

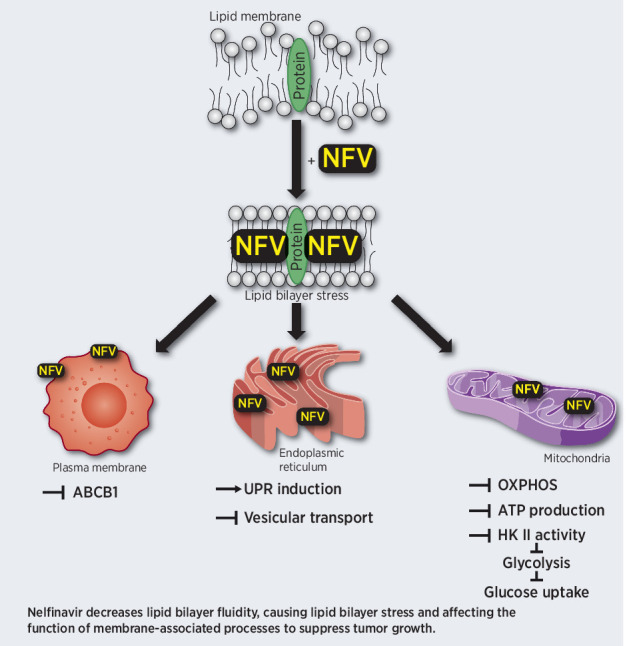
Nelfinavir induces lipid bilayer stress in cellular organelles that disrupts mitochondrial respiration and transmembrane protein transport, resulting in broad anticancer activity via metabolic rewiring and activation of the unfolded protein response.
Abstract
The HIV-protease inhibitor nelfinavir has shown broad anticancer activity in various preclinical and clinical contexts. In patients with advanced, proteasome inhibitor (PI)–refractory multiple myeloma, nelfinavir-based therapy resulted in 65% partial response or better, suggesting that this may be a highly active chemotherapeutic option in this setting. The broad anticancer mechanism of action of nelfinavir implies that it interferes with fundamental aspects of cancer cell biology. We combined proteome-wide affinity-purification of nelfinavir-interacting proteins with genome-wide CRISPR/Cas9–based screening to identify protein partners that interact with nelfinavir in an activity-dependent manner alongside candidate genetic contributors affecting nelfinavir cytotoxicity. Nelfinavir had multiple activity-specific binding partners embedded in lipid bilayers of mitochondria and the endoplasmic reticulum. Nelfinavir affected the fluidity and composition of lipid-rich membranes, disrupted mitochondrial respiration, blocked vesicular transport, and affected the function of membrane-embedded drug efflux transporter ABCB1, triggering the integrated stress response. Sensitivity to nelfinavir was dependent on ADIPOR2, which maintains membrane fluidity by promoting fatty acid desaturation and incorporation into phospholipids. Supplementation with fatty acids prevented the nelfinavir-induced effect on mitochondrial metabolism, drug-efflux transporters, and stress-response activation. Conversely, depletion of fatty acids/cholesterol pools by the FDA-approved drug ezetimibe showed a synergistic anticancer activity with nelfinavir in vitro. These results identify the modification of lipid-rich membranes by nelfinavir as a novel mechanism of action to achieve broad anticancer activity, which may be suitable for the treatment of PI–refractory multiple myeloma.
Significance:
Nelfinavir induces lipid bilayer stress in cellular organelles that disrupts mitochondrial respiration and transmembrane protein transport, resulting in broad anticancer activity via metabolic rewiring and activation of the unfolded protein response.
Graphical Abstract
Introduction
The repurposing of established drugs is evolving as a promising, sustainable, cost- and time-saving approach to improve success rate, speed, and cost effectiveness of anticancer drug development (1). Nelfinavir is a first generation HIV-protease inhibitor approved for HIV treatment that by design binds to the viral protease in a competitive manner, based on high enthalpy and entropy (2). To date, nelfinavir has largely been replaced for HIV treatment by next-generation HIV-protease inhibitors (HIV-PI) with increased specificity and efficacy (3). Meanwhile, nelfinavir has shown strong anticancer activity in multiple preclinical models and clinical trials, both as monotherapy (4, 5) and in combination with established antineoplastic drugs and treatment modalities (6, 7).
In particular, nelfinavir sensitizes cancer cells to proteasome inhibitor (PI) treatment, a backbone therapy for multiple myeloma (8, 9). The combination of nelfinavir with the PI bortezomib (BTZ) and carfilzomib (CFZ) overcomes PI resistance in preclinical models of multiple myeloma (10, 11) and has significant activity against solid tumors and hematological malignancies (8, 12, 13). In patients with bortezomib-refractory multiple myeloma, the combination of nelfinavir yielded an overall response rate (ORR, partial response or better) >65% in phase II clinical trial (14), scoring among the highest ORR observed in PI–refractory multiple myeloma in phase II/III trials.
A plethora of individual molecular effects of nelfinavir has been described previously to date: Induction of the unfolded protein response (UPR) through IRE1/XBP1, PERK/eIF2a, and ATF6 signaling (11, 15) inhibition of proteasomal protein degradation (11, 16, 17), inhibition of proteolysis and nuclear translocation of ATF6 and SREBP-1 (18, 19), fatty acid (FA) and cholesterol biosynthesis induction (20), STAT3 and PI3K/Akt signaling inhibition (21,22,23), and transmembrane multidrug transporter protein ABCB1 inhibition (10). It is unclear, however, whether such diverse effects are mediated through direct interaction of nelfinavir with different targets in different cell types, or if they represent downstream responses to a primary effect of nelfinavir on one, so far unknown, target. This uncertainty hampers both, a rational clinical repurposing development of nelfinavir as antineoplastic drug, as well as the design, synthesis and testing of next-generation nelfinavir-like compounds with optimized antineoplastic activity, and improved specificity or pharmacologic properties. Therefore, we aimed to identify direct targets of nelfinavir across different human malignant cell lines and link them with cell biological processes and mechanisms mediating sensitivity or resistance to nelfinavir treatment in cancer.
Materials and Methods
Cell lines and chemicals
Across the study, following cell lines were used: Multiple myeloma cell lines AMO-1 (DSMZ, German Collection of Microorganisms and Cell Cultures GmbH) and its derivatives resistant to proteasome inibitors bortezomib: AMO-BTZ and carfilzomib: AMO-CFZ. Further following cell lines were used: MDA-MB-231 (DSMZ), BT-474 (DSMZ), U-2 OS (ATCC) K562 (ATCC), HeLa (ATCC), HEK293 (ATCC), HEK293T (ATCC), and Caki2 (DSMZ). The cells were authenticated by STR-typing and routinely tested for Mycoplasma contamination (MycoAlert Mycoplasma Detection Kit). For detailed information about cell lines maintenance, see Supplementary Methods. For the complete list of chemicals used across the study, see Supplementary Table S1.
Functionalized photoreactive nelfinavir-mimetics probes and chemical pull-down
To identify proteins that interact with nelfinavir in intact cells, a set of functionalized photoreactive nelfinavir-mimetics probes was synthesized: A functional ether modification of nelfinavir with a linker molecule containing the diazirine as photolabel and the alkyne as click handle (SC-441) and a nonfunctional modification of nelfinavir in the putative active site with diazirine and alkyne (SC-451). To validate functionality of probes, a modification of SC-441 without the diazirine (dummy probe, SC-454) was synthesized. Chemical synthesis of the probes is described previously in Supplementary Methods.
The pull-down experiments were carried out in triplicate in multiple myeloma cells (AMO-1 and AMO-CFZ) and breast cancer cells (MDA-MB-231, BT-474). The general experimental layout is shown in Supplementary Table S2. The whole procedure of chemical pull-down is in detail presented in Supplementary Methods.
