

Summary
Clarifying the evolutionary processes underlying species diversification and adaptation is a key focus of evolutionary biology. Begonia (Begoniaceae) is one of the most species‐rich angiosperm genera with c. 2000 species, most of which are shade‐adapted.
Here, we present chromosome‐scale genome assemblies for four species of Begonia (B. loranthoides, B. masoniana, B. darthvaderiana and B. peltatifolia), and whole genome shotgun data for an additional 74 Begonia representatives to investigate lineage evolution and shade adaptation of the genus.
The four genome assemblies range in size from 331.75 Mb (B. peltatifolia) to 799.83 Mb (B. masoniana), and harbor 22 059–23 444 protein‐coding genes. Synteny analysis revealed a lineage‐specific whole‐genome duplication (WGD) that occurred just before the diversification of Begonia. Functional enrichment of gene families retained after WGD highlights the significance of modified carbohydrate metabolism and photosynthesis possibly linked to shade adaptation in the genus, which is further supported by expansions of gene families involved in light perception and harvesting. Phylogenomic reconstructions and genomics studies indicate that genomic introgression has also played a role in the evolution of Begonia.
Overall, this study provides valuable genomic resources for Begonia and suggests potential drivers underlying the diversity and adaptive evolution of this mega‐diverse clade.
Keywords: Begonia, evolution, genomes, introgression, shade adaptation, whole‐genome duplication
Introduction
The mechanisms underlying the diversification of large clades of closely related species (often designated taxonomically as genera) remain one of the biggest mysteries in plant biology (Frodin, 2004). Although speciose genera have received widespread attention from evolutionary biologists, typically few genomic resources are available for these closely related, species‐rich clades. Representative completely assembled nuclear genomes of only three of the 10 largest angiosperm genera (Frodin, 2004) have been published, namely Solanum (A. Bolger et al., 2014; Song et al., 2019), Dendrobium (Yan et al., 2015) and Begonia (Griesmann et al., 2018). However, these genomic studies either focused on the specific characteristics of the reference species (A. Bolger et al., 2014; Yan et al., 2015; Song et al., 2019) or were part of a large comparative genomic project (Griesmann et al., 2018); none of them used genomic data to explore the evolutionary patterns in these mega‐diverse clades.
Begonia L. (Begoniaceae, Cucurbitales) is well known for a huge diversity of leaf shapes, patterns and textures (Fig. 1). The genus is pantropical and comprises more than 2000 currently accepted species (Hughes et al., 2015) of herbs and occasionally subshrubs; it thus represents an excellent evolutionary study system for processes that generate numerous closely related species. Species of Begonia are mostly narrow endemics occupying specific microhabits. Begonia has high species diversity in the New World and Asia and relatively low species numbers in Africa, the continent of its putative origin (Neale et al., 2006). This high species diversity forms a stark contrast with its sister genus, the monotypic Hillebrandia Oliv. comprising the rare Haiwaiian endemic H. sandwicensis. Previous studies have suggested that the overall patterns of speciation in Begonia may be driven by local speciation in fragmented habitats (Hughes & Hollingsworth, 2008), hybridization and polyploidization (Dewitte et al., 2011).
Fig. 1.
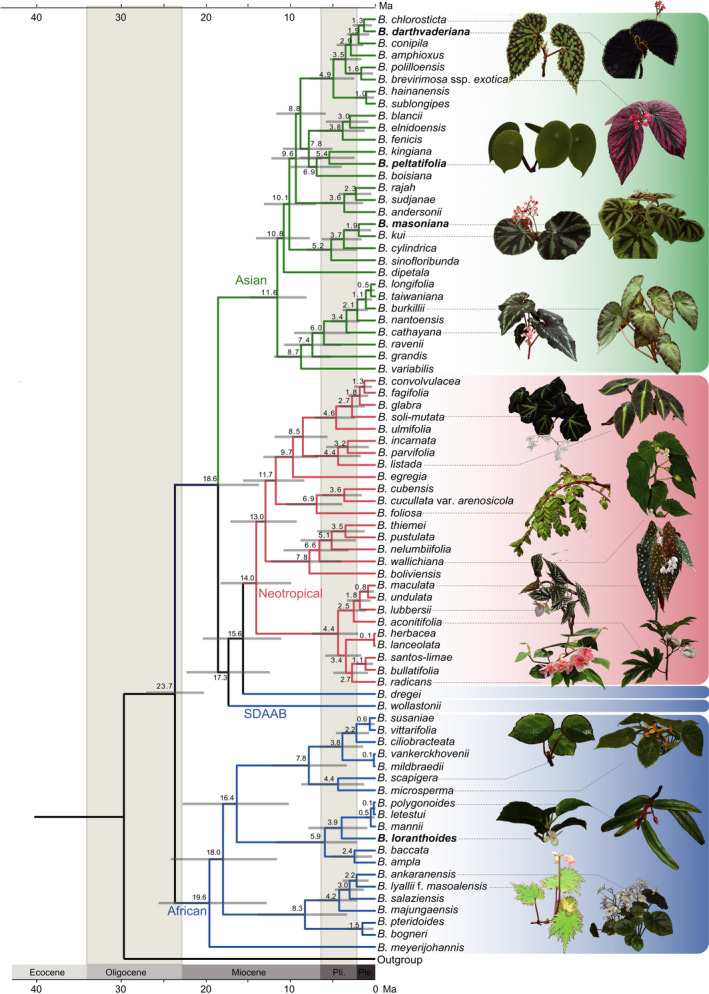
Phylogenetic tree showing the topology and divergence times for 78 newly sequenced species of Begonia. Maximum‐likelihood tree inferred with RAxML based on SNPs within regions of nuclear single‐copy genes. Divergence times are indicated at the internodes; the range of the blue bars indicates the 95% confidence interval of the divergence times. Representative images on the right show Begonia diversity. Three major geographically circumscribed clades are colored blue, red and green for the African, Neotropical and Asian clade, respectively. Taxa in bold highlight the four species with chromosome‐scale genomes generated in this study. Branches are maximally supported unless otherwise indicated.
Most begonias are shade‐adapted and become sun‐damaged when exposed to full sun. However, exceptions and intermediates regarding habitat preferences also exist. Begonia species exhibit a continuum of light adaptation ability ranging from deep shade to full sun, affording us the opportunity to unravel mechanisms of adaptation to cope with variable levels of light. Understanding how shade‐adapted species optimize photosynthesis and physical defense, while suppressing the shade‐avoidance syndrome (SAS; strong elongation growth away from shaded microconditions and accelerated flowering), will be valuable for crop improvement (Gommers et al., 2013). However, the genetic footprints and molecular basis of shade adaptation on the genome level remain elusive.
We de novo sequenced and assembled chromosome‐scale genomes of four Begonia species, and generated whole genome shotgun (WGS) data for an additional 74 Begonia species, representing 37 of the 70 recognized sections of Begonia (Moonlight et al., 2018) across all three major continental clades (Supporting Information Fig. S1). We compared the four Begonia genomes and reconstructed the paleogenome of Begonia, explored the evolutionary impact of a Begonia‐specific whole genome duplication (WGD) event (Brennan et al., 2012), analyzed the content of transposable elements (TEs), and analyzed their potential impact on the genomic landscape and potentially on species adaptation. We also examined cytonuclear incongruences detected in our study, and investigated the molecular basis of shade adaptation in Begonia.
The interpretation of Begonia genomic diversity in an evolutionary context will not only contribute to a better understanding of the origin, evolution and shade adaptation of this mega‐diverse clade, but also provide valuable reference genomes for molecular breeding of these highly valued ornamental plants.
Materials and Methods
Sample collections and DNA/RNA extraction
All Begonia samples were collected from the glasshouse in Fairy Lake Botanical Garden (Shenzhen, China) where plants were cultivated at 26°C : 18°C (day : night) with a relative humidity of 65–80%. Specimens have been deposited in the Herbarium of Fairy Lake Botanical Garden. For WGS, genomic DNA from young leaves of each individual was extracted using the cetyl trimethylammonium bromide (CTAB) method (Porebski et al., 1997). For single tube long fragment read (stLFR) sequencing, high‐molecular‐weight genomic DNA was isolated using the IrysPrep® Plant Tissue DNA Isolation kit (RE‐014‐05; Bionano Genomics, San Diego, CA, USA) following the manufacturer’s instructions. DNA quality and quantity were evaluated using pulsed field gel electrophoresis and Qubit® 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). For transcriptome sequencing, total RNA from different tissues (root, stem/rhizome, leaf, peduncle and flower) from four Begonias were isolated using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA), respectively. All the RNA samples were quality controlled using a NanoDrop™ One UV‐Vis spectrophotometer (Thermo Fisher Scientific) and a Qubit® 3.0 Fluorometer.
Library preparation and sequencing
All DNA libraries for WGS were constructed using the MGIEasy FS DNA Library Prep Set (1000006988) with 300–500 bp fragment sizes, and sequenced on an Illumina Hiseq2000 platform to generate paired‐end (PE) reads of 150 bp. Transcriptome libraries were constructed with a MGIEasy RNA Library Prep Kit (1000006384) with inserts of 200–400 bp and sequenced with PE reads of 100 bp. More than 5 Gb of sequence data were generated for each library. The stLFR library was prepared with the MGIEasy stLFR Library Prep Kit (1000005622) (Wang et al., 2019) and sequenced with PE reads of 100 + 42 bp, generating > 150 Gb of raw sequence data for each library. 10 × Genomics Chromium™ Genome libraries with insert sizes of 350–500 bp were prepared with Chromium Genome Reagent Kit (v2 Chemistry, Pleasanton, CA, USA) following the manufacturer’s protocols with modified PCR primers to introduce sequencing primers suitable for the BGISEQ‐500 platform and then sequenced with PE reads of 150 bp. SMART library preparation and sequencing details are given in Methods S1.
