

Abstract
α-Amino acids are essential for life as building blocks of proteins and components of diverse natural molecules. In both industry and academia, the incorporation of unnatural amino acids is often desirable for modulating chemical, physical, and pharmaceutical properties. We here report a protocol for the economical and practical synthesis of optically active α-amino acids based on an unprecedented stereocontrolled 1,3-nitrogen shift. Our method employs abundant and easily accessible carboxylic acids as starting materials, which are first connected to a nitrogenation reagent, followed by a highly regio- and enantioselective ruthenium- or iron-catalyzed C(sp3)−H amination. This straightforward method displays a very broad scope, providing rapid access to optically active α-amino acids with aryl, allyl, propargyl, and alkyl side chains, and also permits stereocontrolled late-stage amination of carboxylic acid-containing drugs and natural products. 
A direct and straightforward strategy for the synthesis of optically active α-amino acids is the catalytic enantioselective introduction of an amino group in α-position of readily available carboxylic acids.1,2 A number of methods for direct asymmetric C(sp3)−H aminations have been reported and typically exploit the acidity of the C−H group next to a carbonyl functionality for electrophilic aminations via enolate intermediates.3–7 However, most existing methods use aldehydes, ketones or dicarbonyl compounds as starting materials instead of more desirable but less acidic carboxylic acid derivatives. To further complicate matters, the electrophilic amination reagents employed are usually diazo compounds, which lead to amination products that cannot be easily converted to the target α-amino acids.
The insertion of nitrenes into C−H bonds provides a more tunable alternative platform for C(sp3)−H aminations as the reactivity of nitrenes can be controlled by transition metal coordination. In addition, milder reaction conditions can often be used (Figure 1a).8 Much progress has been made employing chiral transition metal catalysts for the enantioselective conversion of prochiral C(sp3)−H bonds into C-N bonds by nitrene insertion.9,10 However, intermolecular nitrene insertion reactions suffer from problems with regioselectivity and enantioselectivity (Figure 1b).11–19 Although this is not the case for intramolecular C(sp3)−H amination reactions, in which well-defined cyclic transition states allow exquisite regio- and stereocontrol, such intramolecular C−H nitrene insertions are typically ring-closing reactions and therefore lack general applicability.20–26 Thus, the catalytic enantioselective synthesis of acyclic amines by catalytic enantioselective C(sp3)−H nitrene insertion remains a challenge, and its application to the synthesis of chiral α-amino acids would be highly desirable.
Fig. 1. Stereocontrolled nitrene C(sp3)−H insertions for the synthesis of α-amino acids.

a, Strategy for the straightforward synthesis of α-amino acids by nitrene C−H insertion. b, Intramolecular nitrene C−H insertion goes via a cyclic transition state to form a cyclic product, while intermolecular nitrene C−H insertion forms acyclic products. c, Proposed 1,3-nitrogen migration which combines the advantages of intramolecular (high regio- and stereocontrol via cyclic TS) and intermolecular (acyclic product formation) C−H insertion chemistry. TS = transition state, PG = protecting group.
We here introduce a strategy that combines the advantages of intramolecular (regio- and stereocontrol via cyclic transition state) and intermolecular C−H nitrene insertion chemistry (more general, acyclic products) by covalently connecting a nitrene precursor to a carboxylic acid functionality. After O−N bond cleavage and binding of both fragments to the catalyst, a cyclic transition state facilitates a stereocontrolled C(sp3)−H amination (Figure 1c). This reaction constitutes an unprecedented stereocontrolled 1,3-nitrogen shift and is applied to the catalytic asymmetric synthesis of α-amino acids.
Results and discussion
Initial experiments and optimization
We commenced our study with carboxylic acid derivatives 1a-e in which the nitrogen bears different electron-withdrawing protecting groups, which is established to be beneficial for generating electrophilic nitrene intermediates. The catalysts tested were “chiral-at-metal” ruthenium complexes in which two bidentate N-(2-pyridyl)-substituted N-heterocyclic carbenes and two acetonitrile ligands are coordinated to a central ruthenium in a C2-symmetric fashion (Table 1).27 Despite all ligands being achiral, the overall chirality required for asymmetric catalysis originates from a stereogenic ruthenium center with Λ or Δ configuration, resulting in a left-handed or right-handed helical topology, respectively. We have previously demonstrated such complexes to be capable of catalyzing enantioselective ring-closing C−H aminations.28,29 Importantly, this class of catalysts feature two vacant coordination sites adjacent to each other (coordination sites of the two labile acetonitrile, highlighted in red), which is essential for the envisioned mechanistic design.
Table 1. Initial experiments and optimization of enantioselective 1,3-nitrogen shift.

| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Protecting group (PG) | Conditionsa | Conversion (%)b | Yield of 2(%)b | Yield of PAA (%)b | e.e. of 2(%)c |
| 1 | Λ-RuDMP | COCF3 (a) | Standard | 0 | - | - | - |
| 2 | Λ-RuDMP | Ts(b) | Standard | 37 | <5 | 23 | n.d. |
| 3 | Λ-RuDMP | Ms (c) | Standard | 50 | <5 | 25 | n.d. |
| 4 | Λ-RuDMP | CO2Me (d) | Standard | 100 | 86 | 12 | 89 |
| 5 | Λ-RuDMP | Troc (e) | Standard | 100 | 93 (91)d | 4 | 95 |
| 6 | Λ-RuTMS | Troc (e) | Standard | 98 | 80 | 9 | 92 |
| 7 | Λ-RuCF3 | Troc (e) | Standard | 19 | 6 | 5 | 90 |
| 8 | Λ-RuH | Troc (e) | Standard | 100 | 85 | 9 | 92 |
| 9 | Λ-RuDMP | Troc (e) | Et3N as base | 100 | 25 | 73 | 95 |
| 10 | Λ-RuDMP | Troc (e) | Na2CO3 as base | 99 | 87 | 5 | 95 |
| 11 | Λ-RuDMP | Troc (e) | THF as solvent | 94 | 15 | 75 | 82 |
| 12 | Λ-RuDMP | Troc (e) | MeOH as solvent | 96 | 0 | 64 | - |
Shown are the deviations from the standard reaction conditions. Standard conditions: substrate 1 (0.1 mmol), Ru catalyst (1 mol%) and the indicated base (3 equiv.) in the indicated solvent (2 mL, c 0.05 M) were stirred at room temperature (25 °C) for 16 hours.
