

Abstract
Previous studies had limited power to assess the associations of testosterone with aggressive disease as a primary endpoint. Further, the association of genetically predicted testosterone with aggressive disease is not known. We investigated the associations of calculated free and measured total testosterone and sex hormone‐binding globulin (SHBG) with aggressive, overall and early‐onset prostate cancer. In blood‐based analyses, odds ratios (OR) and 95% confidence intervals (CI) for prostate cancer were estimated using conditional logistic regression from prospective analysis of biomarker concentrations in the Endogenous Hormones, Nutritional Biomarkers and Prostate Cancer Collaborative Group (up to 25 studies, 14 944 cases and 36 752 controls, including 1870 aggressive prostate cancers). In Mendelian randomisation (MR) analyses, using instruments identified using UK Biobank (up to 194 453 men) and outcome data from PRACTICAL (up to 79 148 cases and 61 106 controls, including 15 167 aggressive cancers), ORs were estimated using the inverse‐variance weighted method. Free testosterone was associated with aggressive disease in MR analyses (OR per 1 SD = 1.23, 95% CI = 1.08‐1.40). In blood‐based analyses there was no association with aggressive disease overall, but there was heterogeneity by age at blood collection (OR for men aged <60 years 1.14, CI = 1.02‐1.28; P het = .0003: inverse association for older ages). Associations for free testosterone were positive for overall prostate cancer (MR: 1.20, 1.08‐1.34; blood‐based: 1.03, 1.01‐1.05) and early‐onset prostate cancer (MR: 1.37, 1.09‐1.73; blood‐based: 1.08, 0.98‐1.19). SHBG and total testosterone were inversely associated with overall prostate cancer in blood‐based analyses, with null associations in MR analysis. Our results support free testosterone, rather than total testosterone, in the development of prostate cancer, including aggressive subgroups.
Keywords: aggressive prostate cancer, Mendelian randomisation, prostate cancer, SHBG, testosterone
What's new?
The Endogenous Hormones, Nutritional Biomarkers and Prostate Cancer Collaborative Group (EHNBPCCG) is a pooled dataset of prospective studies of prostate cancer risk. Using this data, the authors conducted blood‐based analysis and Mendelian randomisation analysis to determine the association between circulating testosterone and overall risk of prostate cancer, as well as looking at risk of aggressive disease and early‐onset cancer separately. They found strong evidence that higher concentrations of circulating testosterone increases the risk of prostate cancer, including aggressive subtypes. This is the largest collection of prospective blood‐based observational and genetic data on sex hormones and prostate cancer risk to date.
Abbreviations
- BMI
body mass index
- CI
confidence interval
- EHNBPCCG
The Endogenous Hormones, Nutritional Biomarkers and Prostate Cancer Collaborative Group
- IGF
insulin‐like growth factor
- IGFBP
insulin‐like growth factor binding protein
- MR
Mendelian randomisation
- MR‐PRESSO
MR residual sum and outlier
- MR‐RAPS
MR robust adjusted profile score
- OR
odds ratio
- PRACTICAL
Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome
- PSA
prostate‐specific antigen
- SHBG
sex hormone binding globulin
1. INTRODUCTION
Prostate cancer is the second most common cancer in men worldwide and a leading cause of cancer death. 1 Blood‐based and genetic epidemiological studies show evidence of an association between circulating concentrations of calculated free testosterone and risk of overall prostate cancer. 2 , 3 , 4 , 5 , 6 The association is biologically plausible because androgens are integral to the maintenance of prostate function. 7 In the circulation, testosterone is bound to sex hormone‐binding globulin (SHBG) and albumin. Approximately 2% of total testosterone circulates unbound or ‘free’, and according to the free hormone hypothesis is more biologically active. 8 Prostate cancer varies in aggressiveness and tumours also vary by age of onset, and risk factors for these subgroups may be different from those for overall prostate cancer, 9 but previous studies have lacked statistical power to assess associations of testosterone with prostate cancer subgroups.
The Endogenous Hormones, Nutritional Biomarkers and Prostate Cancer Collaborative Group (EHNBPCCG) is a pooled individual participant case‐control dataset of prospective studies of risk of prostate cancer and associated risk factors. Previous analyses of the associations of circulating testosterone concentrations using the EHNBPCCG dataset were based on up to 6900 cases and 12 100 controls. 2 We observed that men with very low free testosterone had a lower risk of overall prostate cancer, but we had limited power to investigate the associations with aggressive disease as a primary endpoint. This dataset has since been expanded to include more than double the number of prostate cancer cases, including 1900 aggressive and 600 early‐onset cases.
Mendelian randomisation (MR) analyses, which use genetic instruments to predict average adult exposures, are less likely than blood‐based studies to be affected by confounding factors or reverse causation, and are often considered to be a more reliable method for causal inference. 10 Therefore, we carried‐out two‐sample MR analyses, using instruments identified from UK Biobank (up to 194 500 men) and genetic data from the PRACTICAL consortium (up to 79 000 prostate cancer cases [15 000 aggressive and 7000 early‐onset subgroups] and 61 000 controls). 11 , 12
Using these two international consortia, we aimed to extend our prior study in the EHNBPCCG to assess the associations of circulating concentrations of calculated free testosterone, as well as total testosterone and SHBG which are used to calculate free testosterone, with overall, aggressive and early‐onset prostate cancer risk using blood‐based and genetic methods; using these complementary approaches can provide more robust evidence for causal inference.
2. MATERIALS AND METHODS
2.1. Endogenous Hormones, Nutritional Biomarkers and Prostate Cancer Collaborative Group
2.1.1. Data collection and study designs
Individual participant data were available from up to 25 prospective studies with total testosterone and SHBG measurements. Participating studies are listed in Supplementary Table 1 and further details of data collection and processing are provided in the Supplementary Material (Appendix S2). Matching criteria are shown in Supplementary Table 2. Assay details and hormone measurement data are listed in Supplementary Table 3.
2.1.2. Data processing and outcomes
Free testosterone concentrations were estimated using a formula based on the law of mass action from measured total testosterone and SHBG concentrations, 13 , 14 assuming a constant albumin concentration of 43 g/L.
Disease definitions were as defined by the PRACTICAL consortium. 11 , 12 Aggressive prostate cancer was categorised as ‘yes’ for any of the following: disease metastases at diagnosis (M1), Gleason score 8+ (or equivalent), prostate cancer death (defined as death from prostate cancer) or prostate‐specific antigen (PSA) >100 ng/mL. Early‐onset prostate cancer was defined as a diagnosis aged ≤55 years. Further details can be found in the Supplementary Methods (Appendix S2).
2.1.3. Statistical analysis
Conditional logistic regression was used to estimate prostate cancer risk by free and total testosterone and SHBG concentrations. Analyses were conditioned on the study‐specific matching variables and adjusted for age at blood collection, body mass index (BMI), height, smoking status, alcohol consumption, racial/ethnic group, education, married/cohabiting and diabetes status. Biomarkers were standardised by study and entered into the model as continuous variables, so each increment represents a 1 study‐specific SD increase in biomarker concentration. For categorical analyses, biomarkers were categorised into study‐specific fifths with cut‐points determined in controls. 15 Further details are available in the Supplementary Methods (Appendix S2).
