

Abstract
This manuscript describes the development of a remarkably general palladium catalyzed mono-acylation of carbazoles using toluene derivatives playing the dual role of acyl source and organic solvent. The method uses NHPI as the co-catalyst and oxygen as the sole oxidant. Interestingly, the acylation of monosubstituted N-pyridyl carbazoles takes place regioselectively at the C-8 position. The scope of the method is explored using aldehyde as the acyl source. This highly site-selective acylation proceeds through a radical process.

Introduction
C-H bond functionalization has emerged as one of the most powerful tools for generating new carbon-carbon and carbon-heteroatom bonds.1 However, selective C-H functionalization at a particular position is challenging.1a,1g,2 Carbazoles are one of the most ubiquitous heteroaromatic motifs found in myriad of natural products, photorefractive materials, organic dyes3 and bioactive molecules.4 Despite significant progress achieved in the synthesis and functionalization of carbazoles, only a few methods are known to obtain C1/C8- substituted carbazoles.5 Further, due to the nucleophilic character of C3 and C6 positions of carbazole, electrophilic substitution occurs mostly at the C3 and C6 positions.6 Therefore, regioselective functionalization at the less activated C1 and C8 positions of the carbazole without protecting the C3 and C6 positions5,7 is challenging.
Recently, Pd(II)-catalyzed direct 1-arylation of pyridine protected carbazoles,8 1,8-di-olefination of carbazoles,9 Ru and Cu co-catalyzed dehydrogenative C1-N carbazolation,10 Ru catalyzed regioselective C1 and C8 di-acetoxylation of N-pyridylcarbazoles11 have been reported for the functionalization of carbazoles at C1 and C8 positions. We have recently reported palladium(II) catalyzed 1,8-diacylation of carbazoles using aldehydes as the acyl source and TBHP as the oxidant.12 Most of the oxidative C-H activation processes proceed through a Pd(II)/Pd(IV)13 manifold involving stoichiometric oxidant (TBHP, K2S2O8, Cu(OAc)2, DDQ, PIDA, etc.). Keeping the growing demand for sustainable synthesis, we envisioned to carry out acylation of carbazoles using natural and inexpensive molecular O2 as oxidant.14
Till now, few methods are known for the synthesis of 1-acylated carbazoles. The synthesis of 1-aroylcarbazoles by photolysis of N-aroyl carbazoles in low yields (~15-30%) was reported in 1987.15 The Katritzky group reported the synthesis of 1-aroyl carbazoles by o-lithiation of N-pyrrolidinomethyl carbazole and subsequent addition of electrophiles like aromatic aldehydes to the reaction mixture.16 The Friedel–Crafts acylation of carbazole with benzoyl chloride was reported to obtain a mixture of mono and dibenzoyl carbazole derivatives.17 Bandgar and co-workers reported one example of 1-benzoylcarbazole by Pd catalyzed cross-coupling of (9H-carbazol-1-yl)boronic acid with benzoyl chloride.18 Wu and Yang et al. illustrated the synthesis of pyrimidine protected 1-acylated carbazole derivatives by tandem Rh-catalyzed C-H activation of pyrimidine protected indoles and subsequent brønsted acid-catalyzed cyclization.19 Acylation of a pyrimidine protected carbazole with 4-methylbenzaldehyde using both Pd(II) catalyst and Ru(II)-photocatalyst in the presence of visible light and external oxidant TBHP was recently reported.20 Therefore, site-selective synthesis of 1-acylated carbazole derivatives using easily accessible starting materials, metal catalyst without added oxidant is clearly worth exploring.
Pd (II) catalyzed aerobic oxidative radical processes involving Pd III or IV intermediates have been reported for C-H functionalization.21 N-Hydroxyphthalimide (NHPI) is used as co-catalyst, which is oxidised by O2 to generate PINO (phthalimido-N-oxyl) radical.22 PINO initiates the radical oxidative reaction with the formation of benzoyl radical from toluene. Inspired by these transformations, we employed NHPI and O2 for the Pd (II) catalyzed 1-acylation of carbazole using toluene as the acyl source.22,23 Versatile C-H functionalization of toluene derivatives using metal catalysis has been previously reported.24
Results and Discussion
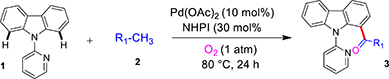
As a model reaction, the acylation of 9-(pyridin-2-yl)-9H-carbazole 1 was performed in the presence of 10 mol% PdCl2, 30 mol% of NHPI as co-catalyst in 1 mL toluene 2a under 1 atm O2 at 80 °C for 12 h (see Table S1; Supporting Information, S.I.). The reaction provided 10% 1-acylated carbazole derivative 3a (Table 1, entry 1). The yield of 3a was only marginally increased even with prolonged reaction time (Table 1, entries 2, 3). The yield of the reaction did not improve on increasing or lowering the reaction temperature (Table 1, entries 4, 5). The reaction did not proceed at room temperature (entry 6). Other Pd sources like Pd(TFA)2 and Pd2(dba)3 provided the product 3a in 55% and 29% isolated yields, respectively (Table 1, entries 7, 8). We were delighted to observe that with 10 mol% Pd(OAc)2, the reaction gave 76% of 3a along with 5% 1,8-diacylated carbazole 4a at 80 °C for 24 h (Table 1, entry 9). Pd(PPh3)2Cl2 or Pd(PPh3)4 failed to give any desired product (Table 1, entries 10, 11). Thus, Pd(OAc)2 was used as the preferred catalyst for this transformation. Next, instead of NHPI, different one-electron oxidants like BQ, TEMPO or Cu(OAc)2 were also investigated. No desired product was formed (Table 1, entries 12, 13, 14). The yield of the reaction was slightly decreased when molecular oxygen was replaced by air (Table 1, entry 15).
Table 1. Optimization of Reaction Conditionsa.

| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Pd catalyst (10 mol%) | Co-cat. (30 mol%) | Oxidant | Time (h) | Temperature (°C) | Yieldb 3a (%) | Yieldb 4a (%) |
| 1 | PdCl2 | NHPI | O2 | 12 | 80 | 10 | - |
| 2 | PdCl2 | NHPI | O2 | 24 | 80 | 27 | trace |
| 3 | PdCl2 | NHPI | O2 | 36 | 80 | 29 | trace |
| 4 | PdCl2 | NHPI | O2 | 24 | 100 | 26 | trace |
| 5 | PdCl2 | NHPI | O2 | 36 | 60 | 10 | trace |
| 6 | PdCl2 | NHPI | O2 | 36 | rt | NRd | - |
| 7 | Pd(TFA)2 | NHPI | O2 | 24 | 80 | 55 | trace |
| 8 | Pd2(dba)3 | NHPI | O2 | 24 | 80 | 29 | trace |
| 9 | Pd(OAc)2 | NHPI | O2 | 24 | 80 | 76 | 5 |
| 10 | Pd(PPh3)2Cl2 | NHPI | O2 | 24 | 80 | NRd | - |
| 11 | Pd(PPh3)4 | NHPI | O2 | 24 | 80 | NRd | - |
| 12 | Pd(OAc)2 | BQ | O2 | 24 | 80 | NRd | - |
| 13 | Pd(OAc)2 | TEMPO | O2 | 24 | 80 | NRd | - |
| 14 | Pd(OAc)2 | Cu(OAc)2 | O2 | 24 | 80 | NRd | - |
| 15 | Pd(OAc)2 | NHPI | air | 24 | 80 | 67 | NDe |
| 16 | - | NHPI | O2 | 24 | 80 | NRd | - |
| 17 | Pd(OAc)2 | - | O2 | 24 | 80 | NRd | - |
| 18 | Pd(OAc)2 | NHPI | Ar | 24 | 80 | NRd | - |
| 19c | Pd(OAc)2 | NHPI | O2 | 24 | 80 | 70 | 10 |
9-(pyridin-2-yl)-9H-carbazole 1 (0.25 mmol), toluene 2a (1 mL);
isolated yields;
preparative scale experiment;
no reaction occurred;
not determined.
In the absence of either NHPI or Pd(OAc)2, the reaction did not proceed (Table 1, entries 16, 17). Furthermore, the reaction did not occur under an argon atmosphere (Table 1, entry 18). It is worth noting that the reaction could be scaled (1.0 g of 1) without a loss of yield under the optimized conditions (entry 19). Thus, the optimization study implied that Pd(OAc)2, NHPI and O2 are respectively the most suitable catalyst, co-catalyst, and oxidant for the site-selective 1-acylation of 9-(pyridin-2-yl)-9H-carbazole 1.
With the optimized reaction condition, we then explored the 1-acylation of 9-(pyridin-2-yl)-9H-carbazole 1 with a variety of toluene derivatives 2. 4-Chlorotoluene 2b showed slightly higher reactivity than 3-chlorotoluene 2c producing the desired products 3b (78%) and 3c (71%) in high yields. 4-Bromotoluene 2d and 3-bromotoluene 2e displayed comparable reactivity, generating the desired products 3d and 3e in 73% and 74% isolated yields, respectively. Similarly, 4-fluorotoluene 2f and 3-fluorotoluene 2g, afforded the desired products 3f and 3g in high yields. However, the reactivity of 2-fluorotoluene 2h reduced considerably, furnishing the desired product 3h in 33% yield. Toluene derivatives bearing other electron withdrawing groups, apart from halogens, also participated well in the 1-acylation reaction. For instance, 4-methylbenzonitrile 2i, 3-methylbenzonitrile 2j and methyl 4-methylbenzoate 2k reacted to provide the desired products 3i, 3j, 3k in 77%, 78% and 79% yields, respectively.
The toluene derivative 4′-methylacetophenone 2l reacted with 1 to furnish the desired product 3l in excellent yield (94%). Toluene derivatives having an electron donating group were also well tolerated. For example, 2-methylanisole 2m afforded 1-acylated carbazole derivative 3m in 71% isolated yield. Also, m-xylene 3n, o-xylene 2o and mesitylene 2p participated in the reaction effectively to provide the desired products in good to moderate yields (Table 2, products 3n, 3o, and 3p). The structure of mono-acylated products 3f (CCDC 1989688) and 3i (CCDC 1989676) were confirmed by single crystal X-ray analysis (Table 2; Figure S1 and S2, S.I.).25 Gram scale experiments using 1g of 1 were turned out to be successful to obtain mono-acylated carbazoles 3a and 3b in good yields (Table 2; Scheme S4 and S5, S.I.).
Table 2. Acylation of Unsubstituted N-pyridyl Carbazoles with Toluene derivativesa,b.

