

Abstract
Background:
Microvascular damage from large artery stiffness (LAS) in pancreatic, hepatic, and skeletal muscle may affect glucose homeostasis. Our goal was to evaluate the association between LAS and the risk of type 2 diabetes mellitus using prospectively collected, carefully phenotyped measurements of LAS as well as Mendelian randomization analyses.
Methods:
Carotid-femoral pulse wave velocity (CF-PWV) and brachial and central pulse pressure were measured in 5,676 participants of the Framingham Heart Study (FHS) without diabetes mellitus. We used Cox proportional hazards regression to evaluate the association of CF-PWV and pulse pressure with incident diabetes mellitus. We subsequently performed two-sample Mendelian randomization analyses evaluating the associations of genetically-predicted brachial pulse pressure with type 2 diabetes mellitus in the United Kingdom Biobank (UKBB).
Results:
In FHS, individuals with higher CF-PWV were older, more often male, and had higher body mass index and mean arterial pressure compared to those with lower CF-PWV. After a median follow-up of 7 years, CF-PWV and central pulse pressure were associated with an increased risk of new-onset diabetes mellitus (per standard deviation increase, multivariable-adjusted CF-PWV hazard ratio [aHR] aHR 1.36, 95% confidence interval [CI] 1.03–1.76, P=0.030; central pulse pressure aHR 1.26, 95% CI 1.08–1.48, P=0.004). In UKBB, genetically-predicted brachial pulse pressure was associated with type 2 diabetes mellitus, independent of mean arterial pressure (adjusted odds ratio 1.16, 95% CI: 1.00–1.35, P=0.049).
Conclusions:
Using prospective cohort data coupled with Mendelian randomization analyses, we found evidence supporting that greater LAS is associated with increased risk of developing diabetes. LAS may play an important role in glucose homeostasis and may serve as a useful marker of future diabetes risk.
Keywords: Large artery stiffness, pulse pressure, pulse wave velocity, diabetes mellitus
Keywords: Clinical Studies, Diabetes, Type 2, Genetics, Association Studies, Hemodynamics, Mechanisms
Graphical Abstract
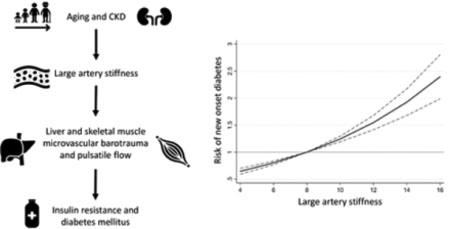
INTRODUCTION
Aortic stiffness occurs as a result of structural and functional changes in the arterial wall. As the aorta stiffens with aging and various disease states, its cushioning function is impaired, leading to increased pressure and flow pulsatility that is preferentially transmitted to the microvasculature of vascular beds that must operate at low-resistance due to high flow needs, such as the brain and the kidney. In these organs, the effects of systemic large artery stiffening (LAS) and the associated pulsatile hemodynamic abnormalities may lead to both increased pulsatile pressure (barotrauma) and pulsatile flow (with excessive shear forces from increased pulsatile flow velocity).1,2 Consistent with these principles, accumulating evidence links increased LAS with development and progression of cerebral microvascular disease and chronic kidney disease.3–7
It has long been known that diabetes mellitus (DM) and obesity are important risk factors for accelerated large artery stiffening. However, LAS may also impact the risk of DM through its hemodynamic effects.8 The endocrine pancreas, which comprises only ~1–2% of the pancreatic mass, receives ~10–20% of its blood supply. Therefore, in contrast to the exocrine pancreatic circulation, pancreatic islet blood flow is amongst the highest in the body when normalized for tissue mass.1,9,10 Like in other high-flow, low-resistance vascular beds, increased LAS has the potential to impact microvascular health in pancreatic islets, leading to dysfunctional or dysregulated endocrine function. Moreover, the liver and post-prandial skeletal muscle may also be vulnerable to barotrauma and shear forces from LAS, resulting in microvascular remodeling and impaired microvascular reactivity.11,12 The hepatic artery is a high-flow vessel that feeds a unique, low-resistance microvascular bed in which the precise regulation of arterial and portal venous flow in hepatic triads is essential to maintain the metabolic function of hepatocytes (including gluconeogenesis and glycogenolysis) and to prevent ischemic hepatic injury.11 Finally, although bulk skeletal muscle vascular resistance is higher than in other vascular beds under resting, fasting conditions,12 the transmission of arterial pulsations to the skeletal muscle microvasculature is well documented,13 and skeletal muscle resistance significantly decreases post-prandially, which is key for insulin effects on skeletal myocytes.12,14,15
We aimed to better understand the longitudinal and causal relationship between LAS and DM. We hypothesized that greater LAS (measured by carotid-femoral pulse wave velocity [CF-PWV], the reference non-invasive measure of LAS) is associated with an increased risk of new-onset DM in the Framingham Heart Study (FHS). We also utilized Mendelian randomization analyses to assess the causal relationship between LAS and DM. Specifically, we assessed the relationship of genetically-predicted brachial pulse pressure with DM risk using data from the United Kingdom Biobank (UKBB).
METHODS
Study Design and Population of FHS
In FHS, arterial tonometry was performed during seventh examination of the Framingham Offspring Cohort (1998–2001; n=3,333) and during first examination of the Third Generation Cohort (2002–2005; n=4,095). The design and baseline characteristics of both cohorts have been described previously.16–18 Participants in the Framingham Offspring Cohort were followed for a total of nine examinations, through 2014; participants in the Third Generation Cohort were followed for a total of two examinations, through 2011. Participants who had missing tonometry data (n=1,207) or who had DM on or before the visit at which tonometry was performed (n=545), were excluded from the current study.
The studies adhered to the principles outlined in the Declaration of Helsinki and were approved by the institutional review boards of all participating sites. All participants provided written informed consent.
Main Exposure in FHS
The primary analyses evaluated the association of LAS using CF-PWV with new-onset DM. Measurement of CF-PWV was described previously in detail.18 Briefly, arterial tonometry was performed after participants rested for at least 5 minutes in the supine position. CF-PWV was calculated using carotid and femoral tonometry waveforms, using the QRS complex of the ECG as a fiducial point to compute the time delay between these locations. To account for parallel transmission along the brachiocephalic artery and carotid artery and around the aortic arch, the carotid-femoral distance was estimated by subtracting the distance from the suprasternal notch to the carotid from the distance between the suprasternal notch and the femoral site. All personnel were trained and certified to perform blood pressure and CF-PWV measurements. In sensitivity analyses, LAS was also evaluated using brachial pulse pressure and central pulse pressure, which are surrogate markers of LAS.19
Brachial artery flow-mediated dilation and hyperemic flow velocity, which evaluate conduit artery endothelial function and microvascular function, are independent predictors of cardiovascular risk,18,20 were also assessed. The approach to assessing flow-mediated dilation and flow velocity in the FHS has been described previously.18,21,22 Flow velocity was determined by using doppler flow at rest and for 15 seconds after cuff deflation following five minutes of forearm cuff occlusion.18
Outcomes and Censoring Events in FHS
DM was defined as a fasting glucose ≥126 mg/dL or self-reported use of hypoglycemic medication, determined at follow-up visits or interval calls. Participants were followed beginning at the date of the first CF-PWV measurement. Participants were censored at 1) the time of development of new-onset DM, 2) death, or 3) the final date of follow-up in the cohort, whichever occurred first.
Covariates in FHS
Baseline characteristics were ascertained at the initial tonometry visit. Covariates included in the analyses were age, sex, body mass index (BMI), brachial mean arterial pressure (MAP), heart rate, fasting blood glucose, and estimated glomerular filtration rate (eGFR).