2.1.4. Further analyses
We examined heterogeneity in the associations of each biomarker with prostate cancer by participant characteristics and study (Supplementary Methods, Appendix S2). Subgroups were defined a priori based on the availability of data and previous analyses using this dataset. 2 , 5 To further investigate the apparent heterogeneity by age at blood collection, we examined associations of free testosterone with overall and aggressive prostate cancer in fifths, stratified by age at blood collection (<60; 60+ years).
We also investigated associations in models conditioned on the matching variables but not further adjusted, associations in tenths, and estimates per 80th percentile increase. Associations were also examined following mutual adjustment for other biomarkers (including insulin‐like growth factors [IGF‐I, II] and IGF binding proteins [IGFBP‐1,2,3]), and we tested for interactions between these biomarkers. Stratified analyses and associations in tenths were not investigated for early‐onset disease due to the limited number of cases.
2.2. Mendelian randomisation analyses
2.2.1. Genetic instruments for hormone concentrations
Summary GWAS results for circulating calculated free and total testosterone and SHBG for men in UK Biobank were extracted from a published analysis (based on up to 194 453 men of white European ancestry) 4 (Supplementary Methods, Appendix S2). UK Biobank participants were aged 40‐69 years at blood collection (mean age = 56.5 years). We pruned single nucleotide polymorphisms (SNPs) by a linkage disequilibrium threshold of r 2 < .001.
2.2.2. Genetic associations with prostate cancer
For each of the SNPs included as an instrument for free testosterone, total testosterone and SHBG, we obtained the association with prostate cancer risk from the PRACTICAL consortium (including GAME‐ON/ELLIPSE). 11 , 12 Individual studies included in these consortia are available from Schumacher et al. 11 Associations with overall prostate cancer risk were generated from 79 148 prostate cancer cases and 61 106 controls, aggressive from 15 167 cases and 58 308 controls, and early‐onset disease from 6988 cases and 44 256 controls. 11 , 12 Genetic data for UK Biobank participants were not included in this dataset.
2.2.3. Statistical analysis
The MR estimation for hormones was conducted using the inverse‐variance weighted (IVW) method. 16 We additionally calculated the I 2 statistic to assess measurement error in SNP‐exposure associations 17 and Cochran's Q statistic for heterogeneity between the MR estimates for each SNP. 18 PhenoScanner was used to assess pleiotropy of the genetic instruments. 19 As sensitivity analyses, we used the MR residual sum and outlier (MR‐PRESSO) and MR robust adjusted profile score (MR‐RAPS) to investigate the role of SNP outliers, 20 and the weighted median, MR‐Egger and the contamination mixture method to investigate horizontal pleiotropy. 21 , 22 , 23
For SHBG, we additionally investigated associations of the cis‐SNP with prostate cancer risk, as this cis‐SNP may be less likely to be affected by horizontal pleiotropy than trans‐SNPs. 24 Associations of the cis‐SNP with prostate cancer were assessed using the Wald ratio.
Details of statistical software and packages used are available in the Supplementary Methods (Appendix S2). All tests of significance were two‐sided, and P‐values <.05 were considered statistically significant.
3. RESULTS
3.1. Study and participant characteristics in the blood‐based analyses
A total of 25 studies, contributing up to 14 944 cases and 36 752 controls, were included in these analyses. Prostate cancer was classified as aggressive in 1870 cases and early‐onset in 611 cases. Study participants were predominantly of white European ancestry (90%) (Table 1).
TABLE 1.
Characteristics of prostate cancer cases and controls in the EHNBPCCG participants
| Cases | ||||
|---|---|---|---|---|
| Controls | Overall | Aggressive a | Early‐onset b | |
| N | 36 752 | 14 944 | 1870 | 611 |
| Age at blood collection (yr), mean (SD) | 61.0 (8.4) | 60.8 (8.6) | 62.1 (8.4) | 46.7 (5.7) |
| Height (cm), mean (SD) | 174.6 (7.2) | 174.7 (7.3) | 173.7 (7.8) | 177.1 (6.9) |
| BMI (kg/m2), mean (SD) | 27.4 (4.1) | 26.9 (3.7) | 27.1 (4.0) | 26.5 (3.7) |
| PSA at blood collection (ng/mL), med (IQR) | 0.9 (1.2) | 2.3 (3.2) | 3.0 (5.6) | 1.6 (2.5) |
| Time from blood collection to diagnosis, mean (SD) | – | 6.5 (5.9) | 7.5 (7.1) | 5.8 (5.3) |
| Age at diagnosis, mean (SD) | – | 67.3 (6.7) | 67.0 (6.2) | 52.5 (2.5) |
| Racial/ethnic group, N (%) | ||||
| White | 33 645 (91.5) | 13 586 (90.9) | 1676 (89.6) | 559 (91.5) |
| Black | 1222 (3.3) | 524 (3.5) | 57 (3.0) | 31 (5.1) |
| East Asian | 875 (2.4) | 484 (3.2) | 89 (4.8) | 3 (0.5) |
| Other | 678 (1.8) | 236 (1.6) | 17 (0.9) | 10 (1.6) |
| Not known | 332 (0.9) | 114 (0.8) | 31 (1.7) | 8 (1.3) |
| Smoking status, N (%) | ||||
| Never | 13 868 (37.7) | 5681 (38.0) | 599 (32.0) | 273 (44.7) |
| Ex | 15 548 (42.3) | 6329 (42.4) | 815 (43.6) | 160 (26.2) |
| Current | 5674 (15.4) | 2351 (15.7) | 407 (21.8) | 140 (22.9) |
| Not known | 1662 (4.5) | 583 (3.9) | 49 (2.6) | 38 (6.2) |
| Alcohol consumption (g ethanol/day), N (%) | ||||
| Nondrinker | 2673 (7.3) | 1615 (10.8) | 250 (13.4) | 46 (7.5) |
| <10 | 8189 (22.3) | 3752 (25.1) | 484 (25.9) | 140 (22.9) |
| 10+ | 19 198 (52.2) | 7309 (48.9) | 883 (47.2) | 300 (49.1) |
| Not known | 6692 (18.2) | 2268 (15.2) | 253 (13.5) | 125 (20.5) |
| Diabetes status, N (%) | ||||
| Yes | 2887 (7.9) | 819 (5.5) | 122 (6.5) | 13 (2.1) |
| No | 28 745 (78.2) | 11 913 (79.7) | 1487 (79.5) | 467 (76.4) |
| Not known | 5120 (13.9) | 2212 (14.8) | 261 (14.0) | 131 (21.4) |
| Married/cohabiting, N (%) | ||||
| Yes | 9767 (26.6) | 6790 (45.4) | 1295 (69.3) | 222 (36.3) |
| No | 1461 (4.0) | 958 (6.4) | 183 (9.8) | 39 (6.4) |
| Not known | 25 524 (69.4) | 7196 (48.2) | 392 (21.0) | 350 (57.3) |
Note: Some aggressive disease characterisation data were available from 88% of included studies.
