

Abstract
Host-pathogen interactions impose recurrent selective pressures that lead to constant adaptation and counter-adaptation in both competing species. Here, we sought to study this evolutionary arms-race and assessed the impact of the innate immune system on viral population diversity and evolution, using Drosophila melanogaster as model host and its natural pathogen Drosophila C virus (DCV). We isogenized eight fly genotypes generating animals defective for RNAi, Imd and Toll innate immune pathways as well as pathogen sensing and gut renewal pathways. Wild-type or mutant flies were then orally infected with DCV, and the virus was serially passaged ten times via reinfection in naïve flies. Viral population diversity was studied after each viral passage by high-throughput sequencing, and infection phenotypes were assessed at the beginning and at the end of the evolution experiment. We found that the absence of any of the various immune pathways studied increased viral genetic diversity while attenuating virulence. Strikingly, these effects were observed in a range of host factors described as having mainly antiviral or antibacterial functions. Together, our results indicate that the innate immune system as a whole, and not specific antiviral defense pathways in isolation, generally constrains viral diversity and evolution.
Introduction
Interaction between hosts and pathogens trigger defense and counter-defense mechanisms that often result in reciprocal adaptation and coevolution of both organisms1. Empirical evidence of such arms-races involving both species can be drawn from genome-wide analysis of hosts and pathogens and in experimental evolution settings. For example, evolutionary analysis of mammalian genomes has revealed evidence of host-virus coevolution between different retroviruses and antiviral factors2,3, and in plants, host resistance genes and virulence genes encoded by pathogens have been found to co-evolve4. Likewise, between bacteria and their infecting bacteriophages, experimental co-evolution studies resulted in the occurrence of genetic variants in both a bacterial lipopolysaccharide synthesis gene and the phage tail fiber gene which binds to lipopolysaccharide during adsorption5. In nematodes and their pathogenic bacteria, the number of toxin-expressing plasmids varies during adaptation to the host6.
In insects, analyses of sequences within and between Drosophila species have shown evidence of adaptive evolution in immunity related genes7–10. In a study that deep sequenced siRNAs from mosquitoes infected with West Nile virus, it was found that the regions of the viral genome more intensively targeted by RNA interference (RNAi) contained a higher number of mutations than genomic regions less affected by this pathway, suggesting that this antiviral defense mechanism imposes a selective pressure on the viral population11. Similar observations on the selective pressure imposed by the RNAi pathway on viral evolution have been made in plant- and human-infecting viruses12–16. Drosophila melanogaster is a well-studied insect model to decipher virus-host interactions and therefore the impact of host antiviral immunity on viral diversity and evolution. Different Drosophila immune pathways and mechanisms are involved in antiviral defense17,18. As is the case for all invertebrates, defense against pathogens in Drosophila relies on innate immunity, which constitutes the first and only defense against microbes. Innate immunity is characterized by the recognition of pathogen derived molecules, called pathogen-associated molecular patterns (PAMPs), by host encoded receptors (pathogen recognition receptors – PRRs), which leads to a rapid defense response.
The RNAi mechanism is known to play a central role in Drosophila antiviral defense, mainly through the action of the small interfering (si) RNA pathway19–22. Antiviral RNAi is triggered by virtually all insect-infecting viruses, resulting in targeting of the viral genome in a sequence-specific manner to control infection. Several other pathways have antiviral properties in flies, but their roles against viruses seem to be virus specific. The Toll and Imd (Immune deficiency) pathways, originally described to be involved in antibacterial and antifungal responses, have been shown to play a role in antiviral defense against Drosophila C virus (DCV), cricket paralysis virus (CrPV), Drosophila X virus, Nora virus, and Flock house virus23–26. The Janus kinase signal transducers and activators of transcription (JAK-STAT) pathway can be activated upon DCV or CrPV infection in flies, triggering the expression of antiviral factors27,28.
DCV, a positive-sense single stranded RNA virus from the genus Cripavirus within the Dicistrioviridae family and Picornavirales order29, is a well characterized natural pathogen of the fruit fly that can be found in laboratory and wild populations30. As for many other Drosophila-infecting viruses, defense against DCV depends on the joint action of different innate immune pathways and mechanisms. RNAi, Toll and Imd pathways, but also the protein encoded by the gene Vago, play a role in the defense against this virus20,24–27,31–33. DCV is thought to be naturally acquired by ingestion30,34,35. For orally acquired pathogens, the digestive tract, and the gut in particular, represents the first host defense barrier. Despite many studies using oral bacterial infections36, the role of gut-specific antiviral responses in Drosophila is not fully understood. Responses triggered against bacterial pathogens in the gut include the production of reactive oxygen species and antimicrobial peptides, as well as tissue repair and regeneration mechanisms37. Furthermore, the maintenance of gut homeostasis after tissue damage caused by pathogenic bacteria relies on the activity of JAK-STAT and epidermal growth factor receptor (EGFR) pathways, amongst others37–39. In the hallmark of viral infections, a role of the Imd and extracellular-signal-regulated kinase (ERK) pathways in the antiviral response in the gut has been suggested24,40. It is important to note that, like many other RNA viruses with error-prone polymerases and fast replication kinetics, DCV exists as large populations composed of a cloud of genetically related mutant variants known as viral quasispecies or mutant swarms41. Viral mutant swarms constitute a dynamic repertoire of genetic and phenotypic variability that renders great adaptability.
In this work, we leveraged the vast knowledge on antiviral mechanisms, the extensive genetic tool-box available for D. melanogaster, the intrinsic variability of the DCV mutant swarm, and the great depth power of next generation sequencing to study the impact of innate immune pathways on viral diversity and evolution. We aimed to determine not only if each pathway has a specific impact on the selective pressure imposed to DCV mutant swarms, but also their relative impact. In addition, we investigated possible links between selected viral variants (viral function) and specific defense mechanisms. Our results with infections in flies defective for several immune pathways show that the host genotype has an impact on viral genetic diversity regardless of the immune pathway being affected and this is accompanied by an attenuation of the virulence along evolutionary passages. We also describe complex mutation dynamics, with several examples of clonal interference in which increases in frequency of adaptive mutations have been displaced by other mutations of stronger effect that arose in different genetic backgrounds. Overall, our results highlight that innate immune pathways constrain RNA virus evolution and further demonstrate that antiviral responses in Drosophila are likely polygenic.
Results
Production of fly mutant lines for innate immune pathways
To determine the impact of the innate immune system on virus population diversity and evolution, we selected fly lines with impaired function in genes belonging to most of the Drosophila innate immune pathways: RNAi, Toll and Imd. We selected genes involved both upstream and downstream of the immune pathways, such as receptors or ligands that trigger the immune response, and effectors of the response (Figure 1a). For the RNAi pathway, Dicer 2 (Dcr-2) and Argonaute 2 (Ago-2); for the Toll pathway, the ligand of Toll receptor Spätzle (spz), and the NF-κB transcription factor Dorsal-related immunity factor (Dif); for the Imd pathway, the NF-κB transcription factor Relish (Rel). We also added to the study the host factor Vago (Vago), that is upregulated during viral infections in a Dicer 2-dependent manner. Because DCV is orally acquired, and to explore the impact of gut homeostasis on the antiviral response, a mutant line for Epidermal growth factor receptor (Egfr), a gene involved in gut epithelium renewal, was also included in our panel. With the exception of Egfr and Dif, all of the selected genes were previously described to play an antiviral role against DCV infection19–21,23–25. It is important to mention that, in contrast to the RNAi antiviral mechanism that relies on the direct interaction between the components of the RNAi pathway and the viral genome, the molecular mechanisms underlying the antiviral responses mediated by Toll, Imd, and Vago in Drosophila remain largely unknow.
Figure 1. Experimental design.

a) Simplified scheme of D. melanogaster immune pathways. The siRNA pathway is triggered by virus-derived dsRNA, recognized by Dcr-2 and cleaved into viral siRNAs, which guide the recognition and cleavage of viral RNA by Ago-2 controlling virus infection. The Toll pathway is activated when spz binds to the Toll receptor, leading to the activation of NF-κB transcription factors (e.g. Dif). The Imd pathway is triggered after the recognition of microbial peptidoglycans (PGN) by PGRP-LC, ultimately leading to the activation of Rel. Toll and Imd pathways induce the expression of antimicrobial peptides to control infection. The expression of Vago is induced after infection with DCV. The EGFR pathway is triggered in the gut after bacterial damage and leads to delamination of enterocytes and renewal. Created with BioRender.com. b) Scheme of the DCV evolution experiment. To produce the DCV stock, w1118 female flies were injected with DCV from a stock produced in S2 Drosophila cells (S2 DCV stock), placed in cages containing fresh Drosophila medium, left for 3 days and then removed to place in these DCV contaminated cages N = 500 w1118 or immune deficient males and females. Flies were feed ad libitum for 3 days, moved to a clean cage for 1 day, and further placed into a new clean cage for 4 days, when they were harvested (DCV passage 1, P = 1). A new group of 500 flies was placed in contaminated cages. This procedure was repeated 10 times (10 DCV passages, P = 1 to P = 10) and replicated twice. For each passage and fly genotype, high-throughput sequencing and viral stocks for phenotypic characterization were obtained. c) Scheme of DCV genome and the location of primers used to amplify the genome. The viral genome is composed of single-stranded positive-sense RNA and contains two ORFs. ORF 1 encodes for the non-structural viral proteins, 1A: viral silencing suppressor, 2C: RNA helicase, VPg: viral genome-linked protein, 3C: protease, RdRp: RNA-dependent RNA polymerase, 2B and 3A: assembly of the viral replication complex. ORF 2 encodes for DCV structural proteins VP1 to VP4 which constitute the viral capsid.