In both study samples, a routinely administered medical history questionnaire ascertained clinical and sociodemographic characteristics. At every annual visit, three seated BP measurements were obtained by trained staff following at least five minutes of quiet rest and were averaged. Brachial MAP was calculated as the diastolic blood pressure plus one third of the (systolic−diastolic) blood pressure. Anthropometric measurements were obtained using standardized protocols.
Statistical Analyses in FHS
All analyses were performed separately in the FHS and the UKBB. Cox proportional hazards modeling was performed to estimate the hazard of new-onset DM associated with a standard deviation higher value of CF-PWV (i.e., the standardized hazard ratio [sHR]). The proportional hazards assumption was assessed by evaluating graphical displays of the Schoenfeld and scaled Schoenfeld residuals.23 Cluster-robust standard errors were estimated to account for dependence within cohorts in the FHS.24 Consistent with previous studies,18,25 CF-PWV was inverted to limit heteroskedasticity and multiplied by −1000 to convert units to ms/m and address the directionality of the associated LAS. Covariates were selected a priori for inclusion in the models based on clinical relevance and known associations between LAS and DM (see Covariates section, above).1,12 Continuous variables were assessed for a non-linear association with the outcome using splines. We assessed for effect modification of the relationship between CF-PWV and new-onset DM by age, sex, and prediabetes (defined as fasting blood glucose 100–125 mg/dL26) using the likelihood ratio test.
In sensitivity analyses, we evaluated the association of flow velocity with new-onset DM and if flow velocity is a mediator of the association between CF-PWV and new-onset DM. Additional sensitivity analyses used generalized estimating equations to estimate the association of baseline CF-PWV with new-onset DM accounting for dependence within families across cohorts (i.e., Offspring Cohort and Third Generation Cohort). All analyses were performed using Stata version 16.1 (StataCorp, College Station, TX).
Mendelian Randomization in UKBB
The UKBB is a prospective, population-based cohort study of United Kingdom residents that was established to evaluate biologic risk factors for several chronic diseases.27 We utilized UKBB data to perform two-sample Mendelian randomization analyses. Mendelian randomization is an observational data technique that assesses whether genetic predictors of a given exposure are associated with an outcome. The independent segregation of alleles at conception means that genetically-defined subgroups of the population should not differ systematically with respect to confounding variables, creating a natural experiment analogous to a randomized trial. Therefore, compared with conventional observational analyses, Mendelian randomization analyses provide more reliable insights into causal relationships between risk factors and disease outcomes.28
Univariable Mendelian randomization analyses were performed to assess the associations of genetically-predicted MAP and genetically-predicted brachial pulse pressure with type 2 DM risk in separate models, and a multivariable Mendelian randomization analysis was performed considering the associations of genetically-predicted MAP and genetically-predicted brachial pulse pressure with type 2 DM in a single multivariable model. The rationale for the multivariable analysis was to determine whether any link with the outcome is general to all blood pressure measurements, or whether there is a specific effect of brachial pulse pressure after conditioning on MAP. Finally, since there may be a bidirectional relationship between LAS and DM, we performed bidirectional Mendelian randomization analyses to further assess the causal relationship between brachial pulse pressure and DM.
As genetic instruments for blood pressure traits, 256 uncorrelated variants were selected that were previously associated with blood pressure at a genome-wide level of significance in the International Consortium for Blood Pressure (ICBP) dataset (not including UKBB participants). This choice was made to minimize overlap between the two genetic association datasets, as such overlap can lead to winner’s curse and weak instrument bias.29 Genetic associations with type 2 DM were obtained in up to 898,130 individuals (74,124 type 2 DM cases and 824,006 controls) of European ancestries from a large genome-wide association study that includes UKBB participants.30 Associations published by the DIAGRAM (DIAbetes Genetics Replication And Meta-analysis) consortium that are adjusted for principal components were corrected for inflation by genomic control, but not adjusted for BMI as this can induce collider bias. Genetic associations with blood pressure were obtained in 299,024 European ancestry participants from the ICBP excluding UKBB participants.31
To investigate a possible a bi-directional Mendelian randomization association between pulse pressure and type 2 DM, models were performed addressing genetic liability to type 2 DM as the exposure and brachial pulse pressure as the outcome. Genetic variants selected as instrumental variables for liability to type 2 DM were uncorrelated (r2 <0.001) single-nucleotide polymorphisms associated with type 2 DM at genome-wide significance (p <5×10−8) in a genome-wide association study of 74,124 cases and 824,006 controls, all of European ancestry, as mentioned above.30 Genetic association estimates for brachial pulse pressure were obtained from a genome-wide association study of 318,417 White British UKBB participants.32 The random-effects inverse-variance weighted (IVW) Mendelian randomization method was used for the main analysis; sensitivity analyses were conducted using methods that are more robust to the inclusion of pleiotropic variants and violations of the instrumental variable assumptions: weighted median, MR-PRESSO (Mendelian Randomization Pleiotropy RESidual Sum and Outlier), and MR-LASSO (Mendelian Randomization Least Absolute Shrinkage and Selection Operator).33 Additionally, we searched for specific pleiotropic effects in PhenoScanner, a searchable database of genetic associations.34
RESULTS
FHS Cohort Characteristics
In FHS, 5,676 participants met inclusion criteria for the study. FHS participants had a median CF-PWV of 7.3 (interquartile range [IQR] 6.3–8.7) m/s (Table 1, Figure 1), median age of 46 (IQR 37–55) years at the time of tonometry, median BMI of 26 (IQR 23–29) kg/m2, median MAP of 89 mmHg, and 46% were male. Compared to the lowest tertile of CF-PWV (median CF-PWV 6.0 m/s, IQR 5.6–6.3), participants in the highest tertile of CF-PWV (median CF-PWV 9.5 m/s, IQR 8.7–11.2) were older, more often men, and had larger BMI, higher MAP, and lower eGFR (Table S1). FHS participants without baseline DM who underwent CF-PWV testing were younger and had lower BMI, waist circumference, and MAP and lower eGFR compared with those participants who did not undergo CF-PWV testing (Table S2).
Table 1.
Characteristics of participants of the FHS without DM at the time of CF-PWV testing*
| FHS | |
|---|---|
| N | 5,676 |
| PWV, m/s | 7.3 (6.3–8.7) |
| Age, years | 46 (37–55) |
| Male sex, n (%) | 2,615 (46%) |
| Current smoker, n (%) | 846 (15%) |
| Family history of DM, n (%) | 425 (7%) |
| BMI, kg/m2 | 26 (23–29) |
| Waist circumference, in | 37 (33–40) |
| Fasting blood glucose, mg/dl | 94 (88–100) |
| Total cholesterol, mg/dl | 190 (168–215) |
| Calculated LDL, mg/dl | 113 (92–135) |
| HDL, mg/dl | 53 (43–65) |
| Triglycerides, mg/dl | 109 (77–158) |
| eGFR, ml/min/1.73m2 | 99 (87–109) |
| High-sensitivity c-reactive protein, mg/L | 1.3 (0.6–3.2) |
| SBP, mmHg | 117 (108–128) |
| DBP, mmHg | 75 (68–81) |
| MAP, mmHg | 89 (82–96) |
| Brachial PP, mmHg | 42 (36–50) |
| Central PP, mmHg | 48 (41–57) |
| Treated hypertension, n (%) | 823 (15%) |
| Number of antihypertensive medications, n (%) | |
| 1 | 586 (10%) |
| 2 | 188 (3%) |
| 3 | 38 (1%) |
| ≥4 | 11 (<1%) |
| Antihypertensive class | |
| Renin-angiotensin system blocker, n (%) | 327 (6%) |
| Thiazide diuretic, n (%) | 146 (3%) |
| Calcium channel blocker, n (%) | 178 (3%) |
| Beta blocker, n (%) | 315 (6%) |
Continuous variables are reported as median (IQR) and binary and categorical variables are reported as number (proportion)
Abbreviations: BMI = body mass index; CF-PWV = carotid-femoral pulse wave velocity; DBP = diastolic blood pressure; DM = diabetes mellitus; eGFR = estimated glomerular filtration rate (calculated using the Chronic Kidney Disease Epidemiology Collaboration equation); FHS = Framingham Heart Study; HDL = high density lipoprotein; LDL = low density lipoprotein; MAP = mean arterial pressure; PP = pulse pressure; SBP = systolic blood pressure
Figure 1. Distribution of CF-PWV in the FHS.