Conversion and yield were determined by 1H NMR analysis using hexamethylbenzene as an internal standard.
Enantiomeric excess (e.e.) values were determined by HPLC on chiral stationary phases.
Yield of the isolated α-amino acid. n.d. = not determined. Troc = CO2CH2CCl3.
We first subjected the trifluoroacetamide substrate 1a to the ruthenium complex RuDMP27 (1 mol%) in CH2Cl2 in the presence of the base K2CO3 (3 equiv.) at room temperature (25 °C) for 16 hours, but were disappointed that no conversion occurred (Table 1, entry 1). Upon replacement of the trifluoroacetamide group with a p-toluenesulfonyl (Ts) group (1b), a 37% conversion was observed, although the undesired phenylacetic acid (PAA) was obtained as the main product in 23% yield as measured by 1H NMR (entry 2). Encouragingly, the amination product 2b was also detected, albeit only in small quantities (<5%). A methylsulfonyl (Ms) group did not provide significantly better results with product 2c formed in trace amounts (<5%) (entry 3). Because of the difficulties associated with cleaving sulfonylamides, sulfonyl groups are synthetically undesirable as amine protecting groups. We therefore tested the more practical methoxycarbonyl protecting group and were delighted to find that the substrate 1d was completely consumed under our standard conditions and the chiral amino acid (R)-2d formed in 86% yield with 89% enantiomeric excess (e.e.) (entry 4). Finally, the best results were obtained with the 2,2,2-trichloroethoxycarbonyl (Troc) protecting group (1e), which provided the amino acid (R)-2e in an excellent 93% yield as determined by 1H NMR and with 95% e.e. (entry 5). Other catalysts with different substituents on the pyridine moieties provided inferior results; replacing the 3,5-dimethylphenyl groups with trimethylsilyl (TMS) groups (RuTMS28), CF3 groups (RuCF3329), or just hydrogen (RuH27) resulted in reduced yields and enantioselectivities (entries 6-8). Table 1 also reveals that CH2Cl2 is the optimal solvent and K2CO3 the optimal base for this stereocontrolled 1,3-nitrogen shift (entries 9-12 and Supplementary Table 1).
Substrate scope for ruthenium catalysis
With the optimal reaction conditions in hand, we investigated the scope of this stereocontrolled 1,3-nitrogen shift. N,N’-Dicyclohexylcarbodiimide (DCC)- or 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI)-induced coupling of readily available carboxylic acids with N-Troc-protected hydroxylamine provided rapid access to a variety of azanyl esters, which were then subjected to the rearrangement under the developed standard conditions (Table 2). We started with functionalizing the phenyl moiety of phenylacetic acid azanyl ester 1e and found that the method tolerates a large variety of different substituents in the phenyl ring, affording the corresponding chiral α-amino acids 3-21 in up to 96% yield and with up to 98% e.e.. Electron-donating functional groups in the phenyl moiety such as methyl (α-amino acid 3), methoxy (α-amino acids 5), 1,3-dioxole (α-amino acid 8), a Fmoc-protected alcohol (α-amino acid 9), hydroxymethyl (α-amino acid 10), and N-pivaloylamide (α-amino acid 19) are tolerated, as are electron-withdrawing substituents such as halogens (α-amino acid 11-14), trifluoromethyl (α-amino acid 15), nitro (α-amino acid 16), acetyl (α-amino acid 17), and methoxycarbonyl (α-amino acid 18). The method also enables the synthesis of the azido α-amino acid 20 (95% yield and 97% e.e.) and the alkynyl α-amino acid 21 (88% yield, 94% e.e.), both of which are building blocks of interest for click chemistry (see Supplementary Table 2 regarding the configurational stability of the generated 30 stereocenter).30 The stereocontrolled 1,3-nitrogen shift was also applied to the synthesis of α-aryl amino acids with benzannulated aromatic and heteroaromatic systems (α-amino acids 22-25) in 77-92% yield and with 90-96% e.e.. Notably, the reaction could be readily scaled up to afford the naphthyl α-amino acid 23 in gram quantities with high yield (91%) and enantiopurity (95% e.e., increased to 99% e.e. after crystallization protocol, see Supplementary Figure 1). It is noteworthy that the stereocontrolled 1,3-nitrogen shift can also be applied to the late-stage functionalization of bioactive compounds. For example, isoxepac31 is an arylacetic acid derivative with anti-inflammatory and analgesic activity, and the azanyl ester of which was converted to the corresponding α-amino acid 26 in 87% yield and with 98% e.e.. Diclofenac,32 a pain medication for the treatment of inflammatory diseases, was converted into its azanyl ester and then rearranged to afford α-amino acid 27 in 54% yield and with 96% e.e., which is remarkable considering the presence of an unprotected and sterically hindered aniline moiety in the ortho-position.
Table 2. Substrate scope for ruthenium catalysis.