To reduce genetic variation due to differences in genetic background, mutant flies were isogenized prior to beginning viral evolution experiments. Homozygous loss-of-function lines for Dcr-2 (Dcr-2L811fsX and Dcr-2R416X), Ago-2 (Ago-2414), spz (spz2), Dif (Dif1), Rel (RelE20), and Vago (VagoΔM10) and a hypomorphic mutant line for Egfr (Egfrt1) were produced in the same genetic background by crossing parental lines at least 10 times to w1118 flies. Infection phenotypes of the newly produced fly lines were characterized by following their survival after inoculation with DCV by intrathoracic injection (Supplementary Figure 1a). As previously described, Dcr-2L811fsX/L811fsX, Dcr-2R416X/R416X and Ago-2414/414 mutants infected with DCV died faster than w1118 flies20,21, as well as VagoΔM10/ΔM10mutants33. Toll pathway mutants spz2/2 and Dif1/1 and Imd pathway mutant RelE20/E20 were less sensitive to DCV infection than w1118 flies as they died later than w1118 flies (Supplementary Figure 1a); however, these mutants maintained the previously observed increased susceptibility to infection by Gram + and Gram – bacteria, respectively (Supplementary Figure 1b and 1c). No difference in virus-induced mortality was found between w1118 and Egfrt1/t1 mutant flies (Supplementary Figure 1a). This set of isogenic mutant flies with contrasting phenotypes to DCV infection provided us with the host model system to perform the viral evolution experiment.
Experimental DCV evolution
To study the impact of innate immune pathways on virus population diversity and evolution, DCV from a viral stock was serially passaged (P = 1 to P = 10) in w1118 flies and in the isogenic innate immune deficient fly lines (Figure 1a and 1b). DCV population diversity was studied after each passage by next generation sequencing (NGS) and DCV virulence was analyzed at the beginning and at the end of the evolution experiment.
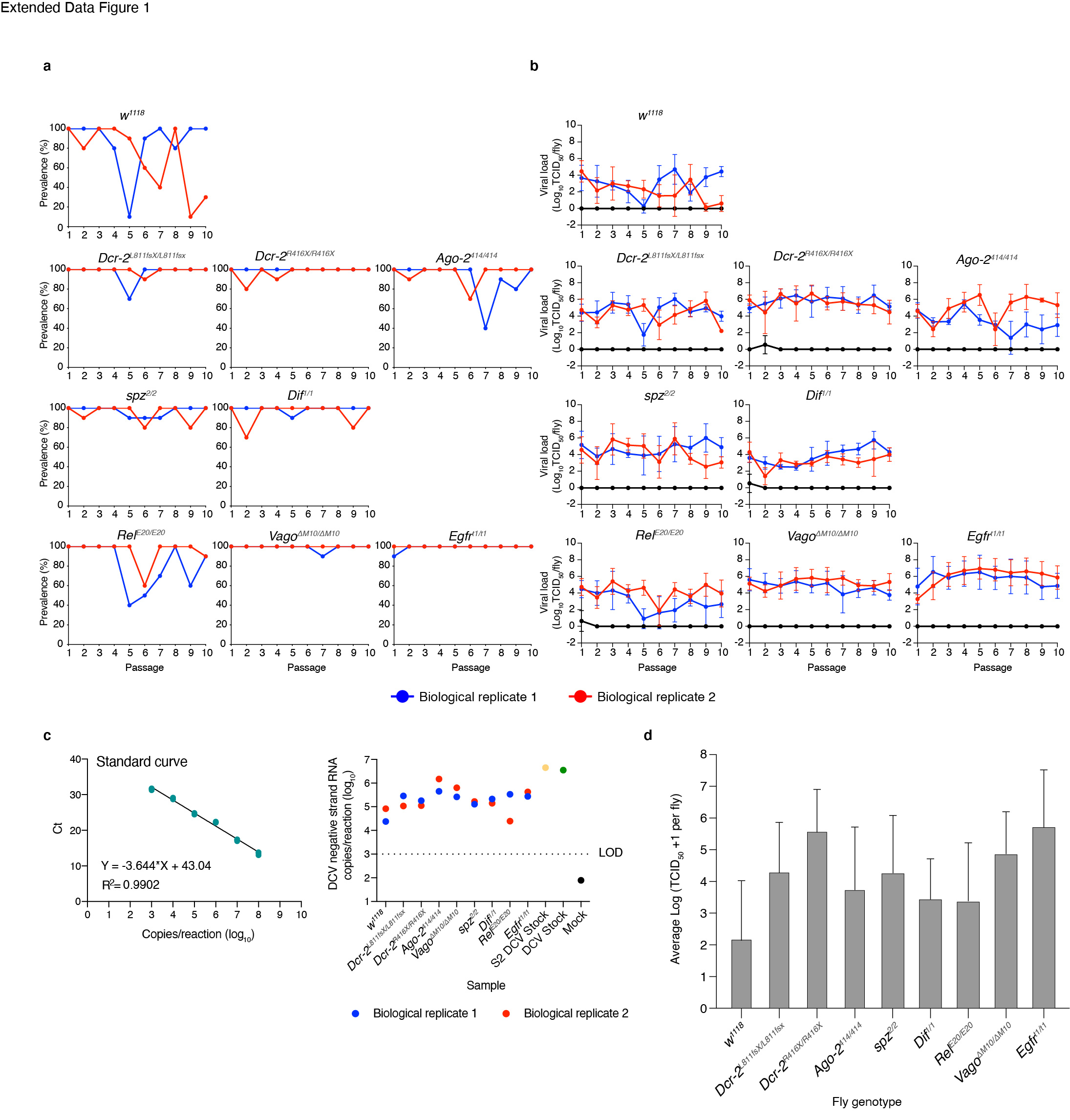
To follow viral infection during the course of the experiment, viral load (TCID50) was determined by end point dilution and prevalence (percentage of flies positive for TCID50) was calculated for all passages in individual flies from DCV contaminated cages. We found that for most fly genotypes and for both biological replicates, DCV infection prevailed along the 10 viral passages (Extended Data Figure 1a and 1b). When considering viral loads along passages, only w1118, Ago-2414/414 and RelE20/E20 fly lines displayed significant temporal dispersion (Durbin-Watson test for outliers < 1.5), consistent among both biological replicates, while viral load in the other fly genotypes remained relatively stable (Durbin-Watson test in the range 1.5 – 2.5) for at least one of the biological replicates (Extended Data Figure 1b). The negative strand of the DCV genome was detected in P = 10 in all genotypes and biological replicates, confirming that active viral replication occurred for the duration of the evolution experiment (Extended Data Figure 1c). Whether remnants of non-replicating virus remained in the fly surface was not assessed. Of note, the DCV stock was experimentally introduced to the system only once, to start the P = 1.
To assess the impact that fly genotype, biological replicate, and viral passage has on viral loads, the log-transformed TCID50 values from each fly genotype (Extended Data Figure 1d) were fitted to the generalized linear model (GLM) described in the Materials and Methods section. In short, the model incorporates fly genotype and experimental block as orthogonal factors and passage as covariable. Highly significant differences were observed in viral load among fly genotypes (test of the intercept: χ2 = 146.734, 8 d.f., p < 0.001) that were of very large magnitude , thus confirming that DCV load strongly varies among host genotypes. A significant effect was also observed for the viral passages (test of the covariable: χ2 = 5.075, 1 d.f., p = 0.024), indicating overall differences in viral accumulation among passages, though the magnitude of this effect was rather small . Regarding second-order interactions among factors and the covariable, a significant interaction exists between fly genotype and experimental block (χ2 = 27.082, 8 d.f., p < 0.001) indicating that some of the differences observed in virus accumulation among host genotypes differed among biological replicates, and between fly genotype and evolutionary passage (χ2 = 52.511, 8 d.f., p < 0.001). However, despite being statistically significant, these two effects were of very small magnitude and , respectively), casting doubts about their biological irrelevant. Likewise, the third-order interaction was statistically significant (χ2 = 86.023, 8 d.f., p < 0.001), suggesting that the differences in viral load among experimental blocks observed for a particular host genotype also depended on the evolutionary passages, although once again the effect could be considered as minor Next, we evaluated whether differences exist in viral load between immune competent (w1118) and the different mutant fly genotypes. In all eight cases, DCV accumulated to significantly higher levels in the immune deficient flies than in the wild-type flies (p < 0.001), with the smallest significant difference corresponding to viral populations replicating in RelE20/E20 and Dif1/1 and the largest to those replicating in Egfrt1/t1 and Dcr-2R416X/R416X (Extended Data Figure 1d).
Overall, these results show that in both immune competent (w1118) and immune deficient flies, DCV oral infection was maintained along passages and confirm that mutant flies are more permissive to DCV infection.
Viral nucleotide diversity increases in the absence of a fully functional immune response
To look into the selective pressure imposed by the Drosophila innate immune pathways on DCV population variation and dynamics, we analyzed virus genome diversity after each passage. Half of the population of infected flies was used to sequence the full-length DCV genome by NGS (Figure 1b and 1c). The viral stocks used to start the experiment, S2 DCV stock and DCV stock, were also sequenced (see Methods section). Analysis for the NGS data was performed using the computational pipeline Viral Variance Analysis (ViVan)42. Sequence coverage was at least 8,000 reads per position on the genome. To determine the error rate of the sequencing procedure, including library preparation, four sequencing technical replicates of the S2 DCV stock were used (Supplementary Figure 2). An allele frequency threshold of 0.0028 was used for all subsequent analyses based on variant detection and frequency correlation between technical replicates (see Methods section). We next calculated the site-averaged nucleotide diversity (π) on all polymorphic sites (n = 1869) across the full-length viral genome and present in the full dataset (Figure 2), with the aim of determining if the lack of activity of a given innate immune pathway had an impact on viral population genetic diversity, in terms of size of the viral mutant swarm.
Figure 2. Viral nucleotide diversity differently evolves in each host genotype.

Trajectory of the site-averaged nucleotide diversity (π) on all polymorphic sites (n = 1869) across the full-length DCV genome found for each fly genotype and in each biological replicate of the evolution experiment. Group: DCV population diversity found in each fly genotype was pairwise compared and grouped by similarity after a Bonferroni post hoc test (Table 1 and Supplementary Table 1). S: S2 DCV stock and D: DCV stock, in grey.