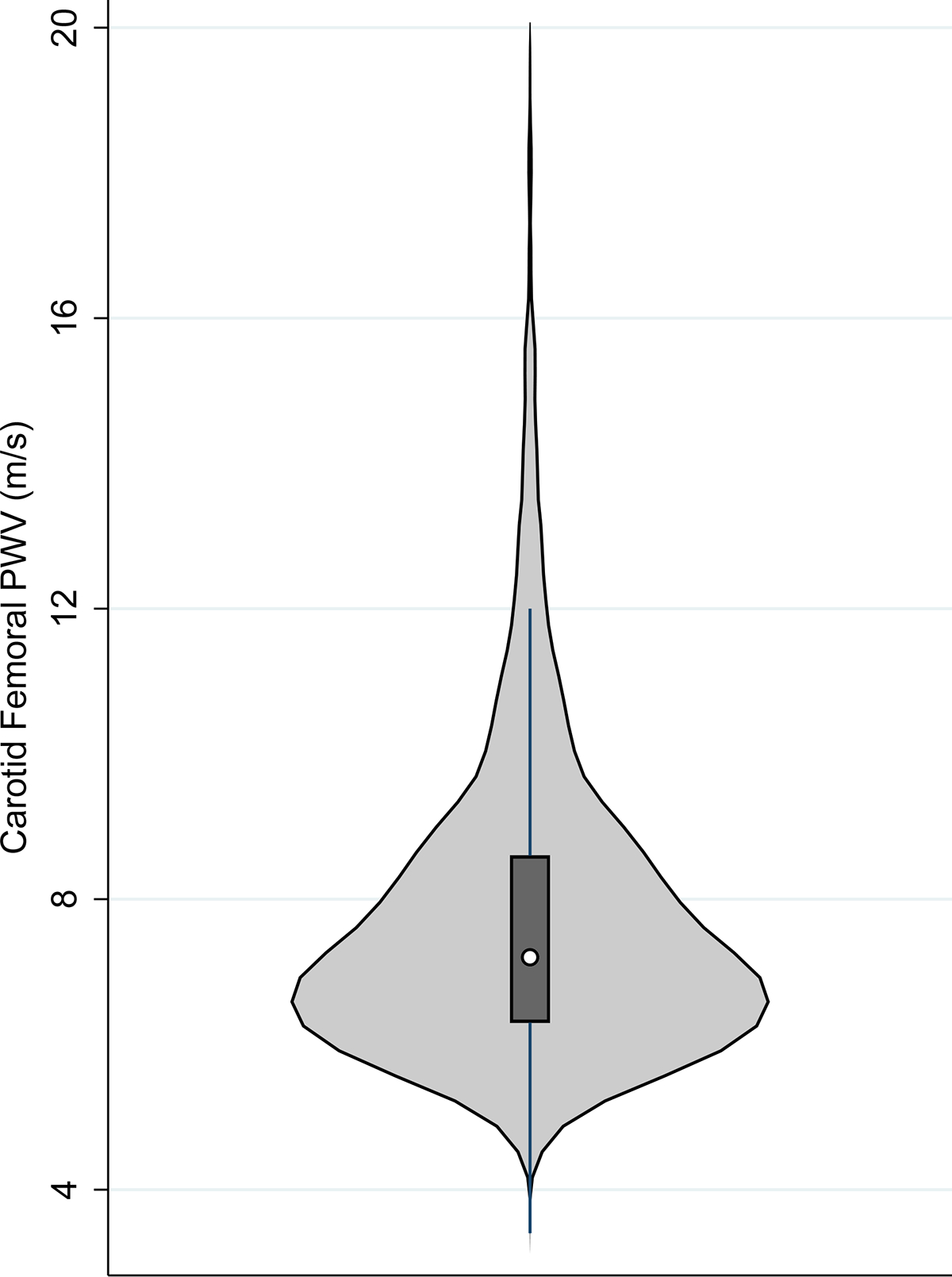
Violin plots depict the frequency distribution (outer histogram), median (white circle), interquartile range (box), and 1.5 multiple of the interquartile range (solid lines) of CF-PWV.
CF-PWV and Risk of Incident DM in FHS
In the FHS, there were 200 cases of incident DM during a median duration of follow-up after arterial tonometry of 7 years. In unadjusted Cox proportional hazards analyses, a standard deviation higher CF-PWV was associated with a 56% greater risk of new-onset DM (sHR 1.56, 95% confidence interval [CI] 1.35–1.80; P<0.001; Table 2, Figure 2). The association of higher CF-PWV with incident DM persisted in various pre-specified multivariable models, including a model that adjusted for age, sex, BMI, MAP, heart rate, and eGFR (sHR 1.47, 95% CI 1.12–1.93; P=0.006) and a model that also adjusted for smoking, total cholesterol, high density lipoprotein, thiazide diuretic, beta-blocker, total antihypertensive medications, C-reactive protein, and family history of DM (sHR 1.36, 95% CI 1.03–1.76; P=0.030). There was no effect modification by age, sex, or prediabetes status with regard to the relationship between CF-PWV and risk of DM.
Table 2.
Risk of new-onset DM by carotid-femoral CF-PWV in the FHS
| Unadjusted | Model 1* | Model 2† | Model 3‡ | Model 4§ | |
|---|---|---|---|---|---|
| sHR (95% CI, P-value) | sHR (95% CI, P-value) | sHR (95% CI, P-value) | sHR (95% CI, P-value) | sHR (95% CI, P-value) | |
| CF-PWV | 1.56 (1.35–1.80, P=1.1×10−9) | 1.78 (1.41–2.23, P=7.4×10−7) | 1.56 (1.19–2.03, P=0.001) | 1.47 (1.12–1.93, P=0.006) | 1.36 (1.03–1.76, P=0.030) |
| Brachial pulse pressure | 1.24 (1.12–1.38, P=3.7×10−5) | 1.28 (1.11–1.47, P=0.001) | 1.17 (0.98–1.39, P=0.082) | 1.15 (0.97–1.38, P=0.108) | 1.18 (0.98–1.41, P=0.075) |
| Central pulse pressure | 1.31 (1.22–1.41, P=1.6×10−13) | 1.37 (1.19–1.57, P=1.3×10−5) | 1.31 (1.12–1.52, P=0.001) | 1.28 (1.10–1.50, P=0.001) | 1.26 (1.08–1.48, P=0.004) |
Abbreviations: CF-PWV = carotid-femoral pulse wave velocity; CI = confidence interval; FHS = Framingham Heart Study; sHR = standardized hazard ratio (calculated using Cox regression, determined for each standard deviation increase in CF-PWV)
Model 1: Adjusted for age, sex, and BMI
Model 2: Adjusted for Model 1 + MAP + HR
Model 3: Adjusted for Model 2 + eGFR
Model 4: Adjusted for Model 3 + smoking + total cholesterol + high density lipoprotein + thiazide diuretic + beta-blocker + total antihypertensive medications + c-reactive protein + family history of DM
Figure 2. Association of CF-PWV and new-onset DM in the FHS.
