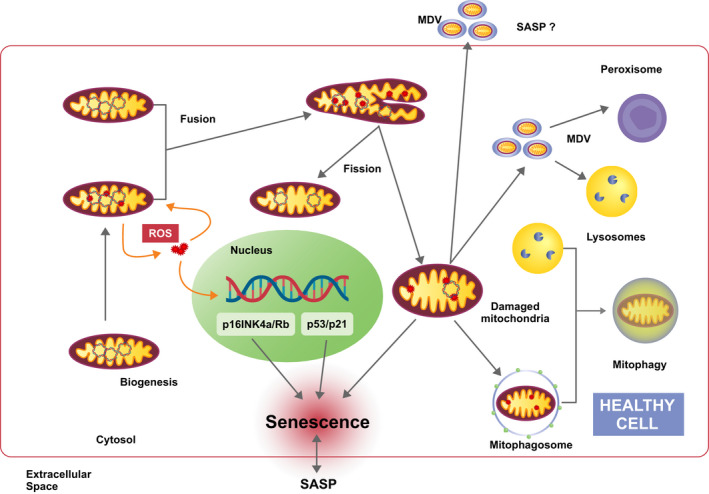
Mitochondrial transitions and interactions in cellular senescence. Mitochondria are dynamic organelles which act as the primary energy‐generating system in most eukaryotic cells. In addition, mitochondria are involved in various other vital processes, such as intermediary metabolism, Ca2+ signaling, and apoptosis. The inevitable production of ROS by mitochondria induces mitochondrial and nuclear DNA damage. Accumulation of damaged DNA and proteins in mitochondria threatens the physiological function of the cell and provokes several defense mechanisms. By fission and fusion processes mitochondrial damage can be partially counteracted and healthy mitochondria might remain functional despite continuous ROS production. Alternatively, damaged mitochondria release so‐called mitochondria‐derived vesicles (MDV), which may be transported to other organelles, such as peroxisomes and lysosomes, in order to restore mitochondrial activity. On the other hand, MDV can be released to the extracellular space as components of the SASP to signal mitochondrial damage to surrounding cells. However, in case of excessive mitochondrial damage, eliminating parts of the organelle is not sufficient and whole mitochondria have to be removed by mitophagy. Dysregulation of mitochondria quality control process leads to accumulation of impaired mitochondria and induction of senescence via various mechanisms.
Mitochondrial DNA is highly vulnerable to oxidative stress due to its proximity to the electron transport chain (ETC) and due to the limited DNA repair capacity within mitochondria [28]. Perturbation of mitochondrial homeostasis induced by genotoxic stress, ROS and defects in the ETC, among other factors, promotes cellular senescence by activating tumor suppressor pathways like p16INK4a/Rb and p53/p21 [29]. Accordingly, senescent cells are characterized by increased mitochondrial ROS production and by metabolic changes related to mitochondrial metabolites and dynamics, mainly attributed to accumulation of dysfunctional mitochondria [30] (Fig. 2).
Senescence caused by mitochondrial dysfunction displays a specific secretory profile, characterized by the absence of factors related to interleukin‐1 (IL‐1), and can be reversed by supplementation with pyruvate [20], suggesting that shortage of oxidized nicotinamide adenosine dinucleotide (NAD+) may impair the synthesis of metabolites required for cell proliferation, thereby inducing a specific subtype of senescence. Thus, manipulation of mitochondrial metabolism through the supplementation of intermediate compounds of the respiration process represents a promising target to be explored in order to counteract senescence.
Mitochondrial quality control is a key process for maintenance of a functional mitochondrial network, which in turn is necessary for adaptive metabolism and survival in response to cellular stress (Fig. 2). Besides fission and fusion cycles, a major component of cellular control of mitochondrial integrity is a specialized form of macroautophagy, known as mitophagy, in which mitochondria are specifically targeted for autophagic degradation [31, 32, 33, 34]. A group of mitophagy receptors, including BCL2/adenovirus E1B 19 kDa protein‐interacting protein 3 (BNIP3), BNIP3‐like (BNIP3L/Nix), PTEN Induced Kinase 1 (PINK1), Parkin, and FUN14 Domain Containing 1 (FUNDC1) has been described in recent years. However, more research will be required to delineate redundant and nonredundant functions of known mitophagy receptors. It also seems conceivable that additional members of this family are still to be discovered. Mitophagy plays an important role in cellular homeostasis by eliminating dysfunctional mitochondria and reducing mitochondrial mass as an adaptive response to stress [35]. In mammals, this process is also important during the differentiation of specific cell types such as erythrocyte maturation, mediated by the mitophagy receptor BNIP3L/Nix [36] and for T‐lymphocyte development [37].
A decline of mitophagic activity is related to aging and neurodegenerative diseases, highlighting the pivotal role of mitochondria quality control in maintenance of longevity [38, 39]. In fact, the potential involvement of dysregulated mitophagy in cellular senescence has been suggested by several studies. For instance, some authors have correlated the accumulation of aberrant mitochondria in senescent cells to insufficient turnover of mitochondrial population due to decreased mitophagic activity and lysosomal dysfunction [30, 40]. Likewise, dysfunctional Dnm1l (dynamin 1 like, also known as Drp1)‐dependent mitophagy triggers senescence and contributes to age‐related hearing loss in mice [41]. Yet, in a model of 3D skin equivalents, pyruvate protected dermal fibroblasts from senescence by improving mitophagic activity and mitochondrial turnover [42]. Thus, mitophagy seems to be an important factor for modulation by senolytic and senostatic compounds.
It is important to note, however, that the reasons for the decline in mitophagic activity during the senescence process are not yet fully understood. In some cases, for example, lysosomal dysfunction appears to be related to the accumulation of damaged mitochondria [40]. Others have reported that stabilization of p53 in the cytoplasm during senescence increases its interaction with Parkin and prevents priming of damaged mitochondria, thereby reducing mitophagic activity [43]. Considering that activation and control of mitophagy depends on the specific stress to which cells are subjected during the senescence process [44], it seems to be crucial to understand the underlying mechanisms of mitophagy in different types of senescence and their contribution to the development of the senescence phenotype in order to clarify whether mitophagy dysfunction is a cause or a consequence of senescence.
Notably, parts of mitochondria are also found in so‐called ‘mitochondrial‐derived vesicles’ (MDVs) (Fig. 2). In addition to mitophagy, which is believed to have evolved as a mechanism for degradation of entire mitochondria, MDVs have been proposed as an additional means of quality control for mitochondria [45, 46]. Although the mechanisms involved in the formation and processing of MDVs are still poorly understood, some studies speculate that these vesicles may be part of the SASP and, thus, participate in the processes of interorganelle and intercellular communication during cellular senescence [45] (Fig. 2). Other aspects related to MDVs and senescence are covered in chapter 3. An overview of mitochondria related senescence pathways is given in Table 1.
Table 1.
Main senescence‐related processes and proteins covered in the review.
| Process | Chapter | Proteins/Protein complexes |
|---|---|---|
| ROS‐driven pathways | 2 | p16/pRB, p53/p21 |
| Mitophagy | 2 | BNIP3, BNIP3L/Nix, PINK/Parkin, FUNDC1 |
| Ca2+ signaling/ROS | 3 | MCU |
| Mitochondria–lysosome | 3 | AMPK, RAB7, vCLAMP, Vps13‐Mcp1, Vps39‐ypt7‐Tom40 |
| Mitophagy, autophagy, and general proteostasis | 4 | Insulin, IGF‐1, DIRAS3, Akt, mTORC1 |
| Ribosome biogenesis and protein synthesis | 4 | Pol I, mTOR, SirT1, eNoSC, METTL5, 4EBP, elF4G, |
| Tumour suppressor loss, oncogene activation | 5 | Sprouty1, Ras, DIRAS3, PI3K, Akt, mTORC1 |
Mechanisms of interorganelle communication in cellular senescence
Mitochondria form a highly complex and dynamic network throughout the cytoplasm. They continuously move along microtubule tracks, alter their shape by fusion and fission, and dynamically establish contact sites with other compartments to meet metabolic requirements of the host cell [47, 48]. Apart from changes in the extra‐ and intramitochondrial concentration of signaling molecules, such as Ca2+ [49] or ROS [50], the interplay between mitochondria and other organelles occurs through physical proximity at membrane contact sites or via vesicular transport [51] (Fig. 3). For instance, there is evidence for regulatory crosstalk between cytosolic and mitochondrial ribosomes leading to coordinated messenger RNA translation in both mitochondria and the cytosol. Furthermore, the insulin/IGF/mTOR (mammalian target of Rapamycin) axis controls both mitochondrial function and protein synthesis (Fig. 3, for details, see chapter 4). The activity of mitochondria is governed by various mechanisms. Among others, Ca2+ ions are necessary for metabolic activation of mitochondria due to the Ca2+ dependency of tricarboxylic acid (TCA) cycle dehydrogenases. However, mitochondrial Ca2+ overload also triggers cell death pathways. Several studies have revealed changes in the protein machinery controlling mitochondrial Ca2+ uptake during aging. For instance, the expression of the mitochondrial Ca2+ uniporter (MCU) was increased in long‐term cultured rat hippocampal neurons, leading to elevated mitochondrial Ca2+ levels [52]. Moreover, an increase in MCU channel activity was found after oxidation of MCU by ROS [53], commonly elevated during aging [54]. In addition, enhanced ER to mitochondrial Ca2+ flux has been shown to boost mitochondrial metabolism in aged endothelial cells, but also bears the risk for mitochondrial Ca2+ overload [55].
Fig. 3.
