

Abstract
Rationale
Chronic obstructive pulmonary disease (COPD) is a disease characterized by persistent airway inflammation and disordered macrophage function. The extent to which alterations in macrophage bioenergetics contribute to impaired antioxidant responses and disease pathogenesis has yet to be fully delineated.
Objectives
Through the study of COPD alveolar macrophages (AMs) and peripheral monocyte-derived macrophages (MDMs), we sought to establish if intrinsic defects in core metabolic processes drive macrophage dysfunction and redox imbalance.
Methods
AMs and MDMs from donors with COPD and healthy donors underwent functional, metabolic, and transcriptional profiling.
Measurements and Main Results
We observed that AMs and MDMs from donors with COPD display a critical depletion in glycolytic- and mitochondrial respiration–derived energy reserves and an overreliance on glycolysis as a source for ATP, resulting in reduced energy status. Defects in oxidative metabolism extend to an impaired redox balance associated with defective expression of the NADPH-generating enzyme, ME1 (malic enzyme 1), a known target of the antioxidant transcription factor NRF2 (nuclear factor erythroid 2–related factor 2). Consequently, selective activation of NRF2 resets the COPD transcriptome, resulting in increased generation of TCA cycle intermediaries, improved energetic status, favorable redox balance, and recovery of macrophage function.
Conclusions
In COPD, an inherent loss of metabolic plasticity leads to metabolic exhaustion and reduced redox capacity, which can be rescued by activation of the NRF2 pathway. Targeting these defects, via NRF2 augmentation, may therefore present an attractive therapeutic strategy for the treatment of the aberrant airway inflammation described in COPD.
Keywords: COPD, macrophage, metabolism, nuclear factor erythroid 2–related factor 2, malic enzyme 1
At a Glance Commentary
Scientific Knowledge on the Subject
The extent to which alterations in macrophage bioenergetics contribute to defective macrophage function in patients with chronic obstructive pulmonary disease (COPD) remains to be fully elucidated.
What This Study Adds to the Field
ln COPD, both monocyte-derived and alveolar macrophages from the inflamed pulmonary niche display defective efferocytosis, which correlates with clinical markers of disease severity. COPD alveolar macrophages and monocyte-derived macrophages exhibit depletion of mitochondrial and glycolytic-derived energy reserves coupled with a skewing toward glycolysis, with consequence for efferocytosis. This lack of metabolic plasticity is associated with loss of expression of the NADPH-generating enzyme malic enzyme 1 and impaired redox buffering capacity. Selective activation of the transcription factor NRF2 (nuclear factor erythroid 2–related factor 2) can be used to recover malic enzyme 1 expression, improve energetic status and redox balance, and overcome defective macrophage function in patients with COPD.
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death globally. To date, we have no therapies that significantly alter the course of this debilitating disease. The histological hallmark of COPD is persistent inflammation of the airways resulting in airflow limitation, measured by a decline in FEV1, chronic bronchitis, and emphysema. This inflammation persists in people with COPD, even after smoking cessation (1).
It has long been established that macrophages play a major role in the pathogenesis of COPD. Alveolar macrophages (AMs) are present in high numbers in the airways and airway secretions of patients with COPD, where their abundance correlates directly with disease severity (2). Despite this abundance, patients with COPD experience high rates of infection, with pathogenic bacteria in the lower airways strongly associated with exacerbation frequency and increased inflammation (3). This major disconnect, whereby excessive cellular inflammation results in ineffective immunity, is now understood to be largely driven by macrophage dysfunction. Work from our group and others has previously shown that COPD macrophages have defective phagocytosis of bacteria that colonize the lungs in COPD, such as nontypeable Haemophilus influenzae and Streptococcus pneumoniae (4–7). Macrophage dysfunction also extends to impaired inflammation resolution, with failure to efferocytose apoptotic cells also described (8). In conjunction with impaired macrophage internalization rates, macrophages in COPD also produce molecules known to induce pulmonary tissue damage, including MMPs (matrix metalloproteinases) (9, 10) and reactive oxygen species (ROS) (11). ROS, in particular, have been linked to mitochondrial dysfunction in COPD (12). Thus, in COPD, macrophages demonstrate functional responses that are ineffective and skew the balance toward “self-injury” in the lung. Despite these well-characterized defects in macrophage function, the mechanisms that drive this aberrant behavior remain poorly defined (7, 13, 14). Therefore, through the study of macrophages isolated from the airways (AMs) and peripheral blood (monocyte-derived macrophages [MDMs]) of donors with COPD and healthy donors, we sought to delineate the intrinsic processes that promote macrophage dysfunction in COPD. We demonstrate that in COPD, AMs and MDMs share an inherent defect in efferocytosis and phagocytosis, driven by the loss of oxidative and glycolytic reserve capacity and impaired metabolic plasticity. Reduced expression of the NADPH-generating enzyme ME1 (malic enzyme 1) recapitulates these metabolic defects and results in a reduction in redox buffering capacity and impaired efferocytosis. By activating the antioxidant transcription factor NRF2 (nuclear factor erythroid 2–related factor 2), we were able to rescue ME1 expression and restore metabolic plasticity and function in COPD macrophages. Some of the results of this study have been previously reported in the form of abstracts (15–18).
