

Summary
The abnormal aggregation of transactive response DNA-binding protein of 43 kDa (TDP-43) in neurons and glia is the defining pathological hallmark of neurodegenerative diseases amyotrophic lateral sclerosis (ALS) and multiple forms of frontotemporal lobar degeneration (FTLD) 1,2 . It is also common in other diseases, including Alzheimer's and Parkinson's. No disease-modifying therapies exist and early diagnosis is not possible. The structures of pathological TDP-43 aggregates are unknown. We used electron cryo-microscopy to determine the structures of aggregated TDP-43 in the frontal and motor cortices of an individual that succumbed to ALS with FTLD, as well as from the frontal cortex of a second individual with the same diagnosis. An identical amyloid-like filament structure comprising a single protofilament was found in both brain regions and individuals. The ordered filament core spans residues 282 to 360 in the TDP-43 low-complexity domain and adopts a novel double-spiral-shaped fold, which shows no similarity to those of TDP-43 filaments formed in vitro 3,4 . An abundance of glycine and neutral polar residues facilitates numerous turns and restricts β-strand length, resulting in an absence of β-sheet stacking associated with cross-β amyloid structure. An uneven distribution of residues gives rise to structurally and chemically distinct surfaces, which face external densities and suggest possible ligand binding sites. This work enhances our understanding of the molecular pathogenesis of ALS and FTLD and informs the development of diagnostic and therapeutic agents targeting aggregated TDP-43.
ALS (Lou Gehrig's disease) is the most common adult-onset motor neuron disease and frontotemporal dementia (FTD), the clinical manifestation of FTLD, is the second most common form of presenile dementia after Alzheimer's disease. Neuropathological staging suggests that, in ALS, abnormal neuronal and oligodendroglial cytoplasmic inclusions of aggregated TDP-43 (NCIs and GCIs) first appear in the motor cortex, brainstem and spinal cord, before progressing to connected regions of the frontal, parietal and temporal lobes 5 , possibly by prion-like propagation mechanisms 6 . Upper and lower motor neuron degeneration leads to rapidly progressive paresis, with an average life expectancy of two to five years. Up to half of individuals with ALS show additional clinical signs of FTD, most commonly the behavioural-variant (bvFTD), resulting from FTLD 7 . Rare mutations in TARDBP, the gene encoding TDP-43, cause dominantly inherited forms of ALS and FTLD 8–10 , demonstrating that TDP-43 dysfunction leads to neurodegeneration. The extent of aggregated TDP-43 pathology is correlated with neuronal loss 5 . Disease mechanisms resulting from TDP-43 aggregation have been linked to a loss of TDP-43 interactions in the nucleus, as well as to the gain of aberrant interactions in the cytoplasm 11 . This genetic, neuropathological and clinical overlap suggests that ALS and FTLD-TDP lie on a disease spectrum.
TDP-43 is a ubiquitously-expressed 414-amino acid heterogeneous ribonucleoprotein (hnRNP) with multiple roles in nucleic acid metabolism. Under physiological conditions, TDP-43 primarily resides in the nucleus and undergoes nucleocytoplasmic shuttling 12 . The N-terminal half of TDP-43 comprises a dishevelled and axin (DIX) domain, which mediates oligomerisation 13 ; a nuclear localization signal (NLS); and tandem RNA recognition motifs (RRMs). The C-terminal half of TDP-43 consists of a prion-like low-complexity (LC) domain, which is involved in liquid-liquid phase separation within RNP granules 14 .
Pathological TDP-43 aggregates are composed of the full-length protein, as well as abnormally-cleaved C-terminal fragments (CTFs) of 18−35 kDa 1,2,15 . The aggregates are insoluble in the detergent Sarkosyl and contain abnormal post-translational modifications, including ubiquitylation and phosphorylation 1,2 . By electron microscopy, the aggregates appear granulo-filamentous and unbranched, with diameters of 10 to 15 nm 6,15–17 . The aggregate cores encompass sequences of 15−25 kDa in the LC domain and are protected from proteolysis, suggesting that they are ordered 18 .
The structures of pathological TDP-43 aggregates are unknown, which limits progress in understanding neurodegenerative disease mechanisms. Structural information is required in order to create accurate disease models of ALS and FTLD, as well as for the development of diagnostic and therapeutic agents. Here, we show that pathological TDP-43 aggregates from individuals that succumbed to ALS with FTLD are amyloid-like filaments and present their structure.
Extraction of pathological TDP-43
We extracted aggregated TDP-43 from the frontal and motor cortices of two individuals with clinical histories of ALS with FTD (Extended Data Table 1). Both individuals had predominantly round NCIs of aggregated TDP-43 in the motor cortex and spinal cord, as well as 'skein-like' inclusions, characteristic of sporadic ALS (Figure 1A, Extended Data Figure 1A, Extended Data Table 1) 1,2 . NCIs and GCIs were also observed throughout all cortical layers in the frontal, temporal and parietal cortices, with few dystrophic neurites (Figure 1A, B, Extended Data Table 1). These findings are consistent with Stage 4 ALS TDP-43 pathology 5 and Type B cortical TDP-43 pathology 19 , both of which are common in individuals that succumb to ALS with FTLD. Immunoblotting confirmed that the extracted aggregates were composed of full-length TDP-43 and CTFs and were phosphorylated at S409 and S410 (Figure 1B, Extended Data Figure 1C), as previously observed in ALS and FTLD 1,2 . Immuno-gold negative-stain electron microscopy revealed that the TDP-43 aggregates were helical filaments with projected widths of 10 to 15 nm (Figure 1D, Extended Data Figure 1D), similar to aggregated TDP-43 extracted from individuals with FTLD 6 , as well as to aggregated TDP-43 imaged in situ in ALS and FTLD brain 15–17 .
Fig. 1. TDP-43 pathology of amyotrophic lateral sclerosis with frontotemporal lobar degeneration.

a, Staining of TDP-43 neuronal cytoplasmic inclusions (brown) in the frontal and motor cortices and spinal cords of individuals 1 and 2 with anti-phosphorylated S409 and S410 TDP-43 antibody. Nuclei were counterstained in blue. Scale bars, 50 μm. b, Immunoblots of the total homogenate, Sarkosyl-soluble fraction and Sarkosyl-insoluble fraction from the frontal cortices of individuals 1 and 2 with anti-TDP-43 (amino acids 203− 209) and anti-phosphorylated S409 and S410 TDP-43 antibodies. The original, uncropped blots are shown in Supplementary Fig. 1. c, Immuno-electron microscopy of the Sarkosyl-insoluble fractions from the frontal and motor cortices of individual 1 using anti-phosphorylated S409 and S410 TDP-43 antibody. Scale bars, 200 nm. a−c, Similar results were obtained in at least three independent experiments.