|

|
|
|

|

|

|
|

|

|

|
|

|

|

|

|

|
|||
N-pyridylcarbazole 1 (0.25 mmol), Pd(OAc)2 (10 mol%), NHPI (30 mol%), toluene 2 (1 mL), O2 (1 atm), 80 °C;
isolated yields;
yield obtained when reaction performed with 1g of 1.
Next, the 1-acylation was examined with various substituted N-pyridylcarbazoles. Symmetrical 3,6-dichloro N-pyridylcarbazole 5 reacted with 4-chlorotoluene 2b to afford 1-acylated product 10b in 83% yield (Table 3). 3,6-Dibromo N-pyridylcarbazole 6 reacted with electron rich 1-methoxy-4-methylbenzene 2q and electron deficient 4-methylbenzoate 2k to give 11q and 11k in 66% and 68% yields, respectively (Table 3); whereas 3,6-diiodo N-pyridylcarbazole 7 reacted with toluenes to provide the desired products 12q and 12k in moderate yields (Table 3).
Table 3. Acylation of Substituted N-pyridyl Carbazoles with Toluene derivativesa,b.

|

|

|

|

|

|

|
|

N-pyridylcarbazole 5-9 (0.25 mmol), Pd(OAc)2 (10 mol%), NHPI (30 mol%), toluene 2 (1 mL), O2 (1 atm), 80 °C;
isolated yields.
It is intriguing to find that when unsymmetrical mono substituted N-pyridylcarbazole was used, acylation occurred exclusively at the ortho position of the unsubstituted ring. Unsymmetrical N-pyridylcarbazole 10 having 3-chloro substituent produced 1-acylated product 13a in 69% yield. The structure of 13a was confirmed by X-ray crystal analysis (Table 3, CCDC 1989702; Figure S3, S.I.).25 N-pyridylcarbazole 12 having 3-methoxy group furnished the 1-acylated product 13b in 60% yield.
To validate our hypothesis about the in-situ generation of the aldehyde from toluene in the reaction media, we performed the reaction by using aldehydes 16 instead of toluenes 2 in chlorobenzene as solvent (Table 4). The desired products were obtained in good isolated yields. The reaction proceeded well with heptaldehyde 16e providing the desired product 3r in 75% yield (Table 4). Inspired by the result, we carried out the acylation of carbazole 15 with heptaldehyde 16e to synthesize the acyl precursor 18 of carbazole alkaloid carazostatin.26 The acylation provided exclusively the regioisomeric acyl derivative 17e in 70% yield (Table 4).
Table 4. Acylation of N-pyridylcarbazoles with Aldehydesa,b.

|

|

|

|

|

|

|
|

N-pyridylcarbazole 1 or 15 (0.25 mmol), aldehyde 16 (0.5 mmol), Pd(OAc)2 (10 mol%), NHPI (30 mol%), chlorobenzene (1 mL), O2 (1 atm), 80 °C;
isolated yields.
To gain insight about the reaction mechanism, we carried out several control experiments. The reaction did not proceed in the presence of argon atmosphere (Table 1, entry 18), suggesting molecular O2 plays a key role in this transformation. The 18O labelling experiment confirms that the oxygen atom of the keto-carbonyl group in the mono-acylated product 3a′ originates from molecular oxygen (Scheme 2; Scheme S4, S.I.).
Scheme 2. 18O Labeling Experiment.

Further, control experiments were performed using dimeric Pd(II) complex CP; prepared following the literature procedure.8 When the acylation of 1 (1 equiv) was carried out using CP (5 mol%) instead of Pd(OAc)2, the desired product 3b was obtained in good yield (Scheme 3, a). However, when complex CP was used as both catalyst and substrate, no desired product was obtained (Scheme 3, b). Interestingly, the reaction of 3-methoxy N-pyridylcarbazole 9 with 2b in the presence of CP (25 mol%) provided 14b in 50% yield and 3b in 28% yield (Scheme 3, c). These experimental observations suggest that decomposition of the strong dimeric Pd(II) complex to the active monomeric Pd(II) intermediate might be essential for this transformation.
Scheme 3. Control Experiments.

To know whether the reaction proceeds through a radical or ionic pathway, the reaction was performed in the presence of a radical inhibitor TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl). The starting material 1 was fully recovered and the acyl adduct 19 was obtained in 70% yield (Scheme 4; Scheme S5, S.I.). This result indicated that acyl radical is trapped by the TEMPO free radical.
Scheme 4. Radical Quenching Experiment.

Based on experimental observations and previous reports,21,22 a possible reaction mechanism is proposed (Scheme 5). The first step involves the generation of PINO radical from NHPI and dioxygen.The in situ generated PINO radical abstracts hydrogen radical from toluene 2a to form the benzylic radical which on reaction with molecular oxygen forms the reactive benzoyl radical via in situ formation of benzaldehyde. Pd(II) catalyzed chelation-directed C–H activation followed by ligand assisted proton abstraction in A leads to the cyclo-palladated complex B. The complex B undergoes oxidative addition to bezoyl radical to generate the intermediate Pd(IV) species C. Reductive elimination of C provides the desired mono-acylated product 3a and regenerates Pd(II) catalyst.
Scheme 5. Probable Catalytic Cycle of the Reaction.