Genome assembly
For assembly of the sequences from 10 × Genomics Chromium and stLFR libraries, the clean reads were obtained using an in‐house script and de novo assembled using Spernova (v.2.1.1) (Weisenfeld et al., 2017) with default parameters. A minimum fasta record size of 100 bp was specified at the ‘mkoutput’ stage for outputting the assembly in the ‘pseudohap’ style. De novo assemblies of the PacBio long reads for B. masoniana and B. darthvaderiana were conducted by Canu (v.0.1) (Koren et al., 2017). Subsequently, two rounds of iterative corrections were performed with PacBio long reads using the software Racon (v.1.2.1) (Vaser et al., 2017), and two rounds of corrections with Pilon (v.1.22) (Walker et al., 2014) using 10 × Genomics reads (see details in Methods S1).
Variant analysis
A total of 468 Gb 150 bp PE Illumina reads were generated, yielding an average coverage of 7× per accession. Raw reads were quality controlled using Trimmomatic (A. M. Bolger et al., 2014) to remove adaptors and low‐quality bases. The clean reads were aligned against the reference genome of B. masoniana using Bwa‐Mem (v.0.7.10) (Li, 2013) with default parameters. Variant detection was performed using the genome analysis toolkit (gatk; v.3.5‐0‐g36282e4) (Mckenna et al., 2010) following the best practices workflow for variant discovery. The resulting BAM files were locally realigned using the IndelRealigner to remove erroneous mismatches around small‐scale insertions and deletions. Variants were called in each accession separately using the HaplotypeCaller and individual gVCF files were merged using GenotypeGVCFs. This two‐step approach includes quality recalibration and regenotyping in the merged vcf file, ensuring variant accuracy. Single nucleotide polymorphisms (SNPs) were filtered based on the following criteria: SNPs in repeat regions; SNPs with read depth > 1000 or < 5; SNPs with missing rate > 40%; SNPs with < 5 bp distance with nearby variant sites; and nonbiallelic SNPs were removed. Phylogenetic reconstruction, admixture analysis, principal component analysis (PCA), diversity statistics and ABBA‐BABA analysis based on the SNP data are detailed in Methods S1.
Phylogenetic analysis
For nuclear phylogenetic analysis, SNPs within 4000 Begonia single‐copy nuclear genes identified using the software OrthoFinder (Emms & Kelly, 2015) with four newly sequenced Begonia genomes with default settings were extracted from vcf files and filtered based on sequence length (> 100 bp) and taxon occurrences (> 50%), aligned with Mafft (Katoh et al., 2005), and trimmed with Gblocks (Talavera & Castresana, 2007). A supermatrix method was used to infer the nuclear phylogeny using RAxML (v.7.2.3) (Stamatakis, 2006). The maximum‐likelihood tree inferred from concatenated nuclear SNPs was used as a starting tree to estimate species divergence time using MCMC Tree as implemented in Paml (Yang, 2007). One calibration point of the Begonia crown group (24 million years ago (Ma) ± 3.57 million years with a normal distribution) was defined following Moonlight et al., 2018). For plastid phylogenetic analysis, we newly generated 78 Begonia plastid genomes with Novoplasty (Dierckxsens et al., 2017) using the seed sequence of rbcL. These plastid genomes were annotated and the conserved 83 plastid protein‐coding genes were extracted for phylogenetic inference in Geneious 10.0.2 (Biomatters, Auckland, New Zealand). The concatenated nucleotide dataset was evaluated with PartitionFinder (Lanfear et al., 2012) for the optimal data partition scheme and the associated nucleotide substitution models, with an initial partitioning strategy by both locus and codon positions, resulting in 13 partitions. The concatenated dataset was analyzed using RAxML (v.7.2.3) (Stamatakis, 2006) with 500 bootstrap replicates.
Genome synteny
The syntenic blocks between two species were defined by Mcscan (Tang et al., 2008) based on core‐orthologous gene sets identified by Blastp (E‐value ≤ 1e‐5; number of gene pairs required to call synteny ≥ 5). The resulting dot plots were inspected to confirm the paleoploidy level of Begonia in relation to the other genomes by counting the syntenic depth in each genomic region. The synonymous Ks value for homologous gene pairs was calculated using the software Paml (Yang, 2007) and a custom perl script (https://ftp.cngb.org/pub/CNSA/data3/CNP0001056/CNS0227982/CNA0013976/), respectively (see details in Methods S1).
Chlorophyll fluorescence analysis
For Chl fluorescence measurement, the plants were dark‐adapted for 30 min before the measurements with the MAXI version of the Imaging‐PAM M‐Series Chl fluorescence system (Heinz‐Walz Instruments, Effeltrich, Germany), as described by Jin et al. (2018). For measurements of the light‐response curves of photosystem II (PSII) quantum yield (ФPSII), plant leaves were illuminated at the following light intensities: 0, 1, 21, 56, 111, 186, 281, 336, 396, 461, 531, 611, 701 and 801 μmol photons·m−2 s−1. The PSI electron transport rate (ETRI) was measured using light gradients of 0, 5, 13, 31, 89, 167, 209, 325, 496, 754 μmol photons m−2 s−1.
Identification and phylogenetic analysis of light‐harvesting Chl a/b‐binding proteins superfamily
For the identification of LHCs, all LHCs previously described from Arabidopsis and Oryza sativa (Umate, 2010) were retrieved from the database of TAIR (www.arabidopsis.org) and NCBI (www.ncbi.nlm.nih.gov/protein/), respectively. Representative members of the subfamilies of Arabidopsis were used as queries to perform Blastp searches against the protein database of each species with an E‐value cutoff of 1e‐5. Candidate sequences identified as LHC orthologs were then aligned using Mafft (Katoh et al., 2005) to remove those that did not contain the intact domain (PF00504). For phylogenetic analysis, sequences of LHC orthologs of four Begonias, Crocus sativus, as well as Arabidopsis thaliana and O. sativa were aligned using Mafft (Katoh et al., 2005), followed by phylogenetic reconstruction with PhyML (v.3.1) (Guindon & Gascuel, 2003).
Results
Genome sequencing and genome characteristics
Seventy‐eight Begonia species were sequenced to acquire genome skim data for comparative genomic studies (Fig. 1; Table S1). As there is already a draft genome for the Neotropical B. fuchsioides (Griesmann et al., 2018), we selected four species, including one from Africa (B. loranthoides, 2n = 38) and three from Asia (B. masoniana, 2n = 30; B. darthvaderiana, 2n = 30; B. peltatifolia, 2n = 30), for reference genome sequencing (Fig. S2). K‐mer analyses based on 10 × Genomics Chromium reads data indicated an estimated genome size of c. 724, c. 806, c. 797 and c. 349 Mb for B. loranthoides, B. masoniana, B. darthvaderiana and B. peltatifolia, respectively (Table S2). The genomes of B. masoniana and B. darthvaderiana had the highest heterozygosity levels (Fig. S3), 0.96% and 0.98%, respectively, compared to 0.19% for B. loranthoides and 0.27% for B. peltatifolia (Table S3). We combined multiple sequencing and assembly technologies (Fig. S4; Table S4), including linked reads from stLFR and 10 × Genomics Chromium for four species, PacBio single‐molecule real‐time (SMRT) to assist in assembly of the more heterozygous B. masoniana and B. darthvaderiana genomes, and Hi‐C scaffolding strategies for chromosome assembly of all the four genomes. For B. loranthoides and B. peltatifolia, the genomes were assembled into 716.44 Mb (scaffold N50: 6.73 Mb) and 334.09 Mb (scaffold N50: 3.20 Mb) using 10 × Genomics data, with c. 88.55 and 87.13% of assembled sequences anchored onto 19 and 15 pseudochromosomes, respectively (Tables S4, S5). For B. masoniana and B. darthvaderiana, the genome assemblies yielded 799.40 Mb (contig N50: 0.44 Mb) and 771.67 Mb (contig N50: 0.32 Mb) using PacBio long reads data, with c. 98.83 and 97.55% of the assembled sequences anchored onto 15 pseudochromosomes, respectively (Table S5).
To evaluate the quality of the assemblies, RNA‐sequencing reads from root, stem/rhizome, flower, peduncle and leaf tissues were mapped to their cognate assemblies (Table S6). About 90.92–98.83% of the reads were aligned to their corresponding genomes (Table S7). The completeness of the assemblies in terms of gene content was assessed with Benchmarking Universal Single‐Copy Orthologs (Busco) analysis. Of the core 1614 conserved plant genes evaluated, 97.00, 91.00, 92.20 and 96.80% were complete in the assemblies for B. loranthoides, B. masoniana, B. darthvaderiana and B. peltatifolia, respectively; c. 0.80–2.50% of the genes were fragmented (Table S5). Collectively, these results demonstrated that our four genome assemblies were of high quality in terms of contiguity, base accuracy and genome completeness.
Repeat annotation and gene prediction
Repetitive elements were estimated to represent 66.52%, 68.40%, 70.33% and 51.47% of the genome assemblies in B. loranthoides, B. masoniana, B. darthvaderiana and B. peltatifolia, respectively (Table S5). Most of these repeats were TEs that were further subclassified into nine groups (Table S8). Long‐terminal repeat retrotransposons (LTR‐RTs) represent 42.80–65.60% of the genome assembiles, with Gypsy elements being the most abundant transposon superfamily in all four Begonia species (30.39–48.60%), followed by the Copia superfamily (7.32–18.36%) (Table S8). The pattern of LTR distribution patterns varied across the genomes for different elements (Fig. 2a). The density of Gypsy scaled negatively with that of the genes whereas Copia was distributed more evenly across the genome and showed no obvious correlation with gene elements (Figs 2a, S5). This is expected since it is known that Gypsy elements accumulate predominantly in heterochromatin and centromeres, whereas Copia elements are normally scattered across the genome (Neumann et al., 2011).
Fig. 2.