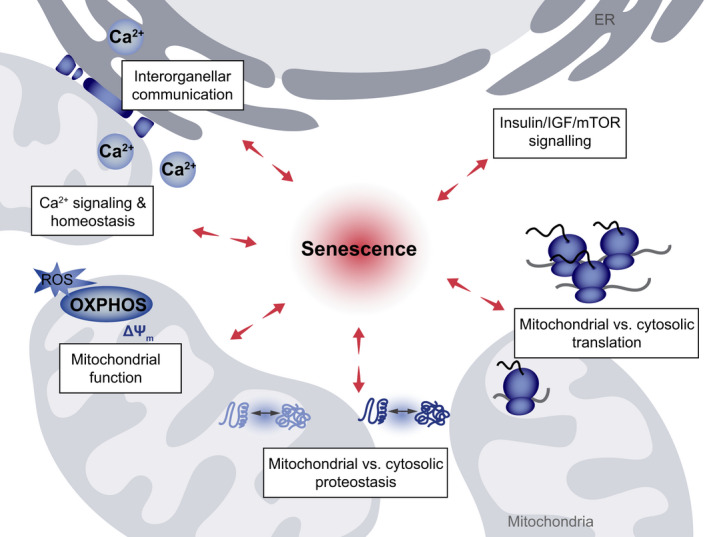
Perturbations of interorganellar connectivity in senescence. Several interconnected processes are affected by cellular senescence, and vice versa are impacting the induction and maintenance of the senescent state. These processes include interorganellar communication at ER–mitochondria contact sites and spatially restricted Ca2+ signaling occurring at these contacts, as well as general Ca2+ homeostasis and transport. The coordination of mitochondrial versus cytosolic translation output as well as the interconnected mitochondrial and cytosolic proteostasis networks, all affected by general mitochondrial function and governed by the Insulin‐, Growth Hormone (GH)/Insulin‐like Growth Factor‐1 (IGF‐1)‐, and mechanistic Target of Rapamycin (mTOR)‐signaling axis, act as determinants of senescence induction.
Consequently, we assume that mitochondrial activity is fine‐tuned by mitochondrial Ca2+ homeostasis. Proper mitochondrial Ca2+ uptake might be essential to maintain cellular function, while elevated levels of mitochondrial Ca2+ might trigger generation of ROS, formation of the permeability transition pore, cytochrome C release [56], and the cell's susceptibility to agents stimulating mitochondrial Ca2+ uptake [55]. However, mitochondrial Ca2+ overload might be beneficial to overcome the resistance of senescent cells to programmed cell death pathways, which is required for normal cell turnover and tissue homeostasis [57].
Spatially separated cellular subdomains facilitate interorganelle communication. For instance, mitochondria‐associated ER membranes (MAMs) stretch closely to mitochondria to ensure locally restricted and protected ion and lipid transfer between these two organelles in eukaryotic cells [58]. Components of these MAM regions are highly conserved throughout different tissues and species [59]. For instance, a counterpart of these contact sites, so‐called ER‐mitochondria encounter structure (ERMES), was also found in yeast [60, 61, 62]. MAM microdomains are established by reversible tethering of proteins and are equipped with highly specialized toolkits to modulate a variety of cellular processes, including lipid transport and synthesis, Ca2+ signaling, autophagy, and energy metabolism [63]. Changes in the ER‐mitochondrial crosstalk have been associated with a reduced adaptive capacity of cells in response to stress and with an increased vulnerability to age‐related diseases [64], including neurodegenerative [65], cardiovascular and metabolic diseases [66], and cancer [67]. Currently, it is a pressing question how substructural composition, dynamics, and function of MAM regions change during the process of aging and which proteins might serve as targets to potentially modulate age‐related cellular dysfunction. We assume that age‐related alterations in the kinetics and dynamics of mitochondrial–ER interaction contribute to the development of cellular dysfunction during senescence. Constant improvement of super‐resolution microscopy techniques may provide the necessary tools to resolve the spatial mitochondrial–ER interplay in real‐time.
Besides ER‐mitochondrial crosstalk, the bidirectional communication between mitochondria and lysosomes has gathered special attention during the last years. As executors and regulators of autophagy, lysosomes are in a crucial position to modulate age‐related pathologies. By preventing the accumulation of damaged mitochondria through mitophagy, lysosomes protect cells from detrimental mitochondria‐derived ROS and pro‐apoptotic factors [68]. Mitochondrial biogenesis was found to be transcriptionally repressed in lysosomal lipid storage diseases, pointing to the immense impact of lysosomal activity on mitochondrial function [69]. In turn, short‐term mitochondrial stress induces lysosomal biogenesis via the regulation of transcription factors and AMP‐activated protein kinase (AMPK) signaling, while chronic mitochondrial stress results in the impairment of lysosomal biogenesis [70]. Moreover, mitochondrial respiratory chain deficiency was found to inhibit lysosomal function via AMPK deactivation and lysosomal Ca2+ accumulation [69]. Besides various bidirectional signaling pathways, direct contact sites between mitochondria and lysosomes do exist too. The GTP‐bound lysosomal protein RAB7 was found to promote the formation of these contact sites in healthy cells. Mitochondria tend to undergo fission at lysosomal contact sites, while lysosomal RAB7, in turn, gets regulated by mitochondria [71]. Lysosome–mitochondria communication is also conserved in yeast cells, where contacts between mitochondria and the vacuole (the yeast equivalent of the lysosome) have been described [72], referred to as vCLAMP (vacuole and mitochondria patch). These contact sites can be established by two distinct tethering pairs: Vps13‐Mcp1 or Vps39‐Ypt7‐Tom40 [73].
There is no doubt regarding the crucial impact of mitochondrial–lysosomal interplay on the process of aging as well as on the development of age‐related diseases. For instance, impaired mitochondria–lysosome crosstalk was associated with neurodegenerative diseases [74]. However, it has to be clarified how the composition and dynamics of the lysosomal–mitochondrial contact and interaction sites change during the process of aging and which proteins function as the key regulators of this interplay. In this regard, the utilization of newly developed lysosomally targeted biosensors [75] might represent the key technology that will allow progress in the respective research. Besides ER and lysosomes, latest reports also suggest a direct communication of mitochondria with other cellular sites and organelles, including plasma membrane [76], peroxisomes [77], and endosomes [78].
Mitochondria are frequently found in close proximity to the plasma membrane and contribute to the ATP supply and Ca2+ signaling in these specific regions [76]. For instance, mitochondria help to maintain and activate store‐operated Ca2+ entry (SOCE), by which depletion of ER Ca2+ stores induces Ca2+ influx through the plasma membrane [79]. Mitochondria and peroxisomes are both characterized by great plasticity and play a major role in cell metabolism and ROS homeostasis [77]. Notably, direct interorganelle crosstalk between mitochondria and peroxisomes has been reported. For instance, enhanced ROS production in peroxisomes disturbs mitochondrial ROS homeostasis and causes mitochondrial fragmentation. Moreover, peroxisomes and mitochondria share key components of their fission machineries [80].
Recent research connects peroxisomal dysfunction to fatal oxidative damage associated with aging‐related diseases. It is now widely accepted that mitochondria and peroxisomes are required to maintain oxidative balance in a cell. However, our understanding of the interdependence of these organelles to maintain cellular homeostasis of ROS is still limited [81]. A direct interaction between mitochondria and endosomes was shown by super‐resolution microscopy, suggesting alterations in the dynamics of endosome–mitochondrial interaction by endosomal cargo and milieu [78]. However, age‐related alterations in mitochondria's interaction with the plasma membrane, peroxisomes and endosomes are still elusive and need to be characterized.
Extracellular vesicles (EVs) are important mediators of intercellular and potentially interorganellar communication. They transport their functional cargo, which might be composed of proteins, lipids, DNA, and various types of RNA to recipient cells, and thereby act in a similar manner as hormones or cytokines [82]. Recently, small EVs containing miRNAs were identified as important components of the SASP, mediating apoptosis and wound closure [83, 84]. It has been reported that MDVs can transport‐specific mitochondrial proteins and lipids to peroxisomes or lysosomes and act as a first line of defense against mitochondrial damage and preventing complete elimination of the organelle by mitophagy [46, 51]. Hence, mitochondria can be even found in the extracellular environment in their free form, or surrounded by a membrane (like in vesicles) [45, 51, 85]. Moreover, circulating cell‐free mitochondrial DNA has been found as well [46]. Notably, all forms of extracellular mitochondria have been found to induce paracrine or endocrine responses in various organisms. However, the effects of extracellular mitochondria just start to be elucidated [46, 51] and their impact on processes of senescence is still elusive. Now, latest developments in super‐resolution microscopy are allowing us to catch a glimpse of mitochondria's versatile interaction with their cellular environment and we might be curious what will be unveiled in the following years. An overview of pathways related to disturbed interorganelle communication during senescence is given in Table 1.
Mechanisms of metabolic control and proteostasis in cellular senescence
The energy status of a cell and the availability of nutrients represent the most important upstream regulators of mitochondrial dynamics and quality control. These, as well as other processes of metabolism and accumulation of biomass, are assured by the mammalian target of rapamycin (mTOR) pathway at the nexus of nutrition, cell growth, and aging [86, 87]. Metabolic interventions known to extend healthy life span, such as caloric/dietary restriction, time‐restricted feeding, and intermittent fasting all affect the main nutrient sensing pathways including mTOR. Therefore, mTOR holds a central position to integrate aging‐associated processes. Over‐activation of mTOR leads to induction of cellular senescence, while repression of mTORC1 by rapamycin or genetic interventions blocks cellular senescence [88, 89], attenuates the SASP [90], and extends the life span of mice [91, 92, 93]. Besides mitophagy [94], mTORC1 also regulates general autophagy, as well as other processes of both the anabolic and catabolic arm of cellular proteostasis. Of interest, previous work from the Jansen‐Dürr laboratory established a tight link between mitochondrial dysfunction and functionality of the ubiquitin‐proteasome system. Thereby, a major mechanism of catabolic proteostasis was found associated with human skin aging, providing a mechanistic link between mitochondrial quality control and proteostasis [95].
Cellular proteostasis is guaranteed by the balance between the synthesis of new proteins, the impact of protein damaging mechanisms versus cellular defence mechanisms, protein refolding by chaperones and related pathways, and the clearance of damaged proteins (Fig. 4). Thus, timely modulation in response to extrinsic stimuli, and correct function of all four interconnected stages of protein turnover and quality control are essential for organismal health.
Fig. 4.