Conclusion
In conclusion, we have developed a palladium catalyzed highly site-selective method for the synthesis of mono-acylated carbazole derivatives using toluene as the acyl source in the presence of molecular O2 as oxidant. For monosubstituted N-pyridyl carbazoles, acylation takes place regioselectively in the unsubstituted benzene ring providing C-1 acylated carbazoles in high yields. Notably, the method does not require any additional organic solvent. Thus, our approach provides a practical and general route for site-selective C−H acylation of carbazoles via an oxidative catalytic transformation. The operational simplicity, substrate compatibility and use of molecular oxygen as sole oxidant make the present protocol synthetically useful and environmentally attractive.
Experimental Section
General Information
All experiments were carried out under an inert atmosphere of argon or under oxygen atmosphere in oven-dried flasks, sealed tubes and schlenk tubes. Solvents were dried according to standard procedures. All starting materials were obtained from commercial suppliers and used as received. Products were purified by column chromatography on silica gel (100–200 mesh, Merck). Unless otherwise stated, yields refer to analytically pure samples. NMR samples were dissolved in CDCl3 and DMSO-d6. The 1H NMR spectroscopic data were recorded with 500, 400 MHz instruments at 298 K. Signals are reported as δ values in ppm by using the residual signal of the protonated solvent as the internal standard (for CHCl3, δ = 7.26 ppm). Data are reported in the order of chemical shift, multiplicity [s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), br. (broad), and m (multiplet)], coupling constant (J) in Hz, and integration values. The 13C NMR spectroscopic data were recorded with complete proton decoupling with either a 100 or 125 MHz spectrometer. Chemical shifts (δ) are reported in ppm downfield from tetramethylsilane with the solvent as the internal reference (for CDCl3, δ = 77.16 ppm). HRMS analyses were performed with Q-TOF high resolution instruments by using positive ion mode electrospray ionization.
General procedure for the synthesis of carbazole derivatives 1, 5 and 8
Using the following literature,27 9H carbazole S1 (167.2 mg, 1.0 mmol) or 3,6-dichloro-9H-carbazole S2 (236.1 mg, 1.0 mmol) or 3-chloro-9H-carbazole S3 (201.6 mg, 1.0 mmol) was taken in an oven-dried 5 mL vial with a magnetic stirrer bar. To this vial, Cs2CO3 (325.8, 1.0 mmol), 2-bromopyridine (105 μL, 1.1 mmol), CuI (19 mg, 10 mol%), 2 mL dry DMF were added. Then the vial was sealed with a cap and irradiated for 45 mins at 220 °C in a microwave reactor and then cooled to room temperature. The reaction mixture was diluted with saturated aqueous ammonium chloride and product was isolated by extraction with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The product was purified by silica gel column chromatography using hexane/ethyl acetate as eluent to give pyridine protected carbazole derivatives 1, 5, 8 as solid (Scheme S1, S.I.).
General procedure for the synthesis of 3,6-dibromo-9-(pyridin-2-yl)-9H-carbazole 6
As reported in the literature,12 9-(pyridin-2-yl)-9H-carbazole 1 (444.6 mg, 2 mmol) in dichlomethane (25 mL), containing silica (100-200 mesh, Merck, 8 gm), a solution of NBS (712 mg, 4 mmol in 25 mL dichloromethane) was added drop wise. The reaction mixture was stirred for 4.5 h in the absence of light at 18 °C until TLC indicated that reaction was completed. The reaction mixture was filtered and the silica was washed with dichloromethane. The combined extracts were washed with water (100 mL) and organic layer was dried and evaporated. The residue was purified by silica gel column chromatography using hexane/ethyl acetate as eluent to give 3,6-dibromo-9-(pyridin-2-yl)-9H-carbazole 6 (563 mg, 70%) as a white solid (Scheme S2, S.I.).
General procedure for the synthesis of 3,6-diiodo-9-(pyridin-2-yl)-9H-carbazole 7
As reported in the literature,12 10 mL glacial acetic acid was taken in a 50 mL round bottom flask equipped with a magnetic stir bar and boiled. To this boiled acetic acid, 9-(pyridin-2-yl)-9H-carbazole 1 (444.6 mg, 2 mmol), KI (431.6 mg, 2.6 mmol) were added. Then the mixture was cooled, potassium iodate (642.0 mg, 3 mmol) was added and the reaction mixture was again boiled until a clear straw-colour was observed. The hot solution mixture was decanted from un-dissolved potassium iodate then diluted with saturated sodium thiosulphate solution and the product was isolated by the extraction into ethyl acetate. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate as eluent to provide 3,6-diiodo-9-(pyridin-2-yl)-9H-carbazole 7 (397.0 mg, 40%) as a white solid (Scheme S3, S.I.).
General procedure for the mono C–H acylation of carbazole derivatives using toluene as the acyl source (GP-1)
Carbazole derivative 1, 5, 6, 7, 8 or 9 (0.25 mmol), Pd(OAc)2 (10 mol %), NHPI (30 mol %), dry toluene 2 (1.0 mL) were added to a 10 mL Schlenk tube with a magnetic bar under O2. The resulting mixture was evacuated and backfilled with O2 for 2-3 times. The reaction mixture was placed in a preheated oil bath at 80 °C for 24 h and monitored by TLC analysis. The solution was then cooled and diluted with dichloromethane (3 x 15 mL), washed with sodium bicarbonate (3 x 10 mL), brine (2 x 10 mL), dried over anhydrous Na2SO4, filtered, and evaporated under vacuum. The crude reaction mixture was purified by column chromatography on silica gel (hexane/ethyl acetate) to obtain products 3, 10, 11, 12, 13, or 14. The structure of the products was confirmed by 1H and 13C NMR spectroscopy and mass spectrometry analyses.
5.0 General procedure for the mono C–H acylation of carbazole derivatives using aldehyde as the acyl source (GP-2)
Carbazole derivative 1 or 15 (0.25 mmol), aldehyde 16 (0.5 mmol), Pd(OAc)2 (10 mol %), NHPI (30 mol %), dry chlorobenzene (1.0 mL) were added to a 10 mL Schlenk tube with a magnetic bar under O2. The resulting mixture was evacuated and backfilled with O2 for 2-3 times. The reaction mixture was placed in a preheated oil bath at 80 °C for 24 h and monitored by TLC analysis. The solution was then cooled and diluted with dichloromethane (3 x 15 mL), washed with sodium bicarbonate (3 x 10 mL), brine (2 x 10 mL), dried over anhydrous Na2SO4, filtered, and evaporated under vacuum. The crude reaction mixture was purified by column chromatography on silica gel (hexane/ethyl acetate) to obtain products 3 or 17. The structure of the products was confirmed by 1H and 13C NMR spectroscopy and mass spectrometry analyses.
Gram scale experiment:
Preparation of Phenyl(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3a)
An oven dried 50 mL round bottom flask equipped with a stirring bar, was charged with 1 (1 g, 4.1 mmol, 1.0 equiv), Pd(OAc)2 (10 mol%, 92.0 mg), NHPI (30 mol%, 200.6 mg) and 15.0 mL of dry toluene 2a. The resulting solution was purged with O2 for 7 minutes and the flask was then fitted with a reflux condenser having O2 (balloon) on top of it and heated at 80 °C (preheated oil bath) for 24 h. After completion of reaction, the solution was cooled to room temperature and diluted with dichloromethane (3 x 35 mL), washed with sodium bicarbonate (3 x 20 mL), brine (3 x 20 mL), dried over anhydrous Na2SO4, filtered, and evaporated under vacuum. The crude reaction mixture was purified by column chromatography on silica gel (hexane/ethyl acetate) to get product 3a (1.02 g, 70%) as white solid.