| ||||
|---|---|---|---|---|
| α-Aryl amino acids | ||||
 3 86%, 97% e.e. |
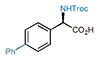 4 86%, 95% e.e. |
 5(para), 96%, 96% e.e. 6(meta), 87%, 96% e.e. 7(ortho), 79%, 85% e.e. |
 8 56%, 92% e.e. |
 9 83%, 93% e.e. |
 10a 64%, 98% e.e. |
 11, X = I, 91%, 94% e.e. 12, X = Br, 93%, 94% e.e. 13, X = CI, 93%, 95% e.e. 14, X = F, 92%, 93% e.e. |
 15 87%, 86% e.e. |
 16 73%, 92% e.e. |
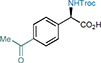 17 79%, 92% e.e. |
 18 91%, 93% e.e. |
 19 93%, 96% e.e. |
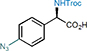 20 95%, 97% e.e. |
 21 88%, 94% e.e. |
 22 92%, 96% e.e. |
 23b 92%, 95% e.e. |
 24 84%, 95% e.e. |
 25 77%, 90% e.e. |
 26(87%, 98% e.e.) from isoxepac |
 27a (54%, 96% e.e.) from diclofenac |
| α-Alkenyl, α-alkynyl & α-alkyl amino acids | ||||
 28 62%, 73% e.e. |
 29, R = Et, 83%, 84% e.e. 30, R = ‘Pr, 88%, 81% e.e. 31, R = nHex, 85%, 82% e.e. 32, R = Ph, 88%, 82% e.e. |
 33 72%, 81% e.e. |
 34c, R = Ph, 46%, 62% e.e. 35c, R = Et, 63%, 64% e.e. |
 36d 20%, 90% e.e. |
| α-Disubstituted amino acids (stereoretentive 1,3-nitrogen shift) | ||||
 Azanyl ester 37 |
 (R)-38e 91%, 98% e.e.f |
 (S)-38 34%, 28% e.e. |
 39f (86%, 99% e.e.) from naproxen |
 40d,f, R = (E)-EtCH=CH, 77%, 87% e.e. 41d,f, R = Et, 57%, 86% e.e. 42d,f, R = cyclohexyl, 25%, 92% e.e. |
Conditions for enantioselective 1,3-nitrogen shift: reactions were carried out with Λ-RuDMP (1-2 mol%), K2CO3 (3 equiv.) and CH2Cl2 (c 0.05 M) at room temperature (25 °C) for 16 h. Enantiomeric excess (e.e.) values were determined by HPLC analysis (see Supplementary Information section 4 for full details).
Isolated after conversion to the methyl ester.
Additional gram-scale reaction performed with 3.5 mmol (1.32 gram) azanyl ester gave 23 in 91% yield with 95% e.e. (see Supplementary Information section 6 for full details).
KHCO3 instead of K2CO3 as a base.
Λ-RuH (2-10 mol%) as the catalyst.
The reaction performed with racemic azanyl ester 37 afforded (R)-38 in 71% yield with 48% e.e.
Reaction performed with enantiopure (S)-azanyl ester.
Reaction performed with enantiopure (R)(S)-38-azanyl ester.
We also investigated C−H aminations at non-benzylic positions, and the stereocontrolled 1,3-nitrogen shift can be applied to the asymmetric synthesis of chiral α-amino acids with alkenyl (28-32) and alkynyl (34 and 35) side chains, albeit with somewhat reduced enantioselectivities (62-84% e.e.). Even the unusual Troc-protected 2-amino-(E)-3,5-hexadienoic acid (33) could be synthesized in 72% yield and with 81% e.e.. These results show that the stereocontrolled 1,3-nitrogen shift is applicable to benzylic, allylic, and propargylic C−H bonds. However, the application to completely non-activated aliphatic methylene groups provides inferior results, with the α-amino acid 36 obtained in merely 20% yield, albeit with a satisfactory 90% e.e..
Next, we investigated the asymmetric synthesis of α-disubstituted α-amino acids by amination of tertiary C−H bonds at a stereogenic carbon center. When we reacted racemic azanyl ester 37 under our standard conditions, the corresponding 2-amino-2-phenylpropanoic acid (R)-38 was obtained in 71% yield but with a modest 48% e.e.. Since the azanyl ester is chiral, we expected high stereodiscrimination between enantiotopic C−H bonds. Indeed, the S-enantiomer (S)-37 (99% e.e.) afforded the α-amino acid (R)-38 smoothly in 91% yield and with 98% e.e. (matched substrate/catalyst combination) while the mirror-imaged substrate (R)-37 reacted sluggishly, providing the α-amino acid (S)-38 in 34% yield with 28% e.e. (mismatched substrate/catalyst combination). The stereospecific C−H amination of chiral non-racemic substrates is general and was applied to the synthesis of the α-disubstituted α-amino acids 39-42 in 25-86% yield and with 86-99% e.e.. For example, the azanyl ester of the nonsteroidal anti-inflammatory drug naproxen, which contains an (S)-configured stereocenter, was converted to the corresponding α-amino acid 39 in 86% yield and 99% e.e., with virtually complete stereoretention. The C−H amination is also applicable to completely nonactivated tertiary C(sp3)−H bonds, as demonstrated by α-amino acids 41 and 42.