Abbreviations: BMI, body mass index; IQR, interquartile range; PSA, prostate‐specific antigen.
Aggressive disease was defined as Gleason Score 8+, death from prostate cancer, metastatic disease or PSA >100 ng/mL.
Early‐onset defined as diagnosed aged ≤55 years.
Prostate cancer characteristics by study are displayed in Supplementary Table 4. Mean age at blood collection for each study ranged from 33.8 to 76.8 years (overall mean = 61.0 years, SD = 8.5 years). Cases were diagnosed a mean of 6.5 years (SD = 5.9) after blood collection, and the mean age at diagnosis was 67.3 years (SD = 6.7) (Table 1). Partial correlations between biomarkers ranged from r = −0.04 (SHBG and PSA) to r = 0.77 (calculated free and total testosterone) (Supplementary Table 5).
3.2. Free testosterone
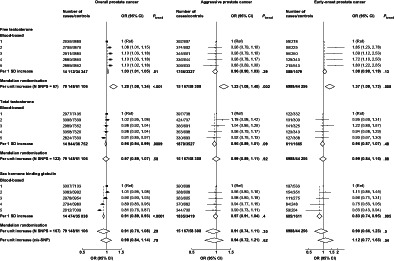
The association of calculated free testosterone with overall prostate cancer risk was significant in both blood‐based (OR per 1 SD increment = 1.03, 95% CI 1.01‐1.05) and MR analyses (OR per genetically predicted 1 SD increment = 1.20, 1.08‐1.34) (Figure 1). Higher free testosterone was associated with a higher risk of aggressive prostate cancer in the MR analysis (1.23, 1.08‐1.40), but there was no evidence of an association in the blood‐based analysis (0.96, 0.90‐1.03) (Figure 1). MR sensitivity analyses generally supported the associations of free testosterone with overall and aggressive prostate cancer, except for MR‐Egger, although the MR‐Egger intercepts did not indicate directional pleiotropy (Table 2).
FIGURE 1.
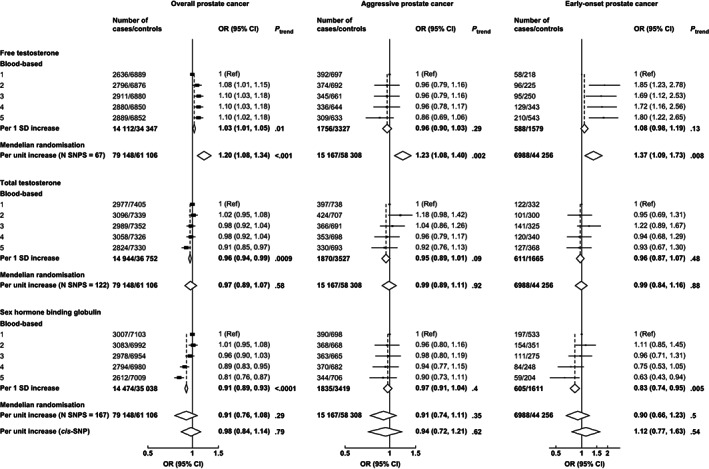
Risks of overall, aggressive* and early‐onset† prostate cancer in by study‐specific fifths of hormone concentrations (blood‐based only) and unit increment (blood‐based and MR). Blood‐based estimates are from logistic regression conditioned on the matching variables and adjusted for age, BMI, height, alcohol intake, smoking status, marital status, education status, racial/ethnic group and diabetes status. The position of each square indicates the magnitude of the odds ratio, and the area of the square is proportional to the inverse of the variance of the logarithm of the OR. The length of the horizontal line through the square indicates the 95% CI. MR risk estimates are estimated using the inverse variance weighted method for the full instrument methods and the Wald ratio in the cis‐SNP analyses (where applicable). In MR analyses, biomarker transformations are outlined in the Supplementary Methods (Appendix S2). *Aggressive cancer defined as Gleason grade 8+, or prostate cancer death or metastases or PSA >100 ng/mL. †Early‐onset defined as diagnosed ≤55 years. BMI, body mass index; CI, confidence interval; OR, odds ratio; PSA, prostate‐specific antigen; SNP, single nucleotide polymorphism
TABLE 2.
Mendelian randomisation estimates between genetically predicted circulating biomarker concentrations and prostate cancer risk
| Overall prostate cancer (79 148 cases, 61 106 controls) | Aggressive prostate cancer a (15 167 cases, 58 308 controls) | Early‐onset prostate cancer b (6988 cases, 44 256 controls) | ||||||
|---|---|---|---|---|---|---|---|---|
| Variance explained | N SNPs | OR per unit increment in biomarker (95% CI) | P | OR per unit increment in biomarker (95% CI) | P | OR per unit increment in biomarker (95% CI) | P | |
| Free testosterone (SD = 59.5 pmol/L) | ||||||||
| Inverse‐variance weighted | 3.8% | 67 | 1.20 (1.08, 1.34) | 0.0006 | 1.23 (1.08, 1.40) | 0.002 | 1.37 (1.09, 1.73) | 0.008 |
| Weighted median | 1.12 (1.01, 1.25) | 0.04 | 1.19 (0.99, 1.43) | 0.07 | 1.16 (0.89, 1.52) | 0.27 | ||
| MR‐Egger | 1.07 (0.87, 1.31) | 0.53 | 1.03 (0.80, 1.32) | 0.84 | 1.09 (0.69, 1.72) | 0.71 | ||
| MR‐Egger intercept | 0.20 | 0.11 | 0.26 | |||||
| MR‐RAPS | 1.16 (1.05, 1.28) | 0.002 | 1.20 (1.05, 1.36) | 0.01 | 1.33 (1.05, 1.67) | 0.02 | ||
| MR‐PRESSO | 1.13 (1.05, 1.22) | 0.002 | 1.23 (1.08, 1.40) c | 0.002 | 1.33 (1.07, 1.65) | 0.01 | ||