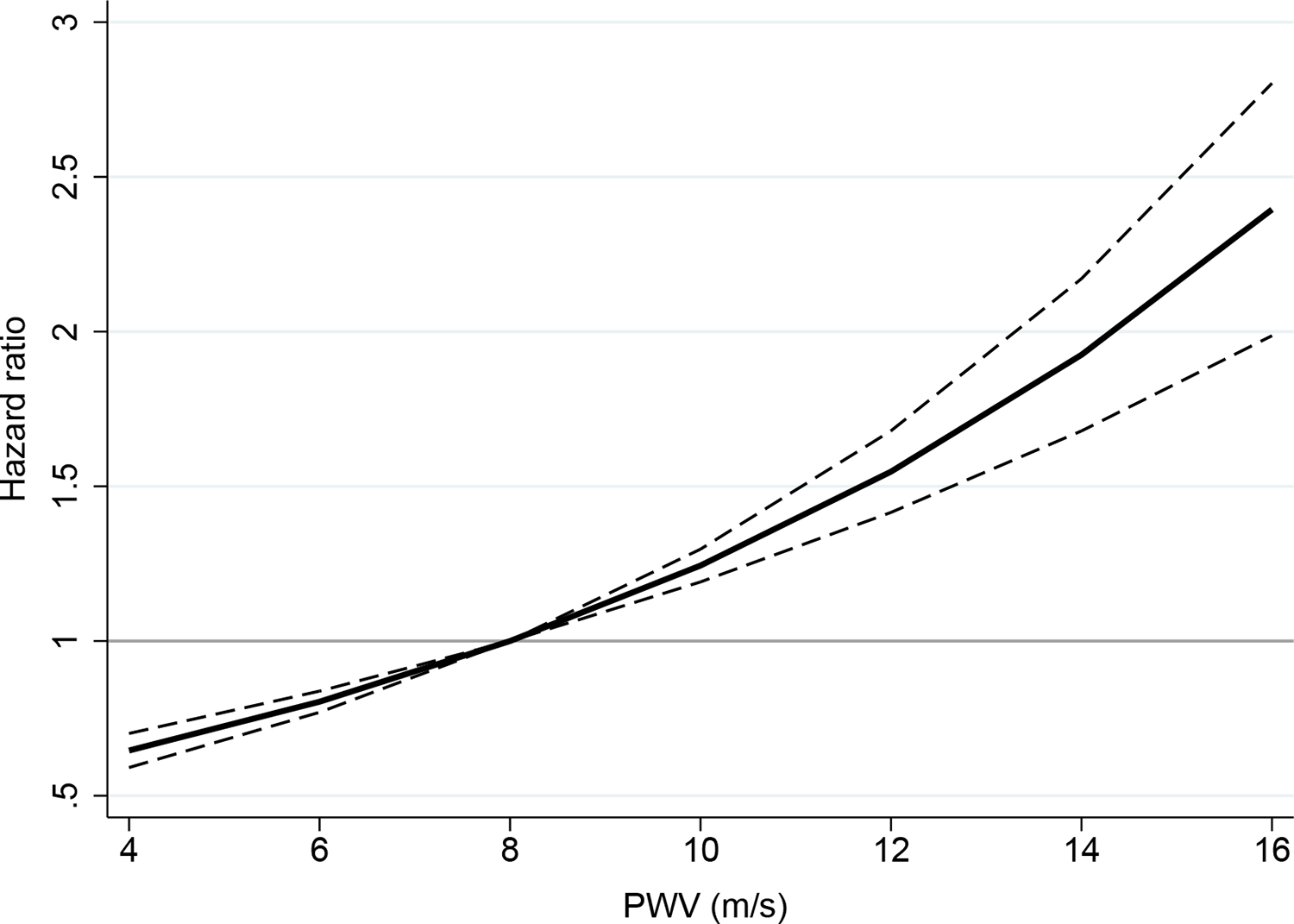
Estimates represent the hazard ratio (solid lines) and 95% confidence intervals of the hazard ratios (dashes) at each value of CF-PWV using Cox regression.
Sensitivity Analyses in FHS
In analyses adjusted for age, sex, BMI, MAP, heart rate, eGFR, smoking, total cholesterol, high density lipoprotein, thiazide diuretic, beta-blocker, total antihypertensive medications, C-reactive protein, and family history of DM, the relationship of central pulse pressure with incident DM was similar to that of CF-PWV (sHR 1.26, 95% CI 1.08–1.48; P=0.004; Table 2). In contrast, the relationship of brachial pulse pressure with incident DM was similar in analyses adjusted for age, sex, and BMI (sHR 1.28, 95% CI 1.11–1.47; P=0.001), but was attenuated after adjustment for MAP and heart rate.
In mediation analyses, fasting blood glucose was a substantial mediator of the association between CF-PWV and new-onset DM (55% mediation, 95% CI 45%−68%, Table S3), and hyperemic forearm flow velocity was a modest mediator of the association between CF-PWV and new-onset DM (9% mediation, 95% CI 7–11%). Sensitivity analyses using generalized estimating equations to evaluate the odds of new-onset DM, accounting for clustering within families across cohorts, demonstrated similar results to the primary analyses (adjusted standardized odds ratio [OR] 1.41, 95% CI 1.10–1.81; Table S4).
Bi-directional Mendelian Randomization Analysis of Brachial Pulse Pressure and Type 2 DM in the UKBB
Estimates from Mendelian randomization are presented in Figure 3 and represent the OR for type 2 DM per 5 mmHg increase in genetically-predicted levels of brachial MAP and pulse pressure, respectively. Genetically-predicted brachial MAP and pulse pressure were positively associated with type 2 DM in univariable analyses (MAP OR 1.11, 95% CI 1.03–1.21; P=0.01; pulse pressure OR 1.19, 95% CI 1.07–1.32; P=0.001). In the multivariable analysis, genetically-predicted brachial pulse pressure remained associated with type 2 DM risk (OR 1.16, 95% CI: 1.00, 1.35; P=0.049), whereas the association for genetically-predicted MAP substantially attenuated (OR 1.03, 95% CI 0.92–1.15; P=0.63).
Figure 3. Associations of genetically-predicted blood pressure measures and type 2 DM risk in univariable and multivariable Mendelian randomization analyses.
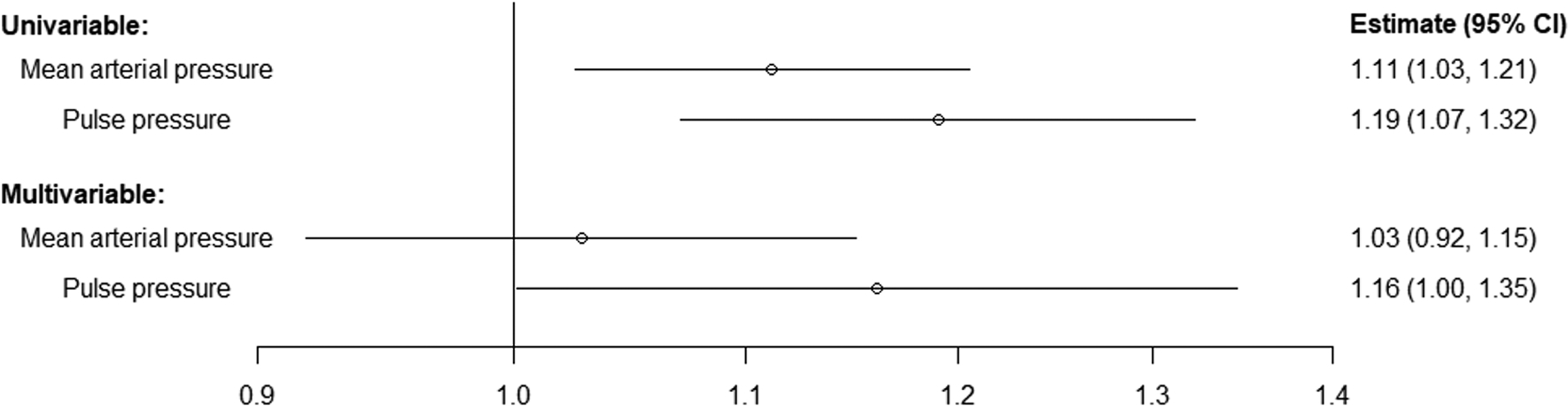
Estimates (95% confidence intervals [CI]) represent the odds ratio for disease per 5 mmHg increase in genetically-predicted levels of the blood pressure trait.