Structure determination
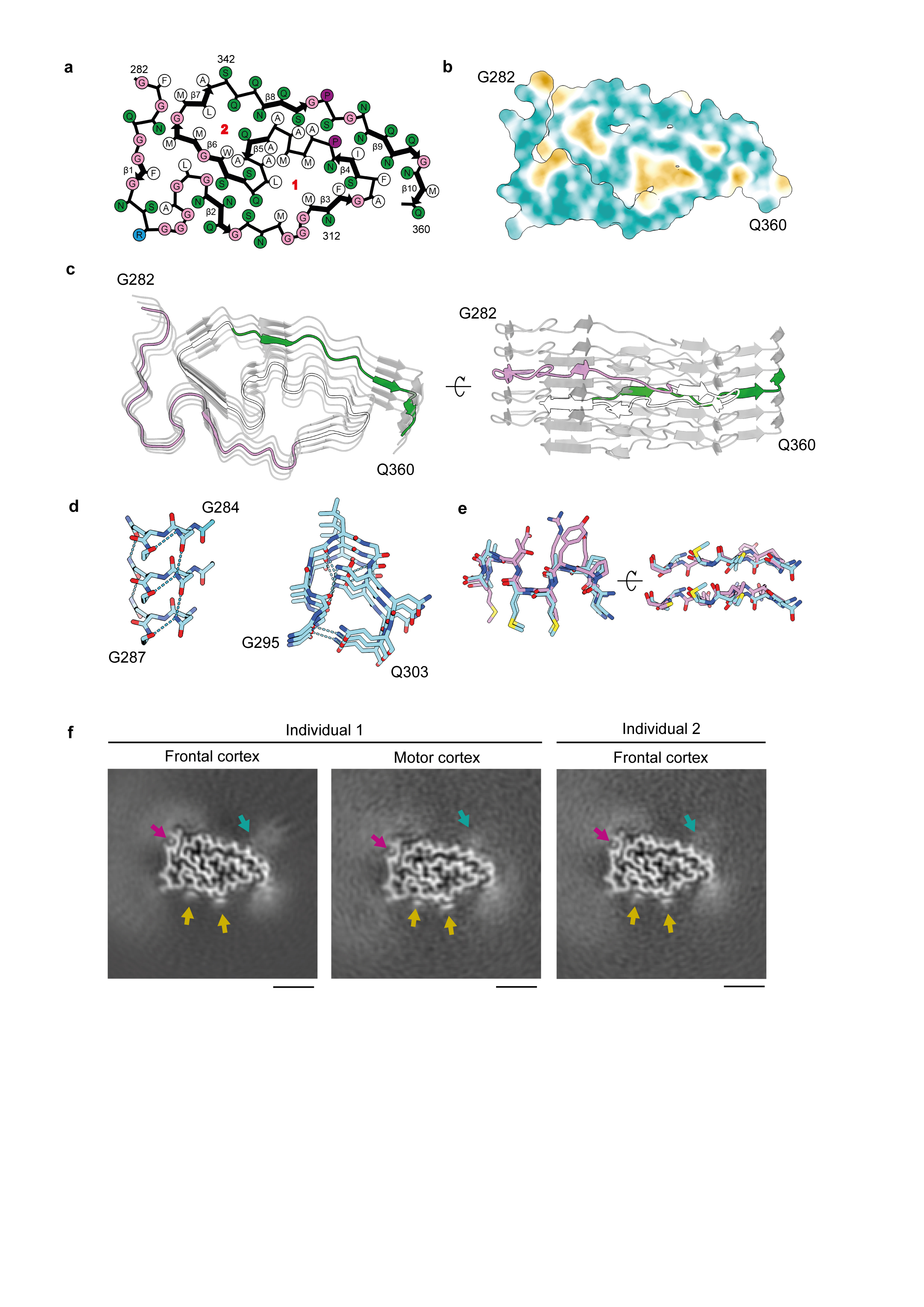
We used cryo-EM and helical reconstruction to determine the structures of the protease-resistant ordered cores of TDP-43 filaments extracted from the frontal and motor cortices of the first individual and the frontal cortex of the second individual, with resolutions up to 2.6 Å (Figure 2A, Extended Data Figure 2, Extended Data Table 2). It was not possible to extract sufficient numbers of filaments from the motor cortex of the second individual for cryo-EM. In both brain regions and individuals, the filaments existed as a single protofilament composed of stacked TDP-43 molecules separated by approximately 4.8 Å, with a helical twist of approximately 1.4 degrees. Perpendicular to the helical axis, TDP-43 formed a novel double-spiral-shaped fold (double-spiral fold), which was identical between brain regions and individuals. This suggests that a single TDP-43 filament structure may characterise ALS and FTLD and offers structural evidence that the two may be on a disease spectrum.
Fig. 2. Cryo-EM maps and atomic model of TDP-43 filaments from amyotrophic lateral sclerosis with frontotemporal lobar degeneration.

a, Cryo-EM maps of TDP-43 filaments from the frontal and motor cortices of individual 1 and from the frontal cortex of individual 2, shown as central slices perpendicular to the helical axis. Scale bars, 25 Å. b, Amino-acid sequence alignment of the secondary structure elements of the double-spiral fold. c, Cryo-EM map (transparent grey) and atomic model of TDP-43 filaments from the frontal cortex of individual 1, shown for a single TDP-43 molecule perpendicular to the helical axis. b and c, The glycine-rich (G282-G310, magenta), hydrophobic (M311-S342, white) and Q/N-rich (Q343-Q360, green) regions are highlighted.
Densities corresponding to amino acid side chains, peptide group oxygen atoms and ordered solvent molecules were resolved in the helical reconstructions, revealing that the filaments had a right-handed helical twist and enabling us to build an accurate atomic model (Figure 2B, C, Extended Data Figure 3). The double-spiral fold is formed by 79 residues encompassing G282−Q360 in the LC domain of TDP-43. Its amino acid composition is skewed toward glycines (20 residues) and neutral polar residues (in total 30 glutamine, asparagine and serine residues). There are also 26 hydrophobic residues, including nine alanines and one tryptophan. The remaining residues consist of two prolines and one arginine, which is the only charged residue in the fold. Cysteine, threonine and tyrosine residues are absent. The fold sequence can be divided into a glycine-rich region (G282−G310); a hydrophobic region (M311−S342), previously shown to form α-helices in solution 20 ; and a glutamine- and asparagine- (Q/N-) rich region (Q343−Q360). The fold sequence is present in both full-length TDP-43 and CTFs, the shortest of which begins at P280 21 . This offers an explanation for the incorporation of these different molecular species within pathological aggregates, with additional sequences flanking the ordered filament core. Using mass spectrometry, we identified partial deamidation of glutamine and asparagine residues, as well as partial oxidation of M311 and M339 (Extended Data Table 3). All confirmed disease-associated phosphorylation sites reside outside the fold and are, therefore, compatible with the structure. With the exception of S342, the serine residues within the fold are buried.
The double-spiral fold
The nucleus of the double-spiral fold is formed by the hydrophobic region (Figure 2B, C, Extended Data Figure 4A, B). Two hydrophobic clusters are located on either side of the main chain. The glycine-rich region towards the N-terminus and the Q/N-rich region towards the C-terminus form spiral branches that wrap around the hydrophobic nucleus and bury the remaining hydrophobic residues, with the exception of M359 at the C-terminal end. The 10 β-strands within the fold (β1-10) are short compared to those of amyloid structures, with only two (β6 in the hydrophobic nucleus and β9 in the Q/N-rich spiral branch) being longer than three residues (Figure 2B, Extended Data Figure 4A, C). Stacking of TDP-43 molecules with this fold gives rise to parallel in-register inter-molecular β-sheets that extend along the helical axis, similar to amyloid structures 22 . The inter-strand segments consist mainly of turns introduced by glycine residues and hydrogen bonds between buried polar side chains, main chain peptide groups and ordered solvent molecules (Extended Data Figure 3D and 4D). Between β4 and β5 there is a peculiar structural motif of three conjugated β-turns that was previously observed in the β-helix domain of glutamate synthase and related structures 23 (Extended Data Figure 4E).