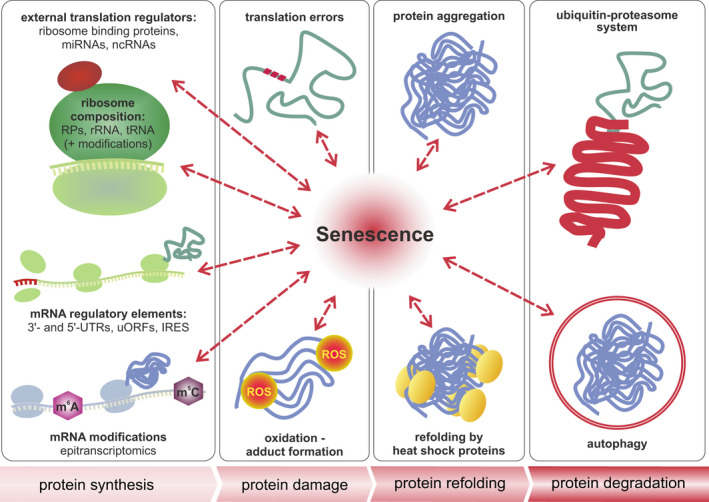
The role of senescence in multilevel proteostasis—the four stages in the life of a protein. Cellular senescence and aging presumably influence all four stages in the life of a protein, namely its synthesis by ribosomes, the accumulation of damage, refolding, and aggregation, as well as its degradation. Protein synthesis is tightly controlled by the composition of ribosomes, association of the core translation machinery with external regulators, mRNA modifications, and specific sequence elements of mRNAs, such as the 3′ and 5′ untranslated regions (UTRs), short upstream open reading frames (uORFs), or internal ribosome entry sites (IRES). Translation errors and oxidation lead to damaged and misfolded proteins, which are prone to aggregation. This process is counteracted by heat shock proteins or molecular chaperones. Damaged and aggregated proteins are degraded by autophagy or via the ubiquitin‐proteasome system. Both refolding and degradation are impaired in cellular and organismal aging and are tightly interconnected with other stages of proteostasis.
Accumulating evidence suggests all levels of proteostasis to be altered when cells enter senescence. The importance of protein misfolding, aggregation, and subsequent clearance by the ubiquitin‐proteasome system or autophagy for healthy aging and senescence‐related pathologies is widely recognized and extensively reviewed elsewhere [96, 97, 98]. Although depletion of ribosomal proteins and other factors involved in translation are known to extend the life span of yeast [99, 100, 101] and C. elegans [102, 103, 104], their potential role in mammalian cell senescence and the underlying molecular mechanisms are by far less understood than other aspects of cellular proteostasis [105].
Current hypotheses postulate that decreased overall translation conserves energy, which can then be invested into maintenance and repair pathways [106] or that reduced elongation speed minimizes translation errors [107]. Both mechanisms, by increasing the overall protein repair capacity, may limit the age‐related accumulation of oxidized, misfolded and aggregated proteins. At the same time, reduced amounts of mis‐translated polypeptides do not unnecessarily block important refolding and degradation pathways [108].
Due to the high energy demands of protein synthesis, it is not surprising that also ribosome biogenesis is tightly controlled by nutrient sensing pathways. Again, mTOR plays a pivotal role by repressing pre‐rRNA transcription by RNA Polymerase I (Pol I) when nutrients are limited. SirT1 is another conserved nutrient sensor linking aging to Pol I activity. Sir2, the yeast orthologue of SirT1, was originally identified as one of the first genes modulating cellular aging by maintaining rDNA integrity [109]. Although the finding that excision of rDNA repeats and the formation of extrachromosomal rDNA circles limit replicative life span is restricted to yeast, a large body of evidence indicates that rRNA transcription by Pol I is also a critical factor in aging of higher eucaryotes [110]. Partial inhibition of Pol I extends life span of the fruit fly and leads to shrinkage of nucleoli, which are clustered around rDNA repeats and represent the sites of ribosome biogenesis [111]. Also, in senescent human cells several small nucleoli fuse to one large nucleolus, indicating profound changes in the chromatin organization of rDNA repeats. Their silencing by the SirT1‐containing eNoSC‐complex and inhibition of Pol I is required for maintaining the senescent state of A549 cancer cells [112]. Interestingly, rDNA integrity is also tightly linked to several human segmental progeroid syndromes. Mutations in distinct RecQ helicases inflict the respective pathologies characterized by high levels of rDNA recombination and rearrangements [110]. Syndrome, for instance, is caused by recessive mutations in five different nucleotide excision repair proteins. Mutations in all five of these proteins cause defects in Pol I‐mediated transcription and translational fidelity, leading to elevated oxidative protein damage [113]. Taken together, these data indicate a striking connection between proteostasis, nutrient sensing, DNA integrity, and mitochondria, which all represent different hallmarks of aging [114].
In addition to the outlined changes in their biogenesis, it is generally believed that ribosomes and other components of the translation machinery deteriorate with age, leading to decreased overall protein synthesis and progressive loss of gene expression regulation at the translation level. Indeed, an increasing number of studies report certain discrepancies between the transcriptome and the proteome, which indicates that profound changes of translational regulation of gene expression occur during aging [115].
We hypothesize that selection of different mRNAs for translation occurs due to alterations in stoichiometry and modification patterns of several components of the translation machinery, which might all be influenced by aging and cellular senescence (Fig. 4). The essential components include the ribosome itself, as well as mRNAs, tRNAs, initiation, and elongation factors. In addition, multiple ribosome‐associated proteins, miRNAs and ncRNAs, such as tRNA fragments and rancRNAs, are described, which can modulate translation by competing with essential components or by allosteric interactions [105]. Indeed, known interventions modulating aging, stress resistance, and cellular senescence alter the translational regulation of gene expression. In flies, it was, for example, shown that the life span extension by deletion of Thor (also known as 4E‐BP) is modulated by the specific translation of mRNAs associated with enhanced mitochondrial activity [116]. Similarly, in worms the translational regulation of stress response genes is required for the life span extension by deletion of eukaryotic translation initiation factor 4G (eIF4G) [117]. In contrast to these protein‐based alterations in ribosome composition and functionality, also lack of a single, conserved N5‐cytosine methylation of ribosomal RNA of the large subunit extends the life span and stress resistance of yeast, worms, and flies. Depletion of the corresponding methyltransferase NSUN5 alters ribosomal structure and thus translational fidelity, resulting in a ‘reprogramming’ of the ribosome towards translation of mRNAs involved in cellular stress response [118]. Similarly, N6‐adenosine methylation of a specific residue of 18S rRNA by methyltransferase like 5 (METTL‐5) modulates heat‐stress resistance in C. elegans by altering translation of a specific mRNA involved in eicosanoid synthesis [119]. With the proof‐of‐principle that already single modifications of rRNA alter the life span and stress tolerance of organisms, it becomes clear that a systematic analysis of global post‐transcriptional modification patterns of rRNA, including pseudouridinylations, as well as base and sugar methylations [120], is of crucial importance to understand the changes of the ribosome in terms of synthesis, composition, structure, and function in the context of aging. In a similar fashion to rRNA modifications, also mRNA modifications influence translation [121] and it is tempting to speculate that these might be regulated in cellular and organismal aging as well.
An additional layer of complexity is added by mitochondrial ribosomes contained in eukaryotic cells in addition to the ribosomes in the cytosol. Both types of ribosomes appear to be coordinated, and this balance between cytosolic and mitochondrial translation may be altered under stress, and in senescent cells in particular [122, 123, 124, 125]. Methionine restriction, which increases the replicative life span of human fibroblasts, differentially affects the translation of selected mitochondrial RNAs [126] and thereby demonstrates the complex interplay between metabolic regulation, proteostasis, and mitochondria. Thus, we hypothesize that certain metabolic interventions might selectively target senescent cells by altering coordinated multilevel proteostasis of both cellular compartments. Moreover, a thorough analysis of age‐related alterations of the protein synthesis machinery might provide us with promising novel targets for anti‐senescence therapies. An overview of senescence pathways related to disturbed proteostasis and metabolic regulation is given in Table 1.
Identification of dietary and pharmacological interventions modulating cellular senescence
Restoring normal cellular and tissue function in an aged organism can be achieved by several strategies, including counteracting senescence‐inducing signals, targeting of specific aging‐related mechanisms, eliminating senescent cells (senolysis), and suppressing or switching senescence‐associated phenotypes including the SASP by senostatics/senomorphics [127, 128, 129].
Simple eukaryotic model organisms as potential tools for senescence effector screening
Simple eukaryotic model organisms, including S. cerevisiae and C. elegans, have been used as conventional tools for genetic screens concerning senescence effectors [130, 131, 132]. In S. cerevisiae, critical telomere shortening upon genetic inactivation of the telomerase, for example, via depletion of one of its subunits (Est1, Est2, Est3, or Tlc1), leads to an abrupt transit into replicative senescence after about 50–80 cell divisions (recently reviewed by [132, 133]). Like in mammalian cells, this is accompanied by a permanent activation of the DNA damage checkpoint, reorganization of chromatin, and prominent changes in gene expression [11, 133, 134]. Interestingly, genes related to mitochondrial energy metabolism, ranging from oxidative phosphorylation to TCA cycle, have been found to be prominently upregulated in senescent yeast [135]. In addition, mitochondrial proliferation is increased, indicating that also in this unicellular eukaryote, mitochondrial metabolism and functionality may contribute to the induction and/or maintenance of the senescent state [135]. Although both yeast and C. elegans lack complex traits associated with senescence, these models contributed substantially to our current understanding of fundamental molecular aspects associated with aging and senescence [11, 12, 15, 132]. Genes identified in such screens were subsequently validated in higher eukaryotic model systems, such as human cultured cells [55, 136, 137], organoids [138, 139], and mice [140, 141], and promoted the identification of promising molecules for translational approaches [55]. It is conceivable that many more senescence effectors from the chemical space can be identified by exploiting existing and newly defined senescence regulators as target proteins in small‐molecule screens.