| Contamination mixture | 1.12 (1.04, 1.22) | 0.007 | 1.20 (1.00, 1.39) | 0.05 | 1.22 (0.94, 1.97) | 0.14 | ||
| Total testosterone (SD = 3.8 nmol/L) | ||||||||
| Inverse‐variance weighted | 7.5% | 122 | 0.97 (0.89, 1.07) | 0.58 | 0.99 (0.89, 1.11) | 0.92 | 0.99 (0.84, 1.16) | 0.88 |
| Weighted median | 0.99 (0.91, 1.08) | 0.89 | 0.99 (0.86, 1.14) | 0.84 | 1.07 (0.86, 1.32) | 0.53 | ||
| MR‐Egger | 0.99 (0.85, 1.15) | 0.92 | 1.06 (0.89, 1.26) | 0.53 | 0.95 (0.73, 1.25) | 0.73 | ||
| MR‐Egger intercept | 0.77 | 0.39 | 0.76 | |||||
| MR‐RAPS | 1.04 (0.94, 1.14) | 0.45 | 1.03 (0.93, 1.14) | 0.62 | 1.00 (0.85, 1.18) | 0.99 | ||
| MR‐PRESSO | 1.02 (0.95, 1.09) | 0.60 | 0.92 (0.79, 1.08) | 0.30 | 1.01 (0.88, 1.17) | 0.88 | ||
| Contamination mixture | 1.06 (0.99, 1.17) | 0.09 | 1.02 (0.94, 1.14) | 0.54 | 1.05 (0.86, 1.21) | 0.72 | ||
| SHBG (SD = 16.5 nmol/L) | ||||||||
| Inverse‐variance weighted | 15.0% | 168 | 0.91 (0.76, 1.08) | 0.29 | 0.91 (0.74, 1.11) | 0.35 | 0.90 (0.66, 1.23) | 0.50 |
| Weighted median | 0.98 (0.85, 1.13) | 0.79 | 0.94 (0.74, 1.18) | 0.58 | 1.12 (0.79, 1.58) | 0.52 | ||
| MR‐Egger | 0.99 (0.76, 1.27) | 0.92 | 1.05 (0.78, 1.40) | 0.76 | 0.97 (0.62, 1.52) | 0.89 | ||
| MR‐Egger intercept | 0.38 | 0.18 | 0.64 | |||||
| MR‐RAPS | 1.01 (0.86, 1.19) | 0.87 | 0.95 (0.79, 1.15) | 0.60 | 0.93 (0.70, 1.25) | 0.63 | ||
| MR‐PRESSO | 0.98 (0.88, 1.10) | 0.76 | 0.93 (0.79, 1.09) | 0.36 | 0.98 (0.77, 1.24) | 0.86 | ||
| Contamination mixture | 0.96 (0.86, 1.07) | 0.55 | 0.90 (0.77, 1.05) | 0.21 | 1.01 (0.70, 1.30) | 0.92 | ||
| cis‐SNP (rs1799941) | 4.2% | 1 | 0.98 (0.84, 1.14) | 0.79 | 0.94 (0.72, 1.21) | 0.62 | 1.12 (0.77, 1.63) | 0.54 |
Note: Biomarker transformations are outlined in the Supplementary Methods (Appendix S2).
Abbreviations: CI, confidence interval; MR, Mendelian randomisation; OR, odds ratio; PRESSO, pleiotropy residual sum and outlier; RAPS, robust adjusted profile score; SHBG, sex hormone‐binding globulin.
Aggressive disease was defined as Gleason Score 8+, death from prostate cancer, metastatic disease or PSA >100 ng/mL.
Early‐onset defined as diagnosed aged ≤55 years.
No statistically significant outliers detected.
In the MR analysis, predicted free testosterone was associated with an increased risk of early‐onset disease (1.37, 1.09‐1.73), and the relationship was directionally consistent in blood‐based analyses (1.08, 0.98‐1.19) (Figure 1). The associations with early‐onset disease were less robust in the MR sensitivity analyses but were directionally consistent (Table 2).
3.3. Total testosterone
The OR for total testosterone in relation to overall prostate cancer was 0.96 (0.94‐0.99) in blood‐based analysis and 0.97 (0.89‐1.07) in MR analysis (Figure 1 and Table 2). Total testosterone was not associated with aggressive or early‐onset disease in blood‐based or in the MR analyses (Figure 1 and Table 2).
3.4. Sex hormone‐binding globulin
SHBG was inversely associated with overall and early‐onset prostate cancer in blood‐based analyses (0.91, 0.89‐0.93 and 0.83, 0.74‐0.95, respectively), but was not associated with aggressive disease risk (0.97, 0.91‐1.04) (Figure 1). In the MR analyses, SHBG was not associated with prostate cancer risk using the full instrument or the cis‐SNP instrument (Figure 1 and Table 2).
3.4.1. Further analyses – blood‐based analysis
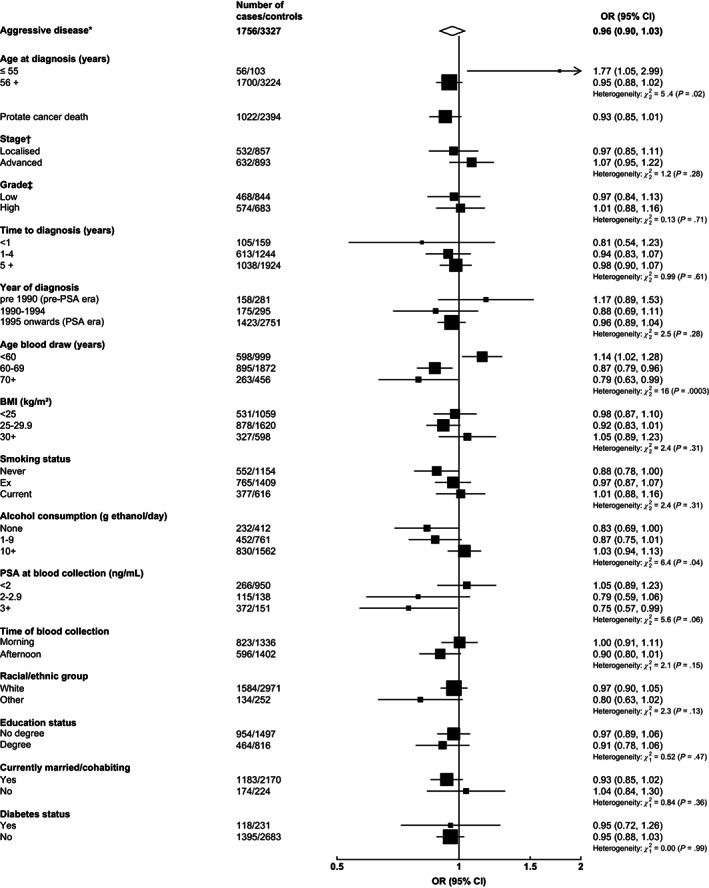
There was significant heterogeneity in the associations of free testosterone with risk according to the prostate cancer aggressiveness; higher free testosterone concentration was associated with an increased risk of nonaggressive (1.07, 1.02‐1.12), but not aggressive disease (0.96, 0.90‐1.03; P het = .02) (Figure 2). Men with diabetes also had a larger magnitude of association of free testosterone with overall prostate cancer than men without diabetes (1.12, 1.04‐1.21 and 1.02, 1.00‐1.05, respectively; P het = .02) (Figure 2).
FIGURE 2.