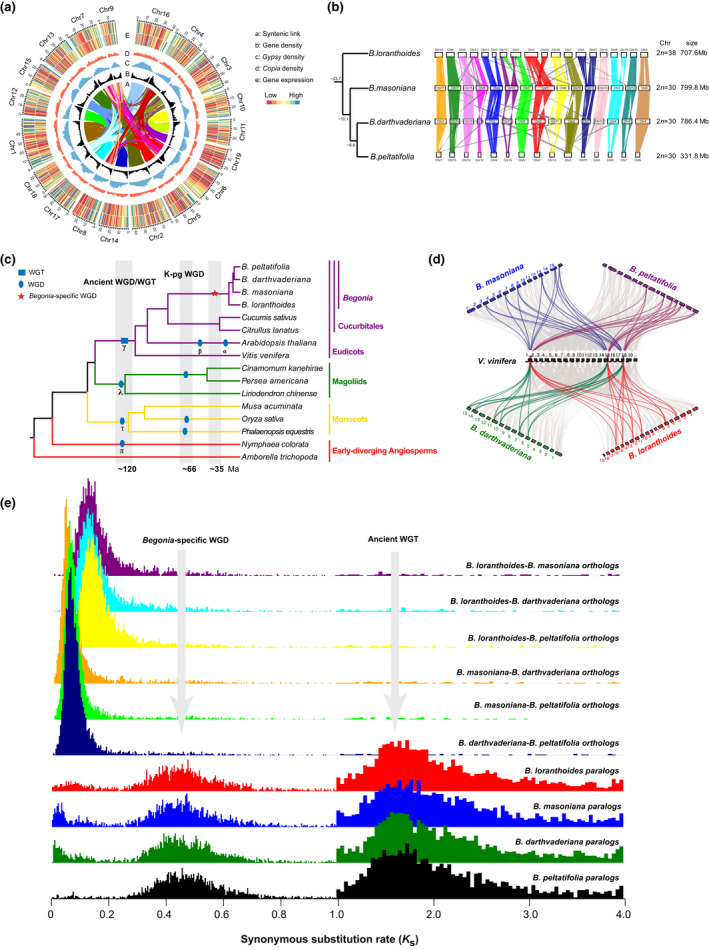
Synteny and lineage‐specific whole‐genome duplication (WGD) in Begonia. (a) Circular view of the Begonia loranthoides genome. a: Lines in the inner circle represent links between synteny‐selected paralogs. b: Gene density, c: Gypsy and d: Copia abundance, e: RNA expression of stem (outer) and leaf (inner). (b) Syntenic blocks in homologous chromosomes between B. loranthoides–B. masoniana, B. masoniana–B. darthvaderiana, and B. darthvaderiana–B. peltatifolia. (c) A simplified phylogenetic tree showing the lineage‐specific WGD in Begonia. The other generally accepted WGDs shown are based on Jiao et al. (2011) and Zhang et al. (2020). (d) Macrosynteny patterns show that three typical ancestral regions in the grape genome can be mapped to six regions in the Begonia genome. Gray wedges in the background highlight major syntenic blocks spanning > 30 genes between the genomes (highlighted by one syntenic set shown in color). (e) Synonymous substitution rate (Ks ) distributions of syntenic blocks for the paralogs of four Begonias and orthologs between either two Begonias are shown in different colors, as indicated. Note the Ks unit in the range 1.0–4.0 is ten‐fold of that in the range 0–1.0.
De novo and homology‐based approaches were combined to predict protein‐coding genes. In total, 22 059, 22 861, 23 444 and 23 010 complete genes were predicted for B. loranthoides, B. masoniana, B. darthvaderiana and B. peltatifolia, respectively, with the highest gene density being 69 genes per Mb in B. peltatifolia, and 28–31 genes per Mb in the other three species (Table S5; Fig. S6), which correlated with the relatively small genome size of B. peltatifolia among the four analyzed Begonia species. The numbers of protein‐coding genes are relatively consistent within Cucurbitales (18 292–32 203), but except for B. peltatifolia the gene densities in Begonia (28–31 genes per Mb) are near two‐fold lower than those in Cucurbitaceae (64–117 genes per Mb), probably due to higher transposon content (Table S9).
Whole genome duplication and gene evolution
The fraction of synonymous substitutions per synonymous site (KS ) distributions of paralogs clearly showed two peaks (Fig. 2e), one around 1.5 representing the γ hexaploidization event shared by the core eudicots (Jaillon et al., 2007; Chanderbali et al., 2017), and the other around 0.5 indicating that a lineage‐specific WGD event occurred in Begonia. By performing a comparative genomic analysis of Begonia with Vitis vinifera, we identified a 2 : 1 syntenic depth ratio (Fig. 2b), which confirms the WGD previously reported in Brennan et al. (2012). We speculate that the WGD event occurred 35 ± 8 Ma (Fig. 2c; Methods S1), and hence before the split of Begonia (median, c. 25 Ma) and its monotypic sister Hillebrandia, the only other genus of Begoniaceae (Moonlight et al., 2018). This is supported by the fact that Hillebrandia has also been found to possess the WGD, probably indeed shared with Begonia (Martínez, 2017). Following the WGD event, 2850 gene families were retained in the common ancestor of the four species of Begonia we sequenced. The retained gene duplicates shared by the four species were considered as core retained genes. This set was enriched for terms such as ‘carbohydrate biosynthetic process’ and ‘nucleotide binding’, and many metabolism pathways such as ‘inositol phosphate metabolism’, ‘starch and sucrose metabolism’, and ‘galactose metabolism’ (Fig. S7).
Individual species retained some specific groups of duplicated genes (Figs S8–S11); for instance, genes annotated as involved in the ‘cutin, suberine and wax biosynthesis’ pathway were differentially retained in B. loranthoides. This gene retention might be associated with the characteristic waxy leaves of this species. Specific retention of genes involved in ‘phenylpropanoid biosynthesis’ and ‘flavonoid biosynthesis’ might be responsible for the colorful leaves of B. masoniana and B. darthvaderiana (Fig. S2). As variegated leaves are commonly found among Begonias and are largely attributed to the accumulation of anthocyanins, we looked more closely at the anthocyanin biosynthesis pathway gene families. We found that in contrast to expansion of gene families of the upstream general phenylpropanoid pathway from Curcubitaceae, Begonia species show significant expansion of gene families related to anthocyanin biosynthesis, especially for Chalcone synthase (CHS) in B. masoniana and Flavanone 3‐hydroxylase (F3H) and Dihydroflavonol 4‐reductase (DFR) in B. darthvaderiana (Fig. S12), and there is recent evidence for relaxed selective constraints and differential expression of paralogs in CHS in Begonia (Emelianova et al., 2021).
Based on a high‐confidence phylogenetic tree reconstructed by 193 single‐copy nuclear gene families of 13 angiosperm species including V. vinifera, Populus trichocarpa, Glycine max, Prunus mume and five species from Cucurbitaceae, we identified gene families that have experienced significant expansions and contractions during the evolution of Begonia and related species (Fig. S13). Twenty gene families, including 1071 genes, were significantly expanded (P < 0.05) in Begonia species compared to the other groups. GO and KEGG enrichment analyses found these to be enriched in terms including ‘zinc ion binding’, ‘transition metal ion binding’ and ‘metal ion binding’ (Table S10), which are primarily involved in the ‘Oxidative phosphorylation’, ‘Endocytosis’ and ‘Pyrimidine metabolism’ pathways (Table S11). Surprisingly, many resistance‐ and defense‐related gene families such as ‘NB‐ARC’ were significantly contracted in the Begonia lineage. The TIR‐NBS‐LRR (TNL) disease resistance gene family (Kim et al., 2009) was completely lost (Fig. S14). We looked for expansion of other disease‐related genes and found that only the Autophagy 17 (APG17) family showed significant expansion in Begonia (Table S12). Other GO terms which were underrepresented in the set of contraction gene families included the ‘protein kinase domain’, ‘Cytochrome p450’ and ‘Terpene synthase’ gene families (Table S13).
Chromosome evolution
To reconstruct the evolutionary events leading to current genome structures in Begonia, homologous chromosome segments between different species were identified. There were 122 shared syntenic blocks in the four species of Begonia sequenced here, which accounted for 74.6%, 78.6%, 67.0% and 74.6% of the B. loranthoides, B. masoniana, B. darthvaderiana and B. peltatifolia genomes, respectively. The lowest percentage of syntenic blocks in B. darthvaderiana among those of the four species was consistent with the low TE proportion in these regions compared to B. masoniana and B. loranthoides (Fig. S15). Synteny analyses between them showed that each chromosome had a nearly one‐to‐one syntenic relationship with chromosomes from other species (Fig. 2b); the relationship was especially strong for those three species with the same chromosome number. Some large inversions could be inferred for each species. One translocation was detected in chromosomes 2 and 17 in B. loranthoides. Chromosome fissions and fusions were identified in the genomes of B. loranthoides and B. masoniana. We suggest that chr9 and chr12, chr1 and chr18, chr3 and chr11, and chr8 and chr19 in B. loranthoides experienced breakage with fusion to chr4, chr1, chr11 and chr15 in B. masoniana, respectively (Fig. 2b).
Conserved gene adjacencies suggest that the ancestral Begonia karyotype reconstructed based on the four species noted above consisted of 22 conserved ancestral regions (CARs), following an ancestral WGD that occurred early in the history of Begoniaceae characterizing all extant members (Fig. S16). From the 22 CARs of the ancestral karyotype, the 15 chromosomes of B. masoniana might be derived by three fusions, and four deletions, while the 15 chromosomes of B. darthvaderiana were shaped through one fission, four fusions and three deletions, the 15 chromosomes of B. peltatifolia through four fissions and 11 fusions, and the 19 B. loranthoides chromosomes through seven fissions and 10 fusions (Fig. S16). Although B. peltatifolia has the same chromosome number as B. masoniana and B. darthvaderiana, it appears to have undergone a large number of chromosome fissions and fusions after the split from their common ancestor. This suggests that genomic rearrangements may be even more frequent in Begonia than apparent from the highly variable chromosome numbers (2n = 16–156) (Dewitte et al., 2009, 2011).