Surface interactions
The skewed amino acid compositions of the two spiral branches gives rise to largely smooth surfaces on the filaments (Figure 3). A single prominent groove on the filament surface is formed by the main chain of G282−Q286 and the side chain of Q286. A small density was observed within the groove, which likely represents ordered solvent molecules (Figure 3, Extended Data Figure 4F). Additional surface grooves are few and shallow, predominantly composed of polar side chains fixed to the main chain or to each other by hydrogen bonding. The surface of the N-terminal spiral branch contains two flat strips between R293 and A315, formed by the abundance of glycine residues and the assimilation of the side chains of Q303, N306 and N312 into the surface by hydrogen bonding to the main chain (Figure 3). These flat strips, which resemble the flat ends of nucleic acid G-quadruplexes 24 , are predicted to bind rigid planar molecules by Van der Waals interactions. We observed strong planar densities, approximately one atom-thick, adjacent to these flat strips in our cryo-EM reconstructions, revealing the presence of interacting molecules at near-stoichiometric occupancies (Figure 3, Extended Data Figure 4F).
Fig. 3. Surfaces of TDP-43 filaments from amyotrophic lateral sclerosis with frontotemporal lobar degeneration.

Surface representations of the atomic model of TDP-43 filaments, shown for six molecules. The glycine-rich (G282-G310, magenta), hydrophobic (M311-S342, white) and Q/N-rich (Q343-Q360, green) regions are highlighted. Additional densities are coloured in transparent grey. Additional densities within the prominent groove formed by the main chain of G282-Q286 and the side chain of Q286 are indicated with magenta arrows. Additional planar densities adjacent to flat strips on the surface of the glycine-rich region are indicated with yellow arrows. Polar patches on the surface of the Q/N-rich region are indicated with cyan dashed lines.
In contrast to the N-terminal spiral branch, the abundance of glutamine and asparagine residues in the C-terminal spiral branch gives rise to polar patches on the filament surface (Figure 3). The amide groups of these residues can form hydrogen bonding networks and provide donor and acceptor groups for a wide range of interactions with external macromolecules. Less well-resolved density was visible in our cryo-EM reconstructions adjacent to a large polar patch formed by the side chains of N352, Q354 and Q356 (Extended Data Figure 4F).
These two types of surfaces are not found on other neurodegenerative disease-associated filaments 25–27 . Instead, for tau and α-synuclein filaments, interactions are observed between external densities and positively-charged side chains 25,26 , which are lacking in the TDP-43 filament structure. This suggests that the molecular interactions of TDP-43 filaments may be distinct from those of other neurodegenerative disease-associated filaments, which may have implications for disease mechanisms.
In addition, the surfaces of amyloid filaments contain deeper and less-polar grooves, which are thought to represent the binding sites of amyloid imaging ligands 28,29 . The lack of such grooves in the double-spiral fold may explain why studies aimed at repurposing amyloid imaging ligands, such as the fluorescent ligand thioflavin-S and the PET ligand 18 F-flortaucipir, have reported poor binding to aggregated TDP-43 in ALS and FTLD 30–35 . The lack of imaging ligands for pathological TDP-43 is a major limitation to the understanding and diagnosis of ALS, FTLD and other diseases. Our results suggest that the smooth surfaces of TDP-43 filaments may provide alternative binding sites for imaging ligands.
Comparison to related structures
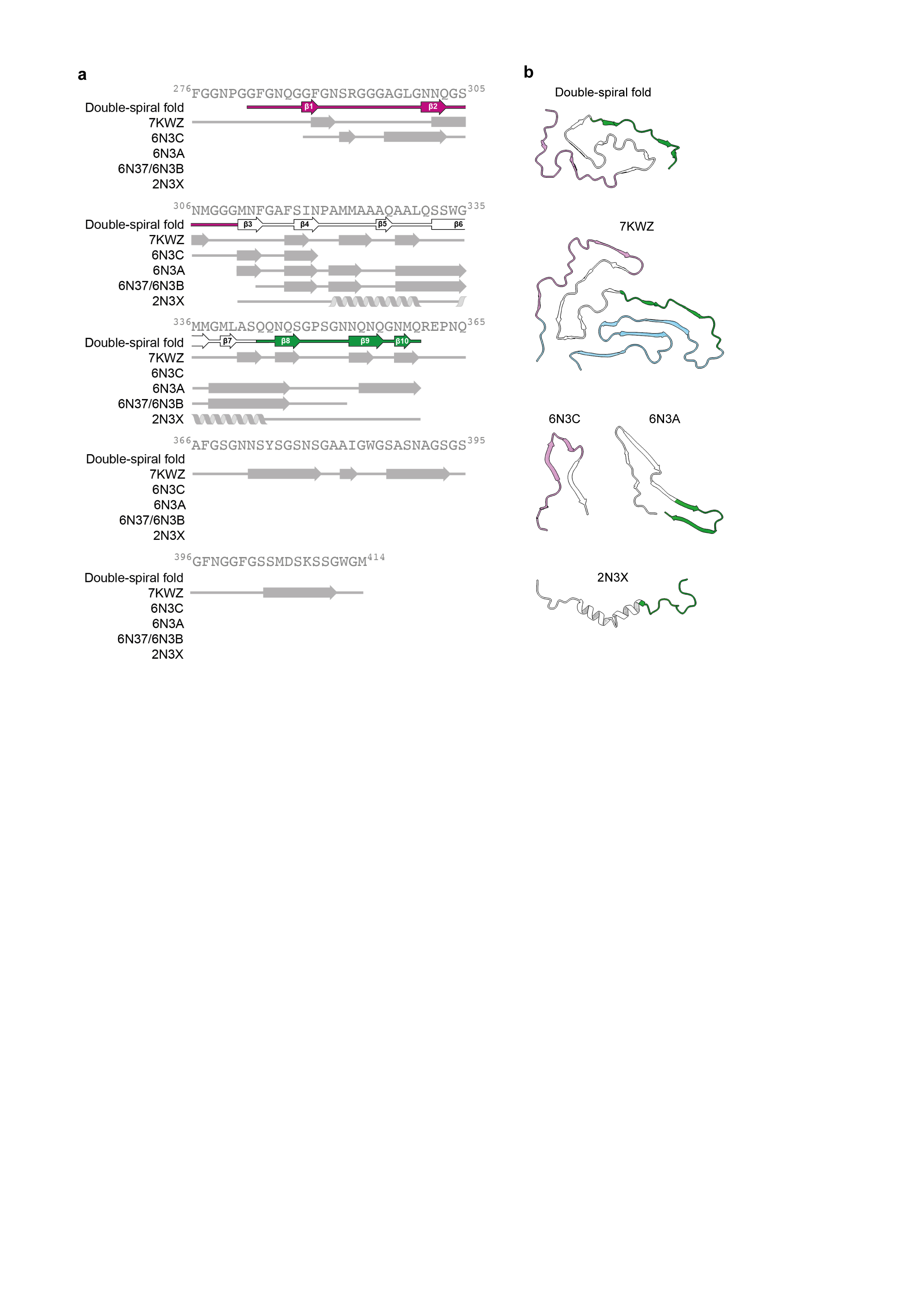
The brain-derived TDP-43 filaments are structurally different from filaments assembled in vitro from its LC domain or fragments thereof (Extended Data Figure 5) 3,4 . They have opposite chirality and differ both in protein fold and secondary structure. In contrast, brain-derived tau and α-synuclein filaments that share similar secondary structure to those produced in vitro, despite having distinct folds 26,36 . This difference may reflect a greater structural plasticity of filaments formed by TDP-43 and other LC domain-containing proteins. Of note, the NMR structure of the α-helices formed in solution by the hydrophobic region shows a long-range contact between Q327 and W334 20 . This contact is also present in the double-spiral fold, but not in the in vitro-assembled filaments. It will be important to produce filaments in vitro that recapitulate the double-spiral fold to accurately model ALS and FTLD.