CRISPR/Cas9 pooled library screen
For the genome-wide CRISPR/Cas9 screen, human Brunello CRISPR/Cas9 knockout pooled library was used. Detailed description of the whole procedure is presented in Supplementary Methods, for the complete list of primers used to amplify the library, see Supplementary Table S3.
Isotope tracing
13C tracer experiments upon nelfinavir treatment were performed as described before (24) and are in detail presented in Supplementary Methods.
Lipidomics
Lipidomic experiments with nelfinavir were performed in HEK293 and AMO-1 cells, global analysis of lipids was performed in AMO-1, MDA-MB-231, and Caki2 cells. For a detailed description, see Supplementary Methods.
Nelfinavir intracellular quantification
LC-MS–based quantification of nelfinavir was performed in AMO-1 cells. For a detailed description, see Supplementary Methods.
Fluorescence recovery after photobleaching experiments
Fluorescence recovery after photobleaching (FRAP) experiments upon nelfinavir treatment were performed with CFP-tagged Rab1A in HeLa cells and with C1-BODIPY-C12 in HEK293 cells. For a detailed description, see Supplementary Methods.
Laurdan dye staining to assess membrane fluidity
Live HEK293 and U-2 OS cells were stained with Laurdan dye at 15 μmol/L in serum-free media for 45 minutes at 37°C. For a detailed description, see Supplementary Methods.
RUSH system and protein secretion assessment
Retention Using Selective Hooks (RUSH) system was used in U-2 OS cells for the visualization of protein trafficking upon nelfinavir or control treatment (25). For a detailed description, see Supplementary Methods.
Generation of cells with various reporter systems
U-2 OS cells were equipped with full length HKII and truncated HKII constructs, AMO-1 and MDA-MB-231 cells were equipped with ratiometric ATP/ADP constructs and AMO-1 and MDA-MB-231 cells were equipped with shRNA constructs that allowed for a decreased ADIPOR2 expression. For a detailed description of generation of respective cell lines, see Supplementary Methods.
Single gene knockout using CRISPR/Cas9
The specific knockout of a gene was performed using two-vector CRISPR/Cas9 system in AMO-1 cells and is described previously in Supplementary Methods. For a detailed information about the sequences of sgRNA used, see Supplementary Table S3.
Mitochondria metabolic activity analysis
For real-time analysis of extracellular acidification rates (ECAR) and oxygen consumption rates (OCR), AMO-1 cells were analyzed using an XF-96e Extracellular Flux Analyzer (Seahorse Bioscience/Agilent Santa Clara) as described previously in detail elsewhere (26, 27).
Flow cytometry
Flow cytometry was used to assess the rate of apoptosis, ABCB1 efflux, glucose flux, and MHC class I expression in AMO-1 cell lines. For a detailed description, see Supplementary Methods.
Quantification and statistical analysis
Statistical evaluation was performed in GraphPad Prism v.5 (GraphPad Software). For group comparison, two-way ANOVA was used with Bonferroni post-test, for comparison of two groups unpaired t test was used, values P < 0.05 were considered as statistically significant. Specific statistical analysis for CRISPR/Cas9 screening, chemical pull-down analysis, lipidomics is presented in respesctive sections in Supplementary Methods.
Flow cytometry data were evaluated using FlowJo v10 Software (FlowJo Company) and are presented as a mean and ±SD of median fluorescence intensity of at least 3 independent experiments.
Results
Conserved binding partners of nelfinavir across different cell types are enriched in mitochondria and the ER membranes
To identify proteins that interact with the active site of nelfinavir in intact living cells, we synthesized photoreactive nelfinavir-mimetics: The nelfinavir active probe (SC-441), the nelfinavir inactive probe with a substitution in the putative active site (SC-451), and the dummy probe (SC-454; Fig. 1A; Supplementary Fig. S1). For the synthesis of SC-451, the available data were analyzed (28) and it was concluded that a modification of C(18) of nelfinavir with a hydrophobic residue could serve to inactivate the molecule. We used a short aliphatic moiety to change the molecular structure as little as possible. A crystal structure of nelfinavir with HIV protease (29) shows the hydroxyl group in the center of the binding pocket, thus a modification of the central hydroxyl may cause enough steric clash to disfavor binding of the inhibitor to the active site. The loss of activity of SC-451 was assessed as the loss of PI-sensitizing activity, in contrast with retained activity of SC-441, so that the probes differentiate between activity-dependent (specific), and activity-independent (nonspecific) interaction partners of nelfinavir. Moreover, a “dummy probe” that carries a terminal alkyne tail similar to the photo-reactive probe just without the photo-active diazirine moiety has been synthesized to confirm that the photo-active moiety has no effect on the SC-441 probe activity (Supplementary Fig. S2A). Three independent sets of experiments were performed (Supplementary Methods and Supplementary Table S1) to identify nelfinavir target proteins, and to verify and validate the hits in multiple myeloma cells (AMO-1), carfilzomib-resistant multiple myeloma cells (AMO-CFZ), as well as the breast cancer cell lines MDA-MB-231 and BT474, which are comparably sensitive to nelfinavir in a low micromolar range (NFV IC50 values: AMO-1 = 10.5 μmol/L, AMO-CFZ = 11.7 μmol/L, MDA-MB-231 = 14.4 μmol/L, BT-474 = 14.9 μmol/L; Supplementary Fig. S2B).
Figure 1.

Nelfinavir binds to targets in organellar membranes and affects ATP transport from mitochondria. A, A set of photoreactive nelfinavir-mimetic probes to identify nelfinavir targets in an activity-dependent fashion. For a detailed scheme illustrating synthesis of the probes and their cytotoxic activity in combination with carfilzomib, see also Supplementary Figs. S1 and S2. The experimental outline to identify candidate proteins binding to the active site of nelfinavir and the identified protein candidates are presented in Supplementary Tables S1 and S4. B, Schematic visualization of the localization of the conserved nelfinavir-binding partners across four different cell lines. C, Assessment of cytosolic ATP/ADP ratio in AMO-1 multiple myeloma cells upon treatment for 6 hours with increasing doses of nelfinavir, oligomycin, and FCCP as a positive control. For the analysis in MDA-MB-231 cells, see Supplementary Fig. S3. D, Assessment of mitochondrial ATP/ADP ratio in AMO-1 multiple myeloma cells upon treatment for 6 hours with increasing doses of nelfinavir, oligomycin, and FCCP. E, Assessment of the JC1 ratio in AMO-1 multiple myeloma cells upon treatment for 6 hours with increasing doses of nelfinavir, oligomycin, and FCCP. F, OCR as a function of mitochondria respiration assessed in AMO-1 cells after incubation with 20 μmol/L nelfinavir for 3 and 6 hours. In all experiments, data represent a mean ±SD from three replicates and statistically significant differences are marked. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Our approach identified 83 binding partners in four tested cell lines, the complete list of identified targets is provided in Supplementary Table S4. The functional impact of nelfinavir, based on all identified proteins, was further investigated by the search for Gene Ontology term enrichment using Enrichr software (30). The most significant GO terms for categories Biological Process, Cellular Component and Molecular Function are included in Supplementary Table S5. On the basis of these, the identified proteins are significantly enriched in lipid droplets, mitochondria, and ER organelles and are associated with processes related to mitochondria and the ER function, protein transport or Ras/Rab-related vesicular transport.