Transposable elements evolution and distribution
Transposable elements generally comprise the bulk of plant genomic DNA and their numbers show a positive correlation with genome size (Wendel et al., 2016). In our Begonia samples, this also appears to be the case: B. peltatifolia has both the smallest genome and the smallest number and proportion of TEs (Fig. S17). Amongst the most abundant superfamilies of TEs, the number of Gypsy and Copia LTR elements were most strongly and positively correlated with genome size (Fig. S17). As the four Begonia species have similar numbers of protein‐coding genes (Table S5), genome size variations between them are essentially attributed to the variation of TE abundance between the different Begonia species.
The investigation of TE representation in our four Begonia genome assemblies showed that they had different compositions of TE superfamilies (Fig. 3a; Table S8), and are quite variable for full ‐length Gypsy and Copia families (Fig. S18). The analysis of full‐length LTR‐RTs indicated several transposon bursts occurred during the last 8 million years, including recent expansions in all species, especially in Gypsy elements compared to Copia (Fig. 3b). When the full‐length Gypsy and Copia families were analyzed in the four Begonia species, they showed an expansion event at 0–2 Ma, with the Reina subgroup of elements expanding 3–4 Ma in B. loranthoides (Fig. 3c).
Fig. 3.
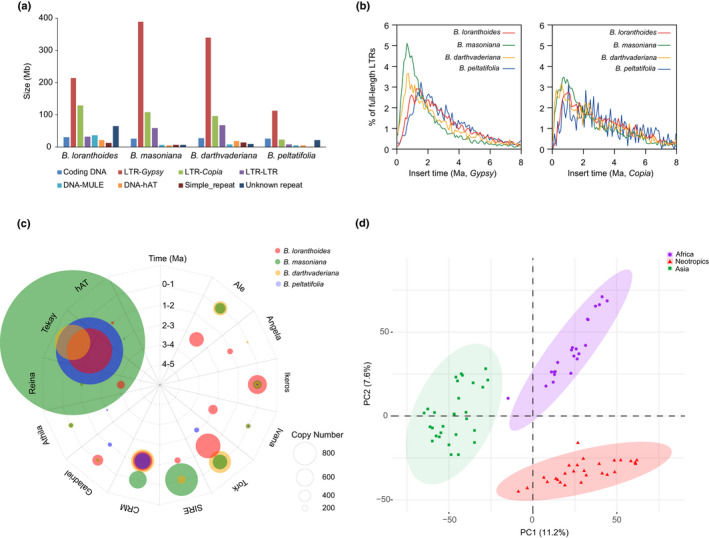
Transposable element (TE) evolution in the Begonia genomes. (a) TE composition of the four Begonia genomes. Stacked histograms represent the contribution of each TE superfamily to the four genomes. The eight most abundant TE superfamilies are shown. (b) Estimation of insertion time of Gypsy and Copia based on analyses of full‐length LTRs in four Begonia species. (c) Activity of 12 prominent full‐length retrotransposon families in four Begonia species. Concentric circles indicate timescale per million years from 6 Ma (center) to the present (outer circle). For each family, the circle size shows the retrotransposon number in four species. Each data point represents the peak activity of that element. (d) Principal component analysis (PCA) based on TE abundances of 74 Begonia species originated from Africa (green), America (orange) and Asia (red).
To determine the historical dynamics of the different lineages of Gypsy and Copia elements in the Begonia genomes, the divergence of their reverse transcriptase (RT) sequences was analyzed. Evolutionary analysis revealed different patterns among different LTR lineages in the four species (Fig. S19). For example, SIRE elements of Copia show a recent activity burst from a few ancestor sequences in B. masoniana, B. loranthoides and B. darthvaderiana, but no burst is observed in B. peltatifolia. A few species‐specific bursts were also observed for the Gypsy Tekay element in all four genomes investigated (Fig. S19). Furthermore, several Copia Ivana and Gypsy CRM copies from the common ancestor of the four Begonia species we investigated have been maintained and are still active (Fig. S19). These findings show that Begonia has a long and ongoing history of active TE elements.
Based on the presence and abundance of TE elements in each species, PCA recovered three well‐circumscribed Begonia lineages, corresponding to the three major geographical groups of the genus, indicating similar TE compositions in geographically restricted clades (Fig. 3d). Closely related species showed similar TE abundance, even in some species that have diverged more than 10 Ma (Fig. S20). Our investigation reveals a congruence of TE abundance with the phylogenetic tree, indicating that TEs are specifically accumulating across clades of species.
To look for effects of TE activity on gene function, we analyzed TE distribution upstream and downstream of genes. The numbers of genes with adjacent Copia and Gypsy elements insertions was very similar for the four Begonia species (Fig. S21a). However, comparison of other enriched TE families surrounding genes indicated different composition patterns in these four Begonia species (Fig. S21b). About 743–2751 (3.23–12.03%) and 1705–2378 (7.41–10.78%) genes have TE insertions in their intron and promoter regions, respectively (Fig. S22). Functional enrichment analysis of those genes with TE insertions identified stress‐related and metabolic process pathways as over‐represented in the set (Tables S14, S15), with distinct differences between the basal African lineage represented by B. loranthoides and the three Asian species (Fig. S23).
Evolution of shade adaptation
As classical shade‐dwelling plants, all Begonias have lower total Chl and lower ratios of Chla/b compared with those of a typical sun‐exposed plant such as Gerbera hybrida (Table S16). Through comparative genomic analysis, we found several gene families belonging to the core components of light perception; that is, Cryptochromes (CRYs), Phototropins (PHOTs), Phytochromes and UV Resistance Locus 8 (UVR8) were obviously expanded in Begonia following the lineage‐specific WGD event compared to other plant species (Figs 4d, S24–S27; Table S17). Furthermore, we found that all these lineage‐specific WGD‐retained photoreceptor genes of B. masoniana displayed differential expression responding to light and dark treatment. Notably, the two retained copies of UVR8 showed divergent expression between light and dark (Fig. 4f). Thus, a higher copy number of these genes might contribute to shade adaptation by increasing the complexity of the light response regulation network (Wu et al., 2019).
Fig. 4.
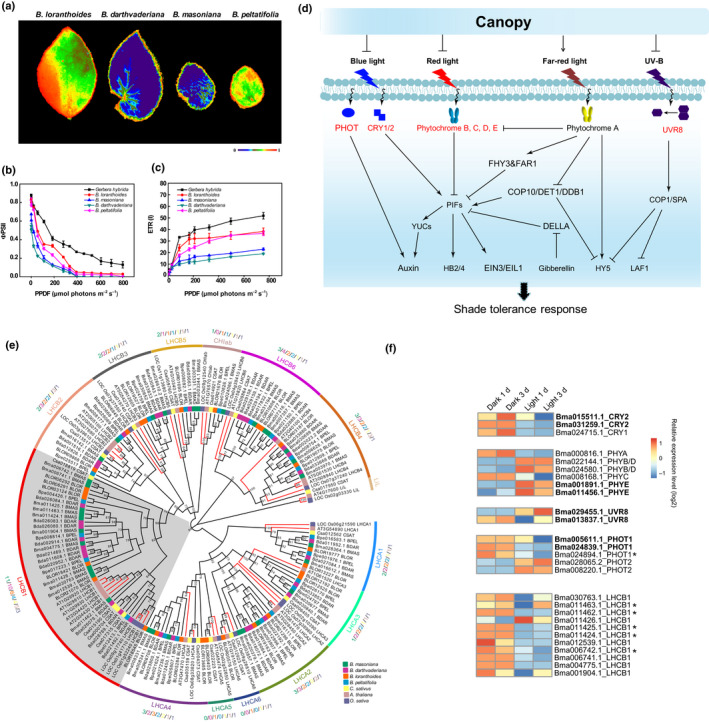
Shade adaptation in Begonia. (a) False‐color images representing maximum photochemical efficiency of PSII (F v/F m) under growth light conditions in four different Begonia species. The false‐color scale ranges from black (0) to red (1) as indicated below the false‐color images. Light‐response curves of PSII quantum yield (ΦPSII) (b) and electron transport rate of PSI (ETRI) (c) of four Begonias and Gerbera hybrida. Four biological replicates were performed in all experiments, and values are given as mean ± SD. (d) Simplified overview of the signal transduction pathway of shade adaptation response modified from Gommers et al. (2013) and Podolec & Ulm (2018). Arrows and blunt arrows indicate positive and negative regulations, respectively. The expansion genes in Begonia are labeled in red. Full names of gene abbreviations are given in Table S17. (e) Phylogenetic tree of light‐harvesting Chla/b‐binding protein (LHC) superfamily show expansion of LHCB in two shade Begonias. Branch of LHCB1 in gray shows prominent expansion. Numbers corresponding to different species are shown beside each subfamily. (f) Expression patterns of photoreceptors and LHCB1 family genes under light and dark treatment. The lineage‐specific whole‐genome duplication retained genes are labeled in bold, and genes marked by an asterisk are derived from tandem duplication.