The high number of glycine-mediated turns in the double-spiral fold prevents β-sheets from stacking perpendicular to the helical axis and forming cross-β structure. Cross-β structure is a prominent feature of amyloid filaments, including those of tau, β-amyloid and α-synuclein 22,25–27 . Instead, the many turns and short β-strands of the double-spiral fold, are reminiscent of filament folds produced in vitro from the LC domains of other hnRNPs, including fused in sarcoma (FUS), hnRNPA1 and hnRNPA2 37–40 . However, the amino acid compositions of these folds differ from that of the double-spiral fold. They are enriched in tyrosine residues, which are distributed approximately equally between the filament interior and surface. In contrast, the double spiral fold lacks tyrosine residues and contains a higher proportion of buried hydrophobic residues, resulting in a higher stability than the other hnRNP LC domain folds. In support of this, we found that TDP-43 filaments from ALS with FTLD were stable upon heating to 65°C (Extended Data Figure 6), a temperature at which hnRNPA2 LC domain filaments disassemble 38 . The hnRNP LC domain structures are thought to represent functional amyloids, although ex vivo structures of functional and pathological aggregates of these proteins, as seen in rare cases of ALS and FTLD 38–40 , are lacking. It remains to be seen if TDP-43 adopts more labile filament structures in physiological RNP granules.
TARDBP mutations
The individuals studied here had wild-type TARDBP sequences, similar to the vast majority of individuals with ALS. There are 24 reported mutations within G282−Q360 that cause ALS, 18 of which are fully compatible with the double-spiral fold (Extended Data Table 4). These include 15 mutations to surface residues and three mutations to interior residues. The remaining mutations would introduce steric clashes and/or uncompensated charged groups in the interior. Of these, only one, S332N, occurs within the hydrophobic nucleus and cannot be accommodated without significant perturbation to the entire fold. The remainder are located at interfaces between the hydrophobic nucleus and the spiral branches and could be accommodated with local rearrangements to the latter, analogous to structural variations of filaments assembled in vitro from fragments of the TDP-43 LC domain 3 . Alternatively, different TDP-43 filament folds may exist in individuals with these mutations.
TARDBP mutations lead to early-onset ALS, possibly through accelerated filament formation. The structure of the double-spiral fold suggests that the A315E mutation might favour filament formation by introducing a carboxyl group that could form a salt bridge with the side chain of R361, one residue outside of the ordered core. The A315T mutation could give rise to similar interactions if the threonine residue was phosphorylated, or else by enabling hydrogen bonding with the C-terminal spiral branch. These interactions may be responsible for the increased in vitro assembly of TDP-43 bearing these mutations compared to the wild-type protein 41,42 .
TDP-43 strains
The presence of the double-spiral fold in both frontal and motor cortices is consistent with the prion-like propagation of TDP-43 filaments during the temporospatial spread and accumulation of aggregated TDP-43 in ALS 5,6 . Neuropathological staging suggests that the motor cortex represents one of the earliest sites of TDP-43 inclusion formation, with the frontal cortex being affected later in disease 5 .
That the fold is shared between individuals suggests that the double-spiral fold may characterise ALS and FTLD with Type B cortical TDP-43 pathology. Aggregated TDP-43 from Type A and Type C FTLD has distinct sensitivities to proteolysis 18,43 , suggesting that different TDP-43 filament folds may characterise these diseases, analogous to disease-specific conformers of filamentous tau 25 . Furthermore, aggregated TDP-43 from these different neuropathological types of FTLD has distinct prion-like seeding activities and toxicities in model systems 6,43,44 , suggesting that prion-like strains of filamentous TDP-43 may exist. This work now paves the way to study the structures of aggregated TDP-43 in additional diseases and in experimental models of prion-like propagation.
Conclusions
The structure of TDP-43 filaments from ALS with FTLD establishes the structural characterisation of aggregated TDP-43 from human brain. It reveals the formation of filaments that are structurally distinct from amyloid filaments in other neurodegenerative diseases. The conserved filament fold of pathological TDP-43 in ALS with FTLD guides the development of accurate disease models, as well as diagnostic and therapeutic agents.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Clinical history and neuropathology
Individual one was previously described as case 7 in 45 . She initially presented at 49-years of age with slowing articulation and a lack of motivation and died 16-months later. Individual two was a man who initially presented at 62-years of age with muscle weakness in the proximal region of the upper limb and weight loss and died 24-months later. Both individuals had neuropathologically-confirmed diagnoses of ALS with FTLD, with round and skein-like TDP-43 inclusions in upper and lower motor neurons and Type B cortical TDP-43 pathology 19 . Both individuals met the criteria for Stage 4 ALS TDP-43 pathology 5 , with NCIs present in the motor system, as well as connected regions of the frontal, parietal and temporal lobes. Individual one had bulbar type ALS, with upper and lower motor neuron involvement, whereas individual two had flail arm syndrome with dominant lower motor neuron involvement. Neither individual had any family history of ALS or FTLD. Further details are given in Extended Data Tables 1 and 2.
Genetic analyses
Genomic DNA was purified from brain tissue using the QIAamp DNA Mini Kit (QIAGEN). Mutations in coding regions and exon−intron boundaries of TARDBP were examined by direct sequencing using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems) and an ABI PRISM 3130/3500 Genetic Analyzer (Applied Biosystems). TARDBP sequence analysis was performed based on RefSeq: NM_007375.4 from the UCSC Genome Browser (https://genome.ucsc.edu/). No mutations in TARDBP were detected in either case. Wild-type numbers of GGGGCC hexanucleotide repeats in intron 1 of C9orf72 were detected in both individuals using a repeat-primed polymerase chain reaction (Asuragen AmplideX PCR/CE C9orf72 kit). Individual one had eight repeats and individual two had two repeats.
Extraction of pathological TDP-43 aggregates
Sarkosyl-insoluble material was extracted from fresh-frozen frontal and motor cortex essentially as described 46 . Tissues were homogenized in 40 volumes (v/w) extraction buffer consisting of 10 mM Tris-HCl pH 7.5, 800 mM NaCl, 10 % sucrose and 1 mM EGTA. The homogenates were brought to 2 % Sarkosyl and incubated for 30 min at 37 °C. Following a 10 min centrifugation at 27,000 g at 25 °C, the supernatants were spun at 166,000 g for 20 min at 25 °C. The pellets were resuspended in 1 mL/g tissue of extraction buffer containing 1 % Sarkosyl by sonication for 5x 1 s at power output 2 (Titec VP-5s), diluted 4-fold with the same buffer, incubated for 20 min at 37 °C, and centrifuged at 21,500 g for 5 min at 25 °C. The supernatants were collected and spun at 166,000 g for 20 min. Sarkosyl-insoluble pellets were resuspended in 100 μL/g tissue of 20 mM Tris-HCl pH 7.4, 150 mM NaCl by sonication for 5x 1 s at power output 2 (Titec VP-5s). 1 mL/g tissue of extraction buffer containing 1 % Sarkosyl and 10 mM dithiothreitol (DTT) was added to the Sarkosyl-insoluble extracts, followed by incubation for 1 h at 37 °C and centrifugation at 100,000 g. Sarkosyl-insoluble pellets were resuspended in 15 μL/g tissue of 20 mM Tris-HCl pH 7.4, 150 mM NaCl by sonication for 2 min at 50% amplitude (Qsonica Q700). 1-2 g tissue was used for each cryo-EM sample.