Preparation of (4-Chlorophenyl)[9-(2-pyridyl)-9H-carbazol-1-yl]methanone (3b)
An oven dried 50 mL round bottom flask equipped with a stirring bar, was charged with 1 (1 g, 4.1 mmol, 1.0 equiv), Pd(OAc)2 (10 mol%, 92.0 mg), NHPI (30 mol%, 200.6 mg) and 15.0 mL of dry toluene 2b. The resulting solution was purged with O2 for 7 minutes and the flask was then fitted with a reflux condenser having O2 (balloon) on top of it and heated at 80 °C (preheated oil bath) for 24 h. After completion of reaction, the solution was cooled to room temperature and diluted with dichloromethane (3 x 35 mL), washed with sodium bicarbonate (3 x 20 mL), brine (3 x 20 mL), dried over anhydrous Na2SO4, filtered, and evaporated under vacuum. The crude reaction mixture was purified by column chromatography on silica gel (hexane/ethyl acetate) to get product 3b (1.4 g, 72%) as yellow solid.
18O labelling experiment
To an oven dried 5.0 mL Schlenk tube equipped with a magnetic stir bar, Pd(OAc)2 (6.5 mg, 0.02 mmol) was placed. The tube was evacuated under vacuum and back refilled with argon (3 times). To the tube, freshly distilled 1 mL toluene 2a, NHPI (9.8 mg, 30 mol%) and 1 (50.0 mg, 0.2 mmol, 1.0 equiv) were added. The mixture was degassed by the freeze-pump-thaw procedure for three times. Finally the tube was evacuated while placing it in a liquid nitrogen bath and filled with 18O2 (provided from a 98% 18O2 cylinder). The solution was heated at 80°C for 24 hours. The solution was allowed to cool to room temperature and diluted with dichloromethane (3 x 15 mL), washed with sodium bicarbonate (3 x 10 mL), brine (2 x 10 mL), concentrated under vacuum. The crude mixture was purified by column chromatography on silica gel (hexane/ethyl acetate) to obtain the desired product 3a′ (48.0 mg, 70%). The incorporation of 18O in the product was confirmed by HRMS of the sample (Scheme S4, S.I.).
Radical quenching experiment
The reaction was performed according to GP-1, combining 1 (50.0 mg, 0.2 mmol, 1.0 equiv), TEMPO (78.0 mg, 0.6 mmol, 3.0 equiv), Pd(II) catalyst (10 mol%, 6.5 mg), NHPI (9.8 mg, 30 mol%), O2-balloon in 1.0 mL of toluene 2a at 80 °C (preheated oil bath) for 24 h. TLC analysis of the crude mixture revealed that the desired mono-acylated product did not form. However, the acyl radical and TEMPO free radical adduct 19 was isolated (70%, 36 mg) and characterized by NMR (Scheme S5, S.I.). 1H NMR (500 MHz, CDCl3): δ = 8.06 (dd, J = 8.3, 1.2 Hz, 2 H), 7.57-7.53 (m, 1 H), 7.45–7.42 (m, 2 H), 1.80-1.66 (m, 3 H), 1.59-1.55 (m, 2 H), 1.46-1.42 (m, 1 H), 1.26 (s, 6 H), 1.11 (s, 6 H) ppm; 13C NMR (125 MHz, CDCl3): δ = 166.4, 132.9, 130.0, 129.6, 128.6, 60.5, 39.3, 32.1, 21.0, 17.2 ppm.
Analytical data of compounds
3,6-dichloro-9-(pyridin-2-yl)-9H-carbazole 5
White solid (85 %). 1H NMR (400 MHz, CDCl3): δ = 8.72 (d, J = 3.6 Hz, 1 H), 8.02 (s, 2 H), 7.95 (t, J = 7.5 Hz, 1 H), 7.75 (d, J = 8.8 Hz, 2 H), 7.58 (d, J = 8.0 Hz, 1 H), 7.41 (d, J = 8.6 Hz, 2 H), 7.36-7.33 (m, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 151.3, 150.1, 138.9, 138.5, 127.1, 126.9, 124.6, 121.9, 120.2, 119.0, 112.6 ppm. HRMS (ESI): calcd. for C17H11Cl2N2 [M + H]+ 313.0294; found 313.0298.
3-chloro-9-(pyridin-2-yl)-9H-carbazole 8
White solid (88 %). 1H NMR (400 MHz, CDCl3): δ = 8.73 (d, J = 4.0 Hz, 1 H), 8.08-8.07 (m, 2 H), 7.94 (t, J = 7.7 Hz, 1 H), 7.80 (t, J = 8.6 Hz, 2 H), 7.62 (d, J = 8.1 Hz, 1 H), 7.47 (t, J = 7.8 Hz, 1 H), 7.39 (dd, J = 8.8, 1.9 Hz, 1 H), 7.33 (t, J = 7.3 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 151.7, 149.9, 140.1, 138.7, 138.1, 127.1, 126.5, 126.4, 125.7, 123.5, 121.6, 121.4, 120.6, 120.1, 119.1, 112.5, 111.4 ppm. HRMS (ESI): calcd. for C17H12ClN2 [M + H]+ 279.0683; found 279.0685.
3,6-dibromo-9-(pyridin-2-yl)-9H-carbazole 6
12 white solid (70 %). 1H NMR (400 MHz, CDCl3): 8.72 (1H, d, J = 4.9 Hz), 8.18 (2H, d, J = 1.8 Hz), 7.96 (1H, dt, J = 7.9, 1.7 Hz), 7.71 (2H, d, J = 8.5 Hz), 7.59-7.53 (3H, m), 7.35 (1H, dd, J = 7.1, 5.1 Hz); 13C NMR (100 MHz, CDCl3): 151.2, 150.0, 138.9, 138.7, 129.8, 125.1, 123.3, 122.0, 119.1, 114.2, 113.0; HRMS (ESI) calcd for C17H11Br2N2 [M+H]+: 402.9269; Found: 402.9262.
3,6-diiodo-9-(pyridin-2-yl)-9H-carbazole 7
12 white solid (40 %). 1H NMR (500 MHz, CDCl3): 8.72 (1H, d, J = 4.0 Hz), 8.37 (2H, d, J = 1.1 Hz), 7.97-7.93 (1H, m), 7.71-7.69 (2H, m), 7.60-7.55 (3H, m), 7.34 (1H, dd, J = 7.1, 5.1 Hz); 13C NMR (100 MHz, CDCl3): 151.3, 150.1, 139.1, 139.0, 135.4, 129.5, 125.6, 122.1, 119.2, 113.5, 84.3; HRMS (ESI) calcd for C17H11I2N2 [M+H]+: 496.9012; Found: 496.9009.
Phenyl(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3a)
White solid (76% using GP-1), mp 200-202 °C. 1H NMR (400 MHz, CDCl3): δ = 8.33 (dd, J = 7.8, 1.0 Hz, 1 H), 8.20 (d, J = 7.7 Hz, 1 H), 8.15 (dd, J = 4.9, 1.3 Hz, 1 H), 7.73 (dt, J = 7.8, 1.9 Hz, 1 H), 7.60-7.29 (m, 11 H), 7.10 (dd, J = 7.7, 4.6 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 195.9, 152.4, 149.3, 140.9, 138.6, 137.9, 137.8, 137.6, 132.2, 129.8, 128.2, 127.8, 126.8, 125.9, 124.4, 123.9, 123.1, 122.0, 121.4, 120.5, 120.4, 110.3 ppm. HRMS (ESI): calcd. for C24H17N2O [M + H]+ 349.1335; found 349.1337.
(4-Chlorophenyl)[9-(2-pyridyl)-9H-carbazol-1-yl]methanone (3b)
12 Yellow solid (76% using GP-1 and 82% using GP-2), mp 214-218 °C. 1H NMR (500 MHz, CDCl3): δ = 8.21 (d, J = 7.6 Hz, 1 H), 8.07 (d, J = 7.7 Hz, 1 H), 8.03–8.02 (m, 1 H), 7.68–7.65 (m, 1 H), 7.44–7.41 (m, 4 H), 7.34–7.24 (m, 4 H), 7.20–7.16 (m, 2 H), 7.01 (dd, J = 5.1, 7.4 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 194.7, 152.4, 149.3, 140.9, 139.1, 138.7, 137.7, 136.0, 134.7, 131.3, 128.9, 128.5, 127.7, 127.0, 126.0, 123.9, 123.7, 122.2, 121.5, 120.5, 120.4, 110.3 ppm. HRMS (ESI): calcd. for C24H16ClN2O [M + H]+ 383.0951; found 383.0954.
(3-Chlorophenyl)(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3c)
White solid (71% using GP-1), mp 218-222 °C. 1H NMR (400 MHz, CDCl3): δ = 8.31 (dd, J = 1.3, 1.0 Hz, 1 H), 8.18-8.15 (m, 2 H), 7.76 (dt, J = 7.7, 1.8 Hz, 1 H), 7.55-7.49 (m, 3 H), 7.45-7.30 (m, 5 H), 7.24 (t, J = 7.8 Hz, 2 H), 7.15 (dd, J = 7.3, 4.9 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 194.6, 152.3, 149.4, 140.9, 139.3, 138.8, 137.7, 134.6, 132.5, 129.5, 129.3, 128.2, 127.9, 127.0, 126.0, 123.9, 123.8, 123.6, 122.2, 121.6, 120.6, 120.5, 110.3 ppm. HRMS (ESI): calcd. for C24H16ClN2O [M + H]+ 383.0946; found 383.0944.
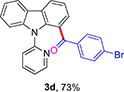
(4-Bromophenyl)(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3d)
Yellow solid (73% using GP-1 and 65% using GP-2), mp 182-184 °C. 1H NMR (400 MHz, CDCl3): δ = 8.30 (d, J = 7.6 Hz, 1 H), 8.17 (d, J = 7.7 Hz, 1 H), 8.12 (d, J = 3.9 Hz, 1 H), 7.77 (dt, J = 7.8, 1.5 Hz, 1 H), 7.46-7.34 (m, 10 H), 7.10 (dd, J = 7.1, 5.1 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 194.9, 152.4, 149.3, 140.9, 138.7, 137.7, 136.4, 131.6, 131.4, 127.8, 127.7, 127.0, 126.0, 124.0, 123.9, 123.4, 122.2, 121.6, 120.5, 120.4, 110.3 ppm. HRMS (ESI): calcd. for C24H16BrN2O [M + H]+ 427.0440; found 427.0442.