Although most species of Begonia are shade‐tolerant plants, the extent of shade adaptation varies: B. masoniana and B. darthvaderiana are deep shade plants from karst limestone cave habitats and the interior of tropical rainforests, respectively, whereas B. loranthoides and B. peltatifolia are acclimated to semishaded and more open environments. Based on phylogenetic relationships, the deep shade adaptations of B. masoniana and B. darthvaderiana are independent events (Fig. 1). As expected, the two deep shade species (B. masoniana and B. darthvaderiana) had significantly lower levels of Chl and lower Chla/b ratios (Table S16), along with lower maximum photochemical efficiency of PSII (F v/F m) and quantum yield than the two semishade species (Fig. 4a–c). Comparative gene family analyses revealed significant expansions of the LHCs family in the two ‘deep shade’ Begonias (Fig. 4e; Table S18). Notably LHCB, and especially the LHCB1 subgroup, show prominent expansions in these two shade‐dwelling species due to parallel tandem duplications (Figs 4e, S28). All the duplicated LHCB1 gene pairs showed upregulation in the dark, and downregulation in the light (Fig. 4f), which may indicate their strengthened ability of light harvesting under low light. Together, these results suggest that both WGD‐ and tandem‐driven photoreceptors and light‐harvesting genes contribute to shade adaptation of Begonia.
Genetic variation and admixture patterns
Begonia originated in Africa and spread across all the tropical regions except Australia (Neale et al., 2006). We selected 78 accessions (Fig. 5a) that cover the full distribution of Begonia, representing 37 out of 70 sections, to investigate patterns of genetic variation across the genus. We detected 1 137 696 SNPs and 66 862 small indel variants (< 10 bp). Phylogenetic analysis using a subset of 926 407 SNPs within regions of putatively single‐copy genes (SCGs) clearly differentiate Begonia accessions into three distinct clades (Fig. 5c). A weakly supported African clade is sister to a clade consisting of two monophyletic lineages including one consisting of largely Neotropical accessions and one consisting of exclusively Asian accessions.
Fig. 5.
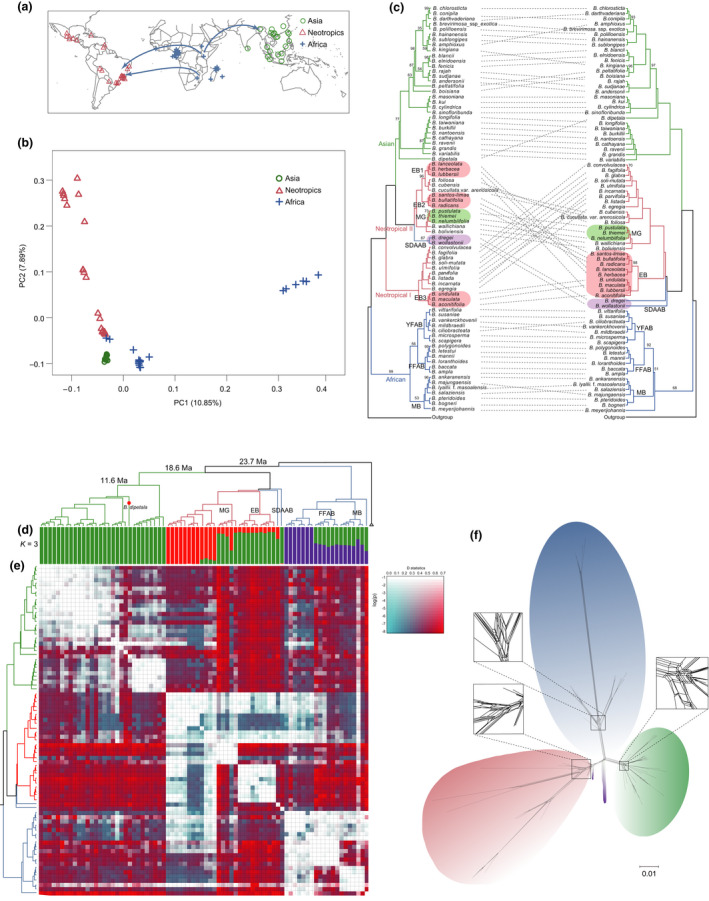
Phylogenomic incongruences and hybridization. (a) Geographic localities of sequenced Begonia individuals and a proposed migration route. (b) Principal component analysis (PCA) of the sequenced 78 Begonia accessions. (c) Cytonuclear conflicts between chloroplast (left) and nuclear (right) phylogenetic trees among 78 sequenced Begonia species with three Cucurbitales species as the outgroup. Branches are maximally supported unless otherwise indicated. (d) Ancestry results from Admixture under the K = 3 model supported by an examination of cross‐validation. (e) Detection of introgression events using Patterson’s D statistics among different Begonia species. (f) SplitsTree network for 78 Begonia individuals. MB, Malagasy Begonia; EB, East Brazil; YFAB, Yellow‐flowered African Begonia; FFAB, Fleshy‐fruited African Begonia; SDAAB, Seasonally dry adapted African Begonia; MG, Mexico section Gireoudia.
Genetic clustering analysis with Admixture showed an optimal value of K = 3 subpopulations (Fig. 5d), which is consistent with the PCA (Fig. 5b). We observed evidence of interspecific admixture within the Neotropical and African Begonia accessions, respectively, and the highest nucleotide diversity (π) in the Neotropical accessions (0.0005755) compared with that of the African (0.0002595) and Asian (0.0002434) accessions (Fig. S29).
Phylogenomic incongruences and hybridizations
Species of Begonia are known to hybridize in nature (Peng & Ku, 2009; Hughes et al., 2018), and previous work (Goodall‐Copestake et al., 2010) identified possible hybridization events early in Begonia evolution. To investigate this further we compared phylogenetic inferences between plastid and nuclear phylogenies. The plastid tree supports the African origin of Begonia and shows successive divergences of four major clades, corresponding to the African, Neotropical I, Neotropical II and Asian clades (Figs 5c, S30, S31). Our plastid phylogeny differs from previous phylogenetic studies based on three plastid markers (Moonlight et al., 2018) in the position of the yellow‐flowered African Begonia (YFAB) clade. The YFAB clade forms a sister group with the Fleshy‐fruited African Begonia (FFAB) clade in our study (Fig. 5c) whereas in previous multilocus studies it diverged at the base of Begonia (Moonlight et al., 2018). The nuclear trees (Figs S32–S34) and TE topology (Fig. S20) in our study consistently recovered a topology with three major geographically restricted clades: the African, Neotropical and Asian clades. Some conspicuous incongruences between nuclear and plastid trees can be identified within the Neotropical clade: the well‐resolved EB (East Brazil) clade containing sections Trachelocarpus, Pereira, Astronthrix, Solananthera, Gaerdtia and Latistigma in the nuclear tree is split into three independent lineages (EB1, EB2, EB3) diffusely distributed between the two Neotropical clades in the plastid tree (Fig. 5c). The position of the two SDAAB (Seasonally dry adapted African Begonia) accessions also show strong cytonuclear incongruence, suggesting hybridization, introgression or incomplete lineage sorting (ILS).
We observed strong discordance for the Neotropical species in the species tree constructed with Astral‐III (Fig. S32). Hybridization or ILS are possible explanations for this and are also suggested by the Splitstree network analysis which revealed a reticulate evolution for these Neotropical accessions (Fig. 5f). To identify possible causes of genetic introgression among Begonia species, we calculated Patterson’s D‐statistics for every triplet (a combination of P1, P2 and P3) in the Begonia phylogeny. The ABBA‐BABA analyses revealed significant introgression in the Neotropical clade between the lineage containing MG (Mexico sect. Gireoudia), and the aforementioned EB clade, the EB clade and the African SDAAB B. wollastonii. We also observed strong genetic introgression between the SDAAB accession B. dregei and the MG clade (Fig. 5e), suggesting that hybridization and introgression might play some roles in the evolution of the Neotropical Begonia. Phylonet network results suggested a hybrid origin of a clade consisting of B. bullatifolia and B. santos‐limae (from the EB clade) from B. radicans and the early Neotropical Begonia colonizers, as well as a hybrid origin of the lineage consisting of Sect. Wageneria and B. soli‐mulata (Fig. S35), lending support for the ABBA‐BABA introgression results.
In contrast to extensive cytonuclear incongruence and putative hybridization and introgression in the Neotropical clade, only a few topological incongruences were detected within Asian and African clades (Fig. 5c). Nonetheless, significant introgression might have occurred between the MB clade and the FFAB clade (Fig. 5e). One independent introgression event was also inferred for the Asian clade, namely the introgression between B. dipetala and the ancestor of the other Asian Begonias (Fig. 5e). These introgressions were also supported by the corresponding Phylonet network results (Fig. S35). Together, these putative introgression and hybridization events are generally in good aggreement with the instances of cytonuclear incongruence and may have played a role in the evolution of Begonia.
Discussion
Putative WGDs have been identified across the eukaryote tree of life, especially in the green plant clade. Many of these WGDs are considered as driving forces contributing to species diversification and evolutionary innovations (Van de Peer et al., 2017; Ren et al., 2018; Leebens‐Mack et al., 2019; Wu et al., 2019). WGD may be followed by lineage‐specific loss of duplicated genes, contributing to adaptation to new niches, survival in response to environmental stress and subsequent rapid accumulations of species diversity (Landis et al., 2018; Ren et al., 2018; Van de Peer et al., 2021). In this study, we confirmed the occurrence of a lineage‐specific WGD event in the common ancestor of Begoniaceae (c. 35 Ma), before the split of cosmopolitan Begonia (median, c. 25 Ma) from the Hawaiian endemic Hillebrandia (Moonlight et al., 2018) (Fig. 2c). As a shade plant, shade adaptation is the key driving force underlying the diversification of Begonia. We provide evidence that the expansion of light signaling pathway genes retained following WGD may have contributed to shade adaptation of Begonia (Fig. 4).
However, WGD is not always associated with species diversification (Landis et al., 2018), as shown in the stark contrast of species diversity between the two genera. The present lack of species diversity in Hillebrandia on the Hawaiian Archipelago is potentially linked to its relict status on the older islands (Clement et al., 2004) and highly homozygous genome (Martínez, 2017). It is tempting to speculate that H. sandwicensis is a dying ember of a once much more species‐rich clade, with diversity having been extinguished in the scramble to colonize the archipelago as islands sank and emerged during its geological evolution.