Odds ratio (95% CIs) for overall prostate cancer per study‐specific 1 SD increment of free testosterone concentration by subgroup. Estimates are from logistic regression conditioned on the matching variables and adjusted for age, BMI, height, alcohol intake, smoking status, marital status, education status, racial/ethnic group and diabetes status. The position of each square indicates the magnitude of the OR, and the area of the square is proportional to the inverse of the variance of the logarithm of the OR). The length of the horizontal line through the square indicates the 95% CI. Tests for heterogeneity for case‐defined factors were obtained by fitting separate models for each subgroup and assuming independence of the ORs using a method analogous to a metaanalysis. Tests for heterogeneity for non‐case‐defined factors were assessed with a χ 2 test of interaction between subgroup and the binary variable. *Aggressive cancer defined as Gleason grade 8+, or prostate cancer death or metastases or PSA >100 ng/mL. †Localised defined as TNM stage <T2 with no reported lymph node involvement or metastases or stage I; other localised stage if TNM stage T2 with no reported lymph node involvement or metastases, stage II or equivalent; advanced stage if they were TNM stage T3 or T4 and/or N1+ and/or M1, stage III‐IV or equivalent. ‡Low grade defined as Gleason score was <7 or equivalent (ie, extent of differentiation good, moderate); medium grade if Gleason score was 7 (ie, poorly differentiated); high grade if the Gleason score was ≥8 or equivalent (ie, undifferentiated). BMI, body mass index; CI, confidence interval; OR, odds ratio; PSA, prostate‐specific antigen
For aggressive disease risk, there was significant heterogeneity in the associations by age at blood collection; free testosterone was positively associated with aggressive prostate cancer (1.14, 1.02‐1.28) for men whose blood was collected at ages <60 years, but the relationship was inverse for men whose blood was collected at older ages (0.87, 0.79‐0.96 and 0.79, 0.63‐0.99 for men whose blood was collected aged 60‐69 and 70+ years, respectively) (P het = .0003) (Figure 3). In analyses based on fifths of free testosterone, there was a positive dose‐response relationship of free testosterone with overall and aggressive prostate cancer for men whose blood was collected at <60 years, while for men whose blood was collected at an older age, the relationship was null with overall prostate cancer and inverse with aggressive prostate cancer (Supplementary Figure 1). Higher free testosterone was also associated with an elevated risk of early‐onset aggressive disease (1.77, 1.05‐2.99) but was not associated with aggressive disease for men diagnosed later in life (0.95, 0.88‐1.02; P het = .02) (Figure 3), although there was a small number of cases of early‐onset aggressive disease (n = 56).
FIGURE 3.
Odds ratio (95% CIs) for aggressive* prostate cancer per study‐specific 1 SD increment of free testosterone concentration by subgroup. Estimates are from logistic regression conditioned on the matching variables and adjusted for age, BMI, height, alcohol intake, smoking status, marital status, education status, racial/ethnic group and diabetes status. The position of each square indicates the magnitude of the OR, and the area of the square is proportional to theinverse of the variance of the logarithm of the OR). The length of the horizontal line through the square indicates the 95% CI. Tests for heterogeneity for case‐defined factors were obtained by fitting separate models for each subgroup and assuming independence of the ORs using a method analogous to a metaanalysis. Tests for heterogeneity for non‐case‐defined factors were assessed with a χ 2 test of interaction between subgroup and the binary variable. *Aggressive cancer defined as Gleason grade 8+, or prostate cancer death, or metastases or PSA >100 ng/mL. †Localised defined as TNM stage <T2 with no reported lymph node involvement or metastases or stage I, or TNM stage T2 with no reported lymph node involvement or metastases, stage II, or equivalent; advanced stage if they were TNM stage T3 or T4 and/or N1+ and/or M1, stage III‐IV or equivalent. ‡Low grade defined as Gleason score was <8 or equivalent (ie, extent of differentiation good, moderate, poor); high grade if the Gleason score was ≥8 or equivalent (ie, undifferentiated). BMI, body mass index; CI, confidence interval; OR, odds ratio; PSA, prostate‐specific antigen
The associations of total testosterone and SHBG with overall and aggressive prostate cancer were generally consistent by subgroups (Supplementary Figures 2‐5). Total testosterone was inversely associated with prostate cancer death (0.90, 0.82‐0.97) and positively associated with early‐onset aggressive prostate cancer (2.40, 1.28‐4.52), while for men diagnosed with aggressive disease aged >55 years the OR was 0.94 (0.88‐1.00; P het = .0004) (Supplementary Figure 3).
There was no statistically significant heterogeneity in the associations with overall and aggressive prostate cancer by study (Supplementary Figures 6‐11), except for free testosterone and aggressive prostate cancer (P het = .02) (Supplementary Figure 7). Associations were broadly similar in unadjusted matched analyses (Supplementary Figure 12), study‐specific tenths (Supplementary Figure 13), per 80%tile increase (Supplementary Table 6) and following mutual adjustment for other biomarkers (Supplementary Table 7).
There were significant interactions in the associations of total testosterone with overall and aggressive prostate cancer by SHBG concentrations (Supplementary Tables 8 and 9). SHBG was positively associated with aggressive disease risk for men with lower IGFBP‐1 concentrations (1.23, 1.00‐1.51), and the relationship was inverse for men with higher IGFBP‐1 concentrations (0.85, 0.71‐1.02; P het = .01) (Supplementary Table 9).
3.4.2. Further analyses – Mendelian randomisation
There was no strong evidence of measurement error in the genetic instruments for the biomarkers (I 2 > 0.96). There was significant heterogeneity in the MR estimates for the SNPs with overall disease, and for aggressive and early‐onset disease (Cochran's Q P < .001), except for the association of free testosterone with aggressive disease (P = .12). Using PhenoScanner, 175, 355 and 358 traits were linked to SNPs for free testosterone, SHBG and total testosterone concentrations, respectively, particularly adiposity and height, and SNPs associated with free testosterone were frequently related to age at puberty (P < 5 × 10−8) (Supplementary Figures 14‐16). Traits linked to the SHBG cis‐SNP (rs1799941) are shown in Supplementary Table 10. MR scatterplots and tables are found in Supplementary Figures 17‐19 and Supplementary Tables 11‐13.
4. DISCUSSION
In this first comprehensive analysis with both blood‐based and genetic data, our results suggest that higher calculated free testosterone is associated with an elevated risk for prostate cancer, including aggressive disease. Neither circulating total testosterone nor SHBG was associated with elevated risks for prostate cancer.
The strong genetic evidence in our MR analyses (which are less likely to be affected by biases such as confounding, reverse causation and detection bias) for a role of free testosterone, alongside the well‐characterised lower risk of prostate cancer in men diagnosed with Klinefelter's syndrome 25 (a genetic abnormality which is characterised by life‐long clinically low total and free testosterone concentrations 26 ), indicates a probable causal relationship of free testosterone with prostate cancer, including with aggressive disease. While in our blood‐based analyses the overall association of free testosterone with aggressive prostate cancer was null, there was evidence of a positive association with aggressive disease for men whose blood who was collected at a younger age. However, we observed inverse associations of free testosterone with aggressive disease for men whose blood was collected at an older age, which warrants further consideration. Differences between the associations of genetically predicted free testosterone and measured blood concentrations with prostate cancer risk may implicate the importance of free testosterone concentrations in younger adulthood. Free testosterone concentrations decline with older age, partly due to cumulative environmental influences, therefore free testosterone concentrations in middle and older age may not be representative of life‐long exposure to free testosterone concentrations, which will attenuate risk estimates. 27 , 28 , 29 , 30 , 31 , 32 There was also some evidence of heterogeneity in the blood‐based association of free testosterone with aggressive disease by study, which may relate to differences in participant and tumour characteristics.