The eight overlapping activity-specific targets of nelfinavir identified in at least three out of four cell lines (Supplementary Table S6) are intramembrane-resident proteins with lipid- and cholesterol-interacting domains (31, 32), embedded predominantly in mitochondria, ER or cellular vesicles, consistent with the identified GO terms. The mitochondrial membrane–embedded proteins are proteins involved in the formation of the multiprotein mitochondria permeability transition pore [mPTP; such as voltage dependent anion channel proteins, VDACs, and adenine nucleotide translocator (ANT) proteins, known as ADP/ATP translocase proteins]. The ER membrane-resident proteins are involved in protein folding (calnexin, CALX), quality control, and export of newly synthesized proteins from the ER to Golgi (B-cell–associated protein, BAP31) or co-translational targeting of secretory and membrane proteins to the ER membrane (Signal recognition particle receptor subunit beta, SRPRB; Fig. 1B). Together, these results suggest that the interacting partners of nelfinavir are partially conserved across different cell types. These conserved binding partners are intra-membrane proteins, suggesting further that irrespectively of the cell type, nelfinavir localizes predominantly to membranous systems of the cells.
Shutdown of mitochondrial respiration and ATP transmembrane transport by nelfinavir
HIV-PIs have been suggested to suppress apoptosis by preserving mitochondrial function via their ability to prevent formation or opening of the mPTP (33, 34). Our data show that nelfinavir directly interacts with several key proteins involved in mPTP formation, such as VDACs and ANT (Fig. 1B). The mPTP has been proposed to form F-ATP synthase dimers in the lipid region that generate ATP during oxidative phosphorylation (35, 36), whereas ANT proteins transport ATP synthesized from oxidative phosphorylation into the cytoplasm (37). To directly assess whether nelfinavir affects ATP generation or transport along the mitochondrial membrane to the cytosol, we determined ATP/ADP ratio using ratiometric ATP/ADP probes located in the cytosol and mitochondria of the cells. Nelfinavir dose-dependently decreased cytosolic ATP/ADP in two independent cell lines (Fig. 1C; Supplementary Fig. S3), whereas it increased mitochondrial ATP/ADP ratio (Fig. 1D), suggesting that it interferes with mitochondrial ATP transport. At the same time, nelfinavir changed mitochondria potential, which was observed by an initial accumulation, followed by consecutive strong dose-dependent decrease of the fluorescence of JC1, a cationic dye that accumulates in energized mitochondria (Fig. 1E). Next, to analyze the functional effect of nelfinavir on mitochondrial respiration (a proxy of oxidative phosphorylation), we measured the OCR. Nelfinavir inhibited mitochondrial respiration in a time-dependent manner (Fig. 1F), confirming the inability of mitochondria to perform oxidative phosphorylation in the presence of nelfinavir. In conclusion, nelfinavir disturbs ATP transport from mitochondria to the cytosol by affecting the function of mitochondria membrane-resident proteins, and thus impairs mitochondria metabolism.
Nelfinavir affects glycolysis by interfering with the VDAC-bound HKII-mediated glucose phosphorylation
HIV-PIs impair glycolysis and cause insulin resistance (38, 39). ATP is critical for the initial step of glycolysis in which glucose is phosphorylated by VDAC-bound hexokinase II (HK II). We hypothesized that nelfinavir may reduce the supply of ATP for VDAC-bound HKII by impairing the ATP translocation from mitochondria. By measuring uptake of fluorescent glucose analogue (2-NDBG) in AMO-1 cells we confirm that nelfinavir inhibits glucose flux in a dose- and time-dependent fashion (Fig. 2A). This finding is accompanied by decreased glycolysis, as determined by a significantly decreased ECAR as a proxy for lactic acid production (Fig. 2B). To further analyze this hypothesis, we followed the metabolism of 13C-glucose in AMO-1 multiple myeloma cells over time (8 and 24 hours) upon nelfinavir treatment. In addition, changes in levels of extracellular glucose metabolites were analyzed. Nelfinavir increased 13C-glucose levels in culture media, consistent with a decreased uptake of extracellular 13C-glucose and with lower glycolytic activity (Supplementary Fig. S4A). At the same time, nelfinavir-treated cells consistently showed a significantly reduced incorporation of 13C into downstream glucose metabolites: Glucose-6 phosphate, pyruvate, and lactate (Fig. 2C and D) and lower lactate production (Supplementary Fig. S4B). This block in downstream glucose metabolites persisted over 24 hours, and the respective metabolites were more significantly reduced over time in nelfinavir-treated cells. This work demonstrates that nelfinavir impairs intracellular glucose metabolism at the level of HKII processing into both the oxidative and non-oxidative pathways, consistent with reduced glycolysis and oxidative phosphorylation (Fig. 2E; Supplementary Fig. S5). Consequently, metabolites of the tricarboxylic acid cycle were also consistently decreased after nelfinavir treatment (Supplementary Fig. S5, relative data).
Figure 2.

Nelfinavir affects glycolysis by interfering with glucose phosphorylation mediated by HKII bound to VDACs. A, Glucose flux in AMO-1 cells was estimated by measuring the uptake of 2-NDBG upon nelfinavir treatment; 10 mmol/L 2-deoxyglucose (2-DG) served as a positive control of glucose flux inhibition. B, ECAR was assessed in AMO-1 cells after incubation with 20 μmol/L nelfinavir for 3 and 6 hours. C, Relative levels of intracellular glucose and glucose-6-phosphate (G6P) after the treatment with nelfinavir. For relative levels of glucose in the cell culture media 8 hours after the treatment, see also Supplementary Fig. S4. D, Relative levels of intracellular levels of lactate and pyruvate after the treatment with nelfinavir. The legend for C and D represents the fractional abundance of 13C isomers in the metabolites. E, A scheme illustrating change in level of metabolites from 13C glucose after 24 hours incubation with 20 μmol/L nelfinavir or DMSO only in AMO-1 cells. The color scale indicates log2-fold change between the metabolites. For a detailed heat map illustrating the changes after 8 and 24 hours with 10 and 20 μmol/L nelfinavir, see also Supplementary Fig. S5. F, Live imaging of single-cell–derived colonies from the U-2 OS cells equipped with FL-HKII (left) and Tr-HKII (right) constructs. ER is visualized with mCherry-ER-3 vector and nuclei by Hoechst staining. G, Dose response curves of U-2 OS cells equipped with FL-HKII and Tr-HKII exposed to increasing concentrations of nelfinavir or 2-deoxyglucose. Data represent a mean ±SD from three replicates and statistically significant differences are marked. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To independently confirm that nelfinavir affects glycolysis via ATP depletion and subsequent impairment of ATP supply to VDAC-bound HKII, rather than VDAC-free HKII, we used U-2 OS cells containing either a full-length HKII (FL-HKII) or a truncated HKII (Tr-HKII), lacking the VDAC-binding sites (40). The FL-HKII localized strictly to rod-type structures in the cells, consistent with the interpretation that it is bound to mitochondria at the VDAC sites, whereas Tr-HKII is dispersed over the entire cytoplasm (Fig. 2F). Nelfinavir did not outcompete the FL-HKII from the mitochondria over the time, suggesting again that it does not impair its VDAC binding, but that it may affect its ATP supply. The FL-HKII–equipped cells were more sensitive to the cytotoxic effect of nelfinavir than the Tr-HKII cells, but equally sensitive to cytotoxicity induced by 2-deoxyglucose, a glucose analog that blocks HKII irrespective of its subcellular location (Fig. 2G). Altogether, these results implicate that nelfinavir causes reduced ATP availability for VDAC-bound HKII that impairs glycolysis and oxidative phosphorylation at the glucose phosphorylation level.
Nelfinavir inhibits ER to Golgi protein trafficking
As nelfinavir targets ER membrane-resident proteins (BAP31, CALX, SRPRB) and proteins required for vesicular protein transport (Rab proteins) between the ER and Golgi compartment, we hypothesized that apart from its known effects on protein homeostasis and induction of the UPR (8, 15), it also affects ER membrane dynamics and protein trafficking from the ER. To test this hypothesis, we used FRAP microscopy of CFP-tagged Rab1A, a GTPase required for vesicular protein transport from the ER to the Golgi compartment. Pretreatment of cells with increasing doses of nelfinavir for 3 hours before FRAP microscopy delayed the recovery of the bleached area in the Golgi starting at the 10 μmol/L dose (Fig. 3A). The alteration of Rab1A dynamics at the Golgi opens the possibility that nelfinavir alters secretory trafficking in the early secretory pathway.