Results from the sensitivity analyses using robust Mendelian randomization methods are presented in Table S5. Univariable estimates from all methods were clearly positive for both brachial MAP and pulse pressure. However, while multivariable estimates were generally positive, it was unclear which was the lead risk factor: compared with results from the IVW method, in the weighted median, MR-PRESSO and MR-LASSO methods, the estimate for brachial pulse pressure attenuated whereas the estimate for MAP increased. A search of PhenoScanner revealed that around three-quarters of genetic variants were associated with a non-BP trait at a genome-wide level of statistical significance (p<5×10−8). While it is possible that some of these associations reflect downstream effects of BP rather than pleiotropic effects, this lack of specificity reflects a limitation of the analysis.
IVW analysis demonstrated that every 1-log OR increase in genetic liability to type 2 DM was associated with 0.598 standard deviation units higher brachial pulse pressure (95% CI 0.413–0.783; P=1×10−10). Similar results were observed in the weighted median sensitivity analysis (0.477, 95% CI 0.233–0.721; P=1×10−4).
DISCUSSION
We studied the relationship between LAS and the risk of DM in two large cohorts. First, in the FHS, we demonstrate that greater LAS (quantified via CF-PWV measurements) is associated with an elevated risk of developing future DM. This association of LAS with incident DM persisted after adjustments for several risk factors for DM, including age, BMI, and family history of DM. Second, we demonstrate that genetically-predicted brachial pulse pressure is associated with increased risk of DM in the UKBB, supporting a causal relationship for this association. Moreover, the effect of genetically-predicted brachial pulse pressure on the risk of DM is independent of MAP, and thus is not simply attributable to higher mean blood pressure. Furthermore, our Mendelian randomization analyses indicate a bidirectional relationship or shared etiology between LAS and type 2 DM, consistent with various hemodynamic and biologic considerations1,8,35,36 as well as with our mediation analyses in the FHS. To our knowledge, this is the largest study to systematically evaluate the association of LAS with DM, and the first to assess its causal association using Mendelian randomization.
Several prior studies have demonstrated a cross-sectional relationship between fasting glucose and insulin resistance with increased LAS in humans.35 In various populations, diabetic individuals have been shown to exhibit higher carotid-femoral PWV than their non-diabetic counterparts.2,37–41 Although the presence of increased LAS in diabetics has long been known, the concept of LAS contributing to the onset of DM is much more recent. Three previous studies have evaluated the relationship of LAS with new-onset DM, with results consistent with our findings in FHS. In 2,450 Swedish participants of the Malmo Diet and Cancer Cardiovascular cohort study, Muhammad et al. demonstrated that participants in the highest tertile of CF-PWV (median CF-PWV 12.3 m/s, IQR 11.5–13.9) had a 3-fold higher risk of developing DM compared to participants in the lowest tertile (median CF-PWV 8.2 m/s).42 In contrast to our study, the participants were markedly older (mean age 72 years) and had substantially higher baseline CF-PWV (median 10 m/s). DM status was determined based on linkage to DM registries, rather than systematic assessment, potentially yielding delayed diagnosis and misclassification of DM. In a post hoc analysis of 2,429 participants of the China Stroke Primary Prevention Trial, a randomized trial comparing enalapril to enalapril plus folic acid for the treatment of hypertension, Zhang et al. reported a 33% increased odds (OR 1.33, 95% 1.13–1.56) of new-onset DM for every standard deviation increase in brachial-ankle CF-PWV.43 Participants had poorly controlled hypertension (mean baseline systolic blood pressure was 167 mmHg), which can significantly impact CF-PWV measurements and potentially limit the generalizability of the study population. Nonetheless, our results reinforced the findings reported by Zhang et al. in a larger, prospective cohort. Additionally, in an analysis of 8,956 participants of the Kailuan Study who underwent repeated measures of brachial-ankle PWV and fasting blood glucose over 3.72 years of follow-up, Zheng et al. observed that elevated LAS was associated with a 2.11-fold (95% CI 1.71–2.61) higher risk of DM, similar to the current study. However, the authors found that a bidirectional relationship between LAS and fasting blood glucose was not present (β=0.00, 95% CI −0.02–0.02).44 Of note, these previous studies were limited by shorter duration of follow-up than FHS, and were not able to explore the association of microvascular function (performed in our analyses of the FHS). More importantly, to our knowledge, the directionality and causality of these associations have not been previously studied with Mendelian randomization, a powerful tool to disentangle causal relationships between phenotypes of interest. Our various Mendelian randomization analyses indeed indicate that: (1) Genetically-predicted brachial pulse pressure (a metric of LAS, although less robust than CF-PWV) is associated with the risk of DM, supporting a causal relationship; (2) This is not simply dependent on genetically-predicted MAP (being specific to pressure pulsatility and not high mean blood pressure per se); (3) The causal relationship appears to be bidirectional, such that LAS can causally contribute to DM and vice-versa, or represents a shared etiology. The primary finding of an association between genetically-predicted brachial pulse pressure and DM risk had greater statistical evidence than the multivariable analysis, which suggests that this link was driven by pulse pressure rather than MAP. However, there are some limitations to the Mendelian randomization analysis. Estimates from some robust methods provided stronger evidence for MAP being the primary risk factor rather than brachial pulse pressure. Additionally, many of the genetic variants were associated with traits other than BP. Nonetheless, estimates were positive in all robust methods, suggesting that evidence for bidirectional causal effects was not overly sensitive to these potentially pleiotropic effects.