Methods
Macrophage Donors
Donors with COPD and healthy donors underwent bronchoscopy and venesection. Patient demographics are outlined in Table 1. Written informed consent was obtained in accordance with local ethics approval, as detailed in the online supplement.
Table 1.
Demographics of Study Participants
| Healthy Nonsmokers | “Healthy” Smokers | COPD | |
|---|---|---|---|
| No. of subjects | 24 | 9 | 52 |
| Age, yr | 54 (31–70) | 53 (36–68) | 61 (39–77) |
| Sex, female/male | 12/12 | 2/7 | 29/23 |
| FEV1, L | 3.1 ± 0.7 | 3.5 ± 0.3 | 1.7 ± 0.5 |
| FEV1% predicted | 105 ± 12.9 | 98 ± 4.1 | 64 ± 10.11 |
| GOLD stage | N/A | N/A | 14 stage 1, 26 stage 2, 11 stage 3, 1 stage 4 |
| Exacerbation per year 0/>1/>2/⩾3 | N/A | N/A | 14/10/20/8 |
| Smoking status, current/ex/never | 0/0/24 | 9/0/0 | 23/29/0 |
| Pack-years | N/A | 30 ± 10 | 37 ± 14 |
| CAT score (max 40) | N/A | N/A | 16 ± 8 |
| Inhaled medications ICS+LABA/LAMA/SABA | N/A | N/A | 25/27/34 |
Definition of abbreviations: CAT = COPD Assessment Test; COPD = chronic obstructive pulmonary disease; GOLD = Global Initiative for Chronic Obstructive Lung Disease; ICS = inhaled corticosteroids; LABA = long-acting beta-2-agonists; LAMA = long-acting muscarinic antagonists; N/A = not applicable; SABA = short-acting beta-agonists.
Healthy nonsmokers are lifelong nonsmokers with normal spirometry. Healthy smokers are current smokers with normal spirometry. Donors with COPD are current or ex-smokers with an FEV1/FVC ratio of <0.70.
Cell Culture
AMs were isolated from BAL as previously described (19), with 93–97% purity as assessed by Cytospin. MDMs were differentiated from peripheral blood mononuclear cells isolated via discontinuous plasma Percoll gradients (Sigma-Aldrich), for 14 days in culture. For Seahorse MDM experiments, monocytes were isolated using the Miltenyi Biotec Pan-Monocyte Isolation kit and then plated into low-adherence flasks (Corning) for culture. THP-1 cells were differentiated to macrophages by treatment with 100 nM phorbol myristol acetate for 3 days, then rested for 3 days before assay. Cells were treated with 0.065 μM KI-696, a selective inhibitor of the KEAP1–NRF2 interaction, or vehicle control ×16 hours before assays. All cells were cultured in RPMI 1640 (Sigma-Aldrich) with 10% heat-inactivated fetal calf serum (Gibco).
Functional Assays
Bacterial internalization assays were performed as previously described (7), using opsonized Serotype 14 S. pneumoniae (National Collection of Type Cultures 11902). For efferocytosis assays, neutrophils isolated via Percoll gradient were stained with a PKH26 labeling kit, as per manufacturer’s guidelines (Sigma-Aldrich), and cultured for 20 hours in RPMI 1640. Apoptotic neutrophils (70–80% apoptotic verified by Annexin-V-Topro3 staining) were added to macrophages at MOI 5:1, with ice control, for 90 minutes before vigorous washing and removal for analysis on a Becton Dickinson FACS Calibur flow cytometer (20).
Quantification and Statistical Analysis
Data represents mean ± SEM. Statistical parametric analyses were performed after confirmation of normal distribution of the data via D’Agostino-Pearson normality tests. All statistical tests were performed with Prism 8 software (GraphPad Software) using unpaired t tests, paired t tests, one-way or two-way ANOVA with Sidak’s multiple comparisons test, or Pearson correlation. Wilcoxon matched-pairs signed-rank test, Mann-Whitney, and Kruskal-Wallis with Dunn’s multiple comparison tests were performed for nonparametric or n ⩽ 6 data, with *P ⩽ 0.05, **P ⩽ 0.01, and ***P ⩽ 0.001.