First, we asked if there was any difference in DCV population diversity and dynamics between the different fly genotypes along the complete evolution experiment. To answer this question, we analyzed if the host genotype, viral passages, biological replicate, or the interactions between these factors had an impact on the evolution of viral population diversity, considering the full-length DCV genome, across all passages. We found that only the fly genotype had a statistically significant impact on π (χ2 = 25.545, 8 d.f., p = 0.001) (Table 1). We then compared the DCV population diversity present in each fly genotype to each other. We found that, except for viral diversity found in Dcr-2L811fs/[L811fsX and Dif1/1 lines, for which no difference was found compared to π in w1118 flies (p ≥ 0.303), DCV population diversity significantly differed from w1118 line in the rest of the innate immune mutants analyzed (p ≤ 0.013) (Supplementary Table 1). A post hoc Bonferroni test further sorted overlapping groups according to their increasing viral nucleotide diversity: group 1 (less diversity): w1118, Dcr-2L811fs/[L811fsX and Dif1/1 fly lines; group 2: Dif1/1, Dcr-2L811fs/[L811fsX, RelE20/E20, spz2/2, and Dcr-2R416X/R416X fly lines; group 3: Dcr-2L811fs/[L811fsX, RelE20/E20, spz2/2, Dcr-2R416X/R416X, and Ago-2414/414 fly lines; group 4 (more diversity): containing spz2/2, Dcr-2R416X/R416X, Ago-2414/414, Egfrt1/t1, and VagoΔM10/ΔM10 fly lines (Extended Data Figure 2 and Supplementary Table 1).
Table 1. Analysis of the impact of each experimental variable on the evolution of DCV nucleotide diversity (mean log10(π) per site).
The site-averaged nucleotide diversity (π) on all polymorphic sites (n = 1869) across the full-length viral genome was determined, the log10-transformed π values were fitted to the generalized linear model (GLM) and the impact of the variables determined by an Analysis of Deviance (Type III tests). BR: biological replicate; VP: viral passage; FG: fly genotype, GR: genomic region.
| Experimental variable | χ2 | d.f. | p | |
|---|---|---|---|---|
| Full length DCV genome | BR | 2.2528 | 1 | 0.1334 |
| VP | 1.6460 | 1 | 0.1995 | |
| FG | 25.5447 | 8 | 0.0013 ** | |
| (BR) × VP | 0.0024 | 1 | 0.9606 | |
| (BR) × FG | 14.2963 | 8 | 0.0744 | |
| VP × FG | 12.1679 | 8 | 0.1439 | |
| (BR) × VP × FG | 10.4253 | 8 | 0.2364 | |
| Each DCV genomic region | BR | 1.2107 | 1 | 0.2712 |
| VP | 2.3528 | 1 | 0.1251 | |
| FG | 27.1779 | 8 | 0.0007 *** | |
| GR | 11.6982 | 3 | 0.0085 ** | |
| (BR) × VP | 0.0001 | 1 | 0.9931 | |
| (BR) × FG | 16.3143 | 8 | 0.0381 * | |
| VP × FG | 8.3498 | 8 | 0.4000 | |
| (BR) × GR | 0.7452 | 3 | 0.8625 | |
| VP × GR | 0.9130 | 3 | 0.8223 | |
| FG × GR | 24.0586 | 24 | 0.4583 | |
| (BR) × VP × FG | 12.8802 | 8 | 0.1160 | |
| (BR) × VP × GR | 0.1274 | 3 | 0.9884 | |
| (BR) × FG × GR | 24.4811 | 24 | 0.4344 | |
| VP × FG × GR | 10.5776 | 24 | 0.9917 | |
| (BR) × VP × FG × GR | 28.3112 | 24 | 0.2471 |
Next, we wondered if the general differences observed in viral nucleotide diversity, between fly genotypes were associated with a particular viral genomic region (i.e., if a determined viral function was affected during the evolution experiment) (Figure 1c). Of note, the intergenic region internal ribosome entry site (IGR IRES) was not included in the analysis because its lack of genetic variation prevented us from determining its nucleotide diversity value. We found that the fly genotype had a statistically significant effect on the nucleotide diversity found in each DCV genomic region (χ2 = 27.178, 8 d.f., p < 0.001), which further differed between each specific viral genomic region (χ2 = 11.698, 8 d.f., p = 0.008). As a second-order interaction, an effect of the fly genotype and the biological replicate was found (χ2 = 16.314, 8 d.f., p = 0.038) (Table 1). Comparison of viral genetic diversity within the genomic regions allowed us to distinguish three main groups: group 1 (less diversity): 3’UTR; group 2: 5’UTR IRES; and group 3 (more diversity): ORF1 and ORF2 (Extended Data Figure 2 and Supplementary Table 1).
Finally, we wondered if viral diversity evolved from the starting viral stock (DCV stock) in each fly genotype. π present in P = 1, P = 5 and P = 10 was compared between fly genotypes and with the diversity present in the DCV stock. We found that pairwise comparisons of viral nucleotide diversity present in each fly genotype in P = 1, between each other and versus DCV stock, yield no statistically significant difference (p = 1.000) (Supplementary Table 1). In P = 5 viral diversity was reduced only in w1118 (Group 1/2; p = 0.026 and p = 0.032) compared to the starting viral stock (Extended Data Figure 2 and Supplementary Table 1). In P = 10, viral nucleotide diversity present in w1118 (Group 1; p = 0.032 and p = 0.041), spz2/2 (Group 1; p = 0.020 and p = 0.025), Dif1/1 (Group 1; p = 0.005 and p = 0.006) and RelE20/E20 (Group 1/2; p = 0.046) mutant flies was reduced when compared to DCV diversity from the DCV stock (Extended Data Figure 2 and Supplementary Table 1).
Altogether, the results show that the absence of a fully functional immune system results in an increase of viral population diversity that remains constant along passages. They also show that the coding regions of the virus are more prone to accumulate variation than the non-coding regions where regulatory elements are present.
Viral population diversity derives from preexisting standing genetic variation
Next, we examined if the levels of viral diversity observed in DCV populations from innate immune mutants compared to the w1118 line were accompanied with the fixation of particular genetic changes in the mutant swarms, and whether (i) whether these changes can be associated with fitness effects and (ii) whether potentially adaptive mutations arose in response to particular immune responses. To do so, we estimated the selection coefficients for each SNP using their variation in frequency across evolutionary time (Figure 3 and Extended Data Figure 3), using a classic population genetics approach43 (Table 2). Thirty-six SNPs yielded significant estimates of selection coefficients (this number reduces to 10 if a stricter FDR correction is applied; Table 2). Twenty-one of them were already detected in the ancestral S2 DCV stock, henceforth a maximum of 15 new SNPs might have arisen during the evolution experiment. Estimated selection coefficients for all these SNPs ranged between −0.304 per passage (synonymous mutation RdRp/C5713U) and 1.204 per passage (VP2/G6311C nonsynonymous change R16P), with a median value of 0.286 per passage (interquartile rank = 0.265). Nine mutations were observed in more than one lineage (range 2 - 7 times), with synonymous mutations VP3/U7824C appearing in seven lineages of six different host genotypes and mutation 5’UTR/A280U in five lineages of five host genotypes (Table 2). These nine SNPs were all present in the S2 DCV stock. Indeed, the frequency of SNPs among evolving lineages is significantly correlated with their frequency in the ancestral S2 DCV stock (Pearson’s r = 0.401, 36 df, p = 0.013), but not with their measured fitness effect (r = −0.091, 36 df, p = 0.588).
Figure 3. Trajectories of DCV variants across passages.

Muller plots illustrating the dynamics of SNPs’ frequencies along evolutionary time. Each color represents the dynamics of a different SNP.
Table 2. Mutations for which significant estimates of fitness effects have been obtained.
For each mutation, we indicate whether it already existed in the DCV starting stocks (and at which frequency) or arose during the evolution experiment. We also provide the estimated selection coefficient, its SEM and statistical significance. Cases significant after FDR correction are marked with an asterisk.