In addition to WGD, hybridization and introgression have also contributed to the species diversity of Begonia. Through population genomic analysis, we detected several putative hybridization events, especially in the Neotropical clade (Fig. 5). These events may have partially contributed to the exceptional species diversity and genetic diversity of Neotropical Begonia though novel combinations of genotypes, introgression and hybridization‐based genome rearrangements or TE activation. Further genomic studies on Neotropical Begonia might help elucidate which factors have contributed to this high species diversity.
Plant genomes tend to accumulate large amounts of LTRs, and these have been shown to create different landscapes across closely related taxa. The presence and activity of TEs in plant genomes has been widely observed in many other plant groups, from largely studied taxa such Brassicaceae (Joly‐Lopez & Bureau, 2014; Rogivue et al., 2019), Solanaceae (Parisod et al., 2012; de Assis et al., 2020) and Poaceae (Ma et al., 2004; Altinkut et al., 2006; Wyler et al., 2020), to nonmodel plant groups such as Quercus (Mascagni et al., 2019), Passiflora (Sader et al., 2021), Anacyclus (Vitales et al., 2019) or Melampodium (McCann et al., 2020), among many others. We show that transposons are also an important source of genetic variation in Begonia. Two thousand genes in Begonia genomes have TE insertions in their promoter regions (Fig. S22). KEGG functional annotations of these genes with TE insertions in the promoter regions revealed a similar pattern for the three Asian Begonias with enrichment in the pathways of carbohydrate and energy metabolism (Fig. S23). This consistency suggested that TE insertions in the promoter regions might be under some selection constraints rather than neutral and random processes (Baduel et al., 2019). Moreover, the GO enrichment analyses found these genes with TE insertions to be specifically enriched in the function of photosynthesis, negative regulation processes, response to biotic stimulus and stress, and defense response (Tables S14, S15). This result suggests that TE insertions into the regulatory regions in Begonia genomes might play some adaptive role, as has been demonstrated in Arabidopsis (Li et al., 2018; Baduel et al., 2019) and maize (Freeling et al., 2015).
In summary, we have assembled for the first time four chromosome‐level genome assemblies of Begonia, and also provide WGS data for 74 representative species within the genus. Through comparative genomics, we confirmed that a lineage‐specific WGD event pre‐dates the radiation of Begonia and may have provided substantial genetic materials for the phenotypic evolution and shade adaptation. Moreover, we found considerable variation in the compositions and abundance of TEs, and strong phylogenetic signal in TE feature clustering. Species‐specific patterns of TE insertions in promoters and introns might have played a role in the adaptative evolution of Begonia. Furthermore, we provide evidence for introgression during the evolution of Begonia, especially for the Neotropical clade. This study not only provides high‐quality genomic resources for Begonia, but also reveals new insights into the evolution mechanisms of a mega‐diverse clade.
Author contributions
SZ and HL conceived and initiated the study. LL, XC, SD and XG designed the major scientific objectives and managed the project with SZ, HL and DT. HY, XX and Xin Liu coordinated the project. XC, CW, Wenguang Wang and LL conducted the sequencing experiments. DF performed the genome assembly and annotation; XC, LL and SD carried out the repeat analysis. LL, NL, DF, XC, MLiu, WM, ZJL, LZ, TY and JS carried out the comparative genomic analysis and analyzed the gene families. DF, LL, NL, XC, SD, YJ and FC were involved in the WGD analysis. DF, LL, XC, SD, LCD, XG and YG performed the gene annotation and transposable element analysis. SD, XG, NL, DF, XZ, AL, EW and Wei Wang coordinated the phylogenetic analysis. SD, YL, DF, SKS and XC annotated the chloroplast genomes. LL, Xiaofei Liu, CZ and XLang performed Chl fluorescence measurements. LL, YP and LY contribute to cytology analysis. SZ, XLang, Wenguang Wang, Suzhou Zhang, JY and LL coordinated and collected the samples. LL, DF, XG, SD and XC drew and modified the figures. LL, XC, SD, XG, DF, NL, CK, LCD, DCT, DES, YVdP, MH and MLisby wrote and edited most of the manuscript. All authors read and approved the final manuscript. LL, XL, DF, SD, XG and NL contributed equally to this work.
Supporting information
Fig. S1 Current whole genome shotgun (WGS) sampling of Begonia accessions (78 individuals in 37 sections, as marked in red) on the sectional‐level Begonia phylogeny by Moonlight et al. (2018).
Fig. S2 Somatic chromosome counts at metaphase in the four sequenced Begonia species.
Fig. S3 K‐mer analyses of the four Begonia species.
Fig. S4 Flowchart of sequencing and assembly for the four Begonia species.
Fig. S5 Scaffold collinear comparisons between two different species (upper: B. masoniana, bottom: B. peltatifolia) show distinct distributions of different transposon elements.
Fig. S6 Distribution of gene density of four Begonia genomes.
Fig. S7 Analyses of post‐whole‐genome duplication (WGD) retained gene families in Begonia.
Fig. S8 Analysis of post‐WGD retained gene families specific to B. loranthoides.
Fig. S9 Analysis of post‐WGD retained gene families specific to B. masoniana.
Fig. S10 Analysis of post‐WGD retained gene families specific to B. darthvaderiana.
Fig. S11 Analysis of post‐WGD retained gene families specific to B. peltatifolia.
Fig. S12 Expansion of gene families in anthocyanin pathway in Begonia.
Fig. S13 Gene family expansions and contractions along a dated angiosperm phylogeny of 13 selected species.
Fig. S14 Contraction and complete loss of the TNL subgroup of the NBS family in Begonia.
Fig. S15 Comparison of TE proportions in 122 shared syntenic blocks across four Begonia species.
Fig. S16 Reconstruction of the paleogenome of four sequenced Begonia species.
Fig. S17 Number of LTR insertions and genome sizes for 13 angiosperm species.
Fig. S18 Number of shared full‐length LTR families across four Begonia species.
Fig. S19 Neighbour‐joining trees built from RT domain sequence similarities among different lineage‐specific copies identified in Begonia genomes.
Fig. S20 Comparison of nuclear ML tree and abundance clustering of TEs.
Fig. S21 The TE landscape surrounding genes in four Begonia species.
Fig. S22 Impacts of TE insertions on the structure of introns and promoters.
Fig. S23 KEGG enrichment of genes with TE insertion either in introns or in promoters.
Fig. S24 Expansion of Cryptochrome (CRYs) genes in Begonia due to WGD.
Fig. S25 Expansion of Phototropin (PHOT) genes in Begonia due to WGD.
Fig. S26 Expansion of Phytochrome (PHY) genes in Begonia due to WGD.
Fig. S27 Expansion of UV Resistance Locus 8 (UVR8) genes in Begonia due to WGD.
Fig. S28 Schematic diagrams show tandem duplication of LHCB1 genes in B. masoniana and B. darthvaderiana.
Fig. S29 Nucleotide diversity (π) and population divergence (F ST) across the three major groups of Begonia.
Fig. S30 Maximum‐likelihood tree inferred from concatenated nucleotide sequences of Begonia plastid protein coding genes using RAxML.
Fig. S31 Maximum‐likelihood tree inferred from Begonia plastome nucleotide alignment of 156 131 bp using RAxML.
Fig. S32 Maximum‐likelihood tree inferred from a concatenated dataset of 1604 nuclear genes using IQtree with individual gene trees mapped.
Fig. S33 Coalescent super tree inferred with Astral‐III using 1604 nuclear single gene trees.
Fig. S34 Coalescent super tree inferred with Astral‐III using SNPs in 1343 nuclear single gene trees.
Fig. S35 Phylonet network results for three geographically delimited Begonia clades.
Methods S1 Supplemental methods.
Table S1 Summary of 78 Begonia species for whole genome shotgun sequencing.
Table S2 Genome size estimation based on K‐mer analysis.
Table S3 Summary of within‐genome heterozygosity of the four Begonia species.
Table S4 Statistics of genome assemblies.
Table S5 Global statistics of genome assembly and annotation of four Begonia species.
Table S6 Statistics of raw data for whole genome sequencing and RNA‐seq.
Table S7 Statistics of reads mapping to genome sequences for RNA‐seq data from different tissues for four Begonia species.
Table S8 Repetitive elements in four Begonia genomes.
Table S9 Summary of genome information across 13 representative angiosperms.
Table S10 Gene ontology (GO) term enrichment analysis of the expanded gene families of Begonia.
Table S11 Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis of the expanded gene families of Begonia.
Table S12 Number of genes in families related to defense in Begonia and other selected genomes.
Table S13 Statistics and annotations of the contracted gene families in Begonia.
Table S14 The significantly enriched GO terms of biological processes for genes with TEs inserting in introns across four Begonia species.
Table S15 The significantly enriched GO terms of biological processes for genes with TEs inserting in promoter across four Begonia species.
Table S16 Chlorophyll data of the sun‐loving plant Gerbera hybrida and four Begonia species.
Table S17 Comparisons of the gene numbers for the light signaling genes in 10 angiosperm genomes.
Table S18 Comparisons of the gene numbers of the light‐harvesting Chla/b‐binding protein (LHCs) family genes in the seven genomes of Begonia and other angiosperms.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We thank Dr Cecilia Koo Botanic Conservation Center (KBCC, Taiwan), Wenke Dong, Kai Xie, Zhongxuan Wang, Taijiu Zhou for providing materials; Hongbin Wang, Qi Chen (Sun Yat‐sen University) for kindly providing equipment for photosynthesis; Jianjun Jin (Kunming Institute of Botany, Chinese Academy of Sciences, China) for assistance with plastome assembly; Xiaoquan Wang, Hongyan Shan, Daming Zhang (Institute of Botany, Chinese Academy of Sciences, China), Zhiqiang Wu, Li Wang (Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences) and Hong Wu (South China Agricultural University) for suggestions and constructive discussions and advice. This work was supported by funding from the National Key R&D Program of China (2019YFC1711000), the Shenzhen Municipal Government of China (JCYJ20170817145512476), the Shenzhen Urban Administration (201917), the Fairy Lake Botanical Garden (FLSF‐2021‐01), the Public Welfare Forestry Industry Project of State Forestry Administration of China (201504322), and the National Natural Science Foundation of China (31601784). This work is part of the 10KP project. YVdP acknowledges funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement 833522) and from Ghent University (Methusalem funding, BOF.MET.2021.0005.01).