Substrate scope for iron catalysis
To expand the substrate scope, we turned our attention to iron catalysis,33 which has additional economic and environmental benefits. Iron-catalyzed C−H bond aminations through nitrenoid intermediates have been reported, including the (racemic) amination of non-activated aliphatic C(sp3)−H bonds.34 We utilized the conversion of azanyl ester 1e to the N-Troc-protected phenylglycine 2e as the initial model reaction to identify a suitable iron catalyst (Table 3). The chiral-at-iron complex Δ-Fe1,35 a lighter homolog of the chiral-at-ruthenium complexes used in this study, provided disappointing results with only trace amounts of product. We next investigated non-heme iron complexes with linear tetradentate N4-donor ligands coordinated in a cis-α-topological configuration. The overall helical topology is similar to the ruthenium catalysts, including the presence of two adjacent labile ligands. Using bis-pyridine complexes (R,R)-Fe2 and (R,R)-Fe3 featuring a rigid chiral 2,2’-bipyrrolidine backbone,36,37 we obtained the desired amino acid (S)-2e in 91% and 75% yield, respectively, albeit with <20% e.e.. Fortunately, replacing the pyridyl moieties with N-methylbenzimidazole38 (BIP ligand) and using labile chloride ligands instead of acetonitrile resulted in the air- and moisture-stable neutral complex (R,R)-[FeCl2(BIP)] ((R,R)-FeBIP), which provided phenylglycine (S)-2e in 95% yield and with a satisfactory 91% e.e.. While the amination of benzylic (2e), allylic (32) and propargylic (34) positions cannot compete with the results obtained for ruthenium catalysis, we found that (R,R)-FeBIP is a superior catalyst for the generation of non-racemic α-amino acids with non-activating aliphatic side chains. Substrates with primary (43-50), secondary (51), and tertiary (36) aliphatic side chains underwent amination in 48-75% yield and with 85-92% e.e.. The method is also suitable for the late-stage amination of more complex molecules. Azanyl ester formation followed by iron-catalyzed stereocontrolled 1,3-nitrogen shift converted lithocholic acid to amino acid 52 (77% yield, 23:1 d.r.). The iron-catalyzed protocol can also be used for the amination of tertiary C(sp3)−H bonds as shown for racemic ibuprofen, which was converted to amino acid 53 in 70% yield and with 85% e.e. in a stereoconvergent transformation.
Table 3. Substrate scope for iron catalysis.

| ||||
|---|---|---|---|---|
 (S)-2e Δ-Fe1 (8 mol%), trace (R,R)-Fe2 (8 mol%), 91%, 15% e.e. (R,R)-Fe3 (8 mol%), 75%, 10% e.e. (R,R)-FeBIP (8 mol%), 95%, 91% e.e. |
 (S)-32 44%, 86% e.e. |
 (S)-34 13%, 44% e.e. |
 43 62%, 91% e.e. |
 44 75%, 92% e.e. |
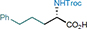 45 60%, 91% e.e. |
 46 70%, 90% e.e. |
 47 53%, 90% e.e. |
 48a,b 51%, 90% e.e. |
 49 57%, 91% e.e. |
 50 65%, 92% e.e. |
 51 48%, 89% e.e. |
 (S)-36 56%, 85% e.e. |
 52a (77%, 23:1 d.r.) from lithocholic acid |
 53 (70%, 85% e.e.) from (±)-ibuprofen |
Reaction conditions: reactions were carried out with (R,R)-FeBIP (8-15 mol%), K2CO3 (3 equiv.) and 1,1,2,2-tetrachloroethane (TCE, c 0.1 M) at 0 °C for 40 h. Enantiomeric excess (e.e.) values were determined by HPLC analysis (see Supplementary Information section 4 for full details).
Reaction performed at room temperature (25 °C) for 16 h.
Isolated after conversion to the methyl ester.
Mechanistic investigations
We performed density functional theory (DFT) calculations to elucidate the reaction mechanism and provide deeper insight (Figure 2a). Ruthenium catalyst RuH and the methylcarbamate substrate 1d were used as the model system. Since the catalyst contains two adjacent labile MeCN ligands and the azanyl ester substrate several suitable coordination sites, it is reasonable to expect that the mechanism proceeds through an initial bidentate coordination. Indeed, the DFT calculations are consistent with this picture. Stepwise displacement of two labile acetonitrile ligands from the Ru center of RuH by substrate 1d gives rise to intermediate I, which proceeds to the chelate coordination mode II with a modest 6.2 kcal/mol increase in free energy. The amide N−H bond in II is now sufficiently acidified to be deprotonated by a weak base such as trimethylamine, leading to the deprotonated chelate intermediate III. A subsequent N−O cleavage occurs through the cyclic transition state TS-I with a free energy barrier of 13.6 kcal/mol to yield Ru nitrene IV in an exergonic fashion. Although the singlet nitrene complex 1IV is calculated to be 2.3 kcal/mol more stable than the triplet nitrene complex 3IV, the C−H cleavage transition state TS-II is significantly lower in energy in the triplet state (3TS-II vs. 1TS-II). This indicates the possibility of a Curtin-Hammett situation in which the presence of the late transition metal ruthenium enhances rapid singlet-triplet spin crossover,39–41 followed by a preferred exergonic hydrogen atom transfer (HAT) from 3IV through the cyclic transition state 3TS-II to afford the diradical 3V. Facile intersystem crossing between intersecting singlet and triplet energy surfaces has also been implicated in other systems involving nitrenoid intermediates, such as Cu- and Ag-catalyzed olefin aziridinations42 and C(sp3)−H amidations promoted by a Ru photocatalyst.43 An experimentally determined kinetic isotope effect (KIE) value of kH/kD = 2.9 indicates that this homolytic C−H cleavage occurs during the rate-determining step and is consistent with the calculated free energy barrier of 13.9 kcal for the hydrogen atom transfer step on the triplet energy surface. A radical mechanism is also supported by an observed Z→E alkene isomerization in an allylic C−H amination reaction (see Supplementary Figure 2 for these mechanistic experiments). Taken together, these computational and experimental results indicate that the dominant mechanistic C−H cleavage pathway involves triplet hydrogen atom transfer, not concerted singlet nitrene insertion. Calculated spin densities of the intermediate 3V, formed in the course of the hydrogen atom transfer, indicate that the single occupied molecular orbitals (SOMOs) are mostly located on the Ru center and the α-carbon of the coordinated substrate so that intermediate 3Vis best represented as a triplet diradical (see framed structure of 3V in Figure 2a). Intermediate 3V undergoes a strongly exergonic radical recombination to form 1VI upon spin crossover to the singlet state through a minimum energy crossing point (MECP). Subsequent O-protonation and release of product2d then completes the catalytic cycle.
Fig. 2. Investigation of the reaction mechanism.