Figure 3.

Nelfinavir impairs intracellular trafficking, plasma membrane deposition, and secretion of ER-resident proteins. A, FRAP of CFP-Rab1A protein in the Golgi of control, untreated cells, or cells pretreated for 3 hours with increasing doses of nelfinavir. B, Representative picture of TNFα–eGFP retained in the ER of the U-2 OS cells after 3 hours treatment with 10 μmol/L brefeldin A or 20 μmol/L nelfinavir. For the movies showing trafficking of TNFα–eGFP after the treatment, see Supplementary Movie S1A–S1C. For the quantification of TNFα–EGFP signal retained in the cell after exposure to nelfinavir and other drugs, see Supplementary Fig. S6. C, IgA secretion in AMO-1 multiple myeloma after treatment for 3 hours with 10 μmol/L brefeldin A or 10 and 20 μmol/L nelfinavir. D, Surface expression of MHC class I on AMO-1 cells after treatment for 3 hours with brefeldin A or 10 and 20 μmol/L nelfinavir. Data for C and D represent means ±SD from three independent replicates, statistically significant differences are marked. ***, P < 0.001.
To test this hypothesis, we generated U-2 OS cells equipped with RUSH system (Str-KDEL_TNF-SBP-EGFP; ref. 25). In this system, the TNFα–EGFP protein, which is initially bound to streptavidin (Str) via a streptavidin-binding peptide (SBP) and thus retained in the ER via the KDEL motif, is released upon biotin treatment, trafficks from the ER to the Golgi, and ultimately to the extracellular space. One hour after biotin treatment, we observed accumulation of the EGFP-tagged TNFα in the ER of the cells pretreated with brefeldin A (positive control) and nelfinavir (Fig. 3B, Movie S1A–C). Importantly, nelfinavir did not completely prevent trafficking of TNFα–EGFP from the ER, in contrast with brefeldin A, but rather delayed it. Flow cytometry–based quantification of the relative amount of EGFP-tagged TNFα that was retained in the cell upon treatment with different drugs (at t = 0 and t = 60 minutes after biotin treatment) showed that only nelfinavir and SC-441 caused retention of TNFα–EGFP in the cell, in contrast with SC-451, other HIV-PIs, the PIs bortezomib and carfilzomib or other known UPR-inducing drugs (tunicamycin and thapsigargin; Supplementary Fig. S6). We subsequently evaluated, whether nelfinavir affects protein transport of secretory and membrane proteins along the secretory pathway, such as MHC class I surface expression or immunoglobulin A secretion in multiple myeloma cells. Nelfinavir significantly decreased both IgA secretion and MHC class I surface expression 3 hours after treatment (Fig. 3C and D). Together, these results show that via interaction with several ER and vesicle membrane-resident proteins, nelfinavir functionally impinges on ER to Golgi vesicular protein trafficking and protein secretion.
Genes involved in vesicular transport and lipid metabolism modulate sensitivity/resistance toward nelfinavir
Genetic knockout of single direct interaction proteins of nelfinavir (BAP31 or MTDH) in AMO-1 cells did not affect nelfinavir cytotoxicity (Supplementary Fig. S7A–S7C), suggesting that either direct interaction of nelfinavir with several of the identified nelfinavir-binding proteins may be critical for its cytotoxicity, or that integration of active nelfinavir into cellular membranes results in its interference with a plethora of intramembrane proteins. To further identify the key functional pathways involved in nelfinavir cytotoxicity in cancer cells, we performed genome-wide CRISPR/Cas9 screening using the Brunello library in the K562 cell line. Both, negative and positive-selection screen with 5 and 10 μmol/L nelfinavir were used to identify genes whose loss sensitizes the cells to a low concentration of nelfinavir or that allow cell survival in the presence of higher concentrations of nelfinavir decreasing the viability to 50%. Overall, we identified 7 candidate sensitivity genes (ACACA, ATG9A, CLUH, MYLIP, VAPA, CSTB, and GOSR2) at an FDR < 0.01, with highest negative fold change, relative to control (log FC < −0.8) and 1 candidate resistance gene at FDR < 0.01 and log FC > 2 (Fig. 4A; Supplementary Table S7A and S7B). The candidate sensitivity genes are particularly involved in FA and cholesterol metabolism, vesicular formation and trafficking and mitochondria biogenesis, whereas the only identified resistance gene, ADIPOR2, encodes a member of the PAQR (Progestin and AdipoQ Receptor) protein family. ADIPOR2 is an integral component of cellular membranes that maintains membrane fluidity and cell viability in the presence of exogenously added saturated FAs, it acts primarily by promoting FAs desaturation and their incorporation into phospholipids, which helps to restore membrane fluidity (41).
Figure 4.

CRISPR/Cas9 library screening suggests involvement of ADIPOR2 and fatty acids in the resistance to nelfinavir and consequently modulation of fatty acids changes nelfinavir-induced effects on cell viability and energetics. A, Genome-wide CRISPR/Cas9 library screening in K562 cells with 5 and 10 μmol/L nelfinavir identified candidate genes involved in nelfinavir sensitivity (red) or in nelfinavir resistance (blue) at the cutoff value of −log10 FDR = 2. For a detailed list of the sensitivity and resistance candidate genes, their log-fold change over the DMSO-treated cells, and FDR value, see Supplementary Table S7A and S7B. B, Apoptosis rate evaluated 24 hours after treatment with 20 μmol/L nelfinavir in the AMO-1 and MDA-MB-231 cells with decreased ADIPOR2 expression. For the efficacy of ADIPOR2 silencing in the two cell lines, see Supplementary Fig. S8. C, Dose response curves of cell lines exposed to increasing doses of nelfinavir alone or in combination with fatty acid (FA) supplement or cholesterol–lipid concentrate. For the cytotoxicity of increasing doses of FA supplement alone, see Supplementary Fig. S9A. D, Cytotoxicity of nelfinavir (N), ezetimibe (E), and their combination (N+E) in AMO-1 and MDA-MB-231 cell lines. For the cytotoxicity of nelfinavir (N), ezetimibe (E), and their combination (N+E) in the presence of 0.1% FA supplement or cholesterol–lipid concentrate, see Supplementary Fig. S9B and S9C. E, OCR (left) and ECAR (right) in AMO-1 cells 6 hours after treatment with 20 μmol/L nelfinavir, 0.1% FA supplement, or their combination. F, Assessment of the cytoplasmic (left) and mitochondrial (right) ATP/ADP ratio in AMO-1 cells 6 hours after treatment with increasing concentrations of nelfinavir (10, 20, and 40 μmol/L) alone or in combination with 0.1% FA supplement. In all experiments, viability was assessed 48 hours after continuous treatment. For the drug combinations, coefficient of drug interaction was calculated. Data of viability assays, flow cytometry, and Seahorse analysis represent a mean ±SD from three replicates and statistically significant differences are marked. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To validate the screening data with an independent approach, we silenced the expression of ADIPOR2 with shRNA in AMO-1 and MDA-MB-231 cells (Supplementary Fig. S8A and S8B). Decreased ADIPOR2 level significantly protected the cells from nelfinavir-induced apoptosis (Fig. 4B). Together, these results suggest that functional pathways involved in nelfinavir's cytotoxicity are conserved across different cell types and center around FA metabolism and membrane fluidity.