| Fly genotype | Biol. Rep. | Mutation | Standing variation (frequency) | Selection coefficient per passage (± SEM) | P |
|---|---|---|---|---|---|
| w1118 | 1 | VP2/G6311C R16P | Yes (0.0104) | 1.2039 ±0.2543 | 0.0418 |
| w1118 | 2 | VP3/U7824C | Yes (0.1457) | 0.4780 ±0.0617 | < 0.0001* |
| Dcr-2L811fsX/L811fsX | 1 | - | |||
| Dcr-2L811fsX/L811fsX | 2 | RpRd/U5302C | No | 0.3877 ±0.0973 | 0.0073 |
| VP1/C8227U H655Y | Yes (0.0147) | 0.3735 ±0.1368 | 0.0258 | ||
| VP1/C8424U | Yes (0.0139) | 0.3880 ±0.1407 | 0.0248 | ||
| Dcr-2R416X/R416X | 1 | VP2/C6932U A223V | Yes (0.0084) | 0.2135 ±0.0169 | < 0.0001* |
| Dcr-2R416X/R416X | 2 | VP2/G6379A A39T | Yes (0.0098) | 0.2074 ±0.0555 | 0.0057 |
| VP3/A7465G I401V | Yes (0.0088) | 0.1185 ±0.0338 | 0.0100 | ||
| VP3/U7824C | Yes (0.1457) | −0.2887 ±0.0884 | 0.0309 | ||
| Ago-2414/414 | 1 | - | |||
| Ago-2414/414 | 2 | 5’UTR/A280U | Yes (0.1176) | −0.1307 ±0.0376 | 0.0084 |
| VP3/U7824C | Yes (0.1457) | 0.5251 ±0.1050 | 0.0024 | ||
| VP1/C8227U H655Y | Yes (0.0147) | 0.6238 ±0.1077 | 0.0007 | ||
| VP1/C8424U | Yes (0.0139) | 0.6206 ±0.1252 | 0.0026 | ||
| Spz2/2 | 1 | 5’UTR/A280U | Yes (0.1176) | −0.2092 ±0.0735 | 0.0215 |
| VP2/G6931A A223T | No | 0.5420 ±0.1477 | 0.0105 | ||
| Spz2/2 | 2 | 2A/A1128C D110A | Yes (0.0041) | −0.0229 ±0.0065 | 0.0246 |
| 3C-Prot/A3787G | No | 0.5238 ±0.0757 | 0.0002* | ||
| 3C-Prot/G4394A V1199I | No | 0.5982 ±0.0764 | 0.0002* | ||
| VP1/G8536A V758I | No | 0.7038 ±0.0915 | 0.0006* | ||
| IGR/A6108G | Yes (0.0044) | 0.4873 ±0.0692 | 0.0002* | ||
| VP3/G8090A R609H | Yes (0.0200) | 0.4947 ±0.0722 | 0.0001* | ||
| Dif 1/1 | 1 | VP3/A7465G I401V | Yes (0.0088) | 0.3213 ±0.1173 | 0.0338 |
| VP3/G7956A | No | 0.2000 ±0.0335 | 0.0094 | ||
| Dif1/1 | 2 | 5’UTR/A280U | Yes (0.1176) | 0.5157 ±0.1289 | 0.0052 |
| VP1/U8629C S5058P | Yes (0.0898) | 0.4864 ±0.1175 | 0.0043 | ||
| RelE20/E20 | 1 | 5’UTR/A280U | Yes (0.1176) | 0.3430 ±0.1017 | 0.0097 |
| RdRp/A5404G | Yes (0.0929) | 0.3993 ±0.1217 | 0.0135 | ||
| VP2/U6303A N13K | Yes (0.0037) | 0.5724 ±0.1409 | 0.0036 | ||
| VP3/U7824C | Yes (0.1457) | −0.2804 ±0.0206 | 0.0467 | ||
| RelE20/E20 | 2 | 5’UTR/A280U | Yes (0.1176) | −0.0917 ±0.0277 | 0.0130 |
| 2B/C1412U | Yes (0.1301) | 0.4554 ±0.0119 | 0.0166 | ||
| VP3/C7760A T499N | No | 0.1340 ±0.0195 | 0.0005 | ||
| VagoΔM10/ΔM10 | 1 | 2B/C1412U | Yes (0.1301) | 0.2386 ±0.0549 | 0.0025 |
| 3C-Prot/A3703G | No | 0.2859 ±0.0537 | 0.0031 | ||
| RdRp/U5188A | Yes (0.1325) | 0.2869 ±0.0705 | 0.0268 | ||
| VP2/C6932U A223V | Yes (0.0084) | 0.1368 ±0.0553 | 0.0426 | ||
| VP1/C8227U H655Y | Yes (0.0147) | 0.1936 ±0.0291 | 0.0002* | ||
| VP1/C8424U | Yes (0.0139) | 0.1915 ±0.0283 | 0.0001* | ||
| VP1/U8697C | No | 0.2053 ±0.0325 | 0.0002* | ||
| 3’UTR/U9163A | No | 0.1473 ±0.0622 | 0.0497 | ||
| VagoΔM10/ΔM10 | 2 | 2C-Hel/G1756A | Yes (0.0059) | 0.3467 ±0.1293 | 0.0364 |
| VP2/A6300U E12D | No | 0.3681 ±0.1297 | 0.0470 | ||
| VP3/U7824C | Yes (0.1372) | 0.1517 ±0.0391 | 0.0060 | ||
| Egfrt1/t1 | 1 | 5’UTR/A280U | Yes (0.1176) | 0.1394 ±0.0364 | 0.0050 |
| 3C-Prot/U3643A | No | -0.2064 ±0.0592 | 0.0399 | ||
| VP1/A8201G Q646R | Yes (0.0045) | 0.3198 ±0.0736 | 0.0225 | ||
| VP2/A6660U | No | −0.1906 ±0.0641 | 0.0409 | ||
| VP2/G6868A V8162I | No | 0.3302 ±0.0389 | 0.0001 | ||
| VP3/A7465G I401V | Yes (0.0088) | −0.1053 ±0.0359 | 0.0261 | ||
| VP3/U7824C | Yes (0.1457) | 0.0997 ±0.0410 | 0.0411 | ||
| Egfrt1/t1 | 2 | 5’UTR/A198G | No | 0.1035 ±0.0363 | 0.0246 |
| RdRp/U4810C | Yes (0.1152) | −0.2635 ±0.0301 | 0.0128 | ||
| RdRp/C5713U | Yes (0.1148) | −0.3036 ±0.0276 | 0.0082 | ||
| VP2/G6379A A39T | Yes (0.0082) | 0.0630 ±0.0254 | 0.0381 | ||
| VP3/U7824C | Yes (0.1457) | −0.1090 ±0.0402 | 0.0421 | ||
| VP3/G8090A R609H | Yes (0.0200) | 0.0764 ±0.0289 | 0.0333 | ||
| VP1/U8250G H662Q | Yes (0.0201) | 0.1734 ±0.0326 | 0.0060 |
An interesting question is whether the fitness effects associated with each of these nine SNPs were the same across all genotypes or, conversely, whether fitness effects were host genotype-dependent. To test this hypothesis, we performed one-way ANOVA tests comparing fitness effects (Table 2) across the corresponding host genotypes. In all cases, significant differences were observed (F ≥ 15.637 and p ≤ 0.001, and ≥ 93.99% of total observed variance in fitness effects explained by true genetic differences among host genotypes), supporting the notion that fitness effects are indeed host-genotype dependent. A pertinent example is the case of the synonymous mutation VP3/U7824C, which was the most prevalent mutation (F6,45 = 158.862, p < 0.001, 99.37% of genetic variance). In this case, a post hoc Bonferroni test shows that host genotypes can be classified into three groups according to the fitness effect of this SNP. In genotypes Dcr-2R416X/R416X and RelE20/E20, the mutation has a deleterious effect (on average, −0.2260 per passage); in genotypes Egfrt1/t1 and VagoΔM10/ΔM10, the mutation is moderately beneficial (on average, 0.1257 per passage; and in genotypes w1118 and Ago-2414/414, the mutation had a strong beneficial effect (on average, 0.502 per passage).
As shown in Figure 3 and Extended Data Figure 3a, some SNPs show a strong parallelism in their temporal dynamics, suggesting they might be linked into haplotypes. This is particularly relevant for mutations shown in Table 2. To test this possibility, we computed all pairwise Pearson correlation coefficients between mutation frequencies along evolutionary time. The results of these analyses are shown in Extended Data Figure 3b to 3k as heatmaps. Again, as an illustrative example, we discuss here the case of the viral population BR2 evolved in Ago-2414/414 (Extended Data Figure 3d). Synonymous mutations VP3/U7824C and VP1/C8424U and nonsynonymous mutation VP1/C8227U (H655Y) are all linked into the same haplotype (r ≥ 0.998, p < 0.001). Since these three mutations already existed in the S2 DCV stock, it is conceivable that the haplotype already existed and has been selected as a unit. Indeed, the fitness effects estimated for these three mutations are indistinguishable (one-way ANOVA: F2, 22 = 1.781, p = 0.192; average fitness effect 0.590 ±0.032 per passage), thus suggesting that the estimated value corresponds to the haplotype as a unit. The absence of this haplotype in Ago-2414/414 BR1 suggests it was lost during the transmission bottleneck from S2 cells to flies. Interestingly, mutations VP1/C8424U VP1/C8227U appear also linked into the same haplotype in population BR2 evolved in Dcr-2L811fsX/L811fsX (Extended Data Figure 3b). These two cases, as well as populations BR1 evolved in RelE20/E20, BR2 evolved in spz2/2 and BR1 and BR2 evolved in vagoΔM10/ΔM10 illustrate some examples of haplotypes (Extended Data Figure 3f, 3e, 3h, and 3i). Other viral populations, especially those evolved in Egfrt1/t1 flies, show much more complex patterns (Extended Data Figure 3j and 3k) in which haplotypes change over time by acquiring de novo mutations.
When mapping the 36 SNPs found to have significant estimates of selection coefficients in the viral genome (Table 2 and Extended Data Figure 4), we found that two mapped to the 5’UTR IRES, twelve to ORF1, one to the IGR IRES, 20 to ORF2, and one to the 3’UTR. Of the twelve mutations observed in ORF1, which encodes the non-structural proteins, four mapped to the 3C viral protease and five to the RdRp. Only one of these mutations in the 3C viral protease was non-synonymous. Of the 20 mutations in ORF2, which encodes the viral structural proteins, eight mapped to VP2, five to VP3, and seven to VP1. These correspond to the three majors predicted DCV capsid proteins.
Taken together, these results show that viral population diversity over these ten in vivo passages mainly derived from preexisting standing genetic variation in the ancestral DCV population. Furthermore, temporal dynamics of population diversity were linked to the fly genotype in which the virus evolved.
DCV virulence decreases along passages in the absence of immune pathways
Finally, we wondered if DCV virulence varied among each lineage in the different fly genotypes. Infectious DCV stocks were produced from viral passages P = 1 and P = 10 and from all fly genotypes. Because the viral evolution experiment was performed by DCV orofecal transmission, we first evaluated DCV virulence by feeding w1118 flies with DCV stocks derived from P = 1 or P = 10; survival was evaluated from each fly genotype. We found that only a small proportion of flies (between 5% and 20%) succumbed to DCV infection, and no statistically significant differences in mortality were found between mock and virus infected flies, regardless of viral passage or fly genotype (Supplementary Figure 3). This is in agreement with previously published works showing that DCV oral infections are cleared in w1118 flies31. We next decided to investigate the evolution of virulence by intrathoracic inoculation of DCV stocks. We found that w1118 flies were less sensitive to viral infection when inoculated with DCV stocks derived from P = 10 since their median survival time was longer than those inoculated with stocks from P = 1 for most DCV stock origins (Figure 4a and Supplementary Table 2). Notable exceptions were DCV stocks from BR2 of VagoΔM10/ΔM10 mutant flies, for which w1118 flies were more sensitive to P = 10 than to P = 1, and stocks from BR1 of spz2/2 and BR2 of Egfrt1/t1 mutant flies, for which no difference in median survival time after infection with DCV between P = 1 and P = 10 was detected.
Figure 4. DCV virulence decreases in the absence of immune pathways.

DCV infectious stocks were prepared from viral passages P = 1 and P = 10 and from each fly genotype. w1118 flies were intrathoracically inoculated with 10 TCID50 units of each DCV stock and survival of the flies was measured daily. a) Survival curves shown in the figure are the combination of the two independent replicates, with three technical replicates each, of a total of at least N = 98 flies per treatment. Error bars indicate ± Standard error of the mean (SEM); n.s., not significant. Survival curves were compared via log-rank (Mantel–Cox) tests. b) Test of the contribution of historical contingency evolved (P = 10) vs ancestral (P = 1) DCV virulence. The dashed red line represents the linear regression, and the black line represents the expected relationship under the null hypothesis of ancestral differences in DCV virulence which are maintained after evolution despite noise introduced by random events (mutation and drift).