Immunolabelling
Histological and immunoblot analyses were performed essentially as described 47 . For histology, brains were fixed with 10 % buffered formalin and embedded in paraffin. 8 μm-thick deparaffinized sections were autoclaved in 10 mM sodium citrate buffer at 105 °C for 10 min and treated with 95 % formic acid for 5 min. After washing, sections were blocked with 10 % calf serum in PBS, and then incubated overnight with a primary antibody to pS409/410 TDP-43 (CosmoBio) at 1:1,000 dilution in 10 % calf serum in PBS. After incubation with biotinylated secondary antibodies for 2 h, labelling was detected using the ABC staining kit (Vector) with DAB. Sections were counterstained with hematoxylin. For immunoblots, total homogenates, Sarkosyl-soluble material in the supernatants of the first 166,000 g spin and Sarkosyl-insoluble material in the pellets after the final ultracentrifugation were resolved using 12% Tris-glycine gels and antibodies to TDP-43 residues 203−209 48 and pS409/410 TDP-43 (CosmoBio) were used at 1:2,000 dilution. Immunogold negative-stain electron microscopy was carried out as previously described 49 .
Sarkosyl-insoluble extracts were deposited onto carbon-coated 300-mesh copper grids (Nissin EM), blocked with 0.3 % gelatin in PBS, and incubated with primary antibody to pS409/410 TDP-43 (CosmoBio) at a dilution of 1:100 in 0.3% gelatin in PBS at 37 °C for 1-2 h. After washing with PBS, the grids were incubated with secondary antibodies conjugated to 10 nm gold particles (Cytodiagnostics) at a dilution of 1:50 in 0.3% gelatin in PBS for 37 °C for 1 h. The grids were then stained with 2% phosphotungstate. Electron micrograph images were acquired using a JEOL JEM-1400 electron microscope operated at 80 keV and equipped with a CCD camera.
Mass spectrometry
Protease digestion and nano-flow liquid chromatography−ion trap tandem mass spectrometry (LC-MS/MS) were used to identify post-translational modifications to the pathological TDP-43 aggregates. Sarkosyl-insoluble extracts were treated with either 4 M urea plus 5% SDS or 70 % formic acid for 10 min at room temperature. Urea plus SDS-treated samples were digested with trypsin and Lys-C (Promega KK) in S-Trap micro spin columns (ProtiFi), according to the manufacturer's manual. Formic acid-treated samples were digested overnight with trypsin and Lys-C. The resulting peptides were analysed by LC-MS/MS (Q Exactive HF, Thomas Scientific) as described 50 .
Cryo-EM
Sarkosyl-insoluble extracts were incubated with 0.4 mg/mL pronase (Sigma) for 1 h at 21 °C and centrifuged at 3,000 g for 15 s. The supernatants were applied to glow-discharged 1.2/1.3 μm holey carbon-coated 300-mesh gold grids (Quantifoil) and plunge-frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher). Images were acquired using a 300 keV Titan Krios microscope (Thermo Fisher) equipped with a K3 detector (Gatan) and a GIF-quantum energy filter (Gatan) operated with a slit width of 20 eV. Aberration-free image shift (AFIS) within the EPU software (Thermo Fisher) was used during image acquisition. Further details are given in Extended Data Table 3. Heat stability experiments were carried out as described 38 . Samples were incubated for 10 min at 65 °C before the centrifugation step. Images were acquired using a 200 keV Glacios microscope (Thermo Fisher) equipped with a Falcon III detector (Thermo Fisher).
Helical reconstruction
Movie frames were gain-corrected, aligned, dose-weighted and summed using the motion correction program in RELION 51 . The motion corrected micrographs were used to estimate the contrast transfer function (CTF) using CTFFIND-4.1 52 . All subsequent image-processing was performed using helical reconstruction methods in RELION 53,54 . The filaments were picked manually and reference-free 2D classification was performed to remove suboptimal segments. Initial 3D reference models were generated de novo by producing sinograms from 2D class averages as described 55 . 3D auto-refinements with optimisation of the helical twist were performed, followed by two cycles of Bayesian polishing 51 and CTF refinement 56 . For filament structures from the motor cortex of individual one and the frontal cortex of individual 2, 3D classification was used to further remove suboptimal segments, after which the 3D auto-refinement, Bayesian polishing and CTF refinement procedures were repeated. The final reconstructions were sharpened using the standard post-processing procedures in RELION and overall resolutions were estimated from Fourier shell correlations at 0.143 between the two independently refined half-maps, using phase-randomisation to correct for convolution effects of a generous, soft-edged solvent mask 57 . Local resolution estimates were obtained using the same phase-randomisation procedure, but with a soft spherical mask that was moved over the entire map. A map sharpened using a B-factor of -80 and low-pass filtered at 2.3 Å was also used to help visualise peptide group oxygen atoms and ordered solvent. Helical symmetry was imposed using the RELION Helix Toolbox. Further details are given in Extended Data Table 3.
Atomic model building and refinement
The atomic model was built manually into the filament structure from the frontal cortex of individual 1 and refined in real-space using COOT 58 . Rebuilding using molecular dynamics was carried out in ISOLDE 59 . The model was refined in Fourier-space using REFMAC5 60 , with appropriate symmetry constraints defined using Servalcat 61 . To confirm the absence of overfitting, the model was shaken, refined in Fourier-space against the first half-map and compared to the second half map (Extended Data Figure 3B). Geometry was validated using MolProbity 62 . Molecular graphics and analyses were performed in ChimeraX 63 . Model statistics are given in Extended Data Table 3.
Extended Data
ED Fig. 1. TDP-43 pathology in individuals that succumbed to amyotrophic lateral sclerosis with frontotemporal lobar degeneration continued.

a, Staining of TDP-43 (brown) in the frontal and motor cortices and spinal cords of individuals 1 and 2 with anti-TDP-43 (amino acids 203−209) antibody. Nuclei were counterstained in blue. Scale bars, 50 μm. b, Staining of TDP-43 glial cytoplasmic inclusions (brown) in the motor cortices of individuals 1 and 2 with anti-phosphorylated S409 and S410 TDP-43 antibody. Nuclei were counterstained in blue. Scale bars, 50 μm. c and d, Immunoblots of the total homogenate (T), Sarkosyl-soluble fraction (S) and Sarkosyl-insoluble fraction (I) from the motor cortices of individuals 1 and 2 with anti-TDP-43 (amino acids 203−209) (c) and anti-phosphorylated S409 and S410 TDP-43 (d) antibodies. The original, uncropped blots are shown in Supplementary Fig. 1. e, Immuno-electron microscopy of the Sarkosyl-insoluble fraction from the frontal cortex of individual 2 using anti-phosphorylated S409 and S410 TDP-43 antibody. Scale bar, 100 nm. a−e, Similar results were obtained in at least three independent experiments.
ED Fig. 2. Cryo-EM and helical reconstruction.

a, Representative cryo-EM images of TDP-43 filaments from the frontal and motor cortices of individual 1 and from the frontal cortex of individual 2. Scale bars, 100 nm. Similar results were obtained in at least three independent experiments. b, Fourier shell correlation (FSC) curves for two independently-refined cryo-EM half-maps. The FSC threshold of 0.143 is shown with red dashed lines. c, Local resolution estimates for the Cryo-EM 3D reconstructions. Scale bars, 10 Å. d, Cryo-EM density maps viewed along the helical axis. Scale bars, 10 Å.
ED Fig. 3. Cryo-EM density map and atomic model comparisons.

a−c, Views of the cryo-EM density map of TDP-43 filaments from the frontal cortex of individual 1 and the corresponding atomic model showing representative densities for amino acid side chains (a), peptide group oxygen atoms (b) and ordered solvent molecules (red arrows) (c). d, Fourier shell correlation (FSC) curves for the refined atomic model against the cryo-EM density map (black); for the atomic model shaken and refined using the first half-map against the first half-map (magenta); and for the same atomic model against the second half-map (green). The FSC threshold of 0.5 is shown with a red dashed line.