Results
Correlation of Defective Macrophage Efferocytosis with Phagocytosis in COPD
To study macrophage efferocytosis and phagocytosis, we recruited patients with COPD with mild (14 patients, 27%). moderate (26 patients, 50%), severe (11 patients, 21%), and very severe disease (1 patient, 2%), who were free from exacerbation for ⩾8 weeks minimum and age matched (±8 yr; mean age) with healthy control subjects (Table 1). In line with disease status, FEV1 was significantly lower in donors with COPD, and 43/52 (82%) of donors with COPD had COPD Assessment Test (CAT) scores of 10 or greater, in keeping with severe symptoms.
AMs and peripheral MDMs were compared with interrogate intrinsic and extrinsic (niche dependent) factors affecting macrophage function. Both AMs and MDMs isolated from patients with COPD demonstrated defective phagocytosis of live opsonized S14 S. pneumoniae (Figures 1A and 1B), and efferocytosis of PKH26-labeled apoptotic neutrophils compared with healthy control subjects (Figures 1C and 1D), as previously described (7, 20). Rates were lowest in current smokers across both processes, as expected (21), but macrophage dysfunction did not recover in patients with COPD who were no longer active smokers (Figures 1A–1D). To establish if there was a direct clinical readout of impaired efferocytosis in COPD, we performed correlation analysis of macrophage efferocytosis against FEV1. There was a significant relationship observed between macrophage efferocytosis and FEV1% predicted (actual donor FEV1 as a percentage of their predicted FEV1, based on height and age) (Figure 1E). Similarly, we observed a negative correlation between COPD macrophage efferocytosis and high CAT scores, representative of increased symptom severity (Figure 1F). Correlation between bacterial phagocytosis in COPD macrophages and both FEV1 and high CAT scores has previously been published by our group (7). Macrophage bacterial internalization (see Figure E1A in the online supplement) and efferocytosis (Figure E1B) did not correlate with study participant age. Crucially, there was a high degree of correlation in both AMs and MDMs between bacterial phagocytosis and efferocytosis within each study participant (Figures 1G and 1H). Together with conservation of phenotype between AM (tissue) and MDM (blood) derived cells, these observations led us to question whether a shared intrinsic defect was driving impaired phagocytosis and efferocytosis in COPD macrophages.
Figure 1.
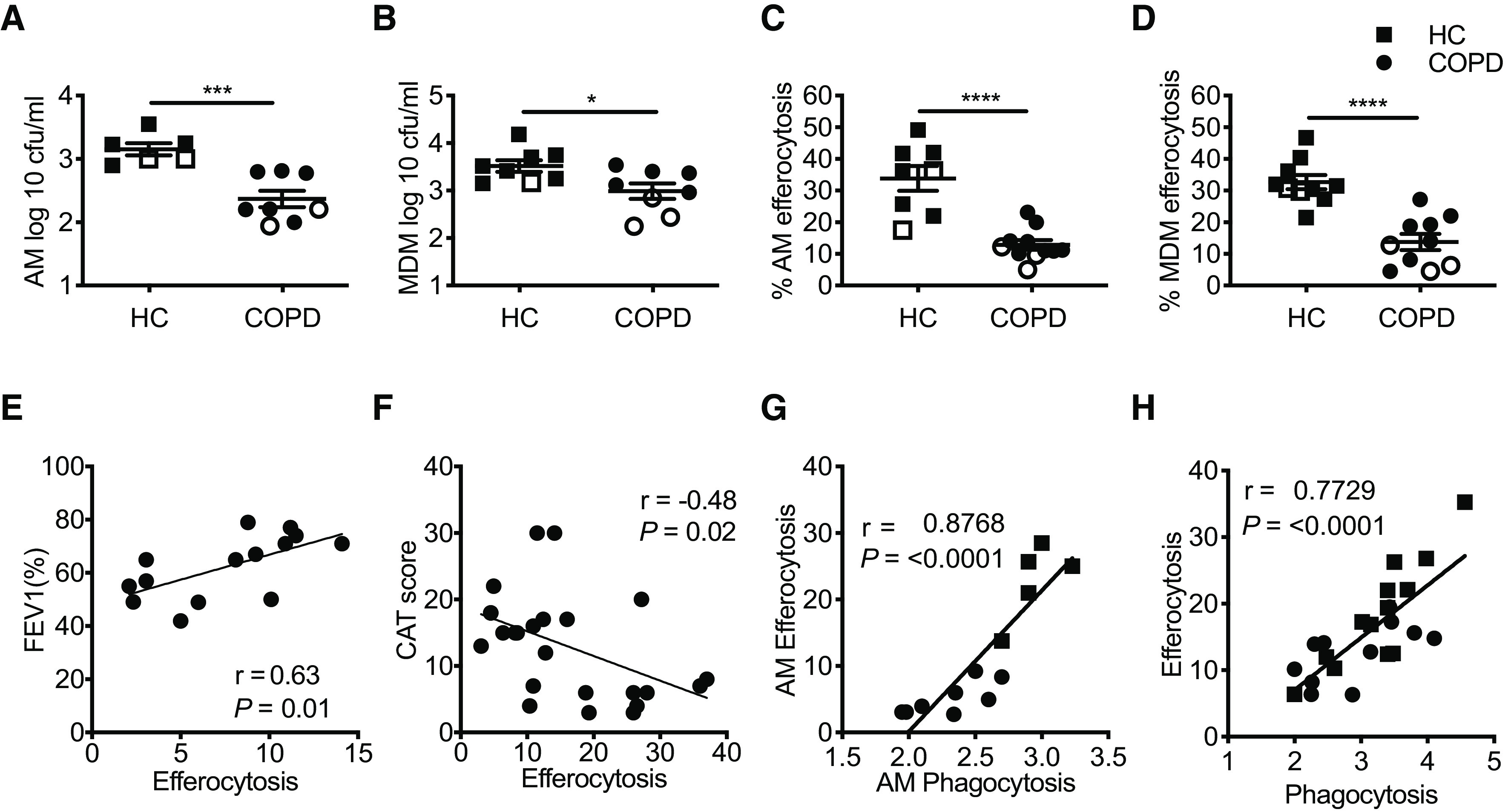