Several potential mechanisms can explain a causal relationship between LAS and the risk of new-onset DM. LAS may contribute to the pathogenesis of DM through its hemodynamic effects. The endocrine pancreas, which comprises only ~1–2% of the pancreatic mass, receives ~10–20% of its blood supply. Therefore, in contrast to the exocrine pancreatic circulation, pancreatic islet blood flow is amongst the highest in the body when normalized for tissue mass.1,9 Like in other high-flow, low-resistance vascular beds, increased LAS has the potential to impact microvascular health in pancreatic islets, leading to dysfunctional or dysregulated endocrine function. Pancreatic islets exhibit a dense glomerular capillary network, which mediates chemical signaling between various endocrine cell types. Vascular integrity may be essential to maintain an order of sequential perfusion of β, δ and α cells, which appears to allow for an adequate inhibition of glucagon secretion by insulin.45 Moreover, hyperglycemia and the associated beta cell activation result in islet arteriolar dilation, capillary dilation and increased islet capillary pressure, suggesting that local hemodynamics participate in endocrine responses46 Therefore, pancreatic islet microvascular dysfunction may contribute to the pathogenesis of type 2 DM via impaired insulin secretion and/or an imbalance between insulin/glucagon secretion. In addition to hemodynamic phenomena in the pancreas, the liver and skeletal muscle microvasculature play important roles in metabolic homeostasis. The liver receives ~25% of the cardiac output and exhibits a high-flow, low-resistance arterial circulation, since the hepatic artery provides approximately one-third of the liver blood flow.1 The hepatic arterial circulation constantly regulates arterial flow to maintain total liver blood flow constant, regardless of changes in portal vein flow (hepatic arterial buffer response), which contributes to metabolic homeostasis.11 Finally, although skeletal muscle exhibits higher resistance at rest and fasting conditions compared to various target organs, skeletal muscle blood flow is highly dynamic, alternating between individual microvascular units (which at a given time may exhibit low local resistance), and increases substantially with muscle activity and in post-prandial states.47 Furthermore, post-prandial vasodilation mediates the effects of insulin on skeletal myocytes,14 and impaired vasodilation has been linked with insulin resistance in animal studies.12,14,15
The above considerations suggest that the ill effects of excess pulsatility on the peripheral microvasculature could also extend to key metabolic organs. Indeed, there is now substantial evidence that normal microvascular function is essential for insulin-mediated glucose disposal in experimental models and humans.48,49 In the FHS, we were able to evaluate the association of LAS with new-onset DM after accounting for flow velocity, a marker of forearm microvascular function.20 We observed that flow velocity was not independently associated with risk of new-onset DM, but was a mediator of a small proportion of (5%) of the relationship of CF-PWV with new-onset DM. The relationship between LAS and incident DM may depend predominantly on effects on the pancreas and liver microvasculature or may depend on skeletal muscle microvasculature dysfunction over time, which may not be captured adequately by post-occlusion flow velocity measurements in the fasting state long before the onset of DM. In particular, high-order resistance vessels which regulate flow to feeding arterioles appear to be more selectively recruited in the post-prandial state by insulin, and to be the most important for mediating the metabolic effects of insulin on skeletal muscle.49
Our study has several strengths. We were able to examine data from over 5,000 participants of a prospective cohort study (FHS), with several years of robust clinical and laboratory information available, as well as CF-PWV measurements, considered the reference non-invasive metric of LAS.1,2 Measurements were performed systematically by trained, experienced investigators, minimizing the potential for misclassification. There were also several limitations. Consistent measurements of glycosylated hemoglobin levels and oral glucose tolerance testing were not available in FHS, precluding the use of these values in the definition of DM and prediabetes. Nonetheless, fasting blood glucose levels and detailed medication lists were obtained at every study visit to confirm the clinical diagnosis of DM. Another limitation of our study is the use of brachial pulse pressure, rather than CF-PWV, in the UKBB since this study did not include CF-PWV measurements. Whereas CF-PWV is a more direct measurement of arterial stiffness, an increased pulse pressure is also a known consequence of arterial stiffness. We note however, that the relationship between brachial pulse pressure and arterial stiffness can be confounded by various factors such as flow rate/stroke volume and pulse pressure amplification. However, our analyses in the UKBB were confirmatory, given the relationship between LAS and DM found in the FHS and previously studied cohorts.42–44 Future additional studies using more direct measurements of LAS in the UKBB and other cohorts are encouraged. Additionally, our primary analyses evaluated the relationship between CF-PWV and new-onset DM in FHS, though several secondary and confirmatory analyses were performed that were not adjusted for multiple comparisons, which may affect the interpretation of the statistical significance of the findings. A further limitation is that the assumptions underlying the MR analysis are not verifiable. Specifically, it is not possible to exclude the possibility that the genetic variants used to predict levels of the exposure under consideration are affecting the outcome through pathways unrelated to the exposure. Such violation of the requisite MR assumptions could potentially bias estimates.
In conclusion, given the growing population-wide burden of type 2 DM worldwide,50 it is critical to better understand the pathogenesis of DM to facilitate early identification of those individuals at risk of developing it, and to develop interventions to reduce the incidence of DM. Our findings demonstrate that increased LAS precedes and predicts the onset of DM, and our Mendelian randomization analyses support a potential causal association. The results provide insight into the potential mechanistic importance of vascular structure and function in the development of insulin resistance and DM. Furthermore, CF-PWV may serve as a useful marker of DM risk. Future studies are needed to better understand the potential role of microvascular dysfunction as a mediator of insulin resistance due to LAS.
Data Availability.
Statistical code for the FHS analyses will be made available upon reasonable request to the corresponding author. Because of the protected nature of the data collected for this study, requests to access the FHS dataset may be sent to the National Institutes of Health Biologic Specimen and Data Repository Information Coordinating Center at https://biolincc.nhlbi.nih.gov/requests/data-request/form/ and requests to access the UKBB dataset may be sent to the UKBB at https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is Known?
Experimental data suggest that elevated LAS plays an important mechanistic role in the development of insulin resistance
What New Information Does this Article Contribute?
In 5,676 participants of the FHS who did not have DM at baseline, we observed that higher CF-PWV was associated with a higher risk of new-onset DM
In Mendelian randomization analyses using data from the UKBB, we found that genetically-predicted pulse pressure is associated with increased risk of DM, independent of MAP
The observed genetic association of LAS and DM supports a causal relationship
Summary
In this study evaluating the temporal relationship of CF-PWV and type 2 DM in the FHS and the genetic relationship of pulse pressure and DM in the UKBB, we observed that LAS precedes and predicts risk of developing new-onset DM and may play a causal role in the risk of DM. Accordingly, LAS may serve as a useful marker of future DM risk.
ACKNOWLEDGEMENTS
We acknowledge the dedication of the FHS and UKBB participants without whom this research would not be possible.
SOURCES OF FUNDING
Dr. Cohen is supported by NIH grants K23-HL133843, R01-HL153646, R01-HL157108, R01-AG074989, U01-HL160277, U01-TR003734, R01-DK123104, U24-DK060990, and an American Heart Association Bugher Award. Dr. Chirinos is supported by NIH grants R01-HL121510, U01-TR003734, U01-TR003734–01S1, UO1-HL-160277, R33-HL-146390, R01-HL153646, K24-AG070459, R01-AG058969, R01-HL104106, P01-HL094307, R03-HL146874, R56-HL136730, R01 HL155599, R01HL157264, R01HL155 and 1R01HL153646. The FHS acknowledges the support of contracts NO1-HC-25195, HHSN268201500001I and 75N92019D00031 from the National Heart, Lung and Blood Institute and grants HL076784, AG028321, HL070100, HL060040, HL080124, HL071039, HL077447, HL107385 R01HL142983, R01HL126136, R01 HL 080124 for this research. Dr. Vasan is supported in part by the Evans Medical Foundation and the Jay and Louis Coffman Endowment from the Department of Medicine, Boston University School of Medicine. Dr. Gill is supported by the British Heart Foundation Centre of Research Excellence (RE/18/4/34215) at Imperial College London and a National Institute for Health Research Clinical Lectureship at St. George’s, University of London (CL–2020–16–001). Dr Burgess is supported by the United Kingdom Research and Innovation Medical Research Council (MC_UU_00002/7) and the National Institute for Health Research Cambridge Biomedical Research Centre (BRC–1215–20014). The views expressed are those of the authors and not necessarily those of the National Institute for Health Research or the Department of Health and Social Care.