CR mimetics as antisenescence drugs
The employment of caloric restriction (CR)/dietary restriction (DR) mimetics [142] may represent one promising strategy for pharmacological targeting of senescent cells. The most robust procedure to counteract aging is CR, the reduction of dietary intake below energy requirements while maintaining adequate nutrition [143, 144]. Although CR is a complex intervention with many experimental variables, including sex, strain, and level of CR, that can alter the CR response in model organisms, it consistently improves health across strains and sexes [145]. Molecular mechanisms underlying CR are not completely understood. According to the current model, the CR response is transduced via modulation of nutrient‐ and energy‐signaling pathways mainly inducing a reduction of Insulin/growth hormone (GH)/insulin‐like growth factor 1 (IGF‐1)/mTOR signaling and an activation of AMPK‐signaling and Sirtuin responses [146, 147]. This drives many downstream changes such as reduced oxidative stress and increasing stress resistance leading to less damage of DNA [148, 149, 150], proteins [151], and lipids [152] (for reviews, see [153, 154]). Most likely these events contribute to the prevention of age‐associated decline in genomic stability, autophagy, and proteostasis. Additional beneficial effects are that CR preserves mitochondrial function with age by increasing mitochondrial biogenesis, reprogramming of metabolism to anaplerotic filling of the TCA cycle and activation of fatty acid oxidation in mitochondria [155]. While there is consensus that CR increases health span in a wide variety of animal models, evidence from observational and randomized controlled clinical studies conducted in the last two decades suggests that CR can not only extend the healthy life span of obese and overweight people [156] but also of normal weight persons and might have the potential to induce longevity in humans under CR [157, 158].
Given the importance of CR for human health, translational research into screening for and developing of CR mimetics, compounds that mimic the positive effects of CR on health and life span without actual food restriction [159], is an appealing strategy. While CR works most likely on several different levels [160], the question whether and how CR antagonizes cellular senescence is one current focus of aging research [161]. Evidence supporting the hypothesis that CR mitigates or reduces cellular senescence comes from studies showing that under CR senescence markers are reduced in mouse and human organs [89, 170]. As it is well known that CR protects against cellular damage induced by oxidative stress and other adverse influences [147, 155], and cellular damage is a major cause for the induction of cellular senescence [171], it is quite likely that CR counteracts the generation of senescent cells by reducing cellular damage. It has been demonstrated that CR ameliorates senescence‐associated DNA damage and induces a decrease of the number of senescent cells in several tissues of mice [162, 163, 164, 169, 172]. The Zwerschke‐lab has shown that weight‐loss (WL) interventions including CR protects human adipose stem/progenitor cells (ASCs) against DNA damage and prolongs their life span by postponing the onset of cellular senescence [164]. Moreover, they identified WL/CR target genes, DIRAS family GTPase 3 (DIRAS3) and Sprouty1, that protect against the induction of cellular senescence [89, 166, 167, 170]. Mechanistically, DIRAS3 and Sprouty1 act by reducing signaling from the two main aging inducing signal transduction pathways, insulin/phosphoinositide 3 kinase (PI3K)/Akt/mTOR signaling, and IGF‐1/ras/mitogen‐activated protein kinase (MAPK) signaling, leading to protection against cellular senescence. Moreover, the activity of the PI3K inhibitor DIRAS3 leads to downregulation of mTOR activity and in turn to the stimulation of autophagy, a process that is well known to recycle damaged cellular components [170]. Thus, one could refer to the CR target gene DIRAS3 as a recycling and/or a cellular rejuvenation gene postponing cellular senescence (Fig. 5). Given that the induction of DIRAS3 and Sprouty1 in response to CR is sufficient to turn off signaling from the central Insulin/IGF‐1/mTOR pro‐aging program, one could speculate that at least some cell types in given tissues or organs are protected by cell intrinsic antisenescence mechanisms, which might act in combination with or independent of Insulin and IGF‐1. It has also been shown that CR in humans protects against cellular deterioration via decreasing oxidative stress and eliminating present damage by increasing the expression of genes encoding for heat shock proteins and proteins involved in autophagy [173, 174]. Moreover, it was previously shown that CR inhibits mTOR activity and hence abrogates the mTOR‐dependent pro‐inflammatory phenotype of senescent cells including factors that induce bystander senescence [90, 175], underscoring that CR could prevent the activation of primary and secondary senescence by inhibiting mTOR.
Fig. 5.
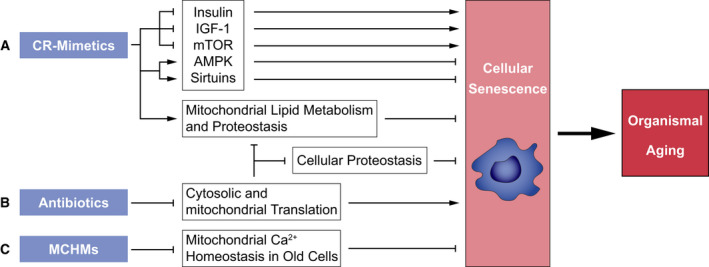
Potential dietary and pharmacological compounds modulating cellular senescence. Mitigating the induction and propagation of cellular senescence by (A) caloric restriction (CR)‐mimetics and (B) antibiotics targeting cytoplasmic and mitochondrial translation. Both compound classes counteract senescence‐inducing signals. (C) Mitochondrial Ca2+ homeostasis manipulators (MCHMs) could act senolytic. AMP‐activated protein kinase (AMPK), ER = Endoplasmic Reticulum, Insulin‐like growth factor‐1 (IGF‐1), mechanistic target of rapamycin (mTOR), Sirtuins (NAD+‐dependent deacetylases).
It is well accepted that genetic or pharmacological inhibition of mTORC1 by rapamycin or rapamycin‐derived compounds (rapalogs), postpones aging and increases the life span of a wide variety of animals including mice [91, 176, 177, 178, 179, 180, 181]; and although there may be exceptions [182], our understanding of the current literature is that rapamycin and the majority of related compounds (rapalogs) are actually dampening the senescence response. Additional mechanisms by which CR can prevent cellular senescence are the elimination of dysfunctional mitochondria by mitophagy [183] and activation of DNA repair mechanisms [184, 185].
Thus, there is overwhelming evidence supporting the hypothesis that CR mimetics can counteract cellular senescence. Our recent studies showing that WL interventions, including CR, postpone the onset of replicative senescence of ASCs in humans [164, 186] and identifying CR target genes in these cells that are involved in the regulation of cellular senescence pathways [89, 166, 167, 170] provided novel candidate molecules for the development of CR mimetics. We hypothesize that CR mimetics identified in such an approach are also active in modulating cellular senescence. Such compounds should have the potential to counteract senescence‐inducing signals and to suppress and/or modulate senescence‐associated signaling, thereby protecting cells and tissues in our body and maintaining physiological functions longer.
Calcium signaling and mitochondria as targets for antisenescence interventions
Another promising strategy to target senescent cells is the manipulation of mitochondrial Ca2+ homeostasis. The tight linkage between mitochondria and the ER strongly impacts the susceptibility of aged endothelial cells to the cytotoxic effects of agents inducing mitochondrial Ca2+ overload, such as polyphenols [55]. Studying this mechanism might lead to a potentially new class of senolytics based on mitochondrial Ca2+ overload. In a similar way, compounds previously selected for regulating mitochondrial activity in various senescence models have the potential to modulate mitochondrial physiology in order to overcome/target specific types of senescence, such as mitochondrial dysfunction‐associated senescence. In such settings, pharmacological modulation of mitochondrial fitness can be evaluated by analysis of mitochondrial fragmentation, mitophagy, and ROS production. Both approaches bear the promise to develop novel senolytic and or senostatic drugs (Fig. 5).
Proteostasis optimizers for antisenescence interventions
Dampening of protein synthesis may represent another promising strategy for pharmacological targeting of senescent cells. As many antibiotics targeting eukaryotic cells selectively inhibit different steps of cytosolic or mitochondrial translation [187, 188], we hypothesize that at least some of these compounds may postpone senescence and mitigate adverse effects of the SASP by reducing protein synthesis (Fig. 5). However, whether promising drug candidates act via repression of global protein synthesis, via promoting the selective translation of specific anti‐senescence mRNAs, or via so far unknown mechanisms, will also remain to be elucidated.
Conclusions and perspectives
The discovery in genetically modified mice that senescent cells drive aging in animal models [189] has spurred huge research attempts to find pharmacological tools that may promote healthy aging by elimination of senescent cells (‘senolysis’) or disabling/switching their function in human tissues (‘senostasis’, ‘senomorphism’). In parallel, enormous research efforts with the goal to understand the biology of cellular senescence have highlighted relevant molecular pathways of senescence. These discoveries may lead the way to unveil potential pharmacological targets. Maintaining cells in various human tissues healthy and fully functional seems to require a sophisticated interplay between various cellular organelles, highly efficient quality control of mitochondria, functional interorganelle communication, and a tight regulation of proteostasis at various levels. This fragile system is challenged by internal and environmental stress factors causing failures of single components of this network that may be sufficient to drive a cell into a pathological state known as cellular senescence. A better understanding of the hierarchy of and synergy between the various signaling pathways of senescence bears the promise to identify new critical targets for antisenescence interventions.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
MC, CTM‐S, SB, MS, WZ, SW, WFG, JG, and PJ‐D collectively wrote the review and read, as well as edited, the entire manuscript.