ED Fig. 4. Double-spiral-shaped fold of TDP-43 filaments from amyotrophic lateral sclerosis with frontotemporal lobar degeneration.

a, Schematic view of the double-spiral fold. The two hydrophobic clusters in the hydrophobic nucleus, composed of the side chains of M307, M311, F313, M322, M323, A326, A329 and L330 (cluster 1); and A328, W334, M336 and L340 (cluster 2), are numbered. b, Hydrophobicity of the double-spiral fold from most hydrophilic (cyan) to most hydrophobic (yellow). c, Secondary structure of the double-spiral fold, depicted as five successive rungs. The glycine-rich (G282-G310, magenta), hydrophobic (M311-S342, white) and Q/N-rich (Q343-Q360, green) regions are highlighted. d, Views of the double-spiral fold, depicted as three successive rungs, showing hydrogen bonds (blue dashed lines) between buried polar side chains and main chain peptide groups. e, Superposition of residues N319-A326 of the double-spiral fold (cyan), depicted as two successive rungs, with residues G88-M95 and G107-A114 of the β-helix domain of glutamate synthase (magenta, PDB ID 1EA0). f, Unmasked cryo-EM 3D reconstructions of TDP-43 filaments from the frontal and motor cortices of individual 1 and from the frontal cortex of individual 2, shown as central slices perpendicular to the helical axis. Additional density within the prominent groove on the filament surface formed by the main chain of G282−Q286 and the side chain of Q286 is indicated with a magenta arrow. Additional densities adjacent to flat strips on the surface of the glycine-rich spiral branch between R293 and A315 are indicated with yellow arrows. Additional less well-resolved density projecting from a polar patch formed by the side chains of N352, Q354 and Q356 on the surface of the Q/N-rich spiral branch is indicated with a cyan arrow. Scale bars, 25 Å.
ED Fig. 5. Comparison of the double-spiral fold with recombinant TDP-43 structures.

a, Amino-acid sequence alignment of the secondary structure elements of the double-spiral fold and recombinant TDP-43 structures with the TDP-43 LC domain. PDB IDs are given for the recombinant TDP-43 structures. The glycine-rich (G282-G310, magenta), hydrophobic (M311-S342, white) and Q/N-rich (Q343-Q360, green) regions are highlighted on the double-spiral fold secondary structure elements. b, Secondary structure of the double-spiral fold and recombinant TDP-43. PDB IDs are given for the recombinant TDP-43 structures. The glycine-rich (G282-G310, magenta), hydrophobic (M311-S342, white) and Q/N-rich (Q343-Q360, green) regions are highlighted.
ED Fig. 6. Heat stability of TDP-43 filaments from amyotrophic lateral sclerosis with frontotemporal lobar degeneration.

Representative cryo-EM images of TDP-43 filaments from the frontal cortex of individual 1 with and without heating at 65°C for 10 min. Scale bars, 100 nm. Similar results were obtained in at least three independent experiments.
ED Table 1. Clinicopathological evaluation and TDP-43 inclusion pathology.
M, male; F, female; DD, disease duration; PMI, post-mortem interval; CERAD, Consortium to Establish a Registry for Alzheimer's Disease; CAA, cerebral amyloid angiopathy; AGD, argyrophilic grains; WT, wild-type; NA, not available; OG, orbital gyrus; AMG, amygdala; MF, middle frontal gyrus; TENT, transentorhinal cortex; DG, dentate gyrus; CG, anterior cingulate gyrus; SMT, superior middle temporal gyrus; NAcc, nucleus accumbens; CP, caudate nucleus/putamen; IO, inferior olivary nucleus; RN, red nucleus; TH, thalamus; MOT, primary motor cortex; ANG, angular gyrus; SEN, primary sensory cortex; XII, hypoglossal nucleus; CSC, cervical spinal cord α-motoneurons; LSC, lumbar spinal cord α-motoneurons; VIS, visual cortex; SN, substantia nigra; LC, locus coeruleus; DN, cerebellar dentate nucleus. The degree of pTDP-43 intraneuronal inclusions (dots, wisps, granular, rounds, skeins) in each region was assessed according to a semiquantitative rating scale (0, not detectable or ≤2 aggregates per region; +, mild; ++, moderate; 3, +++severe).
| Individual 1 | Individual 2 | |
|---|---|---|
| Clinicopathological evaluation | ||
| M/F | F | M |
| Age | 49 | 62 |
| DD (mo) | 16 | 24 |
| PMI (h) | 7 | 12 |
| Clinical diagnosis | ALS, bvFTD | ALS, bvFTD |
| Neuropathological diagnosis | ALS, FTLD | ALS, FTLD |
| ALS TDP-43 stage 15 | 4 | 4 |
| Cortical TDP-43 pathology 32 | Type B | Type B |
| Braaktau stage | 1 | 1 |
| Thai Phase | 0 | 0 |
| CERAD score | 0 | 0 |
| CAA | 0 | 0 |
| Lewy | 0 | 0 |
| AGD | 0 | 0 |
| C9orf repeats | WT | WT |
| TARDBP | WT | WT |
| Reference | Case 7 in 56 | This study |
| TDP-43 inclusion pathology | ||
| OG | +++ | NA |
| AMG | +++ | ++ |
| MF | +++ | NA |
| TENT | ++ | ++ |
| DG | ++ | ++ |
| CG | +++ | + |
| SMT | +++ | + |
| NAcc | +++ | ++ |
| CP | ++ | + |
| IO | ++ | 0 |
| RN | ++ | + |
| Pons | + | 0 |
| TH | ++ | NA |
| MOT | ++ | + |
| ANG | +++ | NA |
| SEN | + | NA |
| XII | ++ | ++ |
| CSC | ++ | ++ |
| LSC | ++ | ++ |
| VIS | + | NA |
| SN | + | + |
| LC | 0 | 0 |
| DN | + | NA |
Extended Data Table 2. Cryo-EM data collection, refinement and validation statistics.
| Individual 1 Frontal Cortex (EMPIAR-10830) (EMDB-13708) (PDB 7PY2) |
Individual 1 Motor Cortex (EMPIAR-10831) (EMDB-13710) |
Individual 2 Frontal Cortex (EMPIAR-10832) (EMDB-13712) |
|
|---|---|---|---|
| Data collection and processing | |||
| Voltage (kV) | 300 | 300 | 300 |
| Electron exposure (e-/Å2) | 39.7 | 39.56 | 39.02 |
| Defocus range (μm) | -2.0 to-1.2 | -2.0 to -1.2 | -2.0 to-1.2 |
| Pixel size (Å) | 0.93 | 0.93 | 0.93 |
| Symmetry imposed | Cl | Cl | Cl |
| Initial particle images (no.) | 308,822 | 65,147 | 84,952 |
| Final particle images (no.) | 308,822 | 22,019 | 28,943 |
| Helical twist (°) | 1.422 | 1.412 | 1.443 |
| Helical rise (Å) | 4.842 | 4.847 | 4.851 |
| Map resolution (Å) | 2.59 | 2.94 | 2.83 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 2.45 to 4.46 | 2.91 to 4.98 | 2.75 to 4.98 |
| Refinement | |||
| Initial model used (PDB code) | − | − | − |
| Model resolution (Å) | 2.60 | ||
| FSC threshold | 0.5 | − | − |
| Map sharpening B factor (Å2) | -61.268 | − | − |
| Model composition | |||
| Non-hydrogen atoms | 538 | − | − |
| Protein residues | 78 | − | − |
| Ligands | 0 | − | − |
| B factors (Å2) | |||
| Protein | 71.482 | − | − |
| Ligand | − | − | − |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.01 | − | − |
| Bond angles (°) | 1.60 | − | − |
| Validation | |||
| MolProbity score | 1.54 | − | − |
| Clashscore | 0.98 | − | − |
| Poor rotamers (%) | 2 | − | − |
| Ramachandran plot | |||
| Favored (%) | 90.91 | − | − |
| Allowed (%) | 100 | − | − |
| Outliers (%) | 0 | − | − |
Extended Data Table 3. Mass spectrometry analyses of post-translational modifications.