Contributor Information
Shouzhou Zhang, Email: shouzhouz@szbg.ac.cn.
Huan Liu, Email: liuhuan@genomics.cn.
Data availability
All of the raw sequence data including whole‐genome sequencing and transcriptome have been deposited in China National GeneBank Sequence Archive (CNSA) database (https://db.cngb.org/cnsa) under accession number CNP0001056 and National Center for Biotechnology Information (NCBI) under accession number PRJNA791490. The assemblies, gene sequences and annotation data are available at the CNSA database with accession nos. CNA0013973, CNA0013974, CNA0013975 and CNA0013976 for B. darthvaderiana, B. loranthoides, B. masoniana and B. peltatifolia, respectively.
References
- Altinkut A, Raskina O, Nevo E, Belyayev A. 2006. En/Spm‐like transposons in Poaceae species: transposase sequence variability and chromosomal distribution. Cellular & Molecular Biology Letters 11: 214–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Assis R, Baba VY, Cintra LA, Goncalves LSA, Rodrigues R, Vanzela ALL. 2020. Genome relationships and LTR‐retrotransposon diversity in three cultivated Capsicum L. (Solanaceae) species. BMC Genomics 21: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baduel P, Quadrana L, Hunter B, Bomblies K, Colot V. 2019. Relaxed purifying selection in autopolyploids drives transposable element over‐accumulation which provides variants for local adaptation. Nature Communications 10: 5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A, Scossa F, Bolger ME, Lanz C, Maumus F, Tohge T, Quesneville H, Alseekh S, Sørensen I, Lichtenstein G et al. 2014. The genome of the stress‐tolerant wild tomato species Solanum pennellii . Nature Genetics 46: 1034–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AC, Bridgett S, Shaukat Ali M, Harrison N, Matthews A, Pellicer J, Twyford AD, Kidner CA. 2012. Genomic resources for evolutionary studies in the large, diverse, tropical genus, Begonia . Tropical Plant Biology 5: 261–276. [Google Scholar]
- Chanderbali AS, Berger BA, Howarth DG, Soltis DE, Soltis PS. 2017. Evolution of floral diversity: genomics, genes and gamma. Philosophical Transactions of the Royal Society B: Biological Sciences 372: 20150509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement WL, Tebbitt MC, Forrest LL, Blair JE, Brouillet L, Eriksson T, Swensen SM. 2004. Phylogenetic position and biogeography of Hillebrandia sandwicensis (Begoniaceae): a rare Hawaiian relict. American Journal of Botany 91: 905–917. [DOI] [PubMed] [Google Scholar]
- Dewitte A, Leus L, Eeckhaut T, Vanstechelman I, Van Huylenbroeck J, Van Bockstaele E. 2009. Genome size variation in Begonia . Genome 52: 829–838. [DOI] [PubMed] [Google Scholar]
- Dewitte A, Twyford A, Thomas D, Kidner C, Van Huylenbroeck J. 2011. The origin of diversity in Begonia: genome dynamism, population processes and phylogenetic patterns. In: Grillo O, Venora G, eds. The dynamical processes of biodiversity‐case studies of evolution and spatial distribution. Rijeka, Croatia: IntechOpen, 27–52. [Google Scholar]
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: d e novo assembly of organelle genomes from whole genome data. Nucleic Acids Research 45: e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emelianova K, Martinez Martinez A, Campos‐Dominguez L, Kidner C. 2021. Multi‐tissue transcriptome analysis of two Begonia species reveals dynamic patterns of evolution in the chalcone synthase gene family. Scientific Reports 11: 17773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology 16: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M, Makarevitch I, Waters AJ, West PT, Stitzer M, Hirsch CN, Ross‐Ibarra J, Springer NM. 2015. Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genetics 11: e1004915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin DG. 2004. History and concepts of big plant genera. Taxon 53: 753–776. [Google Scholar]
- Gommers CM, Visser EJ, St Onge KR, Voesenek LA, Pierik R. 2013. Shade tolerance: when growing tall is not an option. Trends in Plant Science 18: 65–71. [DOI] [PubMed] [Google Scholar]
- Goodall‐Copestake WP, Perez‐Espona S, Harris DJ, Hollingsworth PM. 2010. The early evolution of the mega‐diverse genus Begonia (Begoniaceae) inferred from organelle DNA phylogenies. Biological Journal of the Linnean Society 101: 243–250. [Google Scholar]
- Griesmann M, Chang Y, Liu X, Song Y, Haberer G, Crook MB, Billault‐Penneteau B, Lauressergues D, Keller J, Imanishi L et al. 2018. Phylogenomics reveals multiple losses of nitrogen‐fixing root nodule symbiosis. Science 361: eaat1743. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology 52: 696–704. [DOI] [PubMed] [Google Scholar]
- Hughes M, Hollingsworth PM. 2008. Population genetic divergence corresponds with species‐level biodiversity patterns in the large genus Begonia . Molecular Ecology 17: 2643–2651. [DOI] [PubMed] [Google Scholar]
- Hughes M, Moonlight PW, Jara‐Muñoz A, Tebbitt MC, Pullan M. 2015. Begonia resource centre. [WWW document] URL http://padme.rbge.org.uk/begonia/page/home/ [accessed 16 April 2021]. [Google Scholar]
- Hughes M, Peng CI, Lin CW, Rubite RR, Blanc P, Chung KF. 2018. Chloroplast and nuclear DNA exchanges among Begonia sect. Baryandra species (Begoniaceae) from Palawan Island, Philippines, and descriptions of five new species. PLoS ONE 13: e0194877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C et al. 2007. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449: 463–467. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu YI, Liang H, Soltis PS et al. 2011. Ancestral polyploidy in seed plants and angiosperms. Nature 473: 97–100. [DOI] [PubMed] [Google Scholar]
- Jin H, Fu M, Duan Z, Duan S, Li M, Dong X, Liu B, Feng D, Wang J, Peng L et al. 2018. LOW PHOTOSYNTHETIC EFFICIENCY 1 is required for light‐regulated photosystem II biogenesis in Arabidopsis . Proceedings of the National Academy of Sciences, USA 115: E6075–E6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly‐Lopez Z, Bureau TE. 2014. Diversity and evolution of transposable elements in Arabidopsis . Chromosome Research 22: 203–216. [DOI] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Research 33: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Kwon SI, Saha D, Anyanwu NC, Gassmann W. 2009. Resistance to the Pseudomonas syringae effector HopA1 is governed by the TIR‐NBS‐LRR protein RPS6 and is enhanced by mutations in SRFR1 . Plant Physiology 150: 1723–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Research 27: 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis JB, Soltis DE, Li Z, Marx HE, Barker MS, Tank DC, Soltis PS. 2018. Impact of whole‐genome duplication events on diversification rates in angiosperms. American Journal of Botany 105: 348–363. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SY, Guindon S. 2012. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution 29: 1695–1701. [DOI] [PubMed] [Google Scholar]
- Leebens‐Mack JH, Barker MS, Carpenter EJ, Deyholos MK, Gitzendanner MA, Graham SW, Grosse I, Li Z, Melkonian M, Mirarab S et al. 2019. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574: 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv [WWW document] URL https://arxiv.org/abs/1303.3997v2 [Google Scholar]
- Li ZW, Hou XH, Chen JF, Xu YC, Wu Q, González J, Guo YL. 2018. Transposable elements contribute to the adaptation of Arabidopsis thaliana . Genome Biology and Evolution 10: 2140–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Devos KM, Bennetzen JL. 2004. Analyses of LTR‐retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Research 14: 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez AM. 2017. A draft genome assembly for Hillebrandia sandwicensis . MSc thesis. University of Edinburgh, Edinburgh, UK. [Google Scholar]
- Mascagni F, Vangelisti A, Giordani T, Cavallini A, Natali L. 2019. A computational comparative study of the repetitive DNA in the genus Quercus L. Tree Genetics & Genomes 16: 11. [Google Scholar]
- McCann J, Macas J, Novak P, Stuessy TF, Villasenor JL, Weiss‐Schneeweiss H. 2020. Differential genome size and repetitive DNA evolution in diploid species of Melampodium sect. Melampodium (Asteraceae). Frontiers Plant Science 11: 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonlight PW, Ardi WH, Padilla LA, Chung K‐F, Fuller D, Girmansyah D, Hollands R, Jara‐Muñoz A, Kiew R, Leong W‐C et al. 2018. Dividing and conquering the fastest–growing genus: towards a natural sectional classification of the mega–diverse genus Begonia (Begoniaceae). Taxon 67: 267–323. [Google Scholar]
- Neale S, Goodall‐Copestake W, Kidner CA. 2006. The evolution of diversity in Begonia . In: Teixeira da Silva JA, ed. Floriculture, ornamental and plant biotechnology‐advances and topical issues. Middlesex, UK: Global Science Books, 606–611. [Google Scholar]
- Neumann P, Navrátilová A, Koblížková A, Kejnovský E, Hřibová E, Hobza R, Widmer A, Doležel J, Macas J. 2011. Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mobile DNA 2: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisod C, Mhiri C, Lim KY, Clarkson JJ, Chase MW, Leitch AR, Grandbastien MA. 2012. Differential dynamics of transposable elements during long‐term diploidization of Nicotiana section Repandae (Solanaceae) allopolyploid genomes. PLoS ONE 7: e50352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C‐I, Ku S‐M. 2009. Begonia x chungii (Begoniaceae), a new natural hybrid in Taiwan. Botanical Studies 50: 241–250. [Google Scholar]
- Podolec R, Ulm R. 2018. Photoreceptor‐mediated regulation of the COP1/SPA E3 ubiquitin ligase. Current Opinion in Plant Biology 45: 18–25. [DOI] [PubMed] [Google Scholar]