a, Calculated free-energy profile for the 1,3-nitrogen migration of model substrate 1d with RuH as the catalyst (energies provided in kcal/mol). Ruthenium catalyzes the cleavage of the N−O bond of 1d via transition state TS-I leading to the carboxylate coordinated ruthenium nitrene intermediate IV. This is followed by singlet-triplet spin crossover to the triplet state of IV followed by an exergonic hydrogen atom transfer via cyclic transition state TS-II to generate diradical intermediate V in its triplet state. Next, C−N bond formation occurs upon spin crossover to the singlet state through a minimum energy crossing point (MECP) to generate the chelate complex VI. Protonation of the coordinated carboxylate moiety finally releases the product 2d. b, Geometries and non-covalent interaction plots of calculated transition states for the hydrogen atom transfer step to provide insight into the origin of stereodiscrimination (interatomic distances provided in ångströms). The results reveal that in 3TS-II-major-Ph, a favorable π−π stacking interaction exists between the ligand framework of the catalyst and the phenyl substituent of the substrate 1d (TS-II with Ph substituent). This favorable π−π stacking interaction is absent from 3TS-II-minor-Ph. The calculations also confirm that stereodiscrimination can be achieved with a substrate bearing a tBu instead of a Ph side chain (TS-II with tBu substituent). c, Distortion-interaction analysis performed on 3TS-II-major-Ph and 3TS- II-minor-Ph. The more favorable interaction energy in 3TS-II-major-Ph further supports the contribution of stabilizing interactions such as π−π stacking to the stereoselectivity in the formation of phenyl glycine derivative 2d.
Our calculations also provide insight into the origins of stereocontrol in the 1,3-nitrogen shift. Figure 2b shows the geometries of the calculated cyclic transition state for the hydrogen atom transfer step involving the model substrate 1d bearing a phenyl substituent. Non-covalent interaction (NCI) plots reveal that the transition state leading to the experimentally observed major product enantiomer, 3TS-II-major-Ph, is stabilized by a π-π stacking interaction between a pyridine ligand of the catalyst and the phenyl substituent on the substrate. This π-π stacking interaction is absent from the disfavored minor transition state 3TS-II-minor-Ph, which is 1.5 kcal/mol higher in energy than 3TS-II-major-Ph. To evaluate the effect of solvent, when implicit CH2Cl2 solvent was incorporated in the TS geometry optimizations (see Supplementary Information, Sections 8.4-8.6), the free energy difference was found to be 2.1 kcal/mol favoring 3TS-II-major-Ph consistent with the calculations in the gas phase. Following the stereoselective HAT, the resulting diradical V presumably undergoes spin crossover and recombination faster than C−C bond rotation, allowing the stereoselectivity of the HAT step to be preserved in the nitrogenated product. This is consistent with the C−H amination of chiral substrates such as (S)-37, in which the C−H bond is converted to a C-N bond under retention of configuration. Distortion-interaction analysis44 performed on the two transition states indicates that the distortion energies are 1.4 kcal/mol lower for 3TS-II-minor-Ph, but the interaction energy is 2.9 kcal/mol more favorable in 3TS-II-major-Ph (Figure 2c). These results provide further support that stabilizing interactions such as π-π stacking contribute to the observed stereoselectivity in the formation of phenyl glycine derivative 2d. However, our calculations confirm that stereodiscrimination can also be achieved in the absence of attractive π-π stacking interactions as demonstrated for a substrate bearing a tert-butyl instead of a phenyl side chain, which is consistent with the experimental results (e.g. formation of amino acid 36). The transition state producing the major product enantiomer, 3TS-II-major-tBu, was calculated to be 1.3 kcal/mol more favorable than 3TS-II-minor-tBu. This result is in good agreement with the experimentally observed 90% e.e. value. In 3TS-II-minor-tBu, the tert-butyl substituent on the pyridylketone substrate sterically clashes with the acyl Ru-nitrenoid fragment, creating destabilizing close contacts that are absent from 3TS-II-major-tBu. Such destabilizing close contacts are also absent from the transition states with the phenyl side chain. Taken together, these results support the conclusion that effective stereodiscrimination can either be achieved through π-π stacking (in the case of Ph side chain) or steric effects (in the case of tBu side chain), although contributions from additional attractive interactions, such as C−H-π interactions, cannot be ruled out in 3TS-II-major-tBu.45–47
A simplified mechanism based on the DFT calculations and control experiments discussed above is shown Figure 3. The azanyl ester substrate is first activated by N−O cleavage to provide intermediate A (corresponding to intermediate IV in Figure 2a) after deprotonation. A subsequent stereocontrolled hydrogen atom transfer then affords the diradical B (corresponding to intermediate V in Figure 2a), followed by a rapid radical rebound to yield the chelate complex C (corresponding to intermediate VI in Figure 2a). Protonation of the carboxylate induces product dissociation and concludes the catalytic cycle. The net result of the N−O bond fragmentation and subsequent stepwise C−H insertion is an asymmetric 1,3-nitrogen shift to provide non-racemic chiral α-amino acids.
Fig. 3. Summary of proposed simplified mechanism.

The catalytic cycle of the asymmetric 1,3-nitrogen migration commences with N−O cleavage of azanyl ester in presence of metal catalyst, proceeds through stereocontrolled hydrogen atom transfer and subsequent rebound of the diradical, and concludes with release of the α-amino acid and catalyst regeneration.