Acknowledgements
Work in the PJD/MC laboratory has been supported by the European integrated FP6‐LIFESCIHEALTH project MiMAGE (http://cordis.europa.eu/project/rcn/74075_en.html), by the EU COST Action CA15203 MitoEAGLE as well as by the Austrian Science Fund (FWF, project P31582) to PJD. MC received additional funding by Tiroler Wissenschaftsfonds (ZAP746010), and SW received funding by the University of Innsbruck, Aktion D. Swarovski KG 2018 (Project Nr. 281886, P7460‐030‐011). Research of CMS and WFG is funded by the Austrian Science Fund (FWF), in particular the Doctoral Program Metabolic and Cardiovascular Disease (DK‐MCD W1226 to WFG), Erwin Schroedinger Abroad Fellowship (J4205‐B27 to CMS), by Nikon Austria within the Nikon Center of Excellence, Graz, and MEFO‐Graz. Work in the SB laboratory is supported by the Swedish Research Council Vetenskapsrådet (grants 2015‐05468 and 2019‐05249), the Knut and Alice Wallenberg foundation (grant 2017.0091), and Stiftelsen Olle Engkvist Byggmästare (grant 194‐0681). Work in the MS laboratory was supported by the Austrian Science Fund (FWF) and Herzfelder'sche Familienstiftung [P30623 to MS]. The financial support by the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology and Development, and the Christian Doppler Research Association is gratefully acknowledged. This study also received funding from the European Union's Horizon 2020 research and innovation program under grant agreement 847681—ARDRE—H2020‐MSCA‐COFUND‐2018 and the EUREGIO Environment Food and Health project funded by the European Region Tyrol‐South‐Tyrol‐Trentino (http://euregio‐efh.eu/) both granted to WZ.
Contributor Information
Maria Cavinato, Email: maria.cavinato-nascimento@uibk.ac.at.
Corina T. Madreiter‐Sokolowski, Email: corina.madreiter@hest.ethz.ch.
Sabrina Büttner, Email: sabrina.buettner@su.se.
Markus Schosserer, Email: markus.schosserer@boku.ac.at.
Werner Zwerschke, Email: werner.zwerschke@uibk.ac.at.
Pidder Jansen‐Dürr, Email: pidder.jansen-duerr@uibk.ac.at.
References
- 1.Ogrodnik M, Salmonowicz H, Jurk D & Passos JF (2019) Expansion and cell‐cycle arrest: common denominators of cellular senescence. Trends Biochem Sci 44, 996–1008. [DOI] [PubMed] [Google Scholar]
- 2.Childs BG, Gluscevic M, Baker DJ, Laberge R‐M, Marquess D, Dananberg J & van Deursen JM (2017) Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov 16, 718–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nacarelli T & Sell C (2017) Targeting metabolism in cellular senescence, a role for intervention. Mol Cell Endocrinol 455, 83–92. [DOI] [PubMed] [Google Scholar]
- 4.de Keizer PLJ (2017) The fountain of youth by targeting senescent cells? Trends Mol Med 23, 6–17. [DOI] [PubMed] [Google Scholar]
- 5.Kirkland JL & Tchkonia T (2017) Cellular senescence: a translational perspective. EBioMedicine 21, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grosse L, Wagner N, Emelyanov A, Molina C, Lacas‐Gervais S, Wagner K‐D & Bulavin DV (2020) Defined p16high senescent cell types are indispensable for mouse healthspan. Cell Metab 32, 87–99.e6. [DOI] [PubMed] [Google Scholar]
- 7.Chu X, Wen J & Raju RP (2020) Rapid senescence‐like response after acute injury. Aging Cell 19, e13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Storer M, Mas A, Robert‐Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe Jet al. (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130. [DOI] [PubMed] [Google Scholar]
- 9.Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, VanSteeg H, Dollé METet al. (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell 31, 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lundblad V & Blackburn EH (1993) An alternative pathway for yeast telomere maintenance rescues est1− senescence. Cell 73, 347–360. [DOI] [PubMed] [Google Scholar]
- 11.Teixeira MT (2013) Saccharomyces cerevisiae as a model to study replicative senescence triggered by telomere shortening. Front Oncol 3, 101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Z & Teixeira MT (2019) The many types of heterogeneity in replicative senescence. Yeast 36, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lohr JN, Galimov ER & Gems D (2019) Does senescence promote fitness in Caenorhabditis elegans by causing death? Ageing Res Rev 50, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wood WB (1998) Aging of C. elegans . Cell 95, 147–150. [DOI] [PubMed] [Google Scholar]
- 15.Zhou F, Li SC, Zhu Y, Guo W, Shao L, Nelson J, Simpkins S, Yang D, Liu Q, Yashiroda Yet al. (2019) Integrating yeast chemical genomics and mammalian cell pathway analysis. Acta Pharmacol Sin 40, 1245–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tissenbaum HA (2015) Using C. elegans for aging research. Invertebr Reprod Dev 59, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mavrogonatou E, Pratsinis H, Papadopoulou A, Karamanos NK & Kletsas D (2019) Extracellular matrix alterations in senescent cells and their significance in tissue homeostasis. Matrix Biol 75–76, 27–42. [DOI] [PubMed] [Google Scholar]
- 18.Sikora E & Rattan SIS (2017) The future of ageing: not more of the same. Biogerontology 18, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stöckl P, Hütter E, Zwerschke W & Jansen‐Dürr P (2006) Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp Gerontol 41, 674–682. [DOI] [PubMed] [Google Scholar]
- 20.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan Aet al. (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Passos JF, Saretzki G & von Zglinicki T (2007) DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res 35, 7505–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Correia‐Melo C, Marques FDM, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz Aet al. (2016) Mitochondria are required for pro‐ageing features of the senescent phenotype. EMBO J 35, 724–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dikalov S (2011) Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51, 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weyemi U & Dupuy C (2012) The emerging role of ROS‐generating NADPH oxidase NOX4 in DNA‐damage responses. Mutat Res 751, 77–81. [DOI] [PubMed] [Google Scholar]
- 25.Friguet B, Bulteau A‐L & Petropoulos I (2008) Mitochondrial protein quality control: implications in ageing. Biotechnol J 3, 757–764. [DOI] [PubMed] [Google Scholar]
- 26.Briganti S & Picardo M (2003) Antioxidant activity, lipid peroxidation and skin diseases. What's new. J Eur Acad Dermatol Venereol 17, 663–669. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, McMillan‐Ward E, Kong J, Israels SJ & Gibson SB (2008) Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 15, 171–182. [DOI] [PubMed] [Google Scholar]
- 28.Osiewacz HD & Bernhardt D (2013) Mitochondrial quality control: impact on aging and life span – a mini‐review. Gerontology 59, 413–420. [DOI] [PubMed] [Google Scholar]
- 29.Abate M, Festa A, Falco M, Lombardi A, Luce A, Grimaldi A, Zappavigna S, Sperlongano P, Irace C, Caraglia Met al. (2020) Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin Cell Dev Biol 98, 139–153. [DOI] [PubMed] [Google Scholar]
- 30.Vasileiou P, Evangelou K, Vlasis K, Fildisis G, Panayiotidis M, Chronopoulos E, Passias P‐G, Kouloukoussa M, Gorgoulis V & Havaki S (2019) Mitochondrial homeostasis and cellular senescence. Cells 8, 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashrafi G & Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patergnani S & Pinton P (2015) Mitophagy and mitochondrial balance. Methods Mol Biol 1241, 181–194. [DOI] [PubMed] [Google Scholar]
- 33.Kim I & Lemasters JJ (2011) Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal 14, 1919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8, 3–5. [DOI] [PubMed] [Google Scholar]
- 35.Springer MZ & Macleod KF (2016) In brief: Mitophagy: mechanisms and role in human disease. J Pathol 240, 253–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JLet al. (2007) NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104, 19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pua HH, Guo J, Komatsu M & He Y‐W (2009) Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol 182, 4046–4055. [DOI] [PubMed] [Google Scholar]
- 38.Lionaki E, Markaki M, Palikaras K & Tavernarakis N (2015) Mitochondria, autophagy and age‐associated neurodegenerative diseases: new insights into a complex interplay. Biochim Biophys Acta 1847, 1412–1423. [DOI] [PubMed] [Google Scholar]
- 39.Von Stockum S, Nardin A, Schrepfer E & Ziviani E (2016) Mitochondrial dynamics and mitophagy in Parkinson’s disease: a fly point of view. Neurobiol Dis 90, 58–67. [DOI] [PubMed] [Google Scholar]
- 40.Korolchuk VI, Miwa S, Carroll B & von Zglinicki T (2017) Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine 21, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin H, Xiong H, Su Z, Pang J, Lai L, Zhang H, Jian B, Zhang W & Zheng Y (2019) Inhibition of DRP‐1‐dependent mitophagy promotes cochlea hair cell senescence and exacerbates age‐related hearing loss. Front Cell Neurosci 13, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim JY, Lee SH, Bae I‐HH, Shin DW, Min D, Ham M, Kim K‐HH, Lee TR, Kim H‐JJ, Son EDet al. (2018) Pyruvate protects against cellular senescence through the control of mitochondrial and lysosomal function in dermal fibroblasts. J Invest Dermatol 138, 2522–2530. [DOI] [PubMed] [Google Scholar]
- 43.Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H & Rahman I (2015) Impaired mitophagy leads to cigarette smoke stress‐induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J 29, 2912–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pickles S, Vigié P & Youle RJ (2018) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Picca A, Guerra F, Calvani R, Coelho‐Junior HJ, Bossola M, Landi F, Bernabei R, Bucci C & Marzetti E (2020) Generation and release of mitochondrial‐derived vesicles in health, aging and disease. J Clin Med 9, 1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miliotis S, Nicolalde B, Ortega M, Yepez J & Caicedo A (2019) Forms of extracellular mitochondria and their impact in health. Mitochondrion 48, 16–30. [DOI] [PubMed] [Google Scholar]
- 47.Sukhorukov VM & Meyer‐Hermann M (2015) Structural heterogeneity of mitochondria induced by the microtubule cytoskeleton. Sci Rep 5, 13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmitt K, Grimm A, Dallmann R, Oettinghaus B, Restelli LM, Witzig M, Ishihara N, Mihara K, Ripperger JA, Albrecht Uet al. (2018) Circadian control of DRP1 activity regulates mitochondrial dynamics and bioenergetics. Cell Metab 27, 657–666.e5. [DOI] [PubMed] [Google Scholar]