Intrinsic defects in chronic obstructive pulmonary disease (COPD) alveolar macrophages (AMs) and peripheral blood monocyte-derived macrophages (MDMs). (A and B) AMs and MDMs from healthy control subjects (HC, squares) and donors with COPD (circles) were challenged with opsonized serotype 14 Saccharomyces pneumoniae for 4 hours and numbers of viable bacteria measured or (C and D) coincubated with PKH26-labeled 20-hour apoptotic neutrophils and efferocytosis rates measured by flow cytometry. (A: HC, n = 6; COPD, n = 8; B: HC/COPD, n = 8; C: HC, n = 8; COPD, n = 11; D: HC/COPD, n = 10). (E and F) COPD AM and MDM efferocytosis were correlated with FEV1% (E: n = 14) and symptoms of disease severity, as measured by the CAT score (F: n = 24). (G and H) Efferocytosis of PKH26-labeled apoptotic neutrophils was correlated with phagocytosis of Saccharomyces pneumoniae S14 in both AMs (G: n = 13) and MDMs (H: n = 24) from patients with COPD and healthy donors. Open squares and circles represent current smokers. Data represent individual values with mean ± SEM. P values calculated by unpaired t test; *P ⩽ 0.05, ***P ⩽ 0.001, and ****P ⩽ 0.0001. Pearson’s correlation coefficient (r) as shown. CAT = COPD Assessment Test.
COPD Macrophages Demonstrate a Baseline Defect in Metabolic Processing
To explore intrinsic differences in baseline transcript abundance in AMs isolated from patients with COPD and healthy control subjects, we undertook total RNA-sequencing (RNA-seq) of AMs isolated from BAL, as detailed in the online supplement. Transcriptional analysis revealed a total of 287 genes to be differentially expressed between resting state COPD and healthy AMs (Figure 2A). In keeping with previous studies in patients with COPD, we found metabolic processes to be one of the most significantly suppressed in COPD AMs (Figure 2B) (22, 23). As reprogramming of macrophage metabolism, particularly mitochondrial metabolism (24), is inextricably linked to function and activation states (25–28), we questioned if altered metabolism in COPD macrophages results in defective cellular energetics and impaired effector function. To first define the energetic states of isolated AMs, we undertook liquid chromatography–mass spectrometry (LC-MS) analysis comparing AMs from patients with COPD with AMs from healthy control subjects. COPD AMs revealed a significantly altered abundance of ATP (Figure 2C) and concurrent reduction in energy charge status (ATP:ADP) (Figure 2D). To establish if the altered energy state observed in COPD AMs was consequent upon changes in oxidative or glycolytic metabolism, we used seahorse metabolic