Conclusions
In conclusion, we report the catalytic enantioselective synthesis of chiral α-amino acids by nitrene C(sp3)−H insertion. The method is based on a unique stereocontrolled 1,3-nitrogen shift from one carboxylic acid oxygen to the α-carbon. Our method employs abundant and easily accessible carboxylic acids as starting materials. Ligation to a nitrene precursor, followed by intramolecular C−H cleavage through a cyclic transition state, ensures high regio- and stereocontrol in the synthesis of non-racemic chiral α-amino acids. Chiral ruthenium and iron catalysis jointly provide a very broad scope, enabling rapid access to optically active α-amino acids with aryl, allyl, propargyl (ruthenium catalysis), and non-activated alkyl (iron catalysis) side chains, including α-disubstituted amino acids by stereoretentive (ruthenium catalysis) or enantioconvergent (iron catalysis) C−H amination. The high functional group tolerance of this method also permits the enantioselective late-stage amination of carboxylic acid-containing drugs and natural products. The Troc-protected amino acids obtained through this protocol can be used directly in synthesis with the Troc group being selectively removable under mild conditions via a reductive Grob fragmentation.48 This strategy will expedite the synthesis of unnatural α-amino acids, which are important building blocks of peptidomimetic drugs, as well as engineered proteins and enzymes with modulated properties.49–51
Methods
General
For the 1H NMR, 13C NMR spectra and HPLC traces of compounds in this Article, details of synthetic procedures, as well as details of the computational study, see the Supplementary Information.
General procedure for ruthenium catalyzed 1,3-nitrogen shift
To a Schlenk tube was added the azanyl ester (1 equiv.), K2CO3 (3 equiv.) and the ruthenium catalyst (1-2 mol% of Λ-RuDMP or 2-10 mol% of Λ-RuH). The tube was evacuated and backfilled with N2 for three times. Dichloromethane (c 0.05 M) was added, and the tube was sealed. The reaction mixture was stirred at room temperature (25 °C) for 16 hours. After completion, the reaction was quenched by a mixture of brine and hydrochloric acid, and subsequently extracted with EtOAc. The combined organic layer was dried and was concentrated under reduced pressure, and the residue was purified by column chromatography on silica gel using a mixture of EtOAc and n-hexane (with 0.1% trifluoroacetic acid as the additive) to obtain non-racemic α-amino acids 2-36 and 38-42.
General procedure for iron catalyzed 1,3-nitrogen shift
To a Schlenk tube was added the azanyl ester (1 equiv.), K2CO3 (3 equiv.) and (R,R)-[FeCl2(BIP)] (8-15 mol%). 1,1,2,2-Tetrachloroethane (c 0.1 M) was added, and the mixture was degassed via freeze-pump-thaw for two times. The tube was sealed, and the reaction mixture was stirred at 0 °C for 40 hours or at room temperature (25 °C) for 16 hours. After completion, the reaction was quenched by a mixture of brine and hydrochloric acid, and subsequently extracted with EtOAc. The combined organic layer was dried and was concentrated under reduced pressure, and the residue was purified by column chromatography on silica gel using a mixture of EtOAc and n-hexane (with 0.1% trifluoroacetic acid as the additive) to obtain the non-racemic α-amino acids 43-53.
Supplementary Material
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 883212). Funding was also provided by the Deutsche Forschungsgemeinschaft (Me 1805/15-1). S.C. thanks Oberlin College for financial support. DFT calculations were performed using the SCIURus, the Oberlin College HPC cluster (NSF MRI 1427949), and the startup allocations awarded by Extreme Science and Engineering Discovery Environment (XSEDE TG-CHE200100).
Footnotes
Author contributions
E.M. and S.C. wrote the manuscript. E.M. and C.-X.Y. conceived the project and devised the experiments for the Ru catalysis. E.M. and X.S. devised the experiments for the Fe catalysis. C.-X.Y. carried out the initial experiments and developed the ruthenium catalysis. X.S. developed the iron catalysis. S.C. performed the DFT calculations.
Competing interests
The authors declare no competing interests.
Data availability
All relevant data supporting the findings of this study, including experimental procedures and compound characterization, NMR and HPLC are available within the Article and its Supplementary Information.
References
- 1.Saghyan AS, Langer P. Asymmetric Synthesis of Non-Proteinogenic Amino Acids. Wiley-VCH; Weinheim: 2016. [Google Scholar]
- 2.Nájera C, Sansano JM. Catalytic asymmetric synthesis of α-amino acids. Chem Rev. 2007;107:4584–4671. doi: 10.1021/cr050580o. [DOI] [PubMed] [Google Scholar]
- 3.Janey JM. Recent advances in catalytic, enantioselective α aminations and α oxygenations of carbonyl compounds. Angew Chem Int Ed. 2005;44:4292–4300. doi: 10.1002/anie.200462314. [DOI] [PubMed] [Google Scholar]
- 4.Bøgevig A, Juhl K, Kumaragurubaran N, Zhuang W, Jørgensen KA. Direct organo-catalytic asymmetric α-amination of aldehydes—a simple approach to optically active α-amino aldehydes, α-amino alcohols, and α-amino acids. Angew Chem Int Ed. 2002;41:1790–1793. doi: 10.1002/1521-3773(20020517)41:10<1790::aid-anie1790>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 5.List B. Direct catalytic asymmetric α-amination of aldehydes. J Am Chem Soc. 2002;124:5656–5657. doi: 10.1021/ja0261325. [DOI] [PubMed] [Google Scholar]