A fundamental question in evolutionary biology is the role that past evolutionary events may have in the outcome of evolution44. If ongoing evolution is strongly contingent with past evolutionary events, ancestral phenotypic differences should be retained to some extent, while if other evolutionary forces such as selection and stochastic events (i.e., mutation and genetic drift) dominate, then ancestral differences can be eroded and, in the extreme case, even fully removed. Here, we observed significant differences in the performance of the ancestral DCV across the eight host genotypes. To test whether these differences are still observable in the evolved population, we compared the median survival time (Figure 5a and Supplementary Table 2) for DCV populations isolated at the beginning of the evolution experiment P = 1 and at the end P = 10 (Figure 4b). Under the null hypothesis of strong historical contingency, it is expected that data will fit to a regression line of slope 1 and intercepting the ordinate axis at 0. However, if ancestral differences have been removed, data would fit significantly better to a regression line with a slope smaller than one and with an intercept greater than zero44. Figure 4b shows the data and its fit to the null hypothesis (solid black line) and the alternative hypothesis (dashed red line). A partial F-test shows that adding an intercept to the regression equation significantly improves the fit (F1,16 = 28.437, p < 0.001), thus supporting the notion that ancestral differences among host genotypes have been removed by the action of subsequent adaptation, that is, the fixation of beneficial mutations.
Discussion
In this work we aimed at determining the overall impact of innate immunity on viral evolution. Based on the arms-race hypothesis, we speculated that if a given host defense mechanism imposes a specific selective pressure on a particular pathogen function, the absence of this defense mechanism would result in the relaxation of the selective constraint, which would in turn be detectable in the pathogen at the genomic and phenotypic levels. We found that viral population diversity evolved differently according to each fly genotype; however, viral population diversity mostly derives from ancestral standing genetic variation (i.e., few “new” mutations were selected). Our results further confirm the polygenic nature of antiviral responses; there is not a specific, main immune defense mechanism against a particular virus, but instead a repertoire of defense mechanisms that are triggered after infection and that might interact with each other.
Our results are compatible with a pervasive presence of clonal interference. In the absence of sexual reproduction, clonal interference is the process by which beneficial alleles originated in different clades within a population compete with each other, resulting in one of them reaching fixation. Subsequently, the outcompeted beneficial allele may appear in the new dominant genetic background and, assuming no negative epistasis among both loci, become fixed. As a consequence, beneficial mutations may fix sequentially, thus slowing down the rate of adaptation45. Given their large effective population size and high mutation rates, viral populations are expected to contain considerable amounts of potentially beneficial standing variation, making them prone to clonal interference. Indeed, it has been previously shown to operate in experimental populations of vesicular stomatitis virus adapting to cell cultures46,47, in bacteriophage ϕX174 populations adapting to harsh saline environments48, in tobacco etch virus adapting to novel plant host species49, among HIV-1 escape variants within individual patients50, and also at the epidemiological level among influenza A virus lineages diversifying antigenically51. In our own results, clonal interference can be observed in populations BR1 evolved in Dcr-2L811fsX/L811fsX, BR1 evolved in Ago-2414/414, BR1 evolved in spz2/2, BR2 evolved in RelE20/E20, and BR2 evolved in VagoΔM10/ΔM10. All of these viral populations share similar patterns in which some beneficial allele (or haplotypes) rose in frequency, reached a peak at some intermediate passage, then declined in frequency and were finally outcompeted by a different beneficial mutation (or haplotype) that had lower initial frequency. For example, the nonsynonymous mutation VP2/G6931A (A223T) appeared de novo in population BR1 evolved in spz2/2, and outcompeted several mutations likely linked in a haplotype (Figure 3). Tightly linked to clonal interference is the concept of leap-frogging52, in which the beneficial mutation that ends up dominating the population is less genetically related to the previously dominant haplotype than to the common ancestor of both (Figure 3). The VP2/G6931A mutation illustrates this case well, as it appeared in a genetic background that was minoritarian rather than in the dominant one. Likewise, the mutation VP2/G6311C (R16P), observed in BR1 evolved in w1118 flies, appeared in a low frequency genetic background different from the most abundant one in previous passages. Finally, the haplotype containing five different mutations observed in BR2 evolved in spz2/2 became dominant in frequency after P = 6, outcompeting two other mutations that were dominating the population until then.
The existence and fixation of haplotypes along our evolution experiment deserves further discussion. Linked mutations generate three possible interference effects53. First, all mutations might contribute additively, or may be involved in positive epistasis, to the fitness of the haplotype as a whole, thus increasing its chances to become fixed. Second, hitchhiking and genetic draft may occur, by which deleterious or neutral alleles are driven to fixation along with a linked beneficial allele. Third, there may be background selection by which the spread of a beneficial allele is impeded, or at least delayed, owing to the presence of linked deleterious alleles. For instance, we can hypothesize that haplotype VP3/U7824C-VP1/C8227U-VP1/C8424U, which swept to fixation in population BR2 evolved in Ago-2414/414, may represent a case of genetic draft: two synonymous mutations, potentially neutral, linked to a nonsynonymous one that may be the actual target of selection. Yet, the lack of an infectious clone for DCV does not allow us to test this hypothesis.
Some of the mutations we found to be associated with positive selection coefficients were synonymous changes (Table 3). However, equating synonymous mutations with neutral mutations in compacted RNA genomes has proven to be misleading54,55. Selection operates at different levels of a virus’s infection cycle, and not all these levels necessarily depend on the amino acid sequence of encoded proteins. For instance, a lack of matching between virus and host codon usages would slowdown translational speed and efficiency56; mutations affecting the folding of regulatory secondary structures at noncoding regions would affect the interaction with host and viral factors and thus impact the expression of downstream genes (e.g., mutations 5’UTR/A280U, IGR/A6108G and 3’UTR/U9163A all with significant fitness effects -Table 3)57; or evasion from antiviral RNAi defenses by changing the most important relevant sites in the target of siRNAs12,13.
It is interesting to observe that viral diversity in mutants for antiviral RNAi, whose mode of action relies on a direct interaction with the viral genome, did not display increased diversity when compared to mutants from the other immune pathways. One could expect that the release of the selective pressure that RNAi exerts on the virus genome may allow for the appearance of mutations in the viral suppressor of RNAi. Nonetheless, we did not observe such a change, possibly because the RNAi suppressor in DCV shares the first 99 amino acids of the RdRp58,59 and mutations would affect polymerase activity. The antiviral action of the other immune pathways remains still unknown and may even be indirect; for example the known roles of Imd, Toll, and Egfr pathways in controlling fly microbiota38,39 might possibly affect the prevalence of virus infections. In this regard, it is important to highlight that the diversity of DCV in the Dif1/1 mutant (Toll pathway, already described not to have an impact on DCV defense25), was indistinguishable from w1118, pointing to the specific - although uncharacterized - antiviral functions of these other immune pathways.
Another consideration when interpreting our results is the nature of the virus stock used. This virus stock has been maintained for years in Drosophila S2 cells. The observation that viral population diversity decreased along passages in w1118 flies, highlights the strength of the selective forces that constrain the virus from adapting to a new environment. During the successive passages, in the absence of a given immune response, the capacity of the virus to evolve will be determined by a combination of two factors: the adaptation to the new environment (constraining factor) and the lack of immune response (relaxation factor). Because DCV replication is significantly increased in immune deficient mutants, the potential for population diversification is higher. This effect is clearly observed in w1118 flies where the virus is “only” adapting to the new environment and DCV populations evolved in w1118 flies show less variation than all other lineages. Future experimental evolution studies using viral stocks derived from flies, instead of cell cultures, are warranted to address this topic.
In a study published recently60, Navarro et al. used Arabidopsis thaliana and turnip mosaic virus to carry out experimental virus evolution assays with a similar design to ours. In their work, the authors used plant mutants compromised in their antiviral response (more permissive to viral infection) or with an enhanced antiviral response (less permissive to viral infection) and allowed the virus to evolve for 12 passages. Similarly, to what we found in the D. melanogaster - DCV system, the authors showed that viral population evolutions dynamics, as well as viral loads, depend on host genotype. Interestingly, a reduction of ancestral genetic variation regardless of the immune pathway affected was also clearly observed, in agreement with our observations.
Taken together, our results point to the concerted action of the different immune pathways to limit viral evolution. Response to infection does not simply consist of activating immune pathways, it also encompasses a broad range of physiological consequences including metabolic adaptations, stress responses and tissue repair. Critically, upon infection, the homeostatic regulation of these pathways is altered. However, such alterations do not always result in increased disease severity and in fact can even lead to improved survival (or health) despite active virus replication.
Materials and Methods
Fly strains and husbandry
Flies were maintained on a standard cornmeal diet (Bloomington) at a constant temperature of 25 °C. All fly lines were cleaned of possible chronic infections (viruses and Wolbachia) as described previously61. The presence or absence of these chronic infections was determined by RT-PCR with specific primers for Nora virus, Drosophila A virus, DCV (NoraVfor ATGGCGCCAGTTAGTGCAGACCT, NoraVrev CCTGTTGTTCCAGTTGGGTTCGA DAVfor AGAGTGGCTGTGAGGCAGAT, DAVrev GCCATCTGACAACAGCTTGA, DCVfor GTTGCCTTATCTGCTCTG, DCVrev CGCATAACCATGCTCTTCTG) and by PCR with specific primers Wolbachia sp (wspfor TGGTCCAATAAGTGATGAAGAAAC, wsprev AAAAATTAAACGCTACTCCA and wspBfor TTTGCAAGTGAAACAGAAGG, wspBrev GCTTTGCTGGCAAAATGG).