Fatty acids modulate sensitivity toward nelfinavir and prevent nelfinavir-induced mitochondria shut down
To directly address the role of FAs in nelfinavir-induced cytotoxicity, we exposed nelfinavir-treated AMO-1 and MDA-MB-231 cells to increasing doses of FA supplement, an aqueous mixture of cholesterol-free saturated and unsaturated fatty acids. Initially, we tested the cytotoxicity of FA supplement alone and set the doses of 0.1% and 0.2% to have minimal effect on cell viability (Supplementary Fig. S9A). FA supplement rescued the cells from the cytotoxic activity of nelfinavir in a dose-dependent fashion (Fig. 4C). Moreover, co-treatment of the cells with a cholesterol–lipid concentrate (in a dilution of the commercial product at 1:250, in agreement with the manufacturer's recommendation for cell culture supplementation) prevented toxicity even more effectively (Fig. 4C), suggesting that increasing the supply of membrane components (FA and cholesterol) protects against nelfinavir. In contrast, depletion of FA/cholesterol by ezetimibe, an FDA approved drug reducing lipid and cholesterol uptake, resulted in a highly synergistic cytotoxic effect in combination with nelfinavir against both cell lines (Fig. 4D). This synergistic cytotoxicity could likewise be abolished by the presence of 0.1% FA supplement or the cholesterol–lipid concentrate in both cell lines (Supplementary Fig. S9B and S9C).
Next, we addressed whether the prevention of nelfinavir-induced cell death by FA would likewise restore mitochondrial respiration and glycolysis. Treatment with 0.1% FA supplement abolished the nelfinavir-induced block in mitochondrial respiration (OCR) and glycolysis (ECAR) in AMO-1 cells (Fig. 4E). Likewise, 0.1% FA supplement restored the nelfinavir-induced cytosolic ATP/ADP decrease and mitochondrial ATP/ADP increase (Fig. 4F). Therefore, FA and cholesterol significantly antagonize nelfinavir-induced cytotoxicity and reverse the nelfinavir-induced metabolic shut-down caused by impaired ATP transport through the mitochondrial membranes.
The incorporation of nelfinavir into cellular lipid membranes impairs membrane fluidity
The uniform pattern of intramembrane protein interaction partners of nelfinavir, its highly lipophilic nature (42) and the modulation of the downstream effects of nelfinavir by FA supplementation together suggest that nelfinavir integrates into biomembranes of eukaryotic cells, where it may affect the composition and physical properties of such membranes. To directly test this hypothesis we performed FRAP experiments using C1-BODIPY-C12 recovery. Nelfinavir significantly slows down the recovery of the C1-BODIPY-C12 signal in the bleached area, indicating a significant rigidification of the biomembranes (Fig. 5A and B). This observation was independently confirmed by staining of the cells with laurdan dye, a reporter of membrane penetration by water that correlates with fluidity. Variations in membrane water content cause a shift in the laurdan emission spectrum, which can be quantified by calculating the generalized polarization (GP) index. Nelfinavir-treated HEK293 and U-2 OS cells had significantly more rigid membranes, presented as an increased GP index, including distinct internal structures with significant rigidity (Fig. 5C and D; Supplementary Fig. S10A and S10B). This effect is nelfinavir-specific and is not observed for other drugs, such as the PIs bortezomib and carfilzomib (Supplementary Fig. S10C). To further dissect if nelfinavir co-treatment with FA can prevent membrane rigidification and to address the effect of saturated versus unsaturated FA, we exposed the cells to nelfinavir in combination with saturated (16:0 palmitic acid) and unsaturated (16:1 palmitoleic acid) FA for 6 hours. Only unsaturated FA were able to prevent nelfinavir-induced changes in GP index, whereas loading the cells with saturated FA had the opposite effect and significantly potentiated the effect of nelfinavir on membrane rigidity (Fig. 5E). Interestingly, co-treatment with FA supplement reduced the amount of intracellular nelfinavir, compared with cells treated with nelfinavir alone (Fig. 5F), suggesting that FA may compete with nelfinavir for membrane uptake and thus prevent membrane rigidification. Overall, the data suggest that nelfinavir integrates into lipid-rich membranes of eukaryotic cells and increases membrane rigidity.
Figure 5.

Nelfinavir increases membrane rigidity, which can be reverted by unsaturated FA. A, FRAP results in HEK293 cells challenged with 40 μmol/L nelfinavir for 6 hours. Data of a representative experiment with n = 15–18. B, Quantification of a FRAP experiment in HEK293 treated with increasing concentrations of nelfinavir for 6 hours. Average Thalf values, the time by which half of the maximum fluorescence recovery is reached. Data of a representative experiment with n = 5–15. C, Pseudocolor images showing the laurdan dye GP index at each pixel position in HEK293 cells challenged with 40 μmol/L nelfinavir for 6 hours. Yellow arrow, a spot with very strong rigidity. D, Average GP index from several images as depicted in C (n = 15–19). E, Average GP index from several images of the laurdan dye staining in U-2 OS cells challenged with 40 μmol/L nelfinavir alone or in combination with saturated (Pal) or unsaturated (PalO) FA for 6 hours. For the pseudocolor images showing the laurdan dye staining in U-2 OS cells, see Supplementary Fig. S10A. For the effect of bortezomib and carfilzomib on membrane fluidity, see Supplementary Fig. S10C. F, Intracellular nelfinavir assessment upon treatment of AMO-1 cells for 6 hours with 5 μmol/L nelfinavir alone or in combination with 0.2% FA supplement. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Nelfinavir alters composition of lipids predominantly in lipid membranes
The effect of nelfinavir on lipid-rich membranes led us to hypothesize that it affects the composition of cellular lipids. The quantitative and qualitative analysis of the lipid content 6 hours after nelfinavir treatment in HEK293 cells shows that nelfinavir causes a significant increase in saturated FA (SFA) in membrane phospholipids [both phosphatidylcholines (PC) and phosphatidylethanolamines (PE)], whereas monounsaturated FA (MUFA) were decreased (Fig. 6A and B). SFA increases membrane rigidity, whereas MUFA promotes membrane fluidity (41), consistent with the data indicating a significant loss of membrane fluidity upon nelfinavir treatment. Nelfinavir likewise causes significant changes in the composition of lipid droplets, where we observed a strong decrease in the relative fraction of cholesterol esters, whereas triacylglycerols were increased (Fig. 6C and D). An independent global lipidomics analysis performed in AMO-1 cells confirmed the previous data and shows in more detail that nelfinavir predominantly affects PC and phosphatidylinositols (Fig. 6E and F), two lipid species present predominantly in the membranes. Specifically, nelfinavir increases the unsaturated forms of PC and phosphatidylinositols with low numbers of double bonds, whereas PE with high numbers of double bonds are decreased (Fig. 6G and H). Interestingly, relative resistance of the Caki2 cell line to nelfinavir (NFV IC50 = 20.7 μmol/L) compared with AMO-1 or MDA-MB-231 cells (NFV IC50 = 11.7 and 14.4 μmol/L, respectively), is associated with enrichment of unsaturated PC, PE, and phosphatidylinositols in this cell line (Supplementary Fig. S11A–S11E).
Figure 6.