The numbers of peptides containing the modified residues / the total numbers of peptides containing both the modified and unmodified residues are shown.
| Residue | Modification | Individual 1 | Individual 2 |
|---|---|---|---|
| N285 | Deamidation | 1/21 | 0/12 |
| Q286 | Deamidation | 1/21 | 0/12 |
| N29l | Deamidation | 1/21 | 0/12 |
| M3ll | Oxidation | 1/16 | 0/8 |
| N319 | Deamidation | 2/16 | 0/8 |
| M323 | Oxidation | 4/16 | 0/8 |
| Q327 | Deamidation | 3/16 | 0/8 |
| Q33l | Deamidation | 3/16 | 0/8 |
| M339 | Oxidation | 3/16 | 0/8 |
| Q343 | Deamidation | 3/16 | 0/8 |
| Q344 | Deamidation | 2/16 | 0/8 |
| N345 | Deamidation | 2/16 | 0/8 |
| Q346 | Deamidation | 2/16 | 0/8 |
| N353 | Deamidation | 0/16 | 2/8 |
Extended Data Table 4. Compatibility of ALS-associated TARDBP mutations with the double-spiral fold.
| Mutation | Compatible? | Notes |
|---|---|---|
| G287S | Yes | Surface exposed |
| G290A | Yes | Surface exposed |
| S292N | Yes | With hydrogen bonding to G294 |
| G294A | Yes | Surface exposed |
| G294V | Yes | Surface exposed |
| G295S | Yes | Surface exposed |
| G295R | Yes | Surface exposed |
| G298S | No* | Steric clash |
| M311V | Yes | − |
| A315T | Yes | Surface exposed |
| A315E | Yes | Surface exposed |
| A321G | Yes | − |
| Q331K | No* | Uncompensated charged group in interior |
| S332N | No | Steric clash |
| G335D | No* | Uncompensated charged group in interior |
| M337V | Yes | − |
| Q343R | No* | Uncompensated charged group in interior |
| N345K | No* | Uncompensated charged group in interior |
| G348C | Yes | Surface exposed |
| G348V | Yes | Surface exposed |
| G348R | Yes | Surface exposed |
| N352S | Yes | Surface exposed |
| G357R | Yes | Surface exposed** |
| G357S | Yes | Surface exposed** |
Could be accommodated with local rearrangements within the spiral branches
Would require rotation of the peptide group between residues 357 and 358 to yield a β-arc conformation
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Acknowledgements
We thank the individuals and their families for donating brain tissues; K. Yamada and Y. Itoh for clinical information; A. Akagi for dissecting brain tissues; T. Arai and H. Akiyama for helpful comments on clinical history and neuropathology; R. Otani and M. Takase for technical support with immunohistochemistry; K. Yamashita and G.N. Murshudov for help with Servalcat; the MRC Laboratory of Molecular Biology Electron Microscopy Facility for access to and support with electron microscopy; the MRC Laboratory of Molecular Biology Scientific Computing Facility for access to and support with computing; M. Goedert, S.H.W. Scheres, S.W. Davies, S. Tetter, T.S. Behr and R.R. Chen for discussions. D.A. is a Fellow at Darwin College, University of Cambridge. This work was supported by the Medical Research Council (MRC) grant MC_UP_1201/25 and Alzheimer's Research UK award ARUK-RS2019-001 to B.F.; the Japan Agency for Medical Research and Development (AMED) grants JP20ek0109392 and JP20ek0109391 to M.Y. and JP20dm0207072 to M.H; and the Japan Science and Technology Agency (JST) Core Research for Evolutional Science and Technology (CREST) grant JPMJCR18H3 to M.H.
Footnotes
Author Contributions
M.Y. identified individuals and performed neuropathological examinations; M.A. performed genetic analysis; M.H. extracted TDP-43 filaments and performed immunohistochemistry, immunoblotting and immuno-EM; F.K. performed mass spectrometry; D.A. performed cryo-EM; D.A. and B.F. analysed the cryo-EM data; D.A., B.F. and A.G.M. built and analysed the atomic model; B.F. supervised the study; B.F. M.Y. and M.H. were the senior authors; all authors contributed to writing the manuscript.
Competing interests
The authors declare no competing interests.
Ethical review processes and informed consent
The studies that were performed at Aichi Medical University and at Tokyo Metropolitan Institute of Medical Science were approved the ethics committees and through the ethical review processes at each institution. Informed consent was obtained from the individuals’ next of kin.
Data availability
Cryo-EM datasets have been deposited to the Electron Microscopy Public Image Archive (EMPIAR) under accession numbers 10830 for individual 1, frontal cortex; 10831 for individual 1, motor cortex; and 10832 for individual 2, frontal cortex. Cryo-EM maps have been deposited to the Electron Microscopy Data Bank (EMDB) under accession numbers 13708 for individual 1, frontal cortex; 13710 for individual 1, motor cortex; and 13712 for individual 2, frontal cortex. The atomic model of the double-spiral fold has been deposited to the PDB under accession number 7PY2. The atomic model of the α subunit of glutamate synthase was obtained from the PDB (accession number 1EA0). The atomic model of the α-helices formed in solution by the hydrophobic region of TDP-43 was obtained from the PDB (accession number 2N3X). The atomic models of TDP-43 filaments formed in vitro were obtained from the PDB (accession numbers 7KWZ, 6N3C, 6N3A, 6N37 and 6N3B). Mass spectrometry data have been deposited to the Japan Proteome Standard Repository (jPOSTrepo) under accession number PXD029001. Any other data are available from the corresponding author upon request.