As well as the blood‐based and genetic evidence that we describe here, two randomised controlled trials using 5α‐reductase inhibitors, which aimed to reduce intraprostatic androgen signalling by reducing dihydrotestosterone concentrations by 80‐90%, 33 have reported 23‐25% lower risks of overall prostate cancer. However, these trials also reported 27‐58% increased risks of high‐grade tumours, 34 , 35 possibly due to changes in prostate morphology, function biasing tumour diagnostic grading, and/or the early development of partial androgen insensitivity in more aggressive tumours (in comparison with low‐grade tumours) 36 , 37 ; long‐term follow‐up of these trials does not support an effect on risk of prostate cancer mortality. 38
For total testosterone and SHBG the MR results were null suggesting no direct effect, whereas the blood‐based analyses were inverse for both; it is possible that the inverse results for testosterone are due to reverse causation, but results did not suggest this for SHBG and the explanation for the blood‐based results remains unclear.
These analyses have several strengths. This is the largest collection of prospective blood‐based and genetic data on sex hormones and prostate cancer risk available, representing almost all the available data worldwide. This large sample size maximised power to assess associations robustly and enabled us to investigate associations across subgroups. Further, by incorporating blood‐based and MR methods we were able to use different lines of evidence to inform causal inference. 39
Limitations include that we used calculated rather than directly measured free testosterone concentrations using equilibrium dialysis, 40 we have used a validated formula to estimate concentrations and these are well correlated. 41 , 42 It has also been suggested that the bioavailable fraction of testosterone is the sum of free and albumin‐bound testosterone rather than solely the free fraction, 43 but it is not possible in our data to distinguish between these hypotheses because estimates of these fractions from the formula are perfectly correlated. Furthermore, the predictive value of peripheral free testosterone as an indicator of intraprostatic signalling remains under debate. 44 Our analyses relied on single biomarker measurements, and although these biomarkers have good reproducibility over a 4‐to‐5‐year period (intraclass correlation coefficients 0.54‐0.82), 2 longitudinal studies have shown that free testosterone declines continually throughout adulthood 45 ; this may lead to underestimates of risk. 46 Participants in the EHNBPCCG dataset were predominantly white and therefore we were underpowered to investigate associations for other racial/ethnic groups. Prospective epidemiological studies were generally based on older men, therefore we had more limited power to investigate associations in younger participants.
In the MR analyses of free testosterone, we observed weaker relationships using MR‐Egger. MR‐Egger is less susceptible to confounding from possibly pleiotropic variants that have stronger effects on the outcome than the exposure. However, this approach is also subject to reduced power and therefore does not necessarily imply the absence of a causal effect in the context of consistent sensitivity analyses and balanced pleiotropy. 21 , 47 Further, testosterone is a steroid and therefore no cis‐genetic instruments are available. These limitations mean that we cannot exclude the possibility that the MR results for free testosterone may be influenced by some horizontal pleiotropy. It is also not known whether the performance of the genetic predictors of free testosterone change with age. 48 Future genetic and blood‐based research including younger men with repeat measurements and linkage to detailed medical records will help to clarify associations.
The blood‐based results we report here are an extension of our previous paper, 2 and includes more than double the number of cases, with the incorporation studies including UK Biobank and extended follow‐up from some other studies. Our blood‐based results indicated possible nonlinear relationships with overall prostate cancer (as reported previously) 2 and with early‐onset prostate cancer, with lower risks of overall and early‐onset prostate cancer for men with low free testosterone concentrations. For MR analyses, genetic instruments were based on summary GWAS results, and we were therefore unable to investigate possible nonlinear associations. For overall prostate cancer we also previously reported a possible increased risk of high‐grade disease; however, we have limited additional data for grade, and therefore we do not include an updated detailed grade analysis as reported in the previous paper.
In conclusion, the findings from these blood‐based and genetic analyses implicate free testosterone in the development of prostate cancer, including aggressive and early‐onset disease.
AUTHOR CONTRIBUTIONS
Eleanor L. Watts wrote the original manuscript draft, analysed the data, created the visualisations, acquired funding, and led the conceptualisation of the analysis. Georgina K. Fensom, Urwah Noor and Colm D. Andrews contributed to the data curation, pooling and administration. Karl Smith‐Byrne, Marc J. Gunter, Michael V. Holmes, Richard M. Martin and Konstantinos K. Tsilidis contributed to the conceptualisation of the analysis, methodology and review and drafting of the manuscript. Demetrius Albanes, Aurelio Barricarte, H. Bas Bueno‐de‐Mesquita, Chu Chen, Barbara A. Cohn, Niki L. Dimou, Luigi Ferrucci, Leon Flicker, Neal D. Freedman, Graham G. Giles, Edward L. Giovannucci, Gary E. Goodman, Christopher A. Haiman, Graeme J. Hankey, Jiaqi Huang, Wen‐Yi Huang, Lauren M. Hurwitz, Rudolf Kaaks, Paul Knekt, Tatsuhiko Kubo, Hilde Langseth, Gail Laughlin, Loic Le Marchand, Tapio Luostarinen, Robert J. MacInnis, Hanna O. Mäenpää, Satu Männistö, E. Jeffrey Metter, Kazuya Mikami, Lorelei A. Mucci, Anja W. Olsen, Kotaro Ozasa, Domenico Palli, Kathryn L. Penney, Elizabeth A. Platz, Harri Rissanen, Norie Sawada, Jeannette M. Schenk, Pär Stattin, Akiko Tamakoshi, Elin Thysell, Chiaojung Jillian Tsai, Shoichiro Tsugane, Lars Vatten, Elisabete Weiderpass, Stephanie J. Weinstein, Lynne R. Wilkens, Bu B. Yeap, The PRACTICAL consortium, CRUK, BPC3, CAPS, PEGASUS contributed data resources and reviewed and edited the manuscript. Naomi E. Allen contributed data resources and administration and reviewed and edited the manuscript. Aurora Perez‐Cornago, Ruth C. Travis, Timothy J. Key supervised the project, acquired funding, led the conceptualisation of the analysis, and reviewed and edited the manuscript. The work reported in the paper has been performed by the authors, unless clearly specified in the text.