Nelfinavir impairs the homeostasis of lipid composition in lipid-rich membranes and lipid droplets. A, Relative contents of FAs in membrane phosphatidylcholines (PC) upon treatment with 40 μmol/L nelfinavir for 6 hours. SFA, saturated fatty acids; MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids. B, Relative content of FA in membrane PEs upon treatment with 40 μmol/L nelfinavir for 6 hours. C and D, Cholesterol esters (CE; C) and triacylglycerols (TAG; D) in lipid droplets upon treatment with 40 μmol/L nelfinavir for 6 hours expressed as a ratio between cholesterol ester or triacylglycerol to membrane PC. E, Principal component analysis plot separating control and 20 μmol/L nelfinavir-treated AMO-1 cells for 6 hours, based on their lipid composition. F, Lipid composition separating untreated and nelfinavir-treated AMO-1 cells, where the number of double bonds is indicated for the most differentiated lipids. Note that the main separators are PC containing 0 or 1 double bonds. G and H, Heat maps for PC/PE and PI species. Data show results of five replicates. Data show the mean of a representative experiment ± SEM and statistically significant differences are marked. *, P < 0.05; ***, P < 0.001. PI, phosphatidylinositol.
Perturbation of membrane lipid homeostasis by nelfinavir or ezetimibe induces the UPR, inhibits efflux by ABCB1, and shows synergistic cytotoxicity with PIs
One of the main cellular responses to the perturbation of lipid and cholesterol homeostasis is the induction of the UPR, mainly through activation of the IRE1/XBP1 and ATF3 signaling (43, 44). Nelfinavir was observed previously to activate the IRE1/XBP1 pathway (8, 11). Moreover, it induced rapid and potent expression of ATF3 and CHOP (Fig. 7A). The induction of the UPR was prevented by the co-treatment with increasing, nontoxic concentrations of FA supplement (0.1% and 0.2%), supporting the interpretation that nelfinavir directly induces the UPR by affecting the lipid composition of biomembranes (Fig. 7A). Interestingly, a similar profile of UPR induction observed for nelfinavir was obtained when cells were treated with 40 μmol/L ezetimibe (Fig. 7B), a dose resulting in comparable cytotoxicity with 20 μmol/L nelfinavir (Supplementary Fig. S12A). We previously demonstrated that nelfinavir is a potent modulator of ABCB1 drug export pump (10). We here show that ezetimibe likewise partly inhibits ABCB1 function, and that ABCB1 inhibition by nelfinavir and ezetimibe can be rescued by FA supplement (Supplementary Fig. S12B).
Figure 7.

Nelfinavir induces lipid-bilayer stress by rigidification of lipid-rich membranes, which triggers UPR induction that overcomes proteasome inhibitor (PI) resistance in multiple myeloma in combination with PIs. A, Induction of the UPR in AMO-1 cells assessed as the increase in the splicing of the XBP1, ATF3, and CHOP expression by 20 μmol/L nelfinavir alone, 0.1% and 0.2% fatty acids (FA) supplement or their combination. B, UPR induction by equally cytotoxic doses of nelfinavir (20 μmol/L) and ezetimibe (40 μmol/L) assessed as the increase in the splicing of the XBP1, ATF3, and CHOP expression. For the dose–response curves of cells to nelfinavir and ezetimibe see Supplementary Fig. S12A. C, Dose–response curves of PI-sensitive AMO-1 cells and bortezomib (BTZ) and carfilzomib (CFZ)-resistant cells alone and in combination with 10 μmol/L nelfinavir and 15 μmol/L ezetimibe. For the IC50 values and IC50 fold change between the single drugs and the combination in respective cell lines, see also Supplementary Table S8. The data represent mean ±SD of three independent repeats, statistically significant differences are marked. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Nelfinavir has synergistic activity with PI against myeloma in vitro and in PI-refractory patients with multiple myeloma (11, 14). Myeloma cells adapted to continuous PI treatment in vitro are characterized by altered membrane lipid composition (45), suggesting that the specific membrane properties may be important for cell survival under continuous PI pressure. To address whether the observed synergistic cytotoxicity of nelfinavir with PI against multiple myeloma, and in particular against PI-resistant multiple myeloma is directly linked to perturbation of lipid homeostasis, we compared the cytotoxicity of nelfinavir with the effect of ezetimibe, both combined with PI. Ezetimibe overcame PI resistance in combination with bortezomib and carfilzomib in AMO-BTZ and AMO-CFZ and showed superior synergistic toxicity in PI-adapted cells in comparison with PI-sensitive cells, closely resembling the synergistic cytotoxic activity of nelfinavir (Fig. 7C; Supplementary Table S8). However, the magnitude of the synergistic cytotoxic activity of ezetimibe was lower compared with nelfinavir. Nevertheless, the combination between nelfinavir and ezetimibe showed a strong synergistic cytotoxic effect in PI-resistant cells (Supplementary Fig. S12C), suggesting that both drugs may differentially affect cellular lipid homeostasis, triggering the same effector cascade for cytotoxicity. The manipulation of lipid homeostasis in conjunction with proteasome inhibition is a promising way to overcome PI resistance of multiple myeloma.
Discussion
We here characterize the molecular target and mechanism of action for the antineoplastic activity of nelfinavir. Nelfinavir binds to proteins embedded in lipid-rich cellular membranes, which subsequently alters membrane composition and reduces membrane fluidity of the cell and cellular organelles. These changes in cellular membranes result in UPR induction, defective subcellular, and transmembrane trafficking and interfere with key components of cellular energy supply, including glucose metabolism, cellular respiration and ABCB1 activity.
Our model is supported by multiple lines of evidence. First, we identified a common set of nelfinavir-interacting proteins embedded in intracellular membranes and conserved across multiple cancer cell types, supporting general, rather than cell-type–specific interactions. Next, we identified a key regulator of lipid membrane composition and fluidity, ADIPOR2 (46,47,48), as a unique genetic driver to mediate nelfinavir-induced cytotoxicity. Subsequently, we directly demonstrated the quantitative changes in membrane lipid composition and the induction of increased membrane rigidity upon nelfinavir treatment. On the basis of this, we hypothesized that nelfinavir integrates into lipid-rich membranes due to its very high lipophilicity (42), thereby affecting membrane fluidity in a structural manner, and competing against intramembrane FA and/or cholesterol. The physico-structural alteration of lipid-rich membranes caused by nelfinavir affects the function of membrane-associated processes.
The accurate composition of lipid membranes allowing high membrane fluidity is crucial for cancer cells (49,50,51). Pharmacological targeting of membrane lipid composition and fluidity is emerging as a novel field for potential therapeutic intervention (52). Nelfinavir can therefore be viewed as the first clinically active anticancer drug that acts through targeting structural properties of cellular membranes.
Nelfinavir impairs the function of several membrane-associated protein machineries important for tumor cell survival and growth, that is, glucose uptake and metabolism, oxidative phosphorylation, ATP production/transport, protein and vesicle transport and ABCB1 activity. Altered metabolic and glycolytic activity is a basic hallmark of cancer (53, 54) that represents an important target for specific pharmacological intervention. Nelfinavir significantly decreases glucose metabolism at the level of HKII activity, which matches the reduced glucose uptake and expression of GLUT receptors in patient-derived cells upon treatment with anti-retroviral agents (38), as well as reduced glucose flux and insulin resistance leading to hyperglycemia in patients on the anti-retroviral therapy (39, 55). Nelfinavir thus may be used for targeted disruption of glucose metabolism in diverse cancer types.
Our study suggests a comprehensive model of molecular targets and downstream effects that allows to integrate numerous observations that have been made in the past regarding the activity of nelfinavir on cancer cells. Nelfinavir has been shown to bind to ANT and VDAC proteins embedded in mitochondria (33). We show that nelfinavir directly binds to ANT2 or VDACs proteins. Disruption of the nuclear envelope integrity leading to a release of nuclear DNA into the cytoplasm (56) is consistent with the binding of nelfinavir to ZMPSTE24 (FACE1; Supplementary Table S2) embedded in the nuclear envelope. Our genome-wide screening data reveal candidate genes involved in nelfinavir resistance and sensitivity, such as EIF2AK4 and PPP1R15B, respectively, that play a role in eIF2a signaling. eIF2a signaling, as part of the integrated stress response, has been shown to be modulated by nelfinavir, and PPP1R15B has been proposed as a direct nelfinavir target (15). Moreover, earlier observations of nelfinavir inhibiting the processing and nuclear translocation of ER membrane–embedded transcription factors SREBP-1, ATF6 or TCF11/Nrf1 (17,18,19) may be well explained by our finding that nelfinavir interferes with the functionality of ER membranes and ER-Golgi trafficking.