profiling to define basal metabolic states and metabolic reserve capacity, defined as the difference between basal and peak metabolic rates recorded after injection of metabolic stressor compounds, as outlined in the online supplement. COPD AMs demonstrated equivalent extracellular acidification rate (ECAR) (Figure E2A) and reduced oxygen consumption rate (OCR) (Figure E2B) at baseline compared with healthy control subjects. In addition to the previously reported reduction in spare respiratory capacity (22), we observe a loss of both oxidative (Figure 2E) and glycolytic reserve capacity (Figure 2F) in COPD AMs after exposure to metabolic stressors. Despite equivalent basal respiration and glycolytic rates (Figures E2C and E2D), cellular energetics were also defective in peripherally derived MDMs from donors with COPD, with depleted reserves in both oxidative metabolism (Figure 2G) and glycolysis (Figure 2H). In donors with COPD, both AM and MDM reserves were unaffected by smoking status, suggesting changes in metabolism are hardwired. Moreover, changes in glycolytic enzyme abundance observed in AMs from patients with COPD versus control subjects who were “healthy smokers” (defined as current smokers with normal spirometry), as detected by data-independent acquisition mass spectrometry proteomic analysis, would suggest that skewing toward glycolysis is a disease-specific response and not a consequence of smoking status (Figure 2I). Although healthy AMs increased their maximal respiration rate after coincubation with apoptotic neutrophils, COPD AMs, in contrast, failed to exhibit any uplift in energetic capacity (Figures E2E and E2F). Treatment of healthy donor AMs with established M2 polarization stimuli (28, 29) did not induce a change in respiratory reserve, suggesting the spare respiratory capacity observed in healthy AMs is a fundamental feature of these cells rather than a polarization state (Figure E2G). In addition, we did not observe a predominance of M1 or M2 markers of those detected in the proteome of COPD versus healthy donor AMs (30, 31) (Figure E2H).
Figure 2.
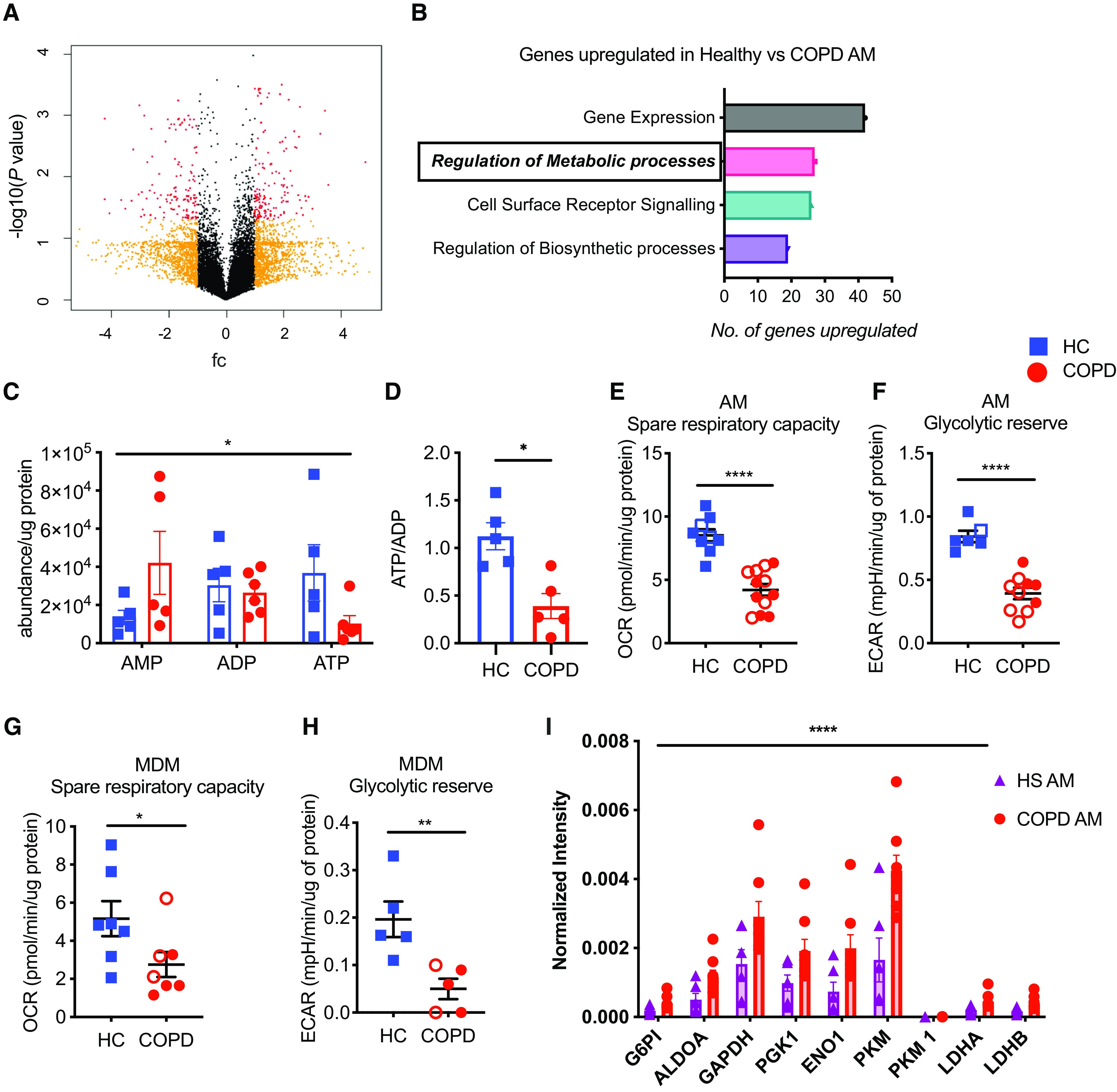
Loss of energy reserves in chronic obstructive pulmonary disease (COPD) macrophages. (A and B) alveolar macrophages (AMs) were isolated from patients with COPD and healthy control (HC) donors via BAL, cultured for 16 hours before collecting RNA for total RNA-sequencing (n = 3). (A) Volcano plots displaying the log2 fold change (FC) between patients with COPD and healthy donors. Red dots (n = 287) represent genes that are