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- BMI
Body mass index
- CF-PWV
Carotid-femoral pulse wave velocity
- CI
Confidence interval
- DM
Diabetes mellitus
- DIAGRAM
DIAbetes Genetics Replication And Meta-analysis
- eGFR
Estimated glomerular filtration rate
- FHS
Framingham Heart Study
- ICBP
International Consortium for Blood Pressure
- IVW
Inverse-variance weighted
- LAS
Large artery stiffness
- MAP
Mean arterial pressure
- OR
Odds ratio
- sHR
Standardized hazard ratio
- UKBB
United Kingdom Biobank
Footnotes
DISCLOSURES
JAC has recently consulted for Bayer, Sanifit, Fukuda-Denshi, Bristol-Myers Squibb, JNJ, Edwards Life Sciences, Merck, NGM Biopharmaceuticals and the Galway-Mayo Institute of Technology. He received University of Pennsylvania research grants from National Institutes of Health, Fukuda-Denshi, Bristol-Myers Squibb, Microsoft and Abbott. He is named as inventor in a University of Pennsylvania patent for the use of inorganic nitrates/nitrites for the treatment of Heart Failure and Preserved Ejection Fraction and for the use of biomarkers in heart failure with preserved ejection fraction. He has received payments for editorial roles from the American Heart Association, the American College of Cardiology and Wiley. He has received research device loans from Atcor Medical, Fukuda-Denshi, Unex, Uscom, NDD Medical Technologies, Microsoft and MicroVision Medical. GFM is the owner of Cardiovascular Engineering, Inc., a small business that designs and develops devices that are used to measure arterial stiffness and has received consulting fees or grants from Novartis, Merck, Bayer and Servier. DG is employed part-time by Novo Nordisk. The remaining authors have nothing to disclose.
Online Tables S1-S5
REFERENCES
- 1.Chirinos JA, Segers P, Hughes T, Townsend R. Large-Artery Stiffness in Health and Disease: JACC State-of-the-Art Review. Journal of the American College of Cardiology. 2019;74:1237–1263. doi: 10.1016/j.jacc.2019.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Townsend RR, Wilkinson IB, Schiffrin EL, Avolio AP, Chirinos JA, Cockcroft JR, Heffernan KS, Lakatta EG, McEniery CM, Mitchell GF, et al. Recommendations for Improving and Standardizing Vascular Research on Arterial Stiffness: A Scientific Statement From the American Heart Association. Hypertension. 2015;66:698–722. doi: 10.1161/HYP.0000000000000033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Townsend RR, Anderson AH, Chirinos JA, Feldman HI, Grunwald JE, Nessel L, Roy J, Weir MR, Wright JT, Bansal N Jr., et al. Association of Pulse Wave Velocity With Chronic Kidney Disease Progression and Mortality: Findings From the CRIC Study (Chronic Renal Insufficiency Cohort). Hypertension. 2018;71:1101–1107. doi: 10.1161/HYPERTENSIONAHA.117.10648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension. 2005;46:200–204. doi: 10.1161/01.HYP.0000168052.00426.65 [DOI] [PubMed] [Google Scholar]
- 5.Nichols WW ORM, Vlachopoulos C. McDonald’s Blood Flow in Arteries: Theoretical, Experimental and Clinical Principles, 6th Edition. Hodder Arnold. 2011. [Google Scholar]
- 6.Mitchell GF, van Buchem MA, Sigurdsson S, Gotal JD, Jonsdottir MK, Kjartansson O, Garcia M, Aspelund T, Harris TB, Gudnason V, et al. Arterial stiffness, pressure and flow pulsatility and brain structure and function: the Age, Gene/Environment Susceptibility--Reykjavik study. Brain. 2011;134:3398–3407. doi: 10.1093/brain/awr253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodard T, Sigurdsson S, Gotal JD, Torjesen AA, Inker LA, Aspelund T, Eiriksdottir G, Gudnason V, Harris TB, Launer LJ, et al. Mediation analysis of aortic stiffness and renal microvascular function. J Am Soc Nephrol. 2015;26:1181–1187. doi: 10.1681/ASN.2014050450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chirinos JA. Large Artery Stiffness and New-Onset Diabetes. Circ Res. 2020;127:1499–1501. doi: 10.1161/CIRCRESAHA.120.318317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsushima Y, Miyazaki M, Taketomi-Takahashi A, Endo K. Feasibility of measuring human pancreatic perfusion in vivo using imaging techniques. Pancreas. 2011;40:747–752. doi: 10.1097/MPA.0b013e318215ac22 [DOI] [PubMed] [Google Scholar]
- 10.Climie RE, van Sloten TT, Bruno RM, Taddei S, Empana JP, Stehouwer CDA, Sharman JE, Boutouyrie P, Laurent S. Macrovasculature and Microvasculature at the Crossroads Between Type 2 Diabetes Mellitus and Hypertension. Hypertension. 2019;73:1138–1149. doi: 10.1161/HYPERTENSIONAHA.118.11769 [DOI] [PubMed] [Google Scholar]
- 11.Eipel C, Abshagen K, Vollmar B. Regulation of hepatic blood flow: the hepatic arterial buffer response revisited. World J Gastroenterol. 2010;16:6046–6057. doi: 10.3748/wjg.v16.i48.6046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serne EH, de Jongh RT, Eringa EC, RG IJ, Stehouwer CD. Microvascular dysfunction: a potential pathophysiological role in the metabolic syndrome. Hypertension. 2007;50:204–211. doi: 10.1161/HYPERTENSIONAHA.107.089680 [DOI] [PubMed] [Google Scholar]
- 13.Lee JJ, Tyml K, Menkis AH, Novick RJ, McKenzie FN. Evaluation of pulsatile and nonpulsatile flow in capillaries of goat skeletal muscle using intravital microscopy. Microvasc Res. 1994;48:316–327. doi: 10.1006/mvre.1994.1058 [DOI] [PubMed] [Google Scholar]
- 14.Emanuel AL, Meijer RI, Muskiet MH, van Raalte DH, Eringa EC, Serne EH. Role of Insulin-Stimulated Adipose Tissue Perfusion in the Development of Whole-Body Insulin Resistance. Arterioscler Thromb Vasc Biol. 2017;37:411–418. doi: 10.1161/ATVBAHA.116.308670 [DOI] [PubMed] [Google Scholar]
- 15.Potenza MA, Marasciulo FL, Chieppa DM, Brigiani GS, Formoso G, Quon MJ, Montagnani M. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am J Physiol Heart Circ Physiol. 2005;289:H813–822. doi: 10.1152/ajpheart.00092.2005 [DOI] [PubMed] [Google Scholar]
- 16.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. American journal of epidemiology. 1979;110:281–290. doi: 10.1093/oxfordjournals.aje.a112813 [DOI] [PubMed] [Google Scholar]
- 17.Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, D’Agostino RB, Sr., Fox CS, Larson MG, Murabito JM, et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. American journal of epidemiology. 2007;165:1328–1335. doi: 10.1093/aje/kwm021 [DOI] [PubMed] [Google Scholar]
- 18.Cooper LL, Palmisano JN, Benjamin EJ, Larson MG, Vasan RS, Mitchell GF, Hamburg NM. Microvascular Function Contributes to the Relation Between Aortic Stiffness and Cardiovascular Events: The Framingham Heart Study. Circ Cardiovasc Imaging. 2016;9. doi: 10.1161/CIRCIMAGING.116.004979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I, Struijker-Boudier H, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27:2588–2605. doi: 10.1093/eurheartj/ehl254 [DOI] [PubMed] [Google Scholar]
- 20.Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, Lloyd JK, Deanfield JE. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340:1111–1115. doi: 10.1016/0140-6736 [DOI] [PubMed] [Google Scholar]
- 21.Mitchell GF, Parise H, Vita JA, Larson MG, Warner E, Keaney JF,Keyes MJ Jr., Levy D, Vasan RS, Benjamin EJ. Local shear stress and brachial artery flow-mediated dilation: the Framingham Heart Study. Hypertension. 2004;44:134–139. doi: 10.1161/01.HYP.0000137305.77635.68 [DOI] [PubMed] [Google Scholar]