- 49.Guerra‐Gomes S, Viana JF, Nascimento DSM, Correia JS, Sardinha VM, Caetano I, Sousa N, Pinto L & Oliveira JF (2018) The role of astrocytic calcium signaling in the aged prefrontal cortex. Front Cell Neurosci 12, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ristow M & Schmeisser K (2014) Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 12, 288–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schrader M, Godinho LF, Costello JL & Islinger M (2015) The different facets of organelle interplay—an overview of organelle interactions. Front Cell Dev Biol 3, 56–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calvo‐Rodríguez M, García‐Durillo M, Villalobos C & Núñez L (2016) In vitro aging promotes endoplasmic reticulum (ER)‐mitochondria Ca2+ cross talk and loss of store‐operated Ca2+ entry (SOCE) in rat hippocampal neurons. Biochim Biophys Acta 1863, 2637–2649. [DOI] [PubMed] [Google Scholar]
- 53.Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan Pet al. (2017) Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell 65, 1014–1028.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ziegler DV, Wiley CD & Velarde MC (2015) Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 14, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Madreiter‐Sokolowski CT, Waldeck‐Weiermair M, Bourguignon M‐P, Villeneuve N, Gottschalk B, Klec C, Stryeck S, Radulovic S, Parichatikanond W, Frank Set al. (2019) Enhanced inter‐compartmental Ca2+ flux modulates mitochondrial metabolism and apoptotic threshold during aging. Redox Biol 20, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brookes PS, Yoon Y, Robotham JL, Anders MW & Sheu S‐S (2004) Calcium, ATP, and ROS: a mitochondrial love‐hate triangle. Am J Physiol Physiol 287, C817–C833. [DOI] [PubMed] [Google Scholar]
- 57.Tower J (2015) Programmed cell death in aging. Ageing Res Rev 23, 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simmen T & Herrera‐Cruz MS (2018) Plastic mitochondria‐endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr Opin Cell Biol 53, 61–69. [DOI] [PubMed] [Google Scholar]
- 59.Wang X, Wen Y, Dong J, Cao C & Yuan S (2018) Systematic in‐depth proteomic analysis of mitochondria‐associated endoplasmic reticulum membranes in mouse and human testes. Proteomics 18, e1700478. [DOI] [PubMed] [Google Scholar]
- 60.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS & Walter P (2009) An ER‐mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kohler V, Aufschnaiter A & Büttner S (2020) Closing the gap: membrane contact sites in the regulation of autophagy. Cells 9, 1184–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eisenberg‐Bord M, Shai N, Schuldiner M & Bohnert M (2016) A tether is a tether is a tether: tethering at membrane contact sites. Dev Cell 39, 395–409. [DOI] [PubMed] [Google Scholar]
- 63.Annunziata I, Sano R & D'Azzo A (2018) Mitochondria‐associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis 9, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fernandez‐Sanz C, Ruiz‐Meana M, Miro‐Casas E, Nuñez E, Castellano J, Loureiro M, Barba I, Poncelas M, Rodriguez‐Sinovas A, Vázquez Jet al. (2014) Defective sarcoplasmic reticulum‐mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis 5, e1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paillusson S, Stoica R, Gomez‐Suaga P, Lau DHW, Mueller S, Miller T & Miller CCJ (2016) There's something wrong with my MAM; the ER‐mitochondria axis and neurodegenerative diseases. Trends Neurosci 39, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thoudam T, Jeon J‐H, Ha C‐M & Lee I‐K (2016) Role of mitochondria‐associated endoplasmic reticulum membrane in inflammation‐mediated metabolic diseases. Mediators Inflamm 2016, 1851420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sassano ML, van Vliet AR & Agostinis P (2017) Mitochondria‐associated membranes as networking platforms and regulators of cancer cell fate. Front Oncol 7, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carmona‐Gutierrez D, Hughes AL, Madeo F & Ruckenstuhl C (2016) The crucial impact of lysosomes in aging and longevity. Ageing Res Rev 32, 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yambire KF, Fernandez‐Mosquera L, Steinfeld R, Mühle C, Ikonen E, Milosevic I & Raimundo N (2019) Mitochondrial biogenesis is transcriptionally repressed in lysosomal lipid storage diseases. Elife 8, e39598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fernández‐Mosquera L, Diogo CV, Yambire KF, Santos GL, Luna Sánchez M, Bénit P, Rustin P, Lopez LC, Milosevic I & Raimundo N (2017) Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci Rep 7, 45076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wong YC, Ysselstein D & Krainc D (2018) Mitochondria‐lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.John Peter AT, Herrmann B, Antunes D, Rapaport D, Dimmer KS & Kornmann B (2017) Vps13‐Mcp1 interact at vacuole‐mitochondria interfaces and bypass ER‐mitochondria contact sites. J Cell Biol 216, 3219–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.González Montoro A, Auffarth K, Hönscher C, Bohnert M, Becker T, Warscheid B, Reggiori F, van der Laan M, Fröhlich F & Ungermann C (2018) Vps39 Interacts with Tom40 to establish one of two functionally distinct vacuole‐mitochondria contact sites. Dev Cell 45, 621–636.e7. [DOI] [PubMed] [Google Scholar]
- 74.Deus CM, Yambire KF, Oliveira PJ & Raimundo N (2020) Mitochondria‐lysosome crosstalk: from physiology to neurodegeneration. Trends Mol Med 26, 71–88. [DOI] [PubMed] [Google Scholar]
- 75.McCue HV, Wardyn JD, Burgoyne RD & Haynes LP (2013) Generation and characterization of a lysosomally targeted, genetically encoded Ca(2+)‐sensor. Biochem J 449, 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Westermann B (2015) The mitochondria‐plasma membrane contact site. Curr Opin Cell Biol 35, 1–6. [DOI] [PubMed] [Google Scholar]
- 77.Demarquoy J & Le Borgne F (2015) Crosstalk between mitochondria and peroxisomes. World J Biol Chem 6, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Das A, Nag S, Mason AB & Barroso MM (2016) Endosome‐mitochondria interactions are modulated by iron release from transferrin. J Cell Biol 214, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Malli R & Graier WF (2017) The role of mitochondria in the activation/maintenance of SOCE: the contribution of mitochondrial Ca2+ uptake, mitochondrial motility, and location to store‐operated Ca2+ entry. Adv Exp Med Biol 993, 297–319. [DOI] [PubMed] [Google Scholar]
- 80.Manivannan S, Scheckhuber CQ, Veenhuis M & van der Klei IJ (2012) The impact of peroxisomes on cellular aging and death. Front Oncol 2, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deori NM, Kale A, Maurya PK & Nagotu S (2018) Peroxisomes: role in cellular ageing and age related disorders. Biogerontology 19, 303–324. [DOI] [PubMed] [Google Scholar]
- 82.Iraci N, Leonardi T, Gessler F, Vega B & Pluchino S (2016) Focus on extracellular vesicles: physiological role and signalling properties of extracellular membrane vesicles. Int J Mol Sci 17, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Terlecki‐Zaniewicz L, Lämmermann I, Latreille J, Bobbili MR, Pils V, Schosserer M, Weinmüllner R, Dellago H, Skalicky S, Pum Det al. (2018) Small extracellular vesicles and their miRNA cargo are anti‐apoptotic members of the senescence‐associated secretory phenotype. Aging (Albany NY) 10, 1103–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Terlecki‐Zaniewicz L, Pils V, Bobbili MR, Lämmermann I, Perrotta I, Grillenberger T, Schwestka J, Weiß K, Pum D, Arcalis Eet al. (2019) Extracellular vesicles in human skin: cross‐talk from senescent fibroblasts to keratinocytes by miRNAs. J Invest Dermatol 139, 2425–2436.e5. [DOI] [PubMed] [Google Scholar]
- 85.Cadete VJJ, Deschênes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, Turnbull D, Picard M, McBride HM & Burelle Y (2016) Formation of mitochondrial‐derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol 594, 5343–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morita M, Prudent J, Basu K, Goyon V, Katsumura S, Hulea L, Pearl D, Siddiqui N, Strack S, McGuirk Set al. (2017) mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol Cell 67, 922–935.e5. [DOI] [PubMed] [Google Scholar]
- 87.de la Cruz López KG, Toledo Guzmán ME, Sánchez EO & García Carrancá A (2019) mTORC1 as a regulator of mitochondrial functions and a therapeutic target in cancer. Front Oncol 9, 1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iglesias‐Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB & Gutkind JS (2012) mTOR inhibition prevents epithelial stem cell senescence and protects from radiation‐induced mucositis. Cell Stem Cell 11, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ejaz A, Mattesich M & Zwerschke W (2017) Silencing of the small GTPase DIRAS3 induces cellular senescence in human white adipose stromal/progenitor cells. Aging (Albany NY) 9, 860–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Laberge R‐M, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson‐Edell KA, Liu Set al. (2015) MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17, 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CSet al. (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fok WC, Zhang Y, Salmon AB, Bhattacharya A, Gunda R, Jones D, Ward W, Fisher K, Richardson A & Pérez VI (2013) Short‐term treatment with rapamycin and dietary restriction have overlapping and distinctive effects in young mice. J Gerontol A Biol Sci Med Sci 68, 108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Selman C, Tullet JMA, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al‐Qassab H, Carmignac D, Ramadani Fet al. (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bartolomé A, García‐Aguilar A, Asahara S‐I, Kido Y, Guillén C, Pajvani UB & Benito M (2017) MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol Cell Biol 37, 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kozieł R, Greussing R, Maier AB, Declercq L & Jansen‐Dürr P (2011) Functional interplay between mitochondrial and proteasome activity in skin aging. J Invest Dermatol 131, 594–603. [DOI] [PubMed] [Google Scholar]