significantly altered. Orange dots represent genes that are altered but do not meet significance. (B) Top four Gene Ontology processes upregulated in healthy AMs at baseline, compared with COPD AMs. (C and D) High-performance liquid chromatography–mass spectrometry analysis was performed to determine the levels of ATP, ADP, and AMP in resting state healthy donor (squares) and COPD (circles) AMs and energy charge ([ATP + 1/2ADP]/(ATP + ADP + AMP]) was calculated (n = 5/6). (E–H) Seahorse mitochondrial (E and G) and glycolytic (F and H) stress testing in COPD AMs and MDMs (E: HC, n = 9; COPD, n = 12; F: HC, n = 6; COPD, n = 10; G: n = 7; H: n = 5). Open squares and circles indicate current smokers. COPD smoker versus ex-smoker, E: P value = 0.103; F: P value = 0.158; G: P value = 0.17. (I) Glycolytic enzyme abundance in COPD (n = 7) versus HS AMs (n = 5) as determined by data-independent acquisition mass spectrometry proteomic analysis. Data represent individual values ± SEM. (A and B) Significance determined at FC > log21.5 and P value ⩽ 0.05. P values calculated via (C and I) two-way ANOVA, (E–G) unpaired t test, or (D and H) Mann-Whitney U test. *P ⩽ 0.05, **P ⩽ 0.01, and ****P ⩽ 0.0001. ECAR = extracellular acidification rate; HS = healthy smoker; MDMs = monocyte-derived macrophages; OCR = oxygen consumption rate.
Defects in Oxidative Metabolism Drive Metabolic Exhaustion in COPD Macrophages
With reductions in both oxidative and glycolytic reserve observed, we next questioned which metabolic processes predominate at baseline and in challenged states. Compared with healthy AMs, COPD AMs had a significantly reduced OCR/ECAR ratio (Figure 3A). This preponderance for glycolysis over oxidative metabolism was again demonstrated by significantly reduced ATP-linked OCR in COPD AMs compared with healthy AMs (Figure 3B). Interestingly, although absolute ATP-linked OCR was reduced, it did represent a significantly higher percentage of maximal respiration in COPD AMs during mitochondrial stress testing (Figure 3C). This suggests a lack of redundancy in mitochondrial units in COPD AMs, whereby these cells are seemingly unable to recruit additional mitochondria or to further increase ATP production in existing mitochondria. Strikingly, this occurred in the presence of comparable protein abundance of the critical ATP-generating complex, Mitochondrial ATP Synthase or Complex V subunits (Figure 3D), conserved mitochondrial-to-nuclear DNA ratios, and comparable abundance of key mitochondrial fusion and fission proteins (Figures 3E and E3A).
Figure 3.