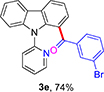
(3-Bromophenyl)(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3e)
Yellow solid (74% using GP-1), mp 192-194 °C. 1H NMR (500 MHz, CDCl3): δ = 8.24 (d, J = 7.4 Hz, 1 H), 8.10 (d, J = 7.1 Hz, 2 H), 7.68 (dt, J = 7.7, 1.8 Hz, 1 H), 7.59 (s, 1 H), 7.51 (d, J = 7.9 Hz, 1 H), 7.47 (d, J = 7.2 Hz, 1 H), 7.42 (d, J = 8.2 Hz, 1 H), 7.39-7.27 (m, 4 H), 7.23-7.18 (m, 2 H), 7.22 (d, J = 8.0 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 194.9, 152.4, 149.3, 140.9, 138.7, 137.7, 136.4, 131.6, 131.4, 127.8, 127.7, 127.0, 126.0, 124.0, 123.9, 123.4, 122.2, 121.6, 120.5, 120.4, 110.3 ppm. HRMS (ESI): calcd. for C24H16BrN2O [M + H]+ 427.0440; found 427.0445.
(4-Fluorophenyl)(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3f)
Yellow solid (83% using GP-1 and 84% using GP-2), mp 204-206 °C. 1H NMR (400 MHz, CDCl3): δ = 8.29 (dd, J = 7.7, 1.0 Hz, 1 H), 8.17 (d, J = 7.6 Hz, 1 H), 8.12 (dd, J = 4.8, 1.1 Hz, 1 H), 7.75 (dt, J = 7.8, 1.9 Hz, 1 H), 7.60 (dd, J = 8.8, 5.5 Hz, 2 H), 7.53-7.50 (m, 2 H), 7.44-7.32 (m, 4 H), 7.09 (dd, J = 7.1, 4.6 Hz, 1 H), 6.97 (t, J = 8.6 Hz, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 194.5, 166.8, 164.2, 152.4, 149.3, 140.9, 138.7, 137.7, 134.6, 134.0, 133.9, 132.5, 132.4, 127.7, 127.0, 126.0, 124.1, 123.9, 123.2, 122.2, 121.5, 120.5, 120.4, 115.5, 115.3, 110.3 ppm. HRMS (ESI): calcd. for C24H15FN2OK [M + K]+ 405.0800; found 405.0804.
(3-Fluorophenyl)(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3g)
Yellow solid (89% using GP-1), mp 198-202 °C. 1H NMR (500 MHz, CDCl3): δ = 8.31 (d, J = 7.6 Hz, 1 H), 8.17 (d, J = 7.7 Hz, 1 H), 8.14 (d, J = 3.6 Hz, 1 H), 7.76 (dt, J = 7.6, 1.5 Hz, 1 H), 7.54-7.50 (m, 2 H), 7.44-7.32 (m, 5 H), 7.30-7.27 (m, 2 H), 7.17 (dt, J = 8.2, 2.2 Hz, 1 H), 7.13-7.11 (m, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 194.6, 163.6, 161.6, 152.4, 149.4, 140.9, 139.8, 137.8, 137.7, 129.8, 127.8, 127.0, 126.0, 125.9, 124.0, 123.9, 123.5, 122.2, 121.6, 120.5, 120.4, 119.7, 119.5, 116.1, 116.0, 110.3 ppm. HRMS (ESI): calcd. for C24H16FN2O [M + H]+ 367.1241; found 367.1243.
(2-Fluorophenyl)(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3h)
Yellow solid (33% using GP-1), mp 210-212 °C. 1H NMR (500 MHz, CDCl3): δ = 8.31 (d, J = 7.0 Hz, 2 H), 8.15 (d, J = 7.7 Hz, 1 H), 7.79 (dt, J = 7.9, 1.9 Hz, 1 H), 7.57-7.54 (m, 2 H), 7.44-7.33 (m, 6 H), 7.16-7.14 (m, 1 H), 7.09 (t, J = 7.6 Hz, 1 H), 7.03-6.99 (m, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 192.2, 162.0, 160.0, 152.8, 149.5, 141.3, 138.8, 137.6, 133.9, 133.8, 132.0, 128.4, 126.9, 126.3, 125.3, 124.0, 123.9, 122.2, 121.6, 120.5, 120.4, 120.1, 116.8, 116.6, 110.7 ppm. HRMS (ESI): calcd. for C24H16FN2O [M + H]+ 367.1241; found 367.1243.
4-(9-(Pyridin-2-yl)-9H-carbazole-1-carbonyl)benzonitrile (3i)
White solid (77% using GP-1), mp 242-244 °C. 1H NMR (500 MHz, CDCl3): δ = 8.33 (d, J = 7.5, Hz, 1 H), 8.17 (d, J = 7.7 Hz, 1 H), 8.09 (d, J = 4.9 Hz, 1 H), 7.87 (dd, J = 5.3, 3.1 Hz, 1 H), 7.79-7.75 (m, 2 H), 7.67 (d, J = 8.3 Hz, 2 H), 7.61 (d, J = 8.3 Hz, 2 H), 7.51 (t, J = 8.1 Hz, 2 H), 7.45-7.33 (m, 2 H), 7.11 (dd, J = 7.3, 5.0 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 194.2, 168.0, 152.3, 149.3, 140.8, 140.7, 138.9, 137.6, 134.4, 132.8, 132.1, 130.1, 127.8, 127.2, 126.2, 123.9, 123.7, 123.2, 122.2, 121.8, 120.6, 120.3, 118.2, 115.7, 110.3 ppm. HRMS (ESI): calcd. for C25H15N3ONa [M + Na]+ 396.1107; found 396.1115.
3-(9-(Pyridin-2-yl)-9H-carbazole-1-carbonyl)benzonitrile (3j)
White solid (78% using GP-1), mp 228-232 °C. 1H NMR (500 MHz, CDCl3): δ = 8.33 (dd, J = 7.7, 0.9 Hz, 1 H), 8.18 (d, J = 7.7 Hz, 1 H), 8.10 (dd, J = 4.8, 1.2 Hz, 1 H), 7.86 (s, 1 H), 7.79-7.73 (m, 3 H), 7.51 (d, J = 7.6 Hz, 2 H), 7.45-7.32 (m, 5 H), 7.16-7.13 (m, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 193.7, 152.4, 149.3, 140.8, 138.9, 138.6, 137.6, 135.4, 134.4, 133.8, 133.1, 129.2, 127.7, 127.2, 126.2, 123.9, 123.7, 123.1, 122.3, 121.8, 120.6, 120.4, 118.0, 112.8, 110.3 ppm. HRMS (ESI): calcd. for C25H16N3O [M + H]+ 374.1288; found 374.1287.
Methyl 4-(9-(pyridin-2-yl)-9H-carbazole-1-carbonyl)benzoate (3k)
White solid (79% using GP-1 and 69% using GP-2), mp 160-162 °C. 1H NMR (500 MHz, CDCl3): δ = 8.31 (d, J = 7.6 Hz, 1 H), 8.17 (d, J = 7.7 Hz, 1 H), 8.09 (d, J = 3.6 Hz, 1 H), 7.97 (d, J = 8.3 Hz, 2 H), 7.87-7.86 (m, 1 H), 7.76-7.73 (m, 2 H), 7.64 (d, J = 8.2 Hz, 2 H), 7.53-7.49 (m, 2 H), 7.43-7.31 (m, 2 H), 7.09-7.07 (m, 1 H), 3.94 (s, 3 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 195.2, 168.0, 166.5, 152.4, 149.3, 141.0, 140.9, 138.8, 137.8, 134.4, 133.4, 132.8, 129.7, 129.4, 127.9, 127.0, 126.1, 124.0, 123.7, 123.6, 122.2, 121.6, 120.5, 120.4, 110.3, 52.5 ppm. HRMS (ESI): calcd. for C26H18N2O3Na [M + Na]+ 429.1210; found 429.1213.
1-(4-(9-(Pyridin-2-yl)-9H-carbazole-1-carbonyl)phenyl)ethanone (3l)
White solid (94% using GP-1), mp 168-172 °C. 1H NMR (400 MHz, CDCl3): δ = 8.31 (d, J = 7.6 Hz, 1 H), 8.16 (d, J = 7.6 Hz, 1 H), 8.07 (d, J = 3.7 Hz, 1 H), 7.88 (d, J = 8.2 Hz, 2 H), 7.74 (dt, J = 7.7, 1.6 Hz, 1 H), 7.68 (d, J = 8.2 Hz, 2 H), 7.51 (d, J = 7.8 Hz, 2 H), 7.44-7.34 (m, 4 H), 7.08 (dd, J = 7.1, 5.1 Hz, 1 H), 2.61 (s, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 197.7, 195.1, 152.3, 149.3, 141.0, 140.9, 139.7, 138.8, 137.7, 130.0, 128.1, 127.8, 127.0, 126.1, 123.9, 123.6, 122.2, 121.6, 120.5, 120.4, 120.3, 110.3, 27.0 ppm. HRMS (ESI): calcd. for C26H19N2O2 [M + H]+ 391.1441; found 391.1442.
(2-Methoxyphenyl)(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3m)
White solid (71% using GP-1), mp 216-220 °C. 1H NMR (500 MHz, CDCl3): δ = 8.37 (dd, J = 4.9, 1.9 Hz, 1 H), 8.27 (d, J = 7.6 Hz, 1 H), 8.14 (d, J = 7.7 Hz, 1 H), 7.77-7.74 (m, 1 H), 7.58 (d, J = 8.3 Hz, 1 H), 7.53 (d, J = 7.3 Hz, 1 H), 7.42-7.37 (m, 3 H), 7.35-7.31 (m, 3 H), 7.14-7.12 (m, 1 H), 6.89-6.84 (m, 2 H), 3.61 (s, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 194.4, 158.4, 153.0, 149.4, 141.5, 138.5, 137.7, 134.5, 134.4, 133.0, 131.7, 129.0, 128.2, 126.8, 126.3, 126.1, 123.9, 123.6, 121.8, 121.4, 120.3, 120.2, 111.8, 110.8, 55.7 ppm. HRMS (ESI): calcd. for C25H19N2O2 [M + H]+ 379.1441; found 379.1445.
(9-(Pyridin-2-yl)-9H-carbazol-1-yl)(m-tolyl)methanone (3n)
White solid (70% using GP-1), mp 200-204 °C. 1H NMR (400 MHz, CDCl3): δ = 8.29 (dd, J = 7.7, 1.1 Hz, 1 H), 8.17 (d, J = 7.6 Hz, 1 H), 8.13 (dd, J = 4.9, 1.2 Hz, 1 H), 7.73 (dt, J = 7.8, 1.9 Hz, 1 H), 7.54 (dd, J = 7.4, 1.1 Hz, 1 H), 7.50 (d, J = 8.3 Hz, 1 H), 7.43-7.27 (m, 7 H), 7.19 (t, J = 7.5 Hz, 1 H), 7.10 (ddd, J = 7.4, 4.9, 0.8 Hz, 1 H), 2.31 (s, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 196.2, 152.4, 149.4, 141.0, 138.6, 138.0, 137.9, 137.7, 133.4, 130.1, 128.1, 128.0, 127.4, 126.8, 125.9, 124.6, 124.0, 123.1, 121.9, 121.2, 120.6, 120.5, 120.3, 110.3, 21.4 ppm. HRMS (ESI): calcd. for C25H19N2O [M + H]+ 363.1492; found 363.1497.
(9-(Pyridin-2-yl)-9H-carbazol-1-yl)(o-tolyl)methanone (3o)