FUNDING INFORMATION
Centralised pooling, checking and data analysis were supported by Cancer Research UK (grant numbers C8221/A19170 and C8221/A29017). Eleanor L. Watts is supported by the Nuffield Department of Population Health Early Career Research Fellowship. Aurora Perez‐Cornago is supported by a Cancer Research UK Population Research Fellowship (C60192/A28516) and by the World Cancer Research Fund (WCRF UK), as part of the World Cancer Research Fund International grant programme (2019/1953). ATBC was supported in part by the Intramural Research Program of the National Institutes of Health and the National Cancer Institute. CARET is funded by the National Cancer Institute, National Institutes of Health through grants U01‐CA063673, UM1‐CA167462, R01‐CA 78812 and U01‐CA167462. CLUE is funded by the American Institute for Cancer Research, NIH Grants IU01AG18033 and IU01CA86308. HIMS was supported by research grants from the National Health and Medical Research Council of Australia. MEC is supported by the US National Institutes of Health grant U01 CA164973. JACC study was supported by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. RMM was supported by the NIHR Biomedical Research Centre at University Hospitals Bristol and Weston NHS Foundation Trust and the University of Bristol. RMM was supported by a Cancer Research UK (C18281/A29019) programme grant (the Integrative Cancer Epidemiology Programme). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
CONFLICT OF INTEREST
Dr Michael V. Holmes declares unpaid consultancy for Boehringer Ingelheim. The other authors have no conflicts to disclose.
ETHICS STATEMENT
This analysis reanalysed data and therefore new ethical approval was not required. Each individual study obtained ethical approval and informed consent from participants.
Supporting information
Appendix S1 PIs from the PRACTICAL (http://practical.icr.ac.uk/), CRUK, BPC3, CAPS, PEGASUS consortia
Appendix S2 Supporting Information
ACKNOWLEDGEMENTS
We thank all participants, researchers and support staff who made the study possible. CARET would like to acknowledge the support of Frank Z. Stanczyk, S. Kay. Lewis, Ruth Etzioni, Matt Barnett, Dante DiTommaso, and the CARET study participants. CHDS would like to acknowledge the support of Alice Whittemore and David Feldman. CLUE thank Kathy J. Helzlsouer for her contributions to the cohort and thank the staff and participants of the CLUE study for their important contributions. Cancer data were provided by the Maryland Cancer Registry, Center for Cancer Prevention and Control, Maryland Department of Health, with funding from the State of Maryland and the Maryland Cigarette Restitution Fund. The collection and availability of cancer registry data is also supported by the Cooperative Agreement NU58DP006333, funded by the Centers for Disease Control and Prevention. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention or the Department of Health and Human Services. The authors would like to thank Paul Appleby for past contributions to establishing the EHNBPCCG dataset and related coding. We additionally thank investigators from EPIC Norfolk, JHCS, and MMAS for contributing data.
Watts EL, Perez‐Cornago A, Fensom GK, et al. Circulating free testosterone and risk of aggressive prostate cancer: Prospective and Mendelian randomisation analyses in international consortia. Int J Cancer. 2022;151(7):1033‐1046. doi: 10.1002/ijc.34116
Members from the PRACTICAL Consortium, CRUK, BPC3, CAPS and PEGASUS are provided in Appendix S1.
Naomi E. Allen, Timothy J. Key and Ruth C. Travis contributed equally to our study.
Disclaimer: Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/World Health Organization.
[Correction added on 26 July 2022, after first online publication: Author name Jeffrey E. Metter has been changed to E. Jeffrey Metter.]
Funding information Cancer Research UK, Grant/Award Numbers: C18281/A29019, C60192/A28516, C8221/A19170, C8221/A29017; National Cancer Institute, Grant/Award Numbers: R01‐CA 78812, U01‐CA063673, U01‐CA167462, UM1‐CA167462; National Institutes of Health, Grant/Award Numbers: IU01AG18033, IU01CA86308, U01 CA164973; World Cancer Research Fund, Grant/Award Number: 2019/1953
DATA AVAILABILITY STATEMENT
For prospective analysis, the authors do not own the rights for the data contained in the EHNBPCCG dataset and therefore cannot redistribute the data. However, researchers can contact individual studies for access requests. UK Biobank individual‐level data are available upon request, while summary genetic data are publicly available (www.ukbiobank.ac.uk). PRACTICAL genetic data for overall prostate cancer are publicly available, while genetic subgroup data are available upon request (http://practical.icr.ac.uk/). Further details and other data that support the findings of our study are available from the corresponding author upon request.
REFERENCES
- 1. Ferlay J, Ervik M, Lam F, et al. Global Cancer Observatory: Cancer Today. Lyon, France: International Agency for Research on Cancer; 2018. [Google Scholar]
- 2. Watts EL, Appleby PN, Perez‐Cornago A, et al. Low free testosterone and prostate cancer risk: a collaborative analysis of 20 prospective studies. Eur Urol. 2018;74:585‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Watts EL, Fensom GK, Smith Byrne K, et al. Circulating insulin‐like growth factor‐I, total and free testosterone concentrations and prostate cancer risk in 200 000 men in UK Biobank. Int J Cancer. 2020; 148:2274‐2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruth KS, Day FR, Tyrrell J, et al. Using human genetics to understand the disease impacts of testosterone in men and women. Nat Med. 2020;26:252‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Travis RC, Appleby PN, Martin RM, et al. A meta‐analysis of individual participant data reveals an association between circulating levels of IGF‐I and prostate cancer risk. Cancer Res. 2016;76:2288‐2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pollak M. The insulin and insulin‐like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159‐169. [DOI] [PubMed] [Google Scholar]
- 7. Dai C, Heemers H, Sharifi N. Androgen signaling in prostate cancer. Cold Spring Harb Perspect Med. 2017;7:a030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goldman AL, Bhasin S, Wu FCW, Krishna M, Matsumoto AM, Jasuja R. A reappraisal of testosterone's binding in circulation: physiological and clinical implications. Endocr Rev. 2017;38:302‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giovannucci E, Liu Y, Platz EA, Stampfer MJ, Willett WC. Risk factors for prostate cancer incidence and progression in the health professionals follow‐up study. Int J Cancer. 2007;121:1571‐1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yarmolinsky J, Wade KH, Richmond RC, et al. Causal inference in cancer epidemiology: what is the role of Mendelian randomization? Cancer Epidemiol Biomarkers Prev. 2018;27:995‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schumacher FR, Al Olama AA, Berndt SI, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet. 2018;50:928‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu L, Wang J, Cai Q, et al. Identification of novel susceptibility loci and genes for prostate cancer risk: a transcriptome‐wide association study in over 140,000 European descendants. Cancer Res. 2019;79:3192‐3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bartsch W. Interrelationships between sex hormone‐binding globulin and testosterone, 5 alpha‐dihydrotestosterone and oestradiol‐17 beta in blood of normal men. Maturitas. 1980;2:109‐118. [DOI] [PubMed] [Google Scholar]
- 14. Vermeulen A, Verdonck L, Kaufman JM. A critical evaluation of simple methods for the estimation of free testosterone in serum. J Clin Endocrinol Metab. 1999;84:3666‐3672. [DOI] [PubMed] [Google Scholar]
- 15. Key TJ, Appleby PN, Allen NE, Reeves GK. Pooling biomarker data from different studies of disease risk, with a focus on endogenous hormones. Cancer Epidemiol Biomark Prev. 2010;19:960‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burgess S, Bowden J. Integrating summarized data from multiple genetic variants in Mendelian randomization: bias and coverage properties of inverse‐variance weighted methods. arXiv Preprint arXiv:151204486; 2015.