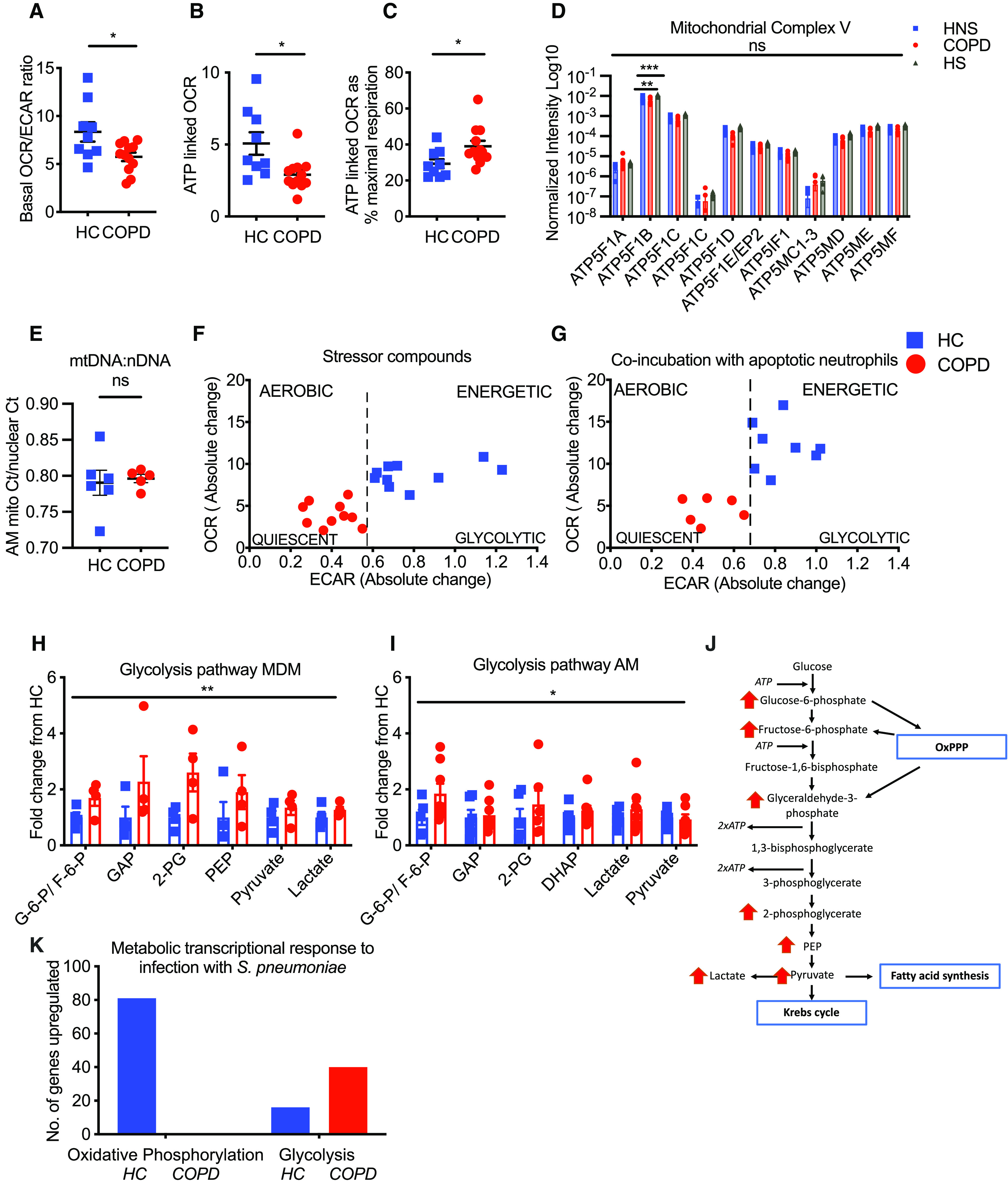
Chronic obstructive pulmonary disease (COPD) macrophages display a preponderance for glycolytic metabolism. (A) Calculated oxygen consumption rate/extracellular acidification rate (OCR/ECAR) ratio in healthy control (HC, squares) and COPD (circles) alveolar macrophages (AMs). (B) OCR consumption linked to ATP generation in healthy and COPD AMs as calculated by the reduction in OCR after oligomycin treatment. (C) ATP-linked OCR as a percentage of maximal OCR in healthy and COPD AMs. HC, n = 9; COPD, n = 12. (D) Relative protein abundance of the 11 detected subunits of Mitochondrial ATP Synthase/Complex V, as measured by data-independent acquisition mass spectrometry proteomic analysis. COPD AMs, n = 7; HNS and HS, n = 5. (E) AM mitochondrial DNA to nuclear DNA ratios, as measured by real-time quantitative PCR. Healthy donor, n = 6; COPD donor, n = 5. (F and G) Absolute change in ECAR was plotted against absolute change in OCR after injection of mitochondrial stressor compounds in resting macrophages (F: n = 21) and after cells were coincubated with 20-hour apoptotic neutrophils for 90 minutes (G: n = 14). (H and I) Glycolytic metabolite abundance determined by high-performance liquid chromatography–mass spectrometry in resting state MDMs (H: n = 4) COPD AMs (I: HC, n = 6; COPD, n = 8), plotted as relative to healthy control. (J) Schematic of glycolytic intermediaries increased throughout the glycolytic pathway in COPD macrophages. (K) The transcriptional response in COPD and healthy control AMs after coincubation with opsonized D39 Streptococcus pneumoniae (n = 3). Data represent individual values and mean ± SEM. P values calculated by (A–C) unpaired t test, (E) Mann-Whitney U test, or (D, H, and I) two-way ANOVA. *P ⩽ 0.05, **P ⩽ 0.01, and ***P ⩽ 0.001. HNS = healthy nonsmoker; HS = healthy smoker; MDMs = monocyte-derived macrophages; ns = not significant.