- 96.Klaips CL, Jayaraj GG & Hartl FU (2018) Pathways of cellular proteostasis in aging and disease. J Cell Biol 217, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaushik S & Cuervo AM (2015) Proteostasis and aging. Nat Med 21, 1406–1415. [DOI] [PubMed] [Google Scholar]
- 98.Labbadia J & Morimoto RI (2015) The biology of proteostasis in aging and disease. Annu Rev Biochem 84, 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Janssens GE, Meinema AC, González J, Wolters JC, Schmidt A, Guryev V, Bischoff R, Wit EC, Veenhoff LM & Heinemann M (2015) Protein biogenesis machinery is a driver of replicative aging in yeast. Elife 4, e08527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Steffen KK, MacKay VL, Kerr EO, Tsuchiya M, Hu D, Fox LA, Dang N, Johnston ED, Oakes JA, Tchao BNet al. (2008) Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell 133, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McCormick MA, Delaney JR, Tsuchiya M, Tsuchiyama S, Shemorry A, Sim S, Chou AC‐Z, Ahmed U, Carr D, Murakami CJet al. (2015) A Comprehensive analysis of replicative lifespan in 4,698 single‐gene deletion strains uncovers conserved mechanisms of aging. Cell Metab 22, 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ & Kapahi P (2007) Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans . Aging Cell 6, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hansen M, Taubert S, Crawford D, Libina N, Lee S‐J & Kenyon C (2007) Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans . Aging Cell 6, 95–110. [DOI] [PubMed] [Google Scholar]
- 104.Syntichaki P, Troulinaki K & Tavernarakis N (2007) eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans . Nature 445, 922–926. [DOI] [PubMed] [Google Scholar]
- 105.Gonskikh Y & Polacek N (2017) Alterations of the translation apparatus during aging and stress response. Mech Ageing Dev 168, 30–36. [DOI] [PubMed] [Google Scholar]
- 106.Kapahi P (2010) Protein synthesis and the antagonistic pleiotropy hypothesis of aging. Adv Exp Med Biol 694, 30–37. [DOI] [PubMed] [Google Scholar]
- 107.Sherman MY & Qian S‐B (2013) Less is more: improving proteostasis by translation slow down. Trends Biochem Sci 38, 585–591. [DOI] [PubMed] [Google Scholar]
- 108.Hipkiss AR (2007) On why decreasing protein synthesis can increase lifespan. Mech Ageing Dev 128, 412–414. [DOI] [PubMed] [Google Scholar]
- 109.Sinclair DA & Guarente L (1997) Extrachromosomal rDNA circles–a cause of aging in yeast. Cell 91, 1033–1042. [DOI] [PubMed] [Google Scholar]
- 110.Sharifi S & Bierhoff H (2018) Regulation of RNA polymerase I transcription in development, disease, and aging. Annu Rev Biochem 87, 51–73. [DOI] [PubMed] [Google Scholar]
- 111.Martínez Corrales G, Filer D, Wenz KC, Rogan A, Phillips G, Li M, Feseha Y, Broughton SJ & Alic N (2020) Partial inhibition of RNA polymerase I promotes animal health and longevity. Cell Rep 30, 1661–1669.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang L, Song T, Chen L, Soliman H & Chen J (2015) Nucleolar repression facilitates initiation and maintenance of senescence. Cell Cycle 14, 3613–3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alupei MC, Maity P, Esser PR, Krikki I, Tuorto F, Parlato R, Penzo M, Schelling A, Laugel V, Montanaro Let al. (2018) Loss of proteostasis is a pathomechanism in cockayne syndrome. Cell Rep 23, 1612–1619. [DOI] [PubMed] [Google Scholar]
- 114.López‐Otín C, Blasco MA, Partridge L, Serrano M & Kroemer G (2013) The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Anisimova AS, Meerson MB, Gerashchenko MV, Kulakovskiy IV, Dmitriev SE & Gladyshev VN (2020) Multifaceted deregulation of gene expression and protein synthesis with age. Proc Natl Acad Sci USA 117, 15581–15590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S & Kapahi P (2009) 4E‐BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila . Cell 139, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rogers AN, Chen D, McColl G, Czerwieniec G, Felkey K, Gibson BW, Hubbard A, Melov S, Lithgow GJ & Kapahi P (2011) Life span extension via eIF4G inhibition is mediated by posttranscriptional remodeling of stress response gene expression in C. elegans . Cell Metab 14, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schosserer M, Minois N, Angerer TB, Amring M, Dellago H, Harreither E, Calle‐Perez A, Pircher A, Gerstl MP, Pfeifenberger Set al. (2015) Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat Commun 6, 6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liberman N, O'Brown ZK, Earl AS, Boulias K, Gerashchenko MV, Wang SY, Fritsche C, Fady P‐E, Dong A, Gladyshev VNet al. (2020) N6‐adenosine methylation of ribosomal RNA affects lipid oxidation and stress resistance. Sci Adv 6, eaaz4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rozenski J, Crain PF & McCloskey JA (1999) The RNA modification database: 1999 update. Nucleic Acids Res 27, 196–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Peer E, Rechavi G & Dominissini D (2017) Epitranscriptomics: regulation of mRNA metabolism through modifications. Curr Opin Chem Biol 41, 93–98. [DOI] [PubMed] [Google Scholar]
- 122.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW & Auwerx J (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Molenaars M, Janssens GE, Williams EG, Jongejan A, Lan J, Rabot S, Joly F, Moerland PD, Schomakers BV, Lezzerini Met al. (2020) A conserved mito‐cytosolic translational balance links two longevity pathways. Cell Metab 31, 549–563.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Suhm T, Kaimal JM, Dawitz H, Peselj C, Masser AE, Hanzén S, Ambrožič M, Smialowska A, Björck ML, Brzezinski Pet al. (2018) Mitochondrial translation efficiency controls cytoplasmic protein homeostasis. Cell Metab 27, 1309–1322.e6. [DOI] [PubMed] [Google Scholar]
- 125.Andréasson C, Ott M & Büttner S (2019) Mitochondria orchestrate proteostatic and metabolic stress responses. EMBO Rep 20, e47865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kozieł R, Ruckenstuhl C, Albertini E, Neuhaus M, Netzberger C, Bust M, Madeo F, Wiesner RJ & Jansen‐Dürr P (2014) Methionine restriction slows down senescence in human diploid fibroblasts. Aging Cell 13, 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Palliyaguru DL, Moats JM, Di Germanio C, Bernier M & de Cabo R (2019) Frailty index as a biomarker of lifespan and healthspan: focus on pharmacological interventions. Mech Ageing Dev 180, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Robbins PD, Jurk D, Khosla S, Kirkland JL, LeBrasseur NK, Miller JD, Passos JF, Pignolo RJ, Tchkonia T & Niedernhofer LJ (2020) Senolytic drugs: reducing senescent cell viability to extend health span. Annu Rev Pharmacol Toxicol. 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Short S, Fielder E, Miwa S & von Zglinicki T (2019) Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 41, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mansfeld J, Urban N, Priebe S, Groth M, Frahm C, Hartmann N, Gebauer J, Ravichandran M, Dommaschk A, Schmeisser Set al. (2015) Branched‐chain amino acid catabolism is a conserved regulator of physiological ageing. Nat Commun 6, 10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lepperdinger G (2008) The use of genetically engineered model systems for research on human aging. Front Biosci. 13, 7022–7031. [DOI] [PubMed] [Google Scholar]
- 132.Laschober GT, Ruli D, Hofer E, Muck C, Carmona‐Gutierrez D, Ring J, Hutter E, Ruckenstuhl C, Micutkova L, Brunauer Ret al. (2010) Identification of evolutionarily conserved genetic regulators of cellular aging. Aging Cell 9, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xu Z, Fallet E, Paoletti C, Fehrmann S, Charvin G & Teixeira MT (2015) Two routes to senescence revealed by real‐time analysis of telomerase‐negative single lineages. Nat Commun 6, 7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Abdallah P, Luciano P, Runge KW, Lisby M, Géli V, Gilson E & Teixeira MT (2009) A two‐step model for senescence triggered by a single critically short telomere. Nat Cell Biol 11, 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nautiyal S, DeRisi JL & Blackburn EH (2002) The genome‐wide expression response to telomerase deletion in Saccharomyces cerevisiae . Proc Natl Acad Sci USA 99, 9316–9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Albertini E, Kozieł R, Dürr A, Neuhaus M & Jansen‐Dürr P (2012) Cystathionine beta synthase modulates senescence of human endothelial cells. Aging (Albany NY) 4, 664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Petit M, Koziel R, Etemad S, Pircher H & Jansen‐Dürr P (2017) Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Exp Gerontol 92, 7–12. [DOI] [PubMed] [Google Scholar]
- 138.Weinmüllner R, Zbiral B, Becirovic A, Stelzer EM, Nagelreiter F, Schosserer M, Lämmermann I, Liendl L, Lang M, Terlecki‐Zaniewicz Let al. (2020) Organotypic human skin culture models constructed with senescent fibroblasts show hallmarks of skin aging. NPJ aging Mech Dis 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cavinato M, Koziel R, Romani N, Weinmüllner R, Jenewein B, Hermann M, Dubrac S, Ratzinger G, Grillari J, Schmuth Met al. (2016) UVB‐induced senescence of human dermal fibroblasts involves impairment of proteasome and enhanced autophagic activity. J Gerontol A Biol Sci Med Sci 72, glw150. [DOI] [PubMed] [Google Scholar]
- 140.Heissenberger C, Liendl L, Nagelreiter F, Gonskikh Y, Yang G, Stelzer EM, Krammer TL, Micutkova L, Vogt S, Kreil DPet al. (2019) Loss of the ribosomal RNA methyltransferase NSUN5 impairs global protein synthesis and normal growth. Nucleic Acids Res 47, 11807–11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Monteforte R, Beilhack GF, Grausenburger R, Mayerhofer B, Bittner R, Grillari‐Voglauer R, Sibilia M, Dellago H, Tschachler E, Gruber Fet al. (2016) SNEV(Prp19/PSO4) deficiency increases PUVA‐induced senescence in mouse skin. Exp Dermatol 25, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ingram DK, Zhu M, Mamczarz J, Zou S, Lane MA, Roth GS & DeCabo R (2006) Calorie restriction mimetics: an emerging research field. Aging Cell 5, 97–108. [DOI] [PubMed] [Google Scholar]
- 143.Weindruch R, Walford RL, Fligiel S & Guthrie D (1986) The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. J Nutr 116, 641–654. [DOI] [PubMed] [Google Scholar]
- 144.Speakman JR & Mitchell SE (2011) Caloric restriction. Mol Aspects Med 32, 159–221. [DOI] [PubMed] [Google Scholar]