- Porebski S, Bailey LG, Bernard RB. 1997. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Molecular Biology Reporter 15: 8–15. [Google Scholar]
- Ren R, Wang H, Guo C, Zhang N, Zeng L, Chen Y, Ma H, Qi J. 2018. Widespread whole genome duplications contribute to genome complexity and species diversity in angiosperms. Molecular Plant 11: 414–428. [DOI] [PubMed] [Google Scholar]
- Rogivue A, Choudhury RR, Zoller S, Joost S, Felber F, Kasser M, Parisod C, Gugerli F. 2019. Genome‐wide variation in nucleotides and retrotransposons in alpine populations of Arabis alpina (Brassicaceae). Molecular Ecology Resources 19: 773–787. [DOI] [PubMed] [Google Scholar]
- Sader M, Vaio M, Cauz‐Santos LA, Dornelas MC, Vieira MLC, Melo N, Pedrosa‐Harand A. 2021. Large vs small genomes in Passiflora: the influence of the mobilome and the satellitome. Planta 253: 86. [DOI] [PubMed] [Google Scholar]
- Song BO, Song Y, Fu Y, Kizito EB, Kamenya SN, Kabod PN, Liu H, Muthemba S, Kariba R, Njuguna J et al. 2019. Draft genome sequence of Solanum aethiopicum provides insights into disease resistance, drought tolerance, and the evolution of the genome. GigaScience 8: giz115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML‐VI‐HPC: maximum likelihood‐based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology 56: 564–577. [DOI] [PubMed] [Google Scholar]
- Tang H, Bowers JE, Wang X, Ming R, Alam M, Paterson AH. 2008. Synteny and collinearity in plant genomes. Science 320: 486–488. [DOI] [PubMed] [Google Scholar]
- Umate P. 2010. Genome‐wide analysis of the family of light‐harvesting chlorophyll a/b‐binding proteins in Arabidopsis and rice. Plant Signaling & Behavior 5: 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer Y, Ashman TL, Soltis PS, Soltis DE. 2021. Polyploidy: an evolutionary and ecological force in stressful times. Plant Cell 33: 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer Y, Mizrachi E, Marchal K. 2017. The evolutionary significance of polyploidy. Nature Reviews Genetics 18: 411–424. [DOI] [PubMed] [Google Scholar]
- Vaser R, Sovic I, Nagarajan N, Sikic M. 2017. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research 27: 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitales D, Álvarez I, Garcia S, Hidalgo O, Nieto Feliner G, Pellicer J, Vallès J, Garnatje T. 2019. Genome size variation at constant chromosome number is not correlated with repetitive DNA dynamism in Anacyclus (Asteraceae). Annals of Botany 125: 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9: e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang OU, Chin R, Cheng X, Wu MKY, Mao Q, Tang J, Sun Y, Anderson E, Lam HK, Chen D et al. 2019. Efficient and unique cobarcoding of second‐generation sequencing reads from long DNA molecules enabling cost‐effective and accurate sequencing, haplotyping, and de novo assembly. Genome Research 29: 798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB. 2017. Direct determination of diploid genome sequences. Genome Research 27: 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel JF, Jackson SA, Meyers BC, Wing RA. 2016. Evolution of plant genome architecture. Genome Biology 17: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Han B, Jiao Y. 2019. Genetic contribution of paleopolyploidy to adaptive evolution in angiosperms. Molecular Plant 13: 59–71. [DOI] [PubMed] [Google Scholar]
- Wyler M, Stritt C, Walser JC, Baroux C, Roulin AC. 2020. Impact of transposable elements on methylation and gene expression across natural accessions of Brachypodium distachyon . Genome Biology and Evolution 12: 1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Wang X, Liu H, Tian Y, Lian J, Yang R, Hao S, Wang X, Yang S, Li Q et al. 2015. The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Molecular Plant 8: 922–934. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wu S, Chang X, Wang X, Zhao Y, Xia Y, Trigiano RN, Jiao Y, Chen F. 2020. The ancient wave of polyploidization events in flowering plants and their facilitated adaptation to environmental stress. Plant, Cell & Environment 43: 2847–2856. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Current whole genome shotgun (WGS) sampling of Begonia accessions (78 individuals in 37 sections, as marked in red) on the sectional‐level Begonia phylogeny by Moonlight et al. (2018).
Fig. S2 Somatic chromosome counts at metaphase in the four sequenced Begonia species.
Fig. S3 K‐mer analyses of the four Begonia species.
Fig. S4 Flowchart of sequencing and assembly for the four Begonia species.
Fig. S5 Scaffold collinear comparisons between two different species (upper: B. masoniana, bottom: B. peltatifolia) show distinct distributions of different transposon elements.
Fig. S6 Distribution of gene density of four Begonia genomes.
Fig. S7 Analyses of post‐whole‐genome duplication (WGD) retained gene families in Begonia.
Fig. S8 Analysis of post‐WGD retained gene families specific to B. loranthoides.
Fig. S9 Analysis of post‐WGD retained gene families specific to B. masoniana.
Fig. S10 Analysis of post‐WGD retained gene families specific to B. darthvaderiana.
Fig. S11 Analysis of post‐WGD retained gene families specific to B. peltatifolia.
Fig. S12 Expansion of gene families in anthocyanin pathway in Begonia.
Fig. S13 Gene family expansions and contractions along a dated angiosperm phylogeny of 13 selected species.
Fig. S14 Contraction and complete loss of the TNL subgroup of the NBS family in Begonia.
Fig. S15 Comparison of TE proportions in 122 shared syntenic blocks across four Begonia species.
Fig. S16 Reconstruction of the paleogenome of four sequenced Begonia species.
Fig. S17 Number of LTR insertions and genome sizes for 13 angiosperm species.
Fig. S18 Number of shared full‐length LTR families across four Begonia species.
Fig. S19 Neighbour‐joining trees built from RT domain sequence similarities among different lineage‐specific copies identified in Begonia genomes.
Fig. S20 Comparison of nuclear ML tree and abundance clustering of TEs.
Fig. S21 The TE landscape surrounding genes in four Begonia species.
Fig. S22 Impacts of TE insertions on the structure of introns and promoters.
Fig. S23 KEGG enrichment of genes with TE insertion either in introns or in promoters.
Fig. S24 Expansion of Cryptochrome (CRYs) genes in Begonia due to WGD.
Fig. S25 Expansion of Phototropin (PHOT) genes in Begonia due to WGD.
Fig. S26 Expansion of Phytochrome (PHY) genes in Begonia due to WGD.
Fig. S27 Expansion of UV Resistance Locus 8 (UVR8) genes in Begonia due to WGD.
Fig. S28 Schematic diagrams show tandem duplication of LHCB1 genes in B. masoniana and B. darthvaderiana.
Fig. S29 Nucleotide diversity (π) and population divergence (F ST) across the three major groups of Begonia.
Fig. S30 Maximum‐likelihood tree inferred from concatenated nucleotide sequences of Begonia plastid protein coding genes using RAxML.
Fig. S31 Maximum‐likelihood tree inferred from Begonia plastome nucleotide alignment of 156 131 bp using RAxML.
Fig. S32 Maximum‐likelihood tree inferred from a concatenated dataset of 1604 nuclear genes using IQtree with individual gene trees mapped.
Fig. S33 Coalescent super tree inferred with Astral‐III using 1604 nuclear single gene trees.
Fig. S34 Coalescent super tree inferred with Astral‐III using SNPs in 1343 nuclear single gene trees.
Fig. S35 Phylonet network results for three geographically delimited Begonia clades.
Methods S1 Supplemental methods.
Table S1 Summary of 78 Begonia species for whole genome shotgun sequencing.
Table S2 Genome size estimation based on K‐mer analysis.
Table S3 Summary of within‐genome heterozygosity of the four Begonia species.
Table S4 Statistics of genome assemblies.
Table S5 Global statistics of genome assembly and annotation of four Begonia species.
Table S6 Statistics of raw data for whole genome sequencing and RNA‐seq.
Table S7 Statistics of reads mapping to genome sequences for RNA‐seq data from different tissues for four Begonia species.
Table S8 Repetitive elements in four Begonia genomes.
Table S9 Summary of genome information across 13 representative angiosperms.
Table S10 Gene ontology (GO) term enrichment analysis of the expanded gene families of Begonia.
Table S11 Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis of the expanded gene families of Begonia.
Table S12 Number of genes in families related to defense in Begonia and other selected genomes.
Table S13 Statistics and annotations of the contracted gene families in Begonia.
Table S14 The significantly enriched GO terms of biological processes for genes with TEs inserting in introns across four Begonia species.
Table S15 The significantly enriched GO terms of biological processes for genes with TEs inserting in promoter across four Begonia species.
Table S16 Chlorophyll data of the sun‐loving plant Gerbera hybrida and four Begonia species.
Table S17 Comparisons of the gene numbers for the light signaling genes in 10 angiosperm genomes.
Table S18 Comparisons of the gene numbers of the light‐harvesting Chla/b‐binding protein (LHCs) family genes in the seven genomes of Begonia and other angiosperms.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
All of the raw sequence data including whole‐genome sequencing and transcriptome have been deposited in China National GeneBank Sequence Archive (CNSA) database (https://db.cngb.org/cnsa) under accession number CNP0001056 and National Center for Biotechnology Information (NCBI) under accession number PRJNA791490. The assemblies, gene sequences and annotation data are available at the CNSA database with accession nos. CNA0013973, CNA0013974, CNA0013975 and CNA0013976 for B. darthvaderiana, B. loranthoides, B. masoniana and B. peltatifolia, respectively.