White solid (66% using GP-1), mp 190-194 °C. 1H NMR (400 MHz, CDCl3): δ = 8.30-8.27 (m, 2 H), 8.16 (d, J = 7.7 Hz, 1 H), 7.84 (dt, J = 7.8, 1.7 Hz, 1 H), 7.52-7.40 (m, 4 H), 7.39-7.31 (m, 4 H), 7.20-7.12 (m, 3 H), 2.34 (s, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 197.2, 152.8, 149.3, 141.4, 138.7, 138.6, 138.0, 137.7, 131.4, 131.2, 129.1, 127.9, 127.6, 126.8, 126.2, 125.9, 125.4, 123.8, 123.7, 122.1, 121.4, 120.4, 120.3, 110.4, 21.0 ppm. HRMS (ESI): calcd. for C25H19N2O [M + H]+ 363.1492; found 363.1495.
(3,5-Dimethylphenyl)(9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (3p)
White solid (43% using GP-1), mp 206-208 °C. 1H NMR (400 MHz, CDCl3): δ = 8.29 (dd, J = 6.7, 1.2 Hz, 1 H), 8.18-8.14 (m, 2 H), 7.75 (dt, J = 7.7, 1.8 Hz, 1 H), 7.53-7.48 (m, 2 H), 7.43-7.33 (m, 4 H), 7.21 (s, 2 H), 7.11-7.09 (m, 2 H), 2.23 (s, 6 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 196.5, 152.4, 149.4, 141.0, 138.6, 137.9, 137.8, 137.7, 134.3, 128.1, 127.6, 126.8, 125.9, 124.7, 124.0, 123.1, 121.9, 121.3, 120.6, 120.4, 120.2, 110.2, 21.2 ppm. HRMS (ESI): calcd. for C26H21N2O [M + H]+ 377.1648; found 377.1649.
(4-Chlorophenyl)(3,6-dichloro-9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (10b)
White solid (83% using GP-1), mp 222-224 °C. 1H NMR (500 MHz, CDCl3): δ = 8.18 (d, J = 1.7 Hz, 1 H), 8.07-8.06 (m, 2 H), 7.80-7.77 (m, 1 H), 7.56 (d, J = 8.4 Hz, 2 H), 7.46 (d, J = 2.2 Hz, 1 H), 7.40-7.39 (m, 2 H), 7.33 (d, J = 8.6 Hz, 3 H), 7.13-7.11 (m, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 193.3, 151.5, 149.4, 139.6, 139.0, 136.5, 135.2, 131.3, 129.5, 128.8, 127.8, 127.7, 127.4, 126.3, 126.2, 125.3, 124.2, 122.8, 122.7, 120.4, 120.3, 111.6 ppm. HRMS (ESI): calcd. for C24H14Cl3N2O [M + H]+ 451.0166; found 451.0162.
(3,6-dibromo-9-(pyridin-2-yl)-9H-carbazol-1-yl)(4-methoxyphenyl)methanone (11q)
12 white solid (72% using GP-1), mp 176-178 °C; 1H NMR (500 MHz): δ = 8.32 (1H, d, J = 1.6 Hz), 8.23 (1H, d, J = 1.6 Hz), 8.08-8.07 (1H, m), 7.77 (1H, dt, J = 1.5 Hz, 7.6 Hz), 7.61-7.58 (3H, m), 7.51 (1H, dd, J = 1.7 Hz, 8.6 Hz), 7.33 (2H, dd, J = 1.7 Hz, 8.6 Hz), 7.11 (1H, dd, J = 4.9 Hz, 7.3 Hz), 6.82 (2H, d, J = 8.7 Hz), 3.86 (3H, s) 13C NMR (100 MHz): δ = 192.9, 163.8, 151.4, 149.5, 139.9, 138.8, 136.8, 132.4, 130.3, 129.9, 126.5, 126.4, 125.4, 124.6, 123.4, 120.7, 120.5, 114.5, 113.7, 113.3, 112.1, 55.7; HRMS (ESI) calcd for C25H16Br2N2O2 [M+H]+: 536.9636; Found: 536.9591.
Methyl 4-(3,6-dibromo-9-(pyridin-2-yl)-9H-carbazole-1-carbonyl)benzoate (11k)
12 white solid (74% using GP-1), mp 204-206 °C; 1H NMR (500 MHz): δ = 8.35 (1H, d, J = 2.5 Hz), 8.22 (1H, d, J = 1.6 Hz,), 8.03-7.98 (3H, m), 7.76 (1H, dt, J = 1.7 Hz, 7.9 Hz), 7.65 (2H, d, J = 8.3 Hz), 7.60 (1H, d, J = 2.5 Hz), 7.52 (1H, dd, J = 1.5 Hz, 9.1 Hz), 7.36 (1H, d, J = 8.35 Hz), 7.30 (1H, d, J = 7.55 Hz), 7.11 (1H, dd, J = 5.0 Hz, 7.3 Hz) 3.95 (3H, s); 13C NMR (100 MHz): δ = 193.5, 188.3, 151.4, 149.4, 140.2, 139.8, 139.1, 136.7, 133.9, 130.5, 130.4, 129.7, 129.6, 126.7, 126.1, 125.5, 124.6, 123.5, 122.8, 120.2, 114.8, 113.4, 111.9, 52.6; HRMS (ESI) calcd for C26H17Br2N2O3 [M+H]+: 564.9585; Found: 564.9595.
(3,6-Diiodo-9-(pyridin-2-yl)-9H-carbazol-1-yl)(4-methoxyphenyl)methanone (12q)
12 white solid (51% using GP-1), mp 184-186 °C; 1H NMR (500 MHz): δ = 8.50 (1H,d, J = 1.5 Hz), 8.41 (1H, d, J = 1.4 Hz), 8.07 (1H, dd, J = 1.1 Hz, 4.7 Hz), 7.77-7.74 (2H, m), 7.67 (1H, dd, J = 1.5 Hz, 8.6 Hz), 7.57 (2H, d, J = 8.8 Hz), 7.30 (1H, d, J = 7.9 Hz), 7.24 (1H, d, J = 8.7 Hz), 7.11 (1H, dd, J = 5.0Hz, 7.0 Hz), 6.82 (2H, d, J = 8.8 Hz), 3.85 (3H, s); 13C NMR (125 MHz): δ = 192.8, 163.7, 151.3, 149.4, 140.2, 138.8, 136.9, 135.8, 135.6, 132.3, 131.4, 129.9, 129.5, 126.7, 124.9, 122.7, 120.5, 113.7, 112.4, 84.4, 82.9, 55.6; HRMS (ESI) calcd for C25H17I2N2O2 [M+H]+: 630.9379; Found: 630.9376.
Methyl 4-(3,6-diiodo-9-(pyridin-2-yl)-9H-carbazole-1-carbonyl)benzoate (12k)
12 white solid (58% using GP-1), mp 194-196 °C; 1H NMR (500 MHz): δ = 8.46 (1H, d, J = 5.3 Hz), 8.34 (1H, d, J = 5.2 Hz), 7.95-7.91 (3H, m), 7.69-7.66 (2H, m), 7.61-7.54 (3H, m), 7.22-7.15 (2H, m), 7.04-7.01 (1H, m), 3.86 (3H, d, J = 5.3 Hz); 13C NMR (125 MHz): δ = 193.4, 166.3, 151.4, 149.4, 140.2, 140.1, 139.0, 136.9, 135.9, 135.8, 133.4, 132.2, 129.7, 129.6, 126.9, 125.9, 124.9, 122.7, 120.2, 112.4, 84.7, 82.9, 52.6; HRMS (ESI) calcd for C26H17I2N2O3 [M+H]+: 658.9329; Found: 658.9325.
(6-Chloro-9-(pyridin-2-yl)-9H-carbazol-1-yl)(phenyl)methanone (13a)
White solid (69% using GP-1), mp 198-200 °C. 1H NMR (400 MHz, CDCl3): δ = 8.24 (dd, J = 6.8, 1.0 Hz, 1 H), 8.12-8.09 (m, 2 H), 7.74 (dt, J = 7.7, 1.6 Hz, 1 H), 7.60-7.55 (m, 3 H), 7.50-7.30 (m, 7 H), 7.10 (dd, J = 7.1, 5.1 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3): δ = 195.7, 152.0, 149.4, 139.3, 138.8, 138.3, 137.4, 132.8, 129.9, 128.6, 128.4, 128.3, 126.9, 125.2, 124.9, 124.6, 123.3, 122.4, 120.7, 120.4, 120.2, 111.5 ppm. HRMS (ESI): calcd. for C24H16ClN2O [M + H]+ 383.0946; found 383.0944.
(4-Chlorophenyl)(6-methoxy-9-(pyridin-2-yl)-9H-carbazol-1-yl)methanone (14b)
White solid (60% using GP-1), mp 224-226 °C. 1H NMR (400 MHz, CDCl3): δ = 8.25 (d, J = 7.2 Hz, 1 H), 8.24-8.10 (m, 1 H), 7.74 (dt, J = 6.0, 1.6 Hz, 1 H), 7.62 (d, J = 2.4 Hz, 1 H), 7.54-7.49 (m, 3 H), 7.43 (d, J = 8.8 Hz, 1 H), 7.38-7.27 (m, 4 H), 7.09-7.03 (m, 2 H), 3.95 (s, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 194.7, 155.4, 152.5, 149.2, 139.0, 138.7, 138.1, 136.0, 135.7, 131.3, 128.6, 127.8, 126.0, 124.6, 124.1, 123.3, 122.0, 120.2, 115.8, 111.2, 103.3, 56.2 ppm. HRMS (ESI): calcd. for C25H18ClN2O2 [M + H]+ 413.1051; found 413.1055.
1-(9-(Pyridin-2-yl)-9H-carbazol-1-yl)heptan-1-one (3r)
Colorless oil (75% using GP-2). 1H NMR (500 MHz, CDCl3): δ = 8.52 (d, J = 3.6 Hz, 1 H), 8.25 (d, J = 7.9 Hz, 1 H), 8.13 (d, J = 7.7 Hz, 1 H), 7.96 (dt, J = 7.6, 1.5 Hz, 1 H), 7.66 (d, J = 7.5 Hz, 1 H), 7.59–7.55 (m, 2 H), 7.43 (t, J = 7.8 Hz, 1 H), 7.36–7.32 (m, 2 H), 7.29 (dd, J = 7.1, 5.0 Hz, 1 H), 2.82 (t, J = 7.6 Hz, 2 H), 1.49–1.45 (m, 2 H), 1.33–1.27 (m, 6 H), 0.90 (t, J = 6.8 Hz, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 203.1, 153.1, 149.3, 141.5, 138.8, 136.8, 126.8, 126.5, 126.3, 126.1, 123.8, 123.4, 122.2, 121.4, 120.7, 120.3, 120.2, 110.5, 41.4, 31.7, 29.1, 24.2, 22.6, 14.1 ppm. HRMS (ESI): calcd. for C24H25N2O [M + H]+ 357.1961; found 357.1964.
1-(6-Methoxy-7-methyl-9-(pyridin-2-yl)-9H-carbazol-1-yl)heptan-1-one (17e)
Colorless oil (75% using GP-2). 1H NMR (400 MHz, CDCl3): δ = 8.51 (d, J = 4.4 Hz, 1 H), 8.16 (d, J = 7.8 Hz, 1 H), 7.95 (ddd, J = 5.8, 1.9, 1.5 Hz, 1 H), 7.59 (d, J = 7.5 Hz, 1 H), 7.53 (d, J = 7.8 Hz, 1 H), 7.48 (s, 1 H), 7.34 (s, 1 H), 7.32-7.27 (m, 2 H), 3.97 (s, 3H), 2.79 (t, J = 7.4 Hz, 2 H), 2.34 (s, 3 H), 1.46–1.41 (m, 2 H), 1.29–1.25 (m, 6 H), 0.88 (t, J = 3.9 Hz, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 203.4, 153.9, 153.4, 149.3, 138.9, 136.8, 136.0, 127.4, 126.8, 126.3, 125.6, 122.9, 122.0, 121.9, 120.5, 120.0, 112.3, 100.8, 56.0, 41.5, 31.8, 29.1, 24.3, 22.6, 17.6, 14.2 ppm. HRMS (ESI): calcd. for C26H29N2O2 [M + H]+ 401.2229; found 401.2227.
Supplementary Material
Scheme 1. Previous Reports and Our Approach for the Synthesis of 1-acyl Carbazoles.