Fly mutant lines for Dcr-2L811fsX and Dcr-2R416X 62, Ago-2414 63, Spz2 64, Dif1 65, RelE20 66, VagoΔM10 33and Egfrt1 67 were isogenized to w1118 fly line genetic background first by replacing the chromosomes not containing the mutation using balancer chromosomes and then by recombination by backcrossing at least ten times to w1118 line. The presence of the mutation was followed during and at the end of the backcrossing procedure by PCR and sequence analysis using specific primers (Dcr2811_3001for TTTGACCCATGACTTTGCGGT, Dcr2811_3294rev CCTTGCAGAGATGCCCCTGTT, Dcr2416_4341for GATTGGCATTACCGTCCCGAA, Dcr2416_4670rev AGCGATTCCTG ATGAGTCTTA, Ago2414_rev TTGTGGATGGCTGTTGTCTCG, Ago251B414_for AGAGTCCCCACTTGAATGGCC, Spz2_for GCCTTTGGCGCTTGCCTAATT, Spz2_rev GCTCCTGCAAAGGAATCGCTC, Dif1_for CTTGGCAATCTTCTCGCACAG, Dif1_rev ATCGTGGTCTCCTGTGTGACG, Rel_Ex4rev AGCTCTCCAGTTTGTGCCGAC, Rel-RD_5’UTRfor CTGGCGTTAGTTTCGGCGTTG, Vagod10_for TTGGCCAACGGAAAGGATGTG, Vagod10_rev TGCCACCGATGATCAATGACA, Egfrt1_for CAAAGCTCGAACCGAAATTA, Egfrt1_rev CTTTCTTAACGTCCACATGA).
Virus production and titration
The S2 DCV stock used to start the experiment was prepared in S2 cells. Cells were maintained in Schneider culture medium and at 25 °C and observed daily. Cells were harvested when cytopathic effects were detected, then frozen at −80 °C, thawed on ice and centrifuged for 15 min at 15,000 g at 4 °C. The supernatant was recovered, aliquoted and stored at −80 °C. Viral stocks were tittered in S2 cells, determined using the end-point dilution method and expressed as fifty-percent tissue culture infective dose (TCID50)68.
To produce the DCV stocks from passages P = 1 and P = 10 from the evolution experiment half of the population of flies infected with DCV from each fly genotype (approx. 250 flies) was homogenized in 1 × PBS, homogenates were frozen at −80 °C, then thawed on ice, centrifuged to discard the tissue debris, supernatant was recovered and filtered to discard bacteria contamination, then aliquoted and stored at −80 °C. Viral stocks were tittered in S2 cells using the end-point dilution method and expressed as fifty-percent tissue culture infective dose (TCID50).
Viral and bacterial infections and survival analysis
To characterize the isogenized fly lines, 4 to 5 day old female flies were intrathoracically injected with a Nanoject II apparatus (Drummond Scientific) with 50 nl of the pathogen suspension. For DCV infections, a suspension of 10 TCID50 units of DCV in 10 mM Tris buffer, pH 7 was used. An injection of the same volume of 10 mM Tris, pH 7 served as a mock-infected control. Infected flies were kept at 25 °C, transferred into fresh vials every 2 days and number of dead flies was scored daily. For bacterial infections, 50 nl of suspensions in 1× PBS buffer, pH 7, of optical density (OD) = 10 for Enterococcus faecalis, and of OD = 200 for Erwinia carotovora carotovora 15 (Ecc15) were used. An injection of the same volume of 1× PBS buffer served as a mock-infected control. Flies infected with E. faecalis were kept at 25 °C, and flies infected with Ecc15 were kept at 29 °C. Flies were transferred into fresh vials every 2 days and number of dead flies was scored daily.
Virus experimental evolution
To produce the starting DCV stock (DCV stock) 5 to 6 days old w1118 female flies were intrathoracically injected with 100 TCID50 of DCV from a stock produced in S2 Drosophila cells (S2 DCV stock) or mock infected. At 4 dpi, N = 90 DCV infected flies (DCV stock) were placed in cages containing fresh medium, left for 3 days and then removed to place in this DCV-or mock-contaminated cages N = 500 5-to-6-day old wild type or mutant flies (males and females). Flies were allowed to feed ad libitum for 3 days (oral inoculation period), then moved to a clean cage for 1 day, and further placed into a new clean cage and left for 4 days, when they were harvested (P = 1). A new group of flies was then placed into the contaminated cages. This procedure was repeated 10 times (10 DCV Passages, P = 1 to P = 10) and replicated twice (biological replicates BR1 and BR2). The total amount of flies from each passage, fly genotype, and biological replicate was collected and randomly divided in halves (approx. 250 flies), one half was used to extract total RNA and produce the NGS libraries and the other half to produce viral stocks to evaluate DCV virulence.
Characterization of infection during passages
Individual flies from each passage were anesthetized and homogenized in 100 ml of 1’ PBS buffer. The tubes containing the homogenates were centrifuged for 5 min at 15,000×g at 4 °C to discard the tissue debris. The supernatant was recovered and used to determine viral load (TCID50) by end point dilution and prevalence (percentage of flies positive for TCID50) for each fly genotype, viral passages and biological replicate.
For statistical analyses, TCID50 data were transformed as T = log(TCID50 + 1) and then fitted to a generalized linear model in which fly genotype (G) and BR (B) were treated as orthogonal factors. G was considered as a fixed effects factor whereas B was considered as a random effects factor. Evolutionary passage (P) was introduced in the model as a fixed effects covariable. We also considered second and third order interactions between the two factors and the covariable. The model equation thus reads:
Where Tijk(P) is the transformed TCID50 observed for a particular titration assay k of BR j of fly genotype i, τ represents the grand mean value and εijk stands for the error assumed to be Gaussian distributed at every P. The significance of each term in the model was evaluated using a likelihood ratio test that follows a χ2 probability distribution. The magnitude of the effects was evaluated using the statistic (proportion of total variability in the traits vector attributable to each factor in the model; conventionally, values of are considered as large effects). These analyses were done using SPSS version 27 (IBM, Armonk, NY).
Detection of negative strand DCV RNA by strand-specific RT-qPCR (ssRT-qPCR)
To determine the amount of negative strand DCV RNA present in the viral stocks produced from each fly genotype in P = 10, S2 DCV stock, and DCV stock, total RNA was extracted from the DCV stocks produced from P = 10 (all fly genotypes, both biological replicates) and from the DCV stocks used to start the experiment. ssRT-qPCR was performed with these RNA samples essentially as described35. We used 800 ng of RNA to perform reverse transcription with SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer’s instructions, with the exception that primer annealing occurred at 70 °C and cDNA synthesis occurred at 50 °C for 30 minutes. Reverse transcription was performed using a forward primer containing a non-target tag sequence (DCV_tag_F: AATTCAAGCTCGTCTTCCTCGAGGCTGTGTTTGCGCGAAG) A standard curve was produced by reverse transcription of a tenfold dilution series (from 108 to 103 copies per reaction) of in vitro transcribed RNA corresponding to a portion of the full-length negative strand DCV RNA. Following reverse transcription, cDNA was diluted 1:10 and used for for qPCR with the Luminaris Color HiGreen low ROX qPCR Master Mix (Thermo Scientific) according to the manufacturer’s instructions. A forward primer containing the non-target tag sequence (Tag_qPCR_F: AATTCAAGCTCGTCTTCCTCG) and a a DCV-specific reverse primer (DCV_qPCR_R: AATGGCAAGCGCACACAATTA) were used for qPCR.
RNA extraction, cDNA synthesis and NGS library production
To produce the NGS libraries from the evolution experiment, half of the total population of flies infected with DCV from each fly genotype, viral passage and biological replicates (approx. 250 flies) was used. To produce the NGS libraries from the viral stock from S2 cells (S2 DCV stock), two different aliquots of the stocks were used. To produce the NGS libraries from the DCV stock (virus infecting w1118 female flies used to contaminate the cages to start the evolution experiment), half of the population of the infected flies (approx. 800 flies: N = 90 flies/cage ’ 9 fly genotypes ’ 2 BR) was used. In all cases, total RNA was extracted using TRIzol reagent (Invitrogen) following the manufacturer’s instructions and the final concentration was determined using a NanoDrop ND-1000 Spectrophotometer. 300 ng of total RNA were used to produce the cDNA using oligo(dT) as primers reverse transcription with the Maxima H Minus Reverse Transcriptase Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. The cDNA obtained served as template to amplify the full-length genome of DCV with specific primers (DCVfor ATATGTACACACGGCTTTTAGGT and DCVrev CAGTAAGCAGGAAAATTGCG) using Phusion High-Fidelity DNA polymerase Kit (Thermo Fisher Scientific) in the following conditions: initial denaturation at 98 °C for 30 seconds; 30 cycles of denaturation at 98 °C for 10 seconds, annealing at 55 °C for 30 seconds and extension at 72 °C for 5 minutes; and final extension at 72 °C for 10 minutes. For both S2 DCV stock and DCV stock, four different DCV PCR amplifications were done to produce a total of four technical replicates of the NGS libraries. The PCR products were gel purified using the NucleoSpin Gel and PCR Clean- up kit (Machery-Nagel) and their concentration was determined using a NanoDrop ND-1000 Spectrophotometer. 200 ng of the purified PCR product were fragmented into 200 to 300 nucleotides long products using an LE220 ultrasonicator (Covaris) following the manufacturer’s instructions. The obtained fragments were used to produce the NGS library using the NEBNext UltraII DNA Library Prep Kit for Illumina (New England BioLabs), according to manufacturer’s instructions. The quality of the libraries was verified using a High Sensitivity DNA Chip (Agilent) and quantified using the Quant-iT DNA assay kit (Thermo Fisher Scientific). A 1 nM dilution of the libraries was used for the sequencing that was performed on a NextSeq sequencer (Illumina) with a NextSeq 500 Mid Output kit v2 (Illumina) (151 cycles). Two of the four technical replicates for S2 DVC stock and DCV stock were included in each run.
Sequencing of DCV populations from Dif1/1 mutant flies from P = 4 and P = 6 from BR1 and P = 8 from BR2 did not work.
Genetic diversity analyses
Variant frequency threshold
To determine the error rate of the sequencing procedure, including library preparation, four sequencing technical replicates from S2 DCV stock were used (Supplementary Figure 3a). First, pairwise comparison was done to identify the variant frequency threshold above which at least 95% of the variants were detected in both considered replicates (highest detection threshold = 0.0028). All variants above detection threshold were then correlated between each technical replicate to ensure good correlation between reported frequency values: the Pearson correlation coefficient between the detected frequency for variants was r ≥ 0.982 for all pairwise correlation (p < 0.001). The R packages used for these analysis were somewhere else described69–72.