- 145.Mitchell SJ, Madrigal‐Matute J, Scheibye‐Knudsen M, Fang E, Aon M, González‐Reyes JA, Cortassa S, Kaushik S, Gonzalez‐Freire M, Patel Bet al. (2016) Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab 23, 1093–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Russell SJ & Kahn CR (2007) Endocrine regulation of ageing. Nat Rev Mol Cell Biol 8, 681–691. [DOI] [PubMed] [Google Scholar]
- 147.Chang H‐C & Guarente L (2014) SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 25, 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sohal RS, Agarwal S, Candas M, Forster MJ & Lal H (1994) Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech Ageing Dev 76, 215–224. [DOI] [PubMed] [Google Scholar]
- 149.Haley‐Zitlin V & Richardson A (1993) Effect of dietary restriction on DNA repair and DNA damage. Mutat Res 295, 237–245. [DOI] [PubMed] [Google Scholar]
- 150.Hofer T, Fontana L, Anton SD, Weiss EP, Villareal D, Malayappan B & Leeuwenburgh C (2008) Long‐term effects of caloric restriction or exercise on DNA and RNA oxidation levels in white blood cells and urine in humans. Rejuvenation Res 11, 793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lewis SE, Goldspink DF, Phillips JG, Merry BJ & Holehan AM (1985) The effects of aging and chronic dietary restriction on whole body growth and protein turnover in the rat. Exp Gerontol 20, 253–263. [DOI] [PubMed] [Google Scholar]
- 152.Rojas C, Cadenas S, Pérez‐Campo R, López‐Torres M, Pamplona R, Prat J & Barja G (1993) Relationship between lipid peroxidation, fatty acid composition, and ascorbic acid in the liver during carbohydrate and caloric restriction in mice. Arch Biochem Biophys 306, 59–64. [DOI] [PubMed] [Google Scholar]
- 153.Weindruch R & Sohal RS (1997) Caloric Intake and Aging. N Engl J Med 337, 986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Masoro EJ (2005) Overview of caloric restriction and ageing. Mech Ageing Dev 126, 913–922. [DOI] [PubMed] [Google Scholar]
- 155.López‐Lluch G (2017) Mitochondrial activity and dynamics changes regarding metabolism in ageing and obesity. Mech Ageing Dev 162, 108–121. [DOI] [PubMed] [Google Scholar]
- 156.Klein S, Sheard NF, Pi‐Sunyer X, Daly A, Wylie‐Rosett J, Kulkarni K, Clark NG, American Diabetes Association, North American Association for the Study of Obesity & American Society for Clinical Nutrition (2004) Weight management through lifestyle modification for the prevention and management of type 2 diabetes: rationale and strategies: a statement of the American Diabetes Association, the North American Association for the Study of Obesity, and the American Society for Clinical Nutrition. Diabetes Care 27, 2067–2073. [DOI] [PubMed] [Google Scholar]
- 157.Most J, Tosti V, Redman LM & Fontana L (2017) Calorie restriction in humans: an update. Ageing Res Rev 39, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dorling JL, Martin CK & Redman LM (2020) Calorie restriction for enhanced longevity: the role of novel dietary strategies in the present obesogenic environment. Ageing Res Rev. 64, 101038–101052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Le Couteur DG, McLachlan AJ, Quinn RJ, Simpson SJ & de Cabo R (2012) Aging biology and novel targets for drug discovery. J Gerontol A Biol Sci Med Sci 67, 168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.de Cabo R, Carmona‐Gutierrez D, Bernier M, Hall MN & Madeo F (2014) The search for antiaging interventions: from elixirs to fasting regimens. Cell 157, 1515–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Blagosklonny MV (2007) An anti‐aging drug today: from senescence‐promoting genes to anti‐aging pill. Drug Discov Today 12, 218–224. [DOI] [PubMed] [Google Scholar]
- 162.Wang C, Maddick M, Miwa S, Jurk D, Czapiewski R, Saretzki G, Langie SAS, Godschalk RWL, Cameron K & von Zglinicki T (2010) Adult‐onset, short‐term dietary restriction reduces cell senescence in mice. Aging (Albany NY) 2, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Jill Saffrey M, Cameron Ket al. (2012) Postmitotic neurons develop a p21‐dependent senescence‐like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mitterberger MC, Mattesich M & Zwerschke W (2014) Bariatric surgery and diet‐induced long‐term caloric restriction protect subcutaneous adipose‐derived stromal/progenitor cells and prolong their life span in formerly obese humans. Exp Gerontol 56, 106–113. [DOI] [PubMed] [Google Scholar]
- 165.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al‐Regaiey K, Su L & Sharpless NE (2004) Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114, 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mandl M, Wagner SA, Hatzmann FM, Mitterberger‐Vogt MC, Zwierzina ME, Mattesich M & Zwerschke W (2019) Sprouty1 is a weight‐loss target gene in human adipose stem/progenitor cells that is mandatory for the initiation of adipogenesis. Cell Death Dis 10, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mandl M, Wagner SA, Hatzmann FM, Ejaz A, Ritthammer H, Baumgarten S, Viertler HP, Springer J, Zwierzina ME, Mattesich Met al. (2020) Sprouty1 prevents cellular senescence maintaining proliferation and differentiation capacity of human adipose stem/progenitor cells. J Gerontol A Biol Sci Med Sci. 10, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fontana L, Mitchell SE, Wang B, Tosti V, van Vliet T, Veronese N, Bertozzi B, Early DS, Maissan P, Speakman JRet al. (2018) The effects of graded caloric restriction: XII. Comparison of mouse to human impact on cellular senescence in the colon. Aging Cell 17, e12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QMet al. (2017) Cellular senescence drives age‐dependent hepatic steatosis. Nat Commun 8, 15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ejaz A, Mitterberger MC, Lu Z, Mattesich M, Zwierzina ME, Hörl S, Kaiser A, Viertler H‐P, Rostek U, Meryk Aet al. (2016) Weight loss upregulates the small GTPase DIRAS3 in human white adipose progenitor cells, which negatively regulates adipogenesis and activates autophagy via Akt–mTOR inhibition. EBioMedicine 6, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.van Deursen JM (2014) The role of senescent cells in ageing. Nature 509, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ishaq A, Schröder J, Edwards N, von Zglinicki T & Saretzki G (2018) Dietary restriction ameliorates age‐related increase in DNA damage, senescence and inflammation in mouse adipose tissue. J Nutr Health Aging 22, 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Il'yasova D, Fontana L, Bhapkar M, Pieper CF, Spasojevic I, Redman LM, Das SK, Huffman KM, Kraus WE& CALERIE Study Investigators (2018) Effects of 2 years of caloric restriction on oxidative status assessed by urinary F2‐isoprostanes: The CALERIE 2 randomized clinical trial. Aging Cell 17, e12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yang L, Licastro D, Cava E, Veronese N, Spelta F, Rizza W, Bertozzi B, Villareal DT, Hotamisligil GS, Holloszy JOet al. (2016) Long‐term calorie restriction enhances cellular quality‐control processes in human skeletal muscle. Cell Rep 14, 422–428. [DOI] [PubMed] [Google Scholar]
- 175.Nelson G, Kucheryavenko O, Wordsworth J & von Zglinicki T (2018) The senescent bystander effect is caused by ROS‐activated NF‐κB signalling. Mech Ageing Dev 170, 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Vellai T, Takacs‐Vellai K, Zhang Y, Kovacs AL, Orosz L & Müller F (2003) Genetics: influence of TOR kinase on lifespan in C. elegans . Nature 426, 620. [DOI] [PubMed] [Google Scholar]
- 177.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V & Benzer S (2004) Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 14, 885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kaeberlein M, Powers RW, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S & Kennedy BK (2005) Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310, 1193–1196. [DOI] [PubMed] [Google Scholar]
- 179.Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JFet al. (2011) Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 66, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, Javors MA, Li X, Nadon NL, Nelson JFet al. (2014) Rapamycin‐mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 13, 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ferrara‐Romeo I, Martinez P, Saraswati S, Whittemore K, Graña‐Castro O, Thelma Poluha L, Serrano R, Hernandez‐Encinas E, Blanco‐Aparicio C, Maria Flores Jet al. (2020) The mTOR pathway is necessary for survival of mice with short telomeres. Nat Commun 11, 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Blagosklonny MV (2018) Rapamycin, proliferation and geroconversion to senescence. Cell Cycle 17, 2655–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, Hara H, Minagawa S, Wakui H, Fujii Set al. (2015) PARK2‐mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 11, 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cabelof DC, Yanamadala S, Raffoul JJ, Guo Z, Soofi A & Heydari AR (2003) Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age‐related decline. DNA Repair (Amst) 2, 295–307. [DOI] [PubMed] [Google Scholar]
- 185.Lee J‐E, Heo J‐I, Park S‐H, Kim J‐H, Kho Y‐J, Kang H‐J, Chung HY, Yoon J‐L & Lee J‐Y (2011) Calorie restriction (CR) reduces age‐dependent decline of non‐homologous end joining (NHEJ) activity in rat tissues. Exp Gerontol 46, 891–896. [DOI] [PubMed] [Google Scholar]
- 186.Mitterberger MC, Lechner S, Mattesich M & Zwerschke W (2014) Adipogenic differentiation is impaired in replicative senescent human subcutaneous adipose‐derived stromal/progenitor cells. J Gerontol A Biol Sci Med Sci 69, 13–24. [DOI] [PubMed] [Google Scholar]
- 187.Shulman E, Belakhov V, Wei G, Kendall A, Meyron‐Holtz EG, Ben‐Shachar D, Schacht J & Baasov T (2014) Designer aminoglycosides that selectively inhibit cytoplasmic rather than mitochondrial ribosomes show decreased ototoxicity: a strategy for the treatment of genetic diseases. J Biol Chem 289, 2318–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cohen BH & Saneto RP (2012) Mitochondrial translational inhibitors in the pharmacopeia. Biochim Biophys Acta 1819, 1067–1074. [DOI] [PubMed] [Google Scholar]
- 189.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki Aet al. (2016) Naturally occurring p16Ink4a‐positive cells shorten healthy lifespan. Nature 530, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]