Acknowledgment
JD thanks Wellcome Trust-DBT India Alliance [Grant Number, IA/S/18/2/503986] and CSIR for funding. SM and TM thank IACS, Kolkata for research fellowship. We thank Mr. Partha Mitra and Mr. Manish Jana, IACS for helping with single crystal X-ray analysis.
References
- 1.(a) Engle KM, Mei TS, Wasa M, Yu J-Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions. Acc Chem Res. 2012;45:788. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Colby DA, Tsai AS, Bergman RG, Ellman JA. Rhodium Catalyzed Chelation-Assisted C–H Bond Functionalization Reactions. Acc Chem Res. 2012;45:814. doi: 10.1021/ar200190g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neufeldt SR, Sanford MS. Controlling Site Selectivity in Palladium-Catalyzed C-H Bond Functionalization. Acc Chem Res. 2012;45:936. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Song G, Wang F, Li X. C–C, C–O and C–N bond formation via rhodium(III)-catalyzed oxidative C-H activation. Chem Soc Rev. 2012;47:3651. doi: 10.1039/c2cs15281a. [DOI] [PubMed] [Google Scholar]; (e) Yeung CS, Dong VM. Catalytic Dehydrogenative Cross-Coupling: Forming Carbon-Carbon Bonds by Oxidizing Two Carbon-Hydrogen Bonds. Chem Rev. 2011;111:1215. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]; (f) Sun C-L, Li B-J, Shi Z-J. Direct C-H Transformation via Iron Catalysis. Chem Rev. 2011;111:1293. doi: 10.1021/cr100198w. [DOI] [PubMed] [Google Scholar]; (g) Lyons TW, Sanford MS. Palladium-Catalyzed Ligand-Directed C-H Functionalization Reactions. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Colby DA, Bergman RG, Ellman JA. Rhodium-Catalyzed C-C Bond Formation via Heteroatom-Directed C-H Bond Activation. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Ackermann L, Vicente R, Kapdi AR. Transition-Metal-Catalyzed Direct Arylation of (Hetero)Arenes by C-H Bond Cleavage. Angew Chem Int Ed. 2009;48:9792. doi: 10.1002/anie.200902996. [DOI] [PubMed] [Google Scholar]; (j) Giri R, Shi B-F, Engle KM, Maugel N, Yu J-Q. Transition metal-catalyzed C-H activation reactions: diastereoselectivity and enantioselectivity. Chem Soc Rev. 2009;38:3242. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]; (k) McGlacken GP, Bataman LM. Recent advances in aryl–aryl bond formation by direct arylation. Chem Soc Rev. 2009;38:2447. doi: 10.1039/b805701j. [DOI] [PubMed] [Google Scholar]; (l) Collet F, Dodd RH, Dauban P. Catalytic C–H amination: recent progress and future directions. Chem Commun. 2009:5061. doi: 10.1039/b905820f. [DOI] [PubMed] [Google Scholar]; (m) Chan C-W, Zhou Z, Yu W-Y. Palladium(II)-Catalyzed Direct ortho -C-H Acylation of Anilides by Oxidative Cross-Coupling with Aldehydes using tert -Butyl Hydroperoxide as Oxidant. Adv Synth Catal. 2011;353:2999. [Google Scholar]; (n) Zhou W, Li H, Wang L. Direct Carbo-Acylation Reactions of 2-Arylpyridines with α-Diketones via Pd-Catalyzed C–H Activation and Selective C(sp2)–C(sp2) Cleavage. Org Lett. 2012;14:4594. doi: 10.1021/ol3020557. [DOI] [PubMed] [Google Scholar]; (o) Liu PM, Frost CG. Ruthenium-Catalyzed C–H Functionalization of Arylpyrazoles: Regioselective Acylation with Acid Chlorides. Org Lett. 2013;15:5862. doi: 10.1021/ol402936c. [DOI] [PubMed] [Google Scholar]
- 2.(a) Daugulis O, Do HQ, Shabashov D. Palladium- and Copper-Catalyzed Arylation of Carbon-Hydrogen Bonds. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Phipps RJ, Gaunt MJ. A Meta-Selective Copper-Catalyzed C–H Bond Arylation. Science. 2009;323:1593. doi: 10.1126/science.1169975. [DOI] [PubMed] [Google Scholar]; (c) Ciana CL, Phipps RJ, Brandt JR, Meyer FM, Gaunt MJ. A Highly Para -Selective Copper(II)-Catalyzed Direct Arylation of Aniline and Phenol Derivatives. Angew Chem Int Ed. 2011;50:458. doi: 10.1002/anie.201004703. [DOI] [PubMed] [Google Scholar]; (d) Leow DS, Li G, Mei TS, Yu JQ. Activation of remote meta-C–H bonds assisted by an end-on template. Nature. 2012;486:518. doi: 10.1038/nature11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Srinivas K, Kumar CR, Reddy MA, Bhanuprakash K, Rao VJ, Giribabu L. D-p-A organic dyes with carbazole as donor for dye-sensitized solar cells. Synth Met. 2011;161:96. [Google Scholar]; (b) Barea EM, Zafer C, Gultekin B, Aydin B, Koyuncu S, Icli S, Santiago FF, Bisquert J. Quantification of the Effects of Recombination and Injection in the Performance of Dye-Sensitized Solar Cells Based on N-Substituted Carbazole Dyes. J Phys Chem C. 2010;114:19840 [Google Scholar]
- 4.(a) Senthilkumar N, Somannavar YS, Reddy SB, Sinha BK, Narayan GKASS, Dandala R, Mukkanti K. Synthesis of Active Metabolites of Carvedilol, an Antihypertensive Drug. Synth Commun. 2010;41:268. [Google Scholar]; (b) Faddeeva MD, Beliaeva TN. Sanguinarine and Ellipticine Cytotoxic Alkaloids Isolated From Well-Known Antitumor Plants. Intracellular Targets of Their Action. Tsitologiia. 1997;39:181. [PubMed] [Google Scholar]; (c) Stafylas PC, Sarafidis PA. Carvedilol in hypertension treatment. Vasc Health Risk Manag. 2008;4:23. doi: 10.2147/vhrm.2008.04.01.23. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mousset D, Rabot R, Bouyssou P, Coudert G, Gillaizeau I. Synthesis and biological evaluation of novel benzoxazinic analogues of ellipticine. Tetrahedron Lett. 2010;51:3987. [Google Scholar]; (e) Rajeshwaran GG, Mohanakrishnan AK. Synthetic Studies on Indolocarbazoles: Total Synthesis of Staurosporine Aglycon. Org Lett. 2011;13:1418. doi: 10.1021/ol200094b. [DOI] [PubMed] [Google Scholar]; (f) Crich D, Rum thao S. Synthesis of carbazomycin B by radical arylation of benzene. Tetrahedron. 2004;60:1513. [Google Scholar]; (g) Knölker H-J, Knoll J. First total synthesis of the neuronal cell protecting carbazole alkaloid carbazomadurin A by sequential transition metal-catalyzed reactions. Chem Commun. 2003:1170. doi: 10.1039/b301979a. [DOI] [PubMed] [Google Scholar]; (h) Zhang A, Lin G. The first synthesis of clausenamine-A and cytotoxic activities of three biscarbazole analogues against cancer cells. Bioorg Med Chem Lett. 2000;10:1021. doi: 10.1016/s0960-894x(00)00158-x. [DOI] [PubMed] [Google Scholar]; (i) Knölker HJ, Reddy KR. In: Chemistry and Biology of Carbazole Alkaloids In The Alkaloids. Cordell GA, editor. Vol. 65. Academic Press; Amsterdam: 2008. p. 195. [DOI] [PubMed] [Google Scholar]
- 5.Niwa T, Nakada M. A Non-Heme Iron(III) Complex with Porphyrin-like Properties That Catalyzes Asymmetric Epoxidation. J Am Chem Soc. 2012;134:13538. doi: 10.1021/ja304219s. [DOI] [PubMed] [Google Scholar]
- 6.(a) Dierschke F, Grimsdale AC, Mullen K. Efficient Synthesis of 2,7-Dibromocarbazoles as Components for Electroactive Materials. Synthesis. 2003:2470. [Google Scholar]; (b) Maegawa Y, Goto Y, Inagaki S, Shimada T. A useful procedure for diiodination of carbazoles and subsequent efficient transformation to novel 3,6-bis(triethoxysilyl)carbazoles giving mesoporous materials. Tetrahedron Lett. 2006;47:6957. [Google Scholar]; (c) Kumar GGKSN, Laali KK. Condensation of propargylic alcohols with N-methylcarbazole and carbazole in [bmim]PF6 ionic liquid; synthesis of novel dipropargylic carbazoles using TfOH or Bi(NO3)3·5H2O as catalyst. Tetrahedron Lett. 2013;54:965. [Google Scholar]
- 7.(a) Huang H, Wang Y, Pan B, Yang X, Wang L, Chen J, Ma D, Yang C. Simple Bipolar Hosts with High Glass Transition Temperatures Based on 1,8-Disubstituted Carbazole for Efficient Blue and Green Electrophosphorescent Devices with “Ideal” Turn-on Voltage. Chem Eur J. 2013;19:1828. doi: 10.1002/chem.201202329. [DOI] [PubMed] [Google Scholar]; (b) Gong W-L, Zhong F, Aldred MP, Fu Q, Chen T, Huang D-K, Shen Y, Qiao X-F, Ma D, Zhu M-Q. Carbazole oligomers revisited: new additions at the carbazole 1- and 8-positions. RSC Advances. 2012;2:10821 [Google Scholar]; (c) Wang H-Y, Liu F, Xie L-H, Tang C, Peng B, Huang W, Wei W. Topological Arrangement of Fluorenyl-Substituted Carbazole Triads and Starbursts: Synthesis and Optoelectronic Properties. J Phys Chem C. 2011;115:6961. [Google Scholar]
- 8.Chu J-H, Wu C-C, Chang D-H, Lee Y-M, Wu M-J. Direct Ortho Arylation of 9-(Pyridin-2-yl)-9H-carbazoles Bearing a Removable Directing Group via Palladium(II)-Catalyzed C-H Bond Activation. Organometallics. 2013;32:272. [Google Scholar]
- 9.Urones B, Arrayas RG, Carretero JC. PdII-Catalyzed Di-o-olefination of Carbazoles Directed by the Protecting A-(2-Pyridyl)sulfonyl Group. Org Lett. 2013;15:1120. doi: 10.1021/ol400206k. [DOI] [PubMed] [Google Scholar]
- 10.Louillat M-L, Patureau FW. Toward Polynuclear Ru–Cu Catalytic Dehydrogenative C–N Bond Formation, on the Reactivity of Carbazoles. Org Lett. 2013;15:164. doi: 10.1021/ol303216u. [DOI] [PubMed] [Google Scholar]
- 11.Okada T, Nobushige K, Satoh T, Miura M. Ruthenium-Catalyzed Regioselective C–H Bond Acetoxylation on Carbazole and Indole Frameworks. Org Lett. 2016;18:1150. doi: 10.1021/acs.orglett.6b00268. [DOI] [PubMed] [Google Scholar]
- 12.Maiti S, Burgula L, Chakraborti G, Dash J. Palladium-Catalyzed Pyridine-Directed Regioselective Oxidative C–H Acylation of Carbazoles by Using Aldehydes as the Acyl Source. Eur J Org Chem. 2017:332. [Google Scholar]