- 6.Kumaragurubaran N, Juhl K, Zhuang W, Bøgevig A, Jørgensen KA. Direct L-proline-catalyzed asymmetric α-amination of ketones. J Am Chem Soc. 2002;124:6254–6255. doi: 10.1021/ja026412k. [DOI] [PubMed] [Google Scholar]
- 7.Morrill LC, Lebl T, Slawin AMZ, Smith AD. Catalytic asymmetric α-amination of carboxylic acids using isothioureas. Chem Sci. 2012;3:2088–2093. [Google Scholar]
- 8.Dequirez G, Pons V, Dauban P. Nitrene chemistry in organic synthesis: still in its infancy? Angew Chem Int Ed. 2012;51:7384–7395. doi: 10.1002/anie.201201945. [DOI] [PubMed] [Google Scholar]
- 9.Park Y, Kim Y, Chang S. Transition metal-catalyzed C−H amination: scope, mechanism, and applications. Chem Rev. 2017;117:9247–9301. doi: 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]
- 10.Ju M, Schomaker JM. Nitrene transfer catalysts for enantioselective C−N bond formation. Nat Rev Chem. 2021;5:580–594. doi: 10.1038/s41570-021-00291-4. [DOI] [PubMed] [Google Scholar]
- 11.Nägeli I, Baud C, Bernardinelli G, Jacquier Y, Moraon M, Müllet P. Rhodium(II)-catalyzed CH insertions with {[(4-nitrophenyl)sulfonyl]imino}phenyl-λ3-iodane. Helv Chim Acta. 1997;80:1087–1105. [Google Scholar]
- 12.Zhou X-G, Yu X-Q, Huang J-S, Che C-M. Asymmetric amidation of saturated C−H bonds catalysed by chiral ruthenium and manganese porphyrins. Chem Commun. 1999:2377–2378. [Google Scholar]
- 13.Kohmura Y, Katsuki T. Mn(salen)-catalyzed enantioselective C−H amination. Tetrahedron Lett. 2001;42:3339–3342. [Google Scholar]
- 14.Yamawaki M, Tsutsui H, Kitagaki S, Anada M, Hashimoto S. Dirhodium(II) tetrakis[N-tetrachlorophthaloyl-(S)-tert-leucinate]: a new chiral Rh(II) catalyst for enantioselective amidation of C−H bonds. Tetrahedron Lett. 2002;43:9561–9564. [Google Scholar]
- 15.Liang C, Robert-Peillard F, Fruit C, Müller P, Dodd RH, Dauban P. Efficient diastereoselective intermolecular rhodium-catalyzed C−H amination. Angew Chem Int Ed. 2006;45:4641–4644. doi: 10.1002/anie.200601248. [DOI] [PubMed] [Google Scholar]
- 16.Nishioka Y, Uchida T, Katsuki T. Enantio- and regioselective intermolecular benzylic and allylic C−H bond amination. Angew Chem Int Ed. 2013;52:1739–1742. doi: 10.1002/anie.201208906. [DOI] [PubMed] [Google Scholar]
- 17.Höke T, Herdtweck E, Bach T. Hydrogen-bond mediated regio- and enantioselectivity in a C−H amination reaction catalysed by a supramolecular Rh(II) complex. Chem Commun. 2013;49:8009–8011. doi: 10.1039/c3cc44197k. [DOI] [PubMed] [Google Scholar]
- 18.Nasrallah A, Boquet V, Hecker A, Retailleau P, Darses B, Dauban P. Catalytic enantioselective intermolecular benzylic C(sp3−H amination. Angew Chem Int Ed. 2019;58:8192–8196. doi: 10.1002/anie.201902882. [DOI] [PubMed] [Google Scholar]
- 19.Jin L-M, Xu P, Xie J, Zhang XP. Enantioselective intermolecular radical C−H amination. J Am Chem Soc. 2020;142:20828–20836. doi: 10.1021/jacs.0c10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang J-L, Yuan S-X, Huang J-S, Yu W-Y, Che C-M. Highly diastereo- and enantioselective intramolecular amidation of saturated C−H bonds catalyzed by ruthenium porphyrins. Angew Chem Int Ed. 2002;41:3465–3468. doi: 10.1002/1521-3773(20020916)41:18<3465::AID-ANIE3465>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 21.Milczek E, Boudet N, Blakey S. Enantioselective C−H amination using cationic ruthenium(II)−pybox catalysts. Angew Chem Int Ed. 2008;47:6825–6828. doi: 10.1002/anie.200801445. [DOI] [PubMed] [Google Scholar]
- 22.Ichinose M, Suematsu H, Yasutomi Y, Nishioka Y, Uchida T, Katsuki T. Enantioselective intramolecular benzylic C−H bond amination: efficient synthesis of optically active benzosultams. Angew Chem Int Ed. 2011;50:9884–9887. doi: 10.1002/anie.201101801. [DOI] [PubMed] [Google Scholar]
- 23.Zalatan DN, Du Bois J. A chiral rhodium carboxamidate catalyst for enantioselective C−H amination. J Am Chem Soc. 2008;130:9220–9221. doi: 10.1021/ja8031955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang K, Torker S, Wojtas L, Zhang XP. Asymmetric induction and enantiodivergence in catalytic radical C−H amination via enantiodifferentiative H-atom abstraction and stereoretentive radical substitution. J Am Chem Soc. 2019;141:12388–12396. doi: 10.1021/jacs.9b05850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park Y, Chang S. Asymmetric formation of γ-lactams via C−H amidation enabled by chiral hydrogen-bond-donor catalysts. Nat Catal. 2019;9:219–227. [Google Scholar]
- 26.van Vliet KM, de Bruin B. Dioxazolones: stable substrates for the catalytic transfer of acyl nitrenes. ACS Catal. 2020;10:4751–4769. [Google Scholar]
- 27.Zheng Y, Tan Y, Harms K, Marsch M, Riedel R, Zhang L, Meggers E. Octahedral ruthenium complex with exclusive metal-centered chirality for highly effective asymmetric catalysis. J Am Chem Soc. 2017;139:4322–4325. doi: 10.1021/jacs.7b01098. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Z, Chen S, Qin J, Nie X, Zheng X, Harms K, Riedel R, Houk KN, Meggers E. Catalytic enantioselective intramolecular C(sp3)−H amination of 2-azidoacetamides. Angew Chem Int Ed. 2019;58:1088–1093. doi: 10.1002/anie.201811927. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Z, Tan Y, Yamahira T, Ivlev S, Xie X, Riedel R, Hemming M, Kimura M, Meggers E. Enantioselective ring-closing C−H amination of urea derivatives. Chem. 2020;6:2024–2034. [Google Scholar]