Clinical activity of nelfinavir-based therapy with PI has yielded a noteworthy >65% ORR in patients with PI-refractory multiple myeloma (14). The cell biology of multiple myeloma cells adapted to PI is highly complementary to the mechanism of action of nelfinavir identified here. PI-resistant multiple myeloma cells show alterations in membrane lipid composition, cellular metabolism, and metabolic reprogramming toward higher oxidative phosphorylation, which leads to increased redox and protein-folding capacity (24, 27, 45). Nelfinavir, as we show here, increases membrane rigidity and decreases the activity of multiple membrane proteins and membrane-associated processes, disrupts ATP transport, and blocks the activity of the ABCB1 transmembrane drug exporter, whose activity is involved in PI resistance, as we have shown previously (10, 12).
In conclusion, we here identify altered lipid homeostasis and membrane lipid composition as the basis for the anticancer activity of nelfinavir. Consequently, drugs that interfere with cellular lipid uptake showed effects similar to nelfinavir and synergized with nelfinavir in vitro. Elevated blood lipids are a major side effect of nelfinavir treatment in patients with HIV (57). The high serum lipids induced by nelfinavir may therefore even have antagonized the anti–multiple myeloma activity of nelfinavir in the clinical setting over time, which might partly explain the limited duration of the clinical responses observed in the clinical trial (14). The addition of lipid lowering drugs like ezetimibe to the nelfinavir-containing regimen is likely feasible and may allow to further improve the clinical effectiveness of the nelfinavir-based treatment for PI-resistant multiple myeloma.
Authors' Disclosures
H. Ludwig reports grants from Amgen, Takeda, and personal fees from Amgen, Takeda, Sanofi, Janssen, Celgene-BMS, Seattle Genetics outside the submitted work. B. Everts reports grants from NWO during the conduct of the study. C. Driessen reports grants from Swiss National Fonds during the conduct of the study. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
The work was supported by Swiss National Science Foundation (SNF; grant 310030_182492 and IZSEZ0_177130), Wilhelm Sander-Stiftung (2016.104.1), and Promedica Stiftung (1438/M).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
#L. Besse and A. Besse contributed equally as co-authors of this article.
Current address for S.C. Stolze: Protein Mass Spectrometry Group, Max Planck Institute for Plant Breeding Research, Cologne, Germany; and current address for M. Sathianathan: Institute of Molecular Biology, University of Innsbruck, Innsbruck, Austria.
Cancer Res 2021;81:4581–93
Authors' Contributions
L. Besse: Conceptualization, supervision, investigation, visualization, writing–original draft. A. Besse: Validation, visualization, methodology. S.C. Stolze: Formal analysis, investigation. A. Sobh: Formal analysis, supervision. E.A. Zaal: Investigation. A.J. van der Ham: Investigation. M. Ruiz: Investigation. S. Phuyal: Investigation. L. Büchler: Validation. M. Sathianathan: Validation. B.I. Florea: Software, supervision. J. Borén: Investigation. M. Ståhlman: Supervision. J. Huber: Investigation, methodology. A. Bolomsky: Formal analysis, investigation. H. Ludwig: Writing–review and editing. J.T. Hannich: Investigation, writing–review and editing. A. Loguinov: Software. B. Everts: Resources, supervision. C.R. Berkers: Resources, supervision. M. Pilon: Resources, supervision, writing–review and editing. H. Farhan: Conceptualization, resources. C.D. Vulpe: Resources, supervision, writing–review and editing. H.S. Overkleeft: Resources, supervision, methodology. C. Driessen: Conceptualization, funding acquisition, project administration, writing–review and editing.
References
- 1. Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 2019;18:41–58. [DOI] [PubMed] [Google Scholar]
- 2. Ohtaka H, Muzammil S, Schon A, Velazquez-Campoy A, Vega S, Freire E. Thermodynamic rules for the design of high affinity HIV-1 protease inhibitors with adaptability to mutations and high selectivity towards unwanted targets. Int J Biochem Cell Biol 2004;36:1787–99. [DOI] [PubMed] [Google Scholar]
- 3. Zhang KE, Wu E, Patick AK, Kerr B, Zorbas M, Lankford A, et al. Circulating metabolites of the human immunodeficiency virus protease inhibitor nelfinavir in humans: structural identification, levels in plasma, and antiviral activities. Antimicrob Agents Chemother 2001;45:1086–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, et al. Nelfinavir, a lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res 2007;13:5183–94. [DOI] [PubMed] [Google Scholar]
- 5. Shim JS, Rao R, Beebe K, Neckers L, Han I, Nahta R, et al. Selective inhibition of HER2-positive breast cancer cells by the HIV protease inhibitor nelfinavir. J Natl Cancer Inst 2012;104:1576–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koltai T. Nelfinavir and other protease inhibitors in cancer: mechanisms involved in anticancer activity. F1000Res 2015;4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rengan R, Mick R, Pryma DA, Lin LL, Christodouleas J, Plastaras JP, et al. Clinical outcomes of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: a phase 1/2 trial. JAMA Oncol 2019;5:1464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kraus M, Muller-Ide H, Ruckrich T, Bader J, Overkleeft H, Driessen C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk Res 2014;38:383–92. [DOI] [PubMed] [Google Scholar]
- 9. Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A, et al. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012;120:947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Besse A, Stolze SC, Rasche L, Weinhold N, Morgan GJ, Kraus M, et al. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018;32:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kraus M, Bader J, Overkleeft H, Driessen C. Nelfinavir augments proteasome inhibition by bortezomib in myeloma cells and overcomes bortezomib and carfilzomib resistance. Blood Cancer J 2013;3:e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abt D, Besse A, Sedlarikova L, Kraus M, Bader J, Silzle T, et al. Improving the efficacy of proteasome inhibitors in the treatment of renal cell carcinoma by combination with the human immunodeficiency virus (HIV)-protease inhibitors lopinavir or nelfinavir. BJU Int 2018;121:600–9. [DOI] [PubMed] [Google Scholar]
- 13. Kawabata S, Gills JJ, Mercado-Matos JR, Lopiccolo J, Wilson W, III, Hollander MC, et al. Synergistic effects of nelfinavir and bortezomib on proteotoxic death of NSCLC and multiple myeloma cells. Cell Death Dis 2012;3:e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Driessen C, Muller R, Novak U, Cantoni N, Betticher D, Mach N, et al. Promising activity of nelfinavir-bortezomib-dexamethasone (NeVd) in proteasome inhibitor-refractory multiple myeloma. Blood 2018;132:2097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Gassart A, Bujisic B, Zaffalon L, Decosterd LA, Di Micco A, Frera G, et al. An inhibitor of HIV-1 protease modulates constitutive eIF2alpha dephosphorylation to trigger a specific integrated stress response. Proc Natl Acad Sci U S A. 2016;113:E117–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piccinini M, Rinaudo MT, Anselmino A, Buccinna B, Ramondetti C, Dematteis A, et al. The HIV protease inhibitors nelfinavir and saquinavir, but not a variety of HIV reverse transcriptase inhibitors, adversely affect human proteasome function. Antivir Ther 2005;10:215–23. [PubMed] [Google Scholar]