Nucleotide diversity (π)
Nucleotide diversity of the viral population was computed using the following formula73:
with D, the sequencing depth and p the frequency of the minority variant at each nucleotide site. For diallelic SNV, π ranges from 0 to 0.5 (both alleles at equal frequency). In the subsequent analyses, π was averaged over all polymorphic nucleotide sites of the DCV genome of each sample74. A site was considered polymorphic if at least one sample showed the presence of a nucleotide variant at said position of the DCV genome. Log10-transformed site-averaged π values were then compared between fly genotypes (orthogonal factor), biological replicates (orthogonal factor), passages (continuous variable) and genomic regions (orthogonal factor) and their interactions using a generalized linear model. The significance of each term in the model was evaluated using a likelihood ratio test that follows a χ2 probability distribution.
Estimation of relative mutational fitness effects
We have followed the classic population genetics method described in Hartl and Clark (1989)43. In short, lets xl(t) be the frequency of a mutant allele (SNP) at genomic position l and passage t and, therefore, 1 − Xl(t) the frequency of the wild-type allele. It holds that where sl is the selection coefficient of the mutant relative to the wild-type allele at locus l. Selection coefficients calculated this way have units of inverse time (per passage in our case). This equation was fitted to the time series data of each locus l shown in Figure 3 by least squares regression, obtaining an estimate of sl and its standard error (SEM).
Haplotype inference was done using two different statistical approaches. First, by assessing the similarity between temporal dynamics of all possible pairs of loci. To this end, Pearson partial correlation coefficients (controlling for passages) were computed and their significance level corrected for multiple tests of the same null hypothesis using Benjamini and Hochberg (1995)75 false discovery rate (FDR) method. Correlation coefficient matrices were visualized as heatmaps in which more similar alleles were clustered together. Second, we confirmed the results from the first method using the longitudinal variant allele frequency factorization problem (LVAFFP) method as implemented in CALDER76. LVAFFP generates spanning trees of a directed graph constructed from the variant allele frequencies. The output of CALDER was used as input of TimeScape77 to generate the Muller plots that illustrate the ancestry of mutations and haplotypes along the evolution experiment (Figure 3).
Statistical analyses described in this section have been done with R version 4.0.2 in RStudio version 1.3.1073.
Extended Data
Extended Data Figure 1.

Extended Data Figure 2.

Extended Data Figure 3.

Extended Data Figure 4.

Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank members of the Saleh Lab, M. Vignuzzi and J. Pfeiffer for fruitful discussions. We thank C. Meignin for RelE20 and VagoΔM10 flies. This work was supported by the European Research Council (FP7/2013–2019 ERC CoG 615220) and the French Government’s Investissement d’Avenir program, Laboratoire d’Excellence Integrative Biology of Emerging Infectious Diseases (grant ANR-10-LABX-62-IBEID) to M.-C.S. Work in S.F.E.’s laboratory was supported by grants BFU2015-65037-P and PID2019-103998GB-I00 (Spain Agencia Estatal de Investigación - FEDER) and PROMETEU2019/012 (Generalitat Valenciana).
Footnotes
Author contributions
V.M. and M.-C.S. conceived the study, and V.M., M.-C.S., A.K. and L.Q-M. established the experimental design. V.M., V.G., H.B, and J.N. performed the investigations. S.L. and S.F.E. performed the formal analyses. V.M., S.F.E. and M.- C.S. wrote the paper and acquired funding.
Competing interests
The authors declare no competing interests.
Data availability
All raw data from high-throughput sequencing were deposited to NCBI BioProjects under accession number PRJNA782868.
Code availability
Scripts are provided in Supplementary Data 1.
References
- 1.Morgan AD, Koskella B. Coevolution of Host and Pathogen in Genetics and Evolution of Infectious Diseases. Elsevier; 2017. pp. 115–140. [DOI] [Google Scholar]
- 2.Daugherty MD, Malik HS. Rules of Engagement: Molecular Insights from Host-Virus Arms Races. Annu Rev Genet. 2012;46:677–700. doi: 10.1146/annurev-genet-110711-155522. [DOI] [PubMed] [Google Scholar]
- 3.Barreiro LB. From evolutionary genetics to human immunology: how selection shapes host defence genes. G E N E Ti C S. 14 doi: 10.1038/nrg2698. [DOI] [PubMed] [Google Scholar]
- 4.Thompson JN, Burdon JJ. Gene-for-gene coevolution between plants and parasites. 1992;360:5. [Google Scholar]
- 5.Buckling A, Rainey PB. Antagonistic coevolution between a bacterium and a bacteriophage. 2002;6 doi: 10.1098/rspb.2001.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masri L, et al. Host-Pathogen Coevolution: The Selective Advantage of Bacillus thuringiensis Virulence and Its Cry Toxin Genes. PLOS Biol. 2015;13:e1002169. doi: 10.1371/journal.pbio.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Obbard DJ, Gordon KHJ, Buck AH, Jiggins FM. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc B Biol Sci. 2009;364:99–115. doi: 10.1098/rstb.2008.0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazzaro BP. Natural selection on the Drosophila antimicrobial immune system. Curr Opin Microbiol. 2008;11:284–289. doi: 10.1016/j.mib.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazzaro BP. Molecular Population Genetics of Inducible Antibacterial Peptide Genes in Drosophila melanogaster. Mol Biol Evol. 2003;20:914–923. doi: 10.1093/molbev/msg109. [DOI] [PubMed] [Google Scholar]
- 10.Sackton TB, et al. Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 2007;39:1461–1468. doi: 10.1038/ng.2007.60. [DOI] [PubMed] [Google Scholar]
- 11.Brackney DE, Beane JE, Ebel GD. RNAi Targeting of West Nile Virus in Mosquito Midguts Promotes Virus Diversification. PLoS Pathog. 2009;5:9. doi: 10.1371/journal.ppat.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin S-S, et al. Molecular Evolution of a Viral Non-Coding Sequence under the Selective Pressure of amiRNA-Mediated Silencing. PLoS Pathog. 2009;5:e1000312. doi: 10.1371/journal.ppat.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lafforgue G, et al. Tempo and Mode of Plant RNA Virus Escape from RNA Interference-Mediated Resistance. J Virol. 2011;85:9686–9695. doi: 10.1128/JVI.05326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martínez F, et al. Ultradeep sequencing analysis of population dynamics of virus escape mutants in RNAi-mediated resistant plants. Mol Biol Evol. 2012;29:3297–3307. doi: 10.1093/molbev/mss135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das AT, et al. Human Immunodeficiency Virus Type 1 Escapes from RNA Interference-Mediated Inhibition. J Virol. 2004;78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gitlin L, Stone JK, Andino R. Poliovirus Escape from RNA Interference: Short Interfering RNA-Target Recognition and Implications for Therapeutic Approaches. J Virol. 2005;79:1027–1035. doi: 10.1128/JVI.79.2.1027-1035.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mondotte JA, Saleh M-C. Advances in Virus Research. Vol. 100. Elsevier; 2018. Antiviral Immune Response and the Route of Infection in Drosophila melanogaster; pp. 247–278. [DOI] [PubMed] [Google Scholar]
- 18.Swevers L, Liu J, Smagghe G. Defense Mechanisms against Viral Infection in Drosophila: RNAi and Non-RNAi. Viruses. 2018;10:230. doi: 10.3390/v10050230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galiana-Arnoux D, Dostert C, Schneemann A, Hoffmann JA, Imler J-L. Essential function in vivo for Dicer-2 in host defense against RNA viruses in drosophila. Nat Immunol. 2006;7:590–597. doi: 10.1038/ni1335. [DOI] [PubMed] [Google Scholar]
- 20.van Rij RP, et al. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes Dev. 2006;20:2985–2995. doi: 10.1101/gad.1482006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X-H, et al. RNA Interference Directs Innate Immunity Against Viruses in Adult Drosophila. Science. 2006;312:452–454. doi: 10.1126/science.1125694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zambon RA, Vakharia VN, Wu LP. RNAi is an antiviral immune response against a dsRNA virus in Drosophila melanogaster. Cell Microbiol. 2006;8:880–889. doi: 10.1111/j.1462-5822.2006.00688.x. [DOI] [PubMed] [Google Scholar]
- 23.Costa A, Jan E, Sarnow P, Schneider D. The Imd Pathway Is Involved in Antiviral Immune Responses in Drosophila. PLoS ONE. 2009;4:e7436. doi: 10.1371/journal.pone.0007436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sansone CL, et al. Microbiota-Dependent Priming of Antiviral Intestinal Immunity in Drosophila. Cell Host Microbe. 2015;18:571–581. doi: 10.1016/j.chom.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferreira ÁG, et al. The Toll-Dorsal Pathway Is Required for Resistance to Viral Oral Infection in Drosophila. PLoS Pathog. 2014;10:e1004507. doi: 10.1371/journal.ppat.1004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zambon RA, Nandakumar M, Vakharia VN, Wu LP. The Toll pathway is important for an antiviral response in Drosophila. Proc Natl Acad Sci. 2005;102:7257–7262. doi: 10.1073/pnas.0409181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dostert C, et al. The Jak-STAT signaling pathway is required but not sufficient for the antiviral response of drosophila. Nat Immunol. 2005;6:946–953. doi: 10.1038/ni1237. [DOI] [PubMed] [Google Scholar]
- 28.Merkling SH, et al. The Epigenetic Regulator G9a Mediates Tolerance to RNA Virus Infection in Drosophila. PLOS Pathog. 2015;11:e1004692. doi: 10.1371/journal.ppat.1004692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christian PD, Johnson KN. The novel genome organization of the insect picorna-like virus Drosophila C virus suggests this virus belongs to a previously undescribed virus family. J Gen Virol. 1998;79:191–203. doi: 10.1099/0022-1317-79-1-191. [DOI] [PubMed] [Google Scholar]