- 13.(a) Stahl SS. Palladium Oxidase Catalysis: Selective Oxidation of Organic Chemicals by Direct Dioxygen-Coupled Turnover. Angew Chem, Int Ed. 2004;43:3400. doi: 10.1002/anie.200300630. [DOI] [PubMed] [Google Scholar]; (b) Gligorich KM, Sigman MS. Recent advancements and challenges of palladiumII-catalyzed oxidation reactions with molecular oxygen as the sole oxidant. Chem Commun. 2009:3854. doi: 10.1039/b902868d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Campbell AN, Stahl SS. Overcoming the “Oxidant Problem”: Strategies to Use O2 as the Oxidant in Organometallic C-H Oxidation Reactions Catalyzed by Pd (and Cu) Acc Chem Res. 2012;45:851. doi: 10.1021/ar2002045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Stoltz BM. Palladium Catalyzed Aerobic Dehydrogenation: From Alcohols to Indoles and Asymmetric Catalysis. Chem Lett. 2004;33:362. [Google Scholar]; (b) Punniyamurthy T, Velusamy S, Iqbal J. Recent Advances in Transition Metal Catalyzed Oxidation of Organic Substrates with Molecular Oxygen. Chem Rev. 2005;105:2329. doi: 10.1021/cr050523v. [DOI] [PubMed] [Google Scholar]; (c) Gligorich MK, Sigman MS. Mechanistic Questions about the Reaction of Molecular Oxygen with Palladium in Oxidase Catalysis. Angew Chem, Int Ed. 2006;45:6612. doi: 10.1002/anie.200602138. [DOI] [PubMed] [Google Scholar]; (d) Sigman MS, Jensen DR. Ligand-Modulated Palladium-Catalyzed Aerobic Alcohol Oxidations. Acc Chem Res. 2006;39:221. doi: 10.1021/ar040243m. [DOI] [PubMed] [Google Scholar]; (e) Muzart J. Molecular Oxygen To Regenerate PdII Active Species. Chem-Asian J. 2006;1:508. doi: 10.1002/asia.200600202. [DOI] [PubMed] [Google Scholar]; (f) Beccalli EM, Broggini G, Martinelli M, Sottocornola S. C-C, C-O, C-N Bond Formation on sp2 Carbon by Pd(II)-Catalyzed Reactions Involving Oxidant Agents. Chem Rev. 2007;107:5318. doi: 10.1021/cr068006f. [DOI] [PubMed] [Google Scholar]; (g) Piera J, Backvall JE. Catalytic Oxidation of Organic Substrates by Molecular Oxygen and Hydrogen Peroxide by Multistep Electron Transfer--A Biomimetic Approach. Angew Chem, Int Ed. 2008;47:3506. doi: 10.1002/anie.200700604. [DOI] [PubMed] [Google Scholar]; (h) Stahl SS. Palladium-catalyzed Oxidation of Organic Chemicals with O2. Science. 2005;309:1824. doi: 10.1126/science.1114666. [DOI] [PubMed] [Google Scholar]; (i) Schultz MJ, Sigman MS. Recent advances in homogeneous transition metal-catalyzed aerobic alcohol oxidations. Tetrahedron. 2006;62:8227. [Google Scholar]; (j) Wendlandt AE, Suess AM, Stahl SS. Copper-Catalyzed Aerobic Oxidative CH Functionalizations: Trends and Mechanistic Insights. Angew Chem, Int Ed. 2011;50:11062. doi: 10.1002/anie.201103945. [DOI] [PubMed] [Google Scholar]; (k) Shi Z, Zhang C, Tang C, Jiao N. Recent advances in transition-metal catalyzed reactions using molecular oxygen as the oxidant. Chem Soc Rev. 2012;41:3381. doi: 10.1039/c2cs15224j. [DOI] [PubMed] [Google Scholar]; (l) Wu W, Jiang H. Palladium-Catalyzed Oxidation of Unsaturated Hydrocarbons Using Molecular Oxygen. Acc Chem Res. 2012;45:1736. doi: 10.1021/ar3000508. [DOI] [PubMed] [Google Scholar]; (m) Allen SE, Walvoord RR, PadillaSalinas R, Kozlowski MC. Aerobic Copper-Catalyzed Organic Reactions. Chem Rev. 2013;113:6234. doi: 10.1021/cr300527g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Ryland BL, Stahl SS. Practical Aerobic Oxidations of Alcohols and Amines with Homogeneous Copper/TEMPO and Related Catalyst Systems. Angew Chem, Int Ed. 2014;53:8824. doi: 10.1002/anie.201403110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh S, Das TK, Bhusan D, Mehta S. Studies on enamides. Part-1: Photochemical rearrangements of N-aroylcarbazoles. Tetrahedron Lett. 1987;39:4611. [Google Scholar]
- 16.Katritzky AR, Rewcastle GW, Vazquez de Miguel LM. Improved syntheses of substituted carbazoles and benzocarbazoles via lithiation of the (dialkylamino)methyl (aminal) derivatives. J Org Chem. 1988;53:794. [Google Scholar]
- 17.Bonesi MS, Erra-Balsells RJ. Recent work on the synthesis of 3,6-dibenzoylcarbazole. Heterocyclic Chem. 1991;28:1035. [Google Scholar]
- 18.Bandgar BP, Patil AV. A rapid, solvent-free, ligandless and mild method for preparing aromatic ketones from acyl chlorides and arylboronic acids via a Suzuki-Miyaura type of coupling reaction. Tetrahedron Lett. 2005;46:7627. [Google Scholar]
- 19.Wu J-Q, Yang Z, Zhang S-S, Jiang C-Y, Li Q, Huang Z-S, Wang H. From Indoles to Carbazoles: Tandem Cp*Rh(III)-Catalyzed C–H Activation/Brønsted Acid-Catalyzed Cyclization Reactions. ACS Catal. 2015;5:6453. [Google Scholar]
- 20.Manna MK, Bairya G, Jana R. Dual visible-light photoredox and palladium(II) catalysis for dehydrogenative C2-acylation of indoles at room temperature. Org Biomol Chem. 2017;15:5899. doi: 10.1039/c7ob01418j. [DOI] [PubMed] [Google Scholar]
- 21.(a) Weinstein AB, Stahl SS. Palladium catalyzed aryl C–H amination with O2 via in situ formation of peroxide-based oxidant(s) from dioxane. Catal Sci Technol. 2014;4:4301. doi: 10.1039/C4CY00764F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stowers KJ, Kubota A, Sanford MS. Nitrate as a redox co-catalyst for the aerobic Pd-catalyzed oxidation of unactivated sp3-C–H bonds. Chem Sci. 2012;3:3192. doi: 10.1039/C2SC20800H. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liang Y-F, Li X, Wang X, Yan Y, Feng P, Jiao N. Aerobic Oxidation of PdII to PdIV by Active Radical Reactants: Direct C–H Nitration and Acylation of Arenes via Oxygenation Process with Molecular Oxygen. ACS Catal. 2015;5:1956. [Google Scholar]
- 22.(a) Sheldon RA, Arends IWCE. Organocatalytic Oxidations Mediated by Nitroxyl Radicals. Adv Synth Catal. 2004;346:1051. [Google Scholar]; (b) Minisci F, Punta C, Recupero F. Mechanisms of the aerobic oxidations catalyzed by N-hydroxyderivatives: Enthalpic, polar and solvent effects, “molecule-induced homolysis” and synthetic involvements. J Mol Catal A: Chem. 2006;251:129. [Google Scholar]; (c) Coseri S. Phthalimide-N-oxyl (PINO) Radical, a Powerful Catalytic Agent: Its Generation and Versatility Towards Various Organic Substrates. Catal Rev. 2009;51:218. [Google Scholar]; (d) Recupero F, Punta C. Free Radical Functionalization of Organic Compounds Catalyzed by N-Hydroxyphthalimide. Chem Rev. 2007;107:3800. doi: 10.1021/cr040170k. [DOI] [PubMed] [Google Scholar]; (e) Ishii Y, Sakaguchi S, Iwahama T. Preparation of Hydroperoxides by N -Hydroxyphthalimide-Catalyzed Aerobic Oxidation of Alkylbenzenes and Hydroaromatic Compounds and Its Application. Adv Synth Catal. 2001;343:393. [Google Scholar]; (f) Wertz S, Studer A. Nitroxide-catalyzed transition-metal-free aerobic oxidation processes. Green Chem. 2013;15:3116. [Google Scholar]; (g) Lin R, Chen F, Jiao N. Metal-Free, NHPI Catalyzed Oxidative Cleavage of C–C Double Bond Using Molecular Oxygen as Oxidant. Org Lett. 2012;14:4158. doi: 10.1021/ol3018215. [DOI] [PubMed] [Google Scholar]; (h) Yan Y, Feng P, Zheng Q-Z, Liang Y-F, Lu J-F, Cui Y, Jiao N. PdCl2 and N-Hydroxyphthalimide Co-catalyzed Csp2-H Hydroxylation by Dioxygen Activation. Angew Chem, Int Ed. 2013;52:5827. doi: 10.1002/anie.201300957. [DOI] [PubMed] [Google Scholar]; (i) Pipaliya BV, Chakraborti AK. Cross Dehydrogenative Coupling of Heterocyclic Scaffolds with Unfunctionalised Aroyl Surrogates by Palladium(II) Catalyzed C(sp2)-H Aroylation through Organocatalytic Dioxygen Activation. J Org Chem. 2017;82:3767. doi: 10.1021/acs.joc.7b00226. [DOI] [PubMed] [Google Scholar]
- 23.(a) Wu Y, Choy PY, Mao F, Kwong FY. Toluene derivatives as simple coupling precursors for cascade palladium-catalyzed oxidative C–H bond acylation of acetanilides. Chem Commun. 2013;49:689. doi: 10.1039/c2cc37352a. [DOI] [PubMed] [Google Scholar]; (b) Xu Z, Xiang B, Sun P. Palladium catalyzed direct ortho C–H acylation of 2-arylpyridines using toluene derivatives as acylation reagents. RSC Adv. 2013;3:1679. [Google Scholar]; (c) Weng J, Yu Z, Liu X, Zhang G. Palladium-catalyzed direct oxidative ortho-acylation of anilides with toluene derivatives. Tetrahedron Lett. 2013;54:1205. [Google Scholar]; (d) Bao Y-S, Zhang D, Jia M, Zhaorigetu B. Replacing Pd(OAc)2 with supported palladium nanoparticles in ortho-directed CDC reactions of alkylbenzenes. Green Chem. 2016;18:2072. [Google Scholar]; (e) Li Z-l, Wu P-y, Sun K-k, Cai C. Nickel-catalyzed regioselective C-H acylation of chelating arenes: a new catalytic system for C–C bond formation via a radical process and its mechanistic explorations. New J Chem. 2019;43:12152 [Google Scholar]
- 24.(a) Xie P, Xie Y, Qian B, Zhou H, Xia C, Huang H. Palladium-Catalyzed Oxidative Carbonylation of Benzylic C–H Bonds via Nondirected C(sp3–H Activation. J Am Chem Soc. 2012;134:9902. doi: 10.1021/ja3036459. [DOI] [PubMed] [Google Scholar]; (b) Xie P, Xia C, Huang H. Palladium-Catalyzed Oxidative Aminocarbonylation: A New Entry to Amides via C-H Activation. Org Lett. 2013;15:3370. doi: 10.1021/ol401419u. [DOI] [PubMed] [Google Scholar]; (c) Qin G, Chen X, Yang L, Huang H. Copper-Catalyzed α-Benzylation of Enones via Radical-Triggered Oxidative Coupling of Two C-H Bonds. ACS Catal. 2015;5:2882. [Google Scholar]
- 25.CCDC 1989688, 1989676 and 1989702 contains the supplementary crystallographic data for 3f 3i and 13a respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre (see also S.I.)
- 26.(a) Chosi T, Sada T, Fujimoto H, Nagayama C, Sugino E, Hibino S. Total syntheses of carazostatin and hyellazole by allene-mediated electrocyclic reaction. Tetrahedron Lett. 1996;37:2593. [Google Scholar]; (b) Chosi T, Sada T, Fujimoto H, Nagayama C, Sugino E, Hibino S. Total Syntheses of Carazostatin, Hyellazole, and Carbazoquinocins B-F. J Org Chem. 1997;62:2535. doi: 10.1021/jo962038t. [DOI] [PubMed] [Google Scholar]; (c) Knölker H-J, Hopfmann T. Transition metal complexes in organic synthesis. Part 65: Iron-mediated synthesis of carazostatin, a free radical scavenger from Streptomyces chromofuscus, and O-methylcarazostatin. Tetrahedron. 2002;58:8937. [Google Scholar]
- 27.Kwon JK, Cho JH, Ryu Y-S, Oh SH, Yum EK. N-Arylation of carbazole by microwave-assisted ligand-free catalytic CuI reaction. Tetrahedron. 2011;67:4820. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.