- 17. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two‐sample Mendelian randomization analyses using MR‐Egger regression: the role of the I 2 statistic. Int J Epidemiol. 2016;45:1961‐1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28:30‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kamat MA, Blackshaw JA, Young R, et al. PhenoScanner V2: an expanded tool for searching human genotype‐phenotype associations. Bioinformatics. 2019;35:4851‐4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Verbanck M, Chen C‐Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR‐Egger method. Eur J Epidemiol. 2017;32:377‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burgess S, Foley CN, Allara E, Staley JR, Howson JMM. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat Commun. 2020;11:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holmes MV, Richardson TG, Ference BA, Davies NM, Davey Smith G. Integrating genomics with biomarkers and therapeutic targets to invigorate cardiovascular drug development. Nat Rev Cardiol. 2021;18:435‐453. [DOI] [PubMed] [Google Scholar]
- 25. Watts EL, Goldacre R, Key TJ, Allen NE, Travis RC, Perez‐Cornago A. Hormone‐related diseases and prostate cancer: an English national record linkage study. Int J Cancer. 2020;147:803‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter's syndrome. Lancet. 2004;364:273‐283. [DOI] [PubMed] [Google Scholar]
- 27. Watts EL, Appleby PN, Albanes D, et al. Circulating sex hormones in relation to anthropometric, sociodemographic and behavioural factors in an international dataset of 12,300 men. PLoS One. 2017;12:e0187741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sutcliffe S, Colditz GA. Prostate cancer: is it time to expand the research focus to early‐life exposures? Nat Rev Cancer. 2013;13:208‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hong‐Yo K. Beyond the male sex hormone: deciphering the metabolic and vascular actions of testosterone. J Endocrinol. 2013;217:C1‐C3. [DOI] [PubMed] [Google Scholar]
- 30. Snyder PJ, Bhasin S, Cunningham GR, et al. Lessons from the testosterone trials. Endocr Rev. 2018;39:369‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Littlejohns TJ, Travis RC, Key TJ, Allen NE. Lifestyle factors and prostate‐specific antigen (PSA) testing in UK Biobank: implications for epidemiological research. Cancer Epidemiol. 2016;45:40‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tangen CM, Goodman PJ, Till C, Schenk JM, Lucia MS, Thompson IM Jr. Biases in recommendations for and acceptance of prostate biopsy significantly affect assessment of prostate cancer risk factors: results from two large randomized clinical trials. J Clin Oncol. 2016;34:4338‐4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van der Sluis TM, Meuleman EJ, van Moorselaar RJ, et al. Intraprostatic testosterone and dihydrotestosterone. Part II: concentrations after androgen hormonal manipulation in men with benign prostatic hyperplasia and prostate cancer. BJU Int. 2012;109:183‐188. [DOI] [PubMed] [Google Scholar]
- 34. Andriole GL, Bostwick DG, Brawley OW, et al. Effect of dutasteride on the risk of prostate cancer. N Engl J Med. 2010;362:1192‐1202. [DOI] [PubMed] [Google Scholar]
- 35. Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215‐224. [DOI] [PubMed] [Google Scholar]
- 36. Cohen YC, Liu KS, Heyden NL, et al. Detection bias due to the effect of finasteride on prostate volume: a modeling approach for analysis of the Prostate Cancer Prevention Trial. J Natl Cancer Inst. 2007;99:1366‐1374. [DOI] [PubMed] [Google Scholar]
- 37. Thompson IM, Chi C, Ankerst DP, et al. Effect of finasteride on the sensitivity of PSA for detecting prostate cancer. J Natl Cancer Inst. 2006;98:1128‐1133. [DOI] [PubMed] [Google Scholar]
- 38. Thompson IM, Goodman PJ, Tangen CM, et al. Long‐term survival of participants in the prostate cancer prevention trial. N Engl J Med. 2013;369:603‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lawlor DA, Tilling K, Davey SG. Triangulation in aetiological epidemiology. Int J Epidemiol. 2017;45:1866‐1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ly LP, Sartorius G, Hull L, et al. Accuracy of calculated free testosterone formulae in men. Clin Endocrinol (Oxf). 2010;73:382‐388. [DOI] [PubMed] [Google Scholar]
- 41. Moreno SA, Shyam A, Morgentaler A. Comparison of free testosterone results by analog radioimmunoassay and calculated free testosterone in an ambulatory clinical population. J Sex Med. 2010;7:1948‐1953. [DOI] [PubMed] [Google Scholar]
- 42. Kacker R, Hornstein A, Morgentaler A. Free testosterone by direct and calculated measurement versus equilibrium dialysis in a clinical population. Aging Male. 2013;16:164‐168. [DOI] [PubMed] [Google Scholar]
- 43. Wheeler MJ. The determination of bio‐available testosterone. Ann Clin Biochem. 1995;32:345‐357. [DOI] [PubMed] [Google Scholar]
- 44. Handelsman DJ. Free testosterone: pumping up the tires or ending the free ride? Endocr Rev. 2017;38:297‐301. [DOI] [PubMed] [Google Scholar]
- 45. Harman SM, Metter EJ, Tobin JD, Pearson J, Blackman MR. Longitudinal effects of aging on serum total and free testosterone levels in healthy men. J Clin Endocrinol Metabol. 2001;86:724‐731. [DOI] [PubMed] [Google Scholar]
- 46. Clarke R, Shipley M, Lewington S, et al. Underestimation of risk associations due to regression dilution in long‐term follow‐up of prospective studies. Am J Epidemiol. 1999;150:341‐353. [DOI] [PubMed] [Google Scholar]
- 47. Burgess S, Davey Smith G, Davies NM, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2019;4:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Labrecque JA, Swanson SA. Interpretation and potential biases of Mendelian randomization estimates with time‐varying exposures. Am J Epidemiol. 2019;188:231‐238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 PIs from the PRACTICAL (http://practical.icr.ac.uk/), CRUK, BPC3, CAPS, PEGASUS consortia
Appendix S2 Supporting Information
Data Availability Statement
For prospective analysis, the authors do not own the rights for the data contained in the EHNBPCCG dataset and therefore cannot redistribute the data. However, researchers can contact individual studies for access requests. UK Biobank individual‐level data are available upon request, while summary genetic data are publicly available (www.ukbiobank.ac.uk). PRACTICAL genetic data for overall prostate cancer are publicly available, while genetic subgroup data are available upon request (http://practical.icr.ac.uk/). Further details and other data that support the findings of our study are available from the corresponding author upon request.