- 30.Jousset FX, Plus N. Study of the vertical transmission and horizontal transmission of ‘Drosophila melanogaster’ and ‘Drosophila immigrans’ picornavirus (author’s transl) Ann Microbiol (Paris) 1975;126:231–249. [PubMed] [Google Scholar]
- 31.Mondotte JA, et al. Immune priming and clearance of orally acquired RNA viruses in Drosophila. Nat Microbiol. 2018;3:1394–1403. doi: 10.1038/s41564-018-0265-9. [DOI] [PubMed] [Google Scholar]
- 32.Torri A, Mongelli V, Mondotte JA, Saleh M-C. Viral Infection and Stress Affect Protein Levels of Dicer 2 and Argonaute 2 in Drosophila melanogaster. Front Immunol. 2020;11:362. doi: 10.3389/fimmu.2020.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deddouche S, et al. The DExD/H-box helicase Dicer-2 mediates the induction of antiviral activity in drosophila. Nat Immunol. 2008;9:1425–1432. doi: 10.1038/ni.1664. [DOI] [PubMed] [Google Scholar]
- 34.Gomariz-Zilber E, Jeune B, Thomas-Orillard M. Limiting conditions of the horizontal transmission of the Drosophila C virus in its host (D. melanogaster) Acta Oecologica. 1998;19:125–137. [Google Scholar]
- 35.Stevanovic AL, Johnson KN. Infectivity of Drosophila C virus following oral delivery in Drosophila larvae. J Gen Virol. 2015;96:1490–1496. doi: 10.1099/vir.0.000068. [DOI] [PubMed] [Google Scholar]
- 36.Royet J. Epithelial homeostasis and the underlying molecular mechanisms in the gut of the insect model Drosophila melanogaster. Cell Mol Life Sci. 2011;68:3651–3660. doi: 10.1007/s00018-011-0828-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buchon N, Broderick NA, Poidevin M, Pradervand S, Lemaitre B. Drosophila Intestinal Response to Bacterial Infection: Activation of Host Defense and Stem Cell Proliferation. Cell Host Microbe. 2009;5:200–211. doi: 10.1016/j.chom.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Buchon N, Broderick NA, Chakrabarti S, Lemaitre B. Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in Drosophila. Genes Dev. 2009;23:2333–2344. doi: 10.1101/gad.1827009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buchon N, Broderick NA, Kuraishi T, Lemaitre B. DrosophilaEGFR pathway coordinates stem cell proliferation and gut remodeling following infection. BMC Biol. 2010;8:152. doi: 10.1186/1741-7007-8-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J, et al. ERK signaling couples nutrient status to antiviral defense in the insect gut. Proc Natl Acad Sci. 2013;110:15025–15030. doi: 10.1073/pnas.1303193110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lauring AS, Andino R. Quasispecies Theory and the Behavior of RNA Viruses. PLoS Pathog. 2010;6:e1001005. doi: 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isakov O, et al. Deep sequencing analysis of viral infection and evolution allows rapid and detailed characterization of viral mutant spectrum. 10 doi: 10.1093/bioinformatics/btv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartl DL, Clark AG. Principles of Population Genetics. (Fourth edition) 7 [Google Scholar]
- 44.Travisano M, Mongold JA, Bennett AF, Lenski RE. Experimental Tests of the Roles of Adaptation, Chance, and History in Evolution. 1995;267:5. doi: 10.1126/science.7809610. [DOI] [PubMed] [Google Scholar]
- 45.Desai MM, Fisher DS. Beneficial Mutation-Selection Balance and the Effect of Linkage on Positive Selection. Genetics. 2007;176:1759–1798. doi: 10.1534/genetics.106.067678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miralles R. Clonal Interference and the Evolution of RNA Viruses. Science. 1999;285:1745–1747. doi: 10.1126/science.285.5434.1745. [DOI] [PubMed] [Google Scholar]
- 47.Miralles R, Moya A, Elena SF. Diminishing Returns of Population Size in the Rate of RNA Virus Adaptation. J Virol. 2000;74:3566–3571. doi: 10.1128/jvi.74.8.3566-3571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pepin KM, Wichman HA. Experimental evolution and genome sequencing reveal variation in levels of clonal interference in large populations of bacteriophage φX174. BMC Evol Biol. 2008;8:85. doi: 10.1186/1471-2148-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Navarro R, Ambrós S, Martínez F, Elena SF. Diminishing returns of inoculum size on the rate of a plant RNA virus evolution. Eurphys Lett. 2017;120:38001. 2017. [Google Scholar]
- 50.Pandit A, de Boer RJ. Reliable reconstruction of HIV-1 whole genome haplotypes reveals clonal interference and genetic hitchhiking among immune escape variants. Retrovirology. 2014;11:56. doi: 10.1186/1742-4690-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strelkowa N, Lässig M. Clonal Interference in the Evolution of Influenza. Genetics. 2012;192:671–682. doi: 10.1534/genetics.112.143396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerrish PJ, Lenski RE. In: Mutation and Evolution. Woodruff RC, Thompson JN, editors. Vol. 7. Springer; Netherlands: 1998. The fate of competing beneficial mutations in an asexual population; pp. 127–144. [PubMed] [Google Scholar]
- 53.Held T, Klemmer D, Lässig M. Survival of the simplest in microbial evolution. Nat Commun. 2019;10:2472. doi: 10.1038/s41467-019-10413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Novella IS, Zárate S, Metzgar D, Ebendick-Corpus BE. Positive Selection of Synonymous Mutations in Vesicular Stomatitis Virus. J Mol Biol. 2004;342:1415–1421. doi: 10.1016/j.jmb.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 55.Zanini F, Neher RA. Quantifying Selection against Synonymous Mutations in HIV-1 env Evolution. J Virol. 2013;87:11843–11850. doi: 10.1128/JVI.01529-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martínez MA, Jordan-Paiz A, Franco S, Nevot M. Synonymous Virus Genome Recoding as a Tool to Impact Viral Fitness. Trends Microbiol. 2016;24:134–147. doi: 10.1016/j.tim.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 57.Kieft JS, Zhou K, Jubin R, Doudna JA. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA. 2001;7:194–206. doi: 10.1017/s1355838201001790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Rij RP, et al. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes Dev. 2006;20:2985–2995. doi: 10.1101/gad.1482006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang L, et al. lncRNA Sensing of a Viral Suppressor of RNAi Activates Non-canonical Innate Immune Signaling in Drosophila. Cell Host Microbe. 2020;27:115–128.:e8. doi: 10.1016/j.chom.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 60.Navarro R, et al. Defects in plant immunity modulate the rates and patterns of RNA virus evolution. 2020. http://biorxiv.org/lookup/doi/10.1101/2020.10.13.337402 . [DOI] [PMC free article] [PubMed]
- 61.Merkling SH, van Rij RP. Analysis of resistance and tolerance to virus infection in Drosophila. Nat Protoc. 2015;10:1084–1097. doi: 10.1038/nprot.2015.071. [DOI] [PubMed] [Google Scholar]
- 62.Lee YS, et al. Distinct Roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA Silencing Pathways. Cell. 2004;117:69–81. doi: 10.1016/s0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- 63.Okamura K. Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev. 2004;18:1655–1666. doi: 10.1101/gad.1210204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Levashina EA, Ohresser S, Lemaitre B, Imler JL. Two distinct pathways can control expression of the gene encoding the Drosophila antimicrobial peptide metchnikowin. J Mol Biol. 1998;278:515–527. doi: 10.1006/jmbi.1998.1705. [DOI] [PubMed] [Google Scholar]
- 65.Rutschmann S, et al. The Rel Protein DIF Mediates the Antifungal but Not the Antibacterial Host Defense in Drosophila. Immunity. 2000;12:569–580. doi: 10.1016/s1074-7613(00)80208-3. [DOI] [PubMed] [Google Scholar]
- 66.Hedengren M, et al. Relish, a Central Factor in the Control of Humoral but Not Cellular Immunity in Drosophila. Mol Cell. 1999;4:827–837. doi: 10.1016/s1097-2765(00)80392-5. [DOI] [PubMed] [Google Scholar]
- 67.Díaz-Benjumea FJ, García-Bellido A. Behaviour of cells mutant for an EGF receptor homologue of Drosophila in genetic mosaics. Proc Biol Sci. 1990;242:36–44. doi: 10.1098/rspb.1990.0100. [DOI] [PubMed] [Google Scholar]
- 68.Reed LJ, Muench H. A SIMPLE METHOD OF ESTIMATING FIFTY PER CENT ENDPOINTS12. Am J Epidemiol. 1938;27:493–497. [Google Scholar]
- 69.Hothorn T, Bretz F, Westfall P. Simultaneous Inference in General Parametric Models. Biom J. 2008;50:346–363. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 70.Weisberg S, F J. An R Companion to Applied Regression. Third edition. Sage; Thousand Oaks CA: 2019. https://socialsciences.mcmaster.ca/jfox/Books/Companion/.in . [Google Scholar]
- 71.Lenth. Estimated Marginal Means, aka Least-Squares Means R package version 1.5.5. 2021 [Google Scholar]
- 72.Wickham H, et al. Welcome to the Tidyverse. J Open Source Softw. 2019;4:1686 [Google Scholar]
- 73.Cornman RS, et al. Population-genomic variation within RNA viruses of the Western honey bee, Apis mellifera, inferred from deep sequencing. BMC Genomics. 2013;14:154. doi: 10.1186/1471-2164-14-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lequime S, Fontaine A, Ar Gouilh M, Moltini-Conclois I, Lambrechts L. Genetic Drift Purifying Selection and Vector Genotype Shape Dengue Virus Intra-host Genetic Diversity in Mosquitoes. PLOS Genet. 2016;12:e1006111. doi: 10.1371/journal.pgen.1006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- 76.Myers MA, Satas G, Raphael BJ. CALDER: Inferring Phylogenetic Trees from Longitudinal Tumor Samples. Cell Syst. 2019;8:514–522.:e5. doi: 10.1016/j.cels.2019.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smith MA, et al. E-scape: interactive visualization of single-cell phylogenetics and cancer evolution. Nat Methods. 2017;14:549–550. doi: 10.1038/nmeth.4303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data from high-throughput sequencing were deposited to NCBI BioProjects under accession number PRJNA782868.
Scripts are provided in Supplementary Data 1.