- 30.Thirumurugan P, Matosiuk D, Jozwiak K. Click chemistry for drug development and diverse chemical−biology applications. Chem Rev. 2013;113:4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- 31.Ueno K, Kubo S, Tagawa H, Yoshioka T, Tsukada W, Tsubokawa M, Kojima H, Kasahara A. 6,11-Dihydro-11-oxodibenz[b,e]oxepinacetic acids with potent antiinflammatory activity. J Med Chem. 1976;19:941–946. doi: 10.1021/jm00229a017. [DOI] [PubMed] [Google Scholar]
- 32.Krupp PJ, Menassé-Gdynia R, Sallmann A, Wilhelmi G, Ziel R, Jaques R. Sodium [o-[(2,6-dichlorophenyl)-amino]-phenyl]-acetate (GP 45 840), a new non-steroidal anti-inflammatory agent. Experientia. 1973;29:450–452. doi: 10.1007/BF01926776. [DOI] [PubMed] [Google Scholar]
- 33.Bauer I, Knölker H-J. Iron catalysis in organic synthesis. Chem Rev. 2015;115:3170–3387. doi: 10.1021/cr500425u. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, You T, Wang H-X, Tang Z, Zhou C-Y, Che C-M. Iron- and cobalt-catalyzed C(sp3)−H bond functionalization reactions and their application in organic synthesis. Chem Soc Rev. 2020;49:5310–5358. doi: 10.1039/d0cs00340a. [DOI] [PubMed] [Google Scholar]
- 35.Hong Y, Jarrige L, Harms K, Meggers E. Chiral-at-iron catalyst: expanding the chemical space for asymmetric earth-abundant metal catalysis. J Am Chem Soc. 2019;141:4569–4572. doi: 10.1021/jacs.9b01352. [DOI] [PubMed] [Google Scholar]
- 36.Chen MS, White MC. A predictably selective aliphatic C−H oxidation reaction for complex molecule synthesis. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- 37.Gormisky PE, White MC. Catalyst-controlled aliphatic C−H oxidations with a predictive model for site-selectivity. J Am Chem Soc. 2013;135:14052–14055. doi: 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- 38.Mitra M, Cusso O, Bhat SS, Sun M, Cianfanelli M, Costas M, Nordlander E. Highly enantioselective epoxidation of olefins by H2O2 catalyzed by a non-heme Fe(II) catalyst of a chiral tetradentate ligand. Dalton Trans. 2019;48:6123–6131. doi: 10.1039/c8dt04449j. [DOI] [PubMed] [Google Scholar]
- 39.Poli R, Harvey JN. Spin forbidden chemical reactions of transition metal compounds. New ideas and new computational challenges. Chem Soc Rev. 2003;32:1–8. doi: 10.1039/b200675h. [DOI] [PubMed] [Google Scholar]
- 40.Harvey JN, Poli R, Smith KM. Understanding the reactivity of transition metal complexes involving multiple spin states. Coord Chem Rev. 2003;238–239:347–361. [Google Scholar]
- 41.Yersin H, Humbs W. Spatial extensions of excited states of metal complexes. Tunability by chemical variation. Inorg Chem. 1999;38:5820–5831. [Google Scholar]
- 42.Maestre L, Sameera WMC, Díaz-Requejo MM, Maseras F, Pérez PJ. A general mechanism for the copper- and silver-catalyzed olefin aziridination reactions: concomitant involvement of the singlet and triplet pathways. J Am Chem Soc. 2013;135:1338–1348. doi: 10.1021/ja307229e. [DOI] [PubMed] [Google Scholar]
- 43.Jung H, Keum H, Kweon J, Chang S. Tuning triplet energy transfer of hydroxamates as the nitrene precursor for intramolecular C(sp3)−H amidation. J Am Chem Soc. 2020;142:5811–5818. doi: 10.1021/jacs.0c00868. [DOI] [PubMed] [Google Scholar]
- 44.Ess DH, Houk KN. Theory of 1,3-dipolar cycloadditions: distortion/interaction and frontier molecular orbital models. J Am Chem Soc. 2008;130:10187–10198. doi: 10.1021/ja800009z. [DOI] [PubMed] [Google Scholar]
- 45.Krenske EH, Houk KN. Aromatic interactions as control elements in stereoselective organic reactions. Acc Chem Res. 2013;46:979–989. doi: 10.1021/ar3000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wheeler SE. Understanding substituent effects in noncovalent interactions involving aromatic rings. Acc Chem Res. 2013;46:1029–1038. doi: 10.1021/ar300109n. [DOI] [PubMed] [Google Scholar]
- 47.Wheeler SE, Bloom JWG. Toward a more complete understanding of noncovalent interactions involving aromatic rings. J Phys Chem A. 2014;118:6133–6147. doi: 10.1021/jp504415p. [DOI] [PubMed] [Google Scholar]
- 48.Isidro-Llobet A, Álvarez M, Albericio F. Amino acid-protecting groups. Chem Rev. 2009;109:2455–2504. doi: 10.1021/cr800323s. [DOI] [PubMed] [Google Scholar]
- 49.Blaskovich MAT. Unusual amino acids in medicinal chemistry. J Med Chem. 2016;59:10807–10836. doi: 10.1021/acs.jmedchem.6b00319. [DOI] [PubMed] [Google Scholar]
- 50.Agostini F, Völler J-S, Koksch B, Acevedo-Rocha CG, Kubyshkin V, Budisa N. Biocatalysis with unnatural amino acids: enzymology meets xenobiology. Angew Chem Int Ed. 2017;56:9680–9703. doi: 10.1002/anie.201610129. [DOI] [PubMed] [Google Scholar]
- 51.Drienovská I, Roelfes G. Expanding the enzyme universe with genetically encoded unnatural amino acids. Nat Catal. 2020;3:193–202. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data supporting the findings of this study, including experimental procedures and compound characterization, NMR and HPLC are available within the Article and its Supplementary Information.