References
- 1.Neumann M, et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 2.Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 3.Cao Q, Boyer DR, Sawaya MR, Ge P, Eisenberg DS. Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nature Struct Mol Biol. 2019;26:619–627. doi: 10.1038/s41594-019-0248-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Q, Babinchak WM, Surewicz WK. Cryo-EM structure of amyloid fibrils formed by the entire low complexity domain of TDP-43. Nature Commun. 2021;12:1620. doi: 10.1038/s41467-021-21912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brettschneider J, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20–38. doi: 10.1002/ana.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nonaka T, et al. Prion-like Properties of Pathological TDP-43 Aggregates from Diseased Brains. Cell Rep. 2013;4:124–134. doi: 10.1016/j.celrep.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6:994–1003. doi: 10.1016/S1474-4422(07)70265-X. [DOI] [PubMed] [Google Scholar]
- 8.Sreedharan J, et al. TDP-43 Mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nature Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 10.Van Deerlin VM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hergesheimer RC, et al. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: a resolution in sight? Brain. 2019;142:1176–1194. doi: 10.1093/brain/awz078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayala YM, et al. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121:3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- 13.Afroz T, et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nature Commun. 2017;8:45. doi: 10.1038/s41467-017-00062-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colombrita C, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111:1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- 15.Hasegawa M, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin W-L, Dickson DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 2008;116:205–213. doi: 10.1007/s00401-008-0408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mori F, et al. Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008;116:193–203. doi: 10.1007/s00401-008-0396-9. [DOI] [PubMed] [Google Scholar]
- 18.Tsuji H, et al. Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain. 2012;135:3380–3391. doi: 10.1093/brain/aws230. [DOI] [PubMed] [Google Scholar]
- 19.Mackenzie IRA, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang L-L, et al. Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J Biol Chem. 2013;288:19614–19624. doi: 10.1074/jbc.M113.463828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kametani F, et al. Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci Rep. 2016;6:23281. doi: 10.1038/srep23281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sunde M, et al. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 23.Binda C, et al. Cross-talk and ammonia channeling between active centers in the unexpected domain arrangement of glutamate synthase. Structure. 2000;8:1299–1308. doi: 10.1016/s0969-2126(00)00540-2. [DOI] [PubMed] [Google Scholar]
- 24.Parkinson GN, Cuenca F, Neidle S. Topology conservation and loop flexibility in quadruplex−drug recognition: crystal structures of inter- and intramolecular telomeric DNA quadruplex−drug complexes. J Mol Biol. 2008;381:1145–1156. doi: 10.1016/j.jmb.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y, et al. Structure-based classification of tauopathies. Nature. 2021 doi: 10.1038/s41586-021-03911-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schweighauser M, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature. 2020;585:464–469. doi: 10.1038/s41586-020-2317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kollmer M, et al. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nature Commun. 2019;10:1–8. doi: 10.1038/s41467-019-12683-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krebs MRH, et al. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J Struct Biol. 2005;149:30–37. doi: 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Shi Y, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta Neuropathol. 2021;141:697–708. doi: 10.1007/s00401-021-02294-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bigio EH, et al. Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid. Acta Neuropathol. 2013;125:463–465. doi: 10.1007/s00401-013-1089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson JL, et al. TDP-43 skeins show properties of amyloid in a subset of ALS cases. Acta Neuropathol. 2013;125:121–131. doi: 10.1007/s00401-012-1055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y, et al. Physiologically important electrolytes as regulators of TDP-43 aggregation and droplet-phase behavior. Biochemistry. 2019;58:590–607. doi: 10.1021/acs.biochem.8b00842. [DOI] [PubMed] [Google Scholar]
- 33.Marquié M, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015;78:787–800. doi: 10.1002/ana.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowe VJ, et al. An autoradiographic evaluation of AV-1451 tau PET in dementia. Acta Neuropathol Commun. 2016;4:58. doi: 10.1186/s40478-016-0315-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sander K, et al. Characterization of tau positron emission tomography tracer [18F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and other dementias. Alzheimers Dement. 2016;12:1116–1124. doi: 10.1016/j.jalz.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 36.Zhang W, et al. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife. 2019;8:e43584. doi: 10.7554/eLife.43584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray DT, et al. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell. 2017;171:615–627.:e16. doi: 10.1016/j.cell.2017.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu J, et al. CryoEM structure of the low-complexity domain of hnRNPA2 and its conversion to pathogenic amyloid. Nature Commun. 2020;11:4090. doi: 10.1038/s41467-020-17905-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Y, et al. The nuclear localization sequence mediates hnRNPA1 amyloid fibril formation revealed by cryoEM structure. Nature Commun. 2020;11:6349. doi: 10.1038/s41467-020-20227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee M, Ghosh U, Thurber KR, Kato M, Tycko R. Molecular structure and interactions within amyloid-like fibrils formed by a low-complexity protein sequence from FUS. Nature Commun. 2020;11:5735. doi: 10.1038/s41467-020-19512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen AK-H, et al. Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J Am Chem Soc. 2010;132:1186–1187. doi: 10.1021/ja9066207. [DOI] [PubMed] [Google Scholar]
- 42.Guo W, et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nature Struct Mol Biol. 2011;18:822–830. doi: 10.1038/nsmb.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laferrière F, et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting diseaseprogression rates. Nature Neurosci. 2019;22:65. doi: 10.1038/s41593-018-0294-y. [DOI] [PubMed] [Google Scholar]
- 44.Porta S, et al. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nature Commun. 2018;9:4220. doi: 10.1038/s41467-018-06548-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshida M. Amyotrophic lateral sclerosis with dementia: The clinicopathological spectrum. Neuropathology. 2004;24:87–102. doi: 10.1111/j.1440-1789.2003.00544.x. [DOI] [PubMed] [Google Scholar]
- 46.Taniguchi-Watanabe S, et al. Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 2016;131:267–280. doi: 10.1007/s00401-015-1503-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasegawa M, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsuji H, et al. Epitope mapping of antibodies against TDP-43 and detection of protease-resistant fragments of pathological TDP-43 in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Biochem Biophys Res Commun. 2012;417:116–121. doi: 10.1016/j.bbrc.2011.11.066. [DOI] [PubMed] [Google Scholar]
- 49.Tarutani A, Hasegawa M. In: Experimental Models of Parkinson’s Disease. Imai Y, editor. Springer; US: 2021. Electron microscopic analysis of α-synuclein fibrils; pp. 17–25. [DOI] [PubMed] [Google Scholar]
- 50.Kametani F, et al. Comparison of common and disease-specific post-translational modifications of pathological tau associated with a wide range of tauopathies. Front Neurosci. 2020;14:581936. doi: 10.3389/fnins.2020.581936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zivanov J, Nakane T, Scheres SHW. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ. 2019;6:5–17. doi: 10.1107/S205225251801463X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He S, Scheres SHW. Helical reconstruction in RELION. J Struct Biol. 2017;198:163–176. doi: 10.1016/j.jsb.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zivanov J, et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 2018;7:e42166. doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scheres SHW. Amyloid structure determination in RELION-3.1. Acta Crystallogr Sect Struct Biol. 2020;76:94–101. doi: 10.1107/S2059798319016577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zivanov J, Nakane T, Scheres SHW. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ. 2020;7:253–267. doi: 10.1107/S2052252520000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen S, et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryo-microscopy. Ultramicroscopy. 2013;135:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casañal A, Lohkamp B, Emsley P. Current developments in Coot for macromolecular model building of electron cryo-microscopy and erystallographic data. Protein Sci. 2020;29:1055–1064. doi: 10.1002/pro.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Croll TI. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr Sect Struct Biol. 2018;74:519–530. doi: 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown A, et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr D Biol Crystallogr. 2015;71:136–153. doi: 10.1107/S1399004714021683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamashita K, Palmer CM, Burnley T, Murshudov GN. Cryo-EM single particle structure refinement and map calculation using Servalcat. bioRxiv. 2021:2021.05.04.442493. doi: 10.1101/2021.05.04.442493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams CJ, et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pettersen EF, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM datasets have been deposited to the Electron Microscopy Public Image Archive (EMPIAR) under accession numbers 10830 for individual 1, frontal cortex; 10831 for individual 1, motor cortex; and 10832 for individual 2, frontal cortex. Cryo-EM maps have been deposited to the Electron Microscopy Data Bank (EMDB) under accession numbers 13708 for individual 1, frontal cortex; 13710 for individual 1, motor cortex; and 13712 for individual 2, frontal cortex. The atomic model of the double-spiral fold has been deposited to the PDB under accession number 7PY2. The atomic model of the α subunit of glutamate synthase was obtained from the PDB (accession number 1EA0). The atomic model of the α-helices formed in solution by the hydrophobic region of TDP-43 was obtained from the PDB (accession number 2N3X). The atomic models of TDP-43 filaments formed in vitro were obtained from the PDB (accession numbers 7KWZ, 6N3C, 6N3A, 6N37 and 6N3B). Mass spectrometry data have been deposited to the Japan Proteome Standard Repository (jPOSTrepo) under accession number PXD029001. Any other data are available from the corresponding author upon request.