- 17. Fassmannova D, Sedlak F, Sedlacek J, Spicka I, Grantz Saskova K. Nelfinavir inhibits the TCF11/Nrf1-mediated proteasome recovery pathway in multiple myeloma. Cancers 2020;12:1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guan M, Fousek K, Chow WA. Nelfinavir inhibits regulated intramembrane proteolysis of sterol regulatory element binding protein-1 and activating transcription factor 6 in castration-resistant prostate cancer. FEBS J 2012;279:2399–411. [DOI] [PubMed] [Google Scholar]
- 19. Guan M, Fousek K, Jiang C, Guo S, Synold T, Xi B, et al. Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin Cancer Res 2011;17:1796–806. [DOI] [PubMed] [Google Scholar]
- 20. Riddle TM, Kuhel DG, Woollett LA, Fichtenbaum CJ, Hui DY. HIV protease inhibitor induces fatty acid and sterol biosynthesis in liver and adipose tissues due to the accumulation of activated sterol regulatory element-binding proteins in the nucleus. J Biol Chem 2001;276:37514–9. [DOI] [PubMed] [Google Scholar]
- 21. Ikezoe T, Saito T, Bandobashi K, Yang Y, Koeffler HP, Taguchi H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-regulated kinase 1/2. Mol Cancer Ther 2004;3:473–9. [PubMed] [Google Scholar]
- 22. Gupta AK, Cerniglia GJ, Mick R, McKenna WG, Muschel RJ. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res 2005;65:8256–65. [DOI] [PubMed] [Google Scholar]
- 23. Yang Y, Ikezoe T, Takeuchi T, Adachi Y, Ohtsuki Y, Takeuchi S, et al. HIV-1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer Sci 2005;96:425–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zaal EA, Wu W, Jansen G, Zweegman S, Cloos J, Berkers CR. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab 2017;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, et al. Synchronization of secretory protein traffic in populations of cells. Nat Methods 2012;9:493–8. [DOI] [PubMed] [Google Scholar]
- 26. Pelgrom LR, van der Ham AJ, Everts B. Analysis of TLR-induced metabolic changes in dendritic cells using the seahorse XF(e)96 extracellular flux analyzer. Methods Mol Biol 2016;1390:273–85. [DOI] [PubMed] [Google Scholar]
- 27. Soriano GP, Besse L, Li N, Kraus M, Besse A, Meeuwenoord N, et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016;30:2198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gaucher B, Rouquayrol M, Roche D, Greiner J, Aubertin AM, Vierling P. Prodrugs of HIV protease inhibitors-saquinavir, indinavir and nelfinavir-derived from diglycerides or amino acids: synthesis, stability and anti-HIV activity. Org Biomol Chem 2004;2:345–57. [DOI] [PubMed] [Google Scholar]
- 29. Johnston E, Winters MA, Rhee SY, Merigan TC, Schiffer CA, Shafer RW. Association of a novel human immunodeficiency virus type 1 protease substrate cleft mutation, L23I, with protease inhibitor therapy and in vitro drug resistance. Antimicrob Agents Chemother 2004;48:4864–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods 2013;10:259–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niphakis MJ, Lum KM, Cognetta AB, III, Correia BE, Ichu TA, Olucha J, et al. A global map of lipid-binding proteins and their ligandability in cells. Cell 2015;161:1668–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weaver JG, Tarze A, Moffat TC, Lebras M, Deniaud A, Brenner C, et al. Inhibition of adenine nucleotide translocator pore function and protection against apoptosis in vivo by an HIV protease inhibitor. J Clin Invest 2005;115:1828–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Phenix BN, Lum JJ, Nie Z, Sanchez-Dardon J, Badley AD. Antiapoptotic mechanism of HIV protease inhibitors: preventing mitochondrial transmembrane potential loss. Blood 2001;98:1078–85. [DOI] [PubMed] [Google Scholar]
- 35. Carraro M, Giorgio V, Sileikyte J, Sartori G, Forte M, Lippe G, et al. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J Biol Chem 2014;289:15980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol 2013;4:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta 2008;1778:1978–2021. [DOI] [PubMed] [Google Scholar]
- 38. Murata H, Hruz PW, Mueckler M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J Biol Chem 2000;275:20251–4. [DOI] [PubMed] [Google Scholar]
- 39. Koster JC, Remedi MS, Qiu H, Nichols CG, Hruz PW. HIV protease inhibitors acutely impair glucose-stimulated insulin release. Diabetes 2003;52:1695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol 2008;28:1007–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Devkota R, Svensk E, Ruiz M, Stahlman M, Boren J, Pilon M. The adiponectin receptor AdipoR2 and its Caenorhabditis elegans homolog PAQR-2 prevent membrane rigidification by exogenous saturated fatty acids. PLoS Genet 2017;13:e1007004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ford J, Khoo SH, Back DJ. The intracellular pharmacology of antiretroviral protease inhibitors. J Antimicrob Chemother 2004;54:982–90. [DOI] [PubMed] [Google Scholar]
- 43. Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hanelt I, et al. Activation of the unfolded protein response by lipid bilayer stress. Mol Cell 2017;67:673–84. [DOI] [PubMed] [Google Scholar]
- 44. Kim J, Di Vizio D, Kim TK, Kim J, Kim M, Pelton K, et al. The response of the prostate to circulating cholesterol: activating transcription factor 3 (ATF3) as a prominent node in a cholesterol-sensing network. PLoS One 2012;7:e39448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Besse L, Besse A, Mendez-Lopez M, Vasickova K, Sedlackova M, Vanhara P, et al. A metabolic switch in proteasome inhibitor-resistant multiple myeloma ensures higher mitochondrial metabolism, protein folding and sphingomyelin synthesis. Haematologica 2019;104:e415–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ruiz M, Stahlman M, Boren J, Pilon M. AdipoR1 and AdipoR2 maintain membrane fluidity in most human cell types and independently of adiponectin. J Lipid Res 2019;60:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vasiliauskaite-Brooks I, Sounier R, Rochaix P, Bellot G, Fortier M, Hoh F, et al. Structural insights into adiponectin receptors suggest ceramidase activity. Nature 2017;544:120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bodhicharla R, Devkota R, Ruiz M, Pilon M. Membrane fluidity is regulated cell nonautonomously by caenorhabditis elegans PAQR-2 and its mammalian homolog AdipoR2. Genetics 2018;210:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deliconstantinos G. Physiological aspects of membrane lipid fluidity in malignancy. Anticancer Res 1987;7:1011–21. [PubMed] [Google Scholar]
- 50. Baritaki S, Apostolakis S, Kanellou P, Dimanche-Boitrel MT, Spandidos DA, Bonavida B. Reversal of tumor resistance to apoptotic stimuli by alteration of membrane fluidity: therapeutic implications. Adv Cancer Res 2007;98:149–90. [DOI] [PubMed] [Google Scholar]
- 51. Sharom FJ. Complex interplay between the P-glycoprotein multidrug efflux pump and the membrane: its role in modulating protein function. Front Oncol 2014;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stoiber K, Naglo O, Pernpeintner C, Zhang S, Koeberle A, Ulrich M, et al. Targeting de novo lipogenesis as a novel approach in anti-cancer therapy. Br J Cancer 2018;118:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 2011;10:671–84. [DOI] [PubMed] [Google Scholar]
- 54. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012;21:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gomez-Vera J, de Alarcon A, Jimenez-Mejias ME, Acosta D, Prados D, Viciana P. Hyperglycemia associated with protease inhibitors in HIV-1–infected patients. Clin Microbiol Infect 2000;6:391–94. [DOI] [PubMed] [Google Scholar]
- 56. Di Micco A, Frera G, Lugrin J, Jamilloux Y, Hsu ET, Tardivel A, et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc Natl Acad Sci U S A 2016;113:E4671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bardsley-Elliot A, Plosker GL. Nelfinavir: an update on its use in HIV infection. Drugs 2000;59:581–620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.