- 22.Benjamin EJ, Larson MG, Keyes MJ, Mitchell GF, Vasan RS, Keaney JF, Jr., Lehman BT, Fan S, Osypiuk E, Vita JA. Clinical correlates and heritability of flow-mediated dilation in the community: the Framingham Heart Study. Circulation. 2004;109:613–619. doi: 10.1161/01.CIR.0000112565.60887.1E [DOI] [PubMed] [Google Scholar]
- 23.Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. In: Statistics for Biology and Health. New York, NY: Springer; 2001. [Google Scholar]
- 24.Lin DY, Wei LJ. The Robust Inference for the Cox Proportional Hazards Model. J Am Statist Assoc. 1989;84:1074–1078. doi: 10.1080/01621459.1989.10478874 [DOI] [Google Scholar]
- 25.Niiranen TJ, Kalesan B, Mitchell GF, Vasan RS. Relative Contributions of Pulse Pressure and Arterial Stiffness to Cardiovascular Disease. Hypertension. 2019;73:712–717. doi: 10.1161/HYPERTENSIONAHA.118.12289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.American Diabetes, Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2020. Diabetes Care. 2020;43:S14–S31. doi: 10.2337/dc20-S002 [DOI] [PubMed] [Google Scholar]
- 27.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanderson E, Glymour MM, Holmes MV, Kang H, Morrison J, Munafò MR, Palmer T, Schooling CM, Wallace C, Zhao Q, et al. Mendelian randomization. Nature Reviews Methods Primers. 2022;2. doi: 10.1038/s43586-021-00092-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016;40:597–608. doi: 10.1002/gepi.21998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50:1505–1513. doi: 10.1038/s41588-018-0241-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, Ntritsos G, Dimou N, Cabrera CP, Karaman I, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50:1412–1425. doi: 10.1038/s41588-018-0205-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carter AR, Gill D, Davies NM, Taylor AE, Tillmann T, Vaucher J, Wootton RE, Munafo MR, Hemani G, Malik R, et al. Understanding the consequences of education inequality on cardiovascular disease: mendelian randomisation study. Bmj. 2019;365:l1855. doi: 10.1136/bmj.l1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slob EAW, Burgess S. A comparison of robust Mendelian randomization methods using summary data. Genet Epidemiol. 2020;44:313–329. doi: 10.1002/gepi.22295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35:4851–4853. doi: 10.1093/bioinformatics/btz469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prenner SB, Chirinos JA. Arterial stiffness in diabetes mellitus. Atherosclerosis. 2015;238:370–379. doi: 10.1016/j.atherosclerosis.2014.12.023 [DOI] [PubMed] [Google Scholar]
- 36.Mitchell GF, Vita JA, Larson MG, Parise H, Keyes MJ, Warner E, Vasan RS, Levy D, Benjamin EJ. Cross-sectional relations of peripheral microvascular function, cardiovascular disease risk factors, and aortic stiffness: the Framingham Heart Study. Circulation. 2005;112:3722–3728. doi: 10.1161/CIRCULATIONAHA.105.551168 [DOI] [PubMed] [Google Scholar]
- 37.Townsend RR, Wimmer NJ, Chirinos JA, Parsa A, Weir M, Perumal K, Lash JP, Chen J, Steigerwalt SP, Flack J, et al. Aortic PWV in chronic kidney disease: a CRIC ancillary study. AmJHypertens. 2010;23:282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Briet M, Boutouyrie P, Laurent S, London GM. Arterial stiffness and pulse pressure in CKD and ESRD. Kidney Int. 2012;82:388–400. [DOI] [PubMed] [Google Scholar]
- 39.Chirinos JA, Bhattacharya P, Kumar A, Proto E, Konda P, Segers P, Akers SR, Townsend RR, Zamani P. Impact of Diabetes Mellitus on Ventricular Structure, Arterial Stiffness, and Pulsatile Hemodynamics in Heart Failure With Preserved Ejection Fraction. J Am Heart Assoc. 2019;8:e011457. doi: 10.1161/JAHA.118.011457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Safar ME, Czernichow S, Blacher J. Obesity, arterial stiffness, and cardiovascular risk. Journal of the American Society of Nephrology : JASN. 2006;17:S109–111. doi: 10.1681/ASN.2005121321 [DOI] [PubMed] [Google Scholar]
- 41.Cruickshank K, Riste L, Anderson SG, Wright JS, Dunn G, Gosling RG. Aortic pulse-wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation. 2002;106:2085–2090. doi: 10.1161/01.cir.0000033824.02722.f7 [DOI] [PubMed] [Google Scholar]
- 42.Muhammad IF, Borne Y, Ostling G, Kennback C, Gottsater M, Persson M, Nilsson PM, Engstrom G. Arterial Stiffness and Incidence of Diabetes: A Population-Based Cohort Study. Diabetes Care. 2017;40:1739–1745. doi: 10.2337/dc17-1071 [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, He P, Li Y, Zhang Y, Li J, Liang M, Wang G, Tang G, Song Y, Wang B, et al. Positive association between baseline brachial-ankle pulse wave velocity and the risk of new-onset diabetes in hypertensive patients. Cardiovasc Diabetol. 2019;18:111. doi: 10.1186/s12933-019-0915-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng M, Zhang X, Chen S, Song Y, Zhao Q, Gao X, Wu S. Arterial Stiffness Preceding Diabetes: A Longitudinal Study. Circ Res. 2020;127:1491–1498. doi: 10.1161/CIRCRESAHA.120.317950 [DOI] [PubMed] [Google Scholar]
- 45.Stagner JI, Samols E. The vascular order of islet cellular perfusion in the human pancreas. Diabetes. 1992;41:93–97. doi: 10.2337/diab.41.1.93 [DOI] [PubMed] [Google Scholar]
- 46.Almaca J, Weitz J, Rodriguez-Diaz R, Pereira E, Caicedo A. The Pericyte of the Pancreatic Islet Regulates Capillary Diameter and Local Blood Flow. Cell Metab. 2018;27:630–644 e634. doi: 10.1016/j.cmet.2018.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vincent MA, Dawson D, Clark AD, Lindner JR, Rattigan S, Clark MG, Barrett EJ. Skeletal muscle microvascular recruitment by physiological hyperinsulinemia precedes increases in total blood flow. Diabetes. 2002;51:42–48. doi: 10.2337/diabetes.51.1.42 [DOI] [PubMed] [Google Scholar]
- 48.Stehouwer CDA. Microvascular Dysfunction and Hyperglycemia: A Vicious Cycle With Widespread Consequences. Diabetes. 2018;67:1729–1741. doi: 10.2337/dbi17-0044 [DOI] [PubMed] [Google Scholar]
- 49.Wagenmakers AJ, Strauss JA, Shepherd SO, Keske MA, Cocks M. Increased muscle blood supply and transendothelial nutrient and insulin transport induced by food intake and exercise: effect of obesity and ageing. J Physiol. 2016;594:2207–2222. doi: 10.1113/jphysiol.2014.284513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou B, Lu Y, Hajifathalian K, Bentham J, Di Cesare M, Danaei G, Bixby H, Cowan MJ, Ali MK, Taddei C, et al. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4·4 million participants. The Lancet. 2016;387:1513–1530. doi: 10.1016/s0140-6736 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Statistical code for the FHS analyses will be made available upon reasonable request to the corresponding author. Because of the protected nature of the data collected for this study, requests to access the FHS dataset may be sent to the National Institutes of Health Biologic Specimen and Data Repository Information Coordinating Center at https://biolincc.nhlbi.nih.gov/requests/data-request/form/ and requests to access the UKBB dataset may be sent to the UKBB at https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access.