To evaluate how cells differentially used either metabolic pathway to meet increased energy demand, absolute change in ECAR was plotted against absolute change in OCR, after injection of stressor compounds. Although healthy AMs increased both ECAR and OCR rates, COPD AMs revealed a defective induction in oxygen consumption rates (Figure 3F), a response magnified when concurrently challenged to undergo normal effector function by efferocytosing apoptotic neutrophils (Figure 3G). Thus, COPD macrophages fail to induce oxidative metabolism, with an overreliance on glycolytic energy.
To more directly identify metabolic blocks in COPD, we undertook high-performance LC-MS analysis of both COPD and healthy donor macrophages at rest. COPD MDMs (Figure 3H) and AMs (Figure 3I) both demonstrated a significant increase in abundance of glycolytic intermediaries compared with healthy control cells, supporting a glycolytic switch in the context of COPD (Figure 3J). This was independent of BAL nutrient availability (Figures E3B and E3C), macrophage glucose uptake (Figures E3E–E3G), or glycogen storage (Figure E3H), suggesting differential substrate availability is not driving this phenomenon. Interestingly, BAL lactate, however, was significantly higher in donors with COPD (Figure E3D). To investigate if the basal reliance on glycolysis and defective induction in oxygen consumption under conditions of activation and stress were regulated at a transcriptional level, we conducted a targeted analysis of a previously published Affymetrix microarray data set from our group (7), in which AMs were cultured in the presence of S. pneumoniae, as detailed in the online supplement. In keeping with a failure to induce oxidative metabolism, COPD AMs failed to induce any genes associated with oxidative phosphorylation in contrast to healthy control AMs after exposure to S. pneumoniae (Figure 3K). Conversely, upregulation of glycolytic genes in COPD AMs represented 10% of the total genes upregulated in response to infection.
Loss of ME1, a Critical Regulator of Macrophage Oxidative Metabolism and Redox Buffering, Recapitulates COPD Macrophage Dysfunction
Further analysis of the metabolic signature detected by total RNA-Seq of COPD versus healthy donor AMs (Figure 2A) revealed a complete downregulation of ME1 transcript (ME1) in COPD AMs (Figure 4A). This was associated with a marked reduction in ME1 protein expression within the lung macrophage compartment in patients with COPD (Figure 4B). ME1 catalyzes the reversible oxidative decarboxylation of malate to pyruvate, resulting in the replenishment of TCA cycle intermediaries and the conversion of NADP+ into NADPH. As a consequence, loss of ME1 has been shown to disrupt redox balance and antioxidant defense, with reduced GSH:GSSG ratios (32) and induction of HO-1 in PC3 and HCT116 cell lines (33). Thus, we sought to assess whether suppression of macrophage ME1 recapitulates the metabolic defects observed in COPD. We used a CRISPR cas9 system to delete ME1 from the THP-1 macrophage cell line (Figure E4A) coupled with a chemical ME1 inhibitor in healthy MDM cells. In keeping with our observed COPD phenotype, ME1 loss resulted in a reduction in OCR:ECAR ratios (Figure 4C) and a reduction in TCA cycle intermediaries (Figure 4D). These metabolic adaptations were associated with a reduction in redox capacity as evidenced by a reduced GSH:GSSG ratio and high basal mitochondrial ROS levels (Figures 4E and 4F). A proportional increase of 13C glucose incorporation into lactate in ME1-deficient cells (Figure 4G), as previously reported in the HCT116 cell line (33), revealed that in the setting of impaired oxidative phosphorylation, macrophages adapted by increasing glycolytic flux. Importantly, this observed increase in glycolysis was, however, insufficient to confer effective macrophage function, as evidenced by reduced efferocytosis rates in ME1 knockout cells, mirroring the defects detected in COPD macrophages (Figure 4H). In keeping with a critical role for oxidative phosphorylation in macrophage efferocytosis, healthy MDMs treated with oligomycin also displayed suppressed efferocytosis (Figure 4I). Glycolysis only contributed to efferocytic capacity when oxidative phosphorylation was blocked (Figure 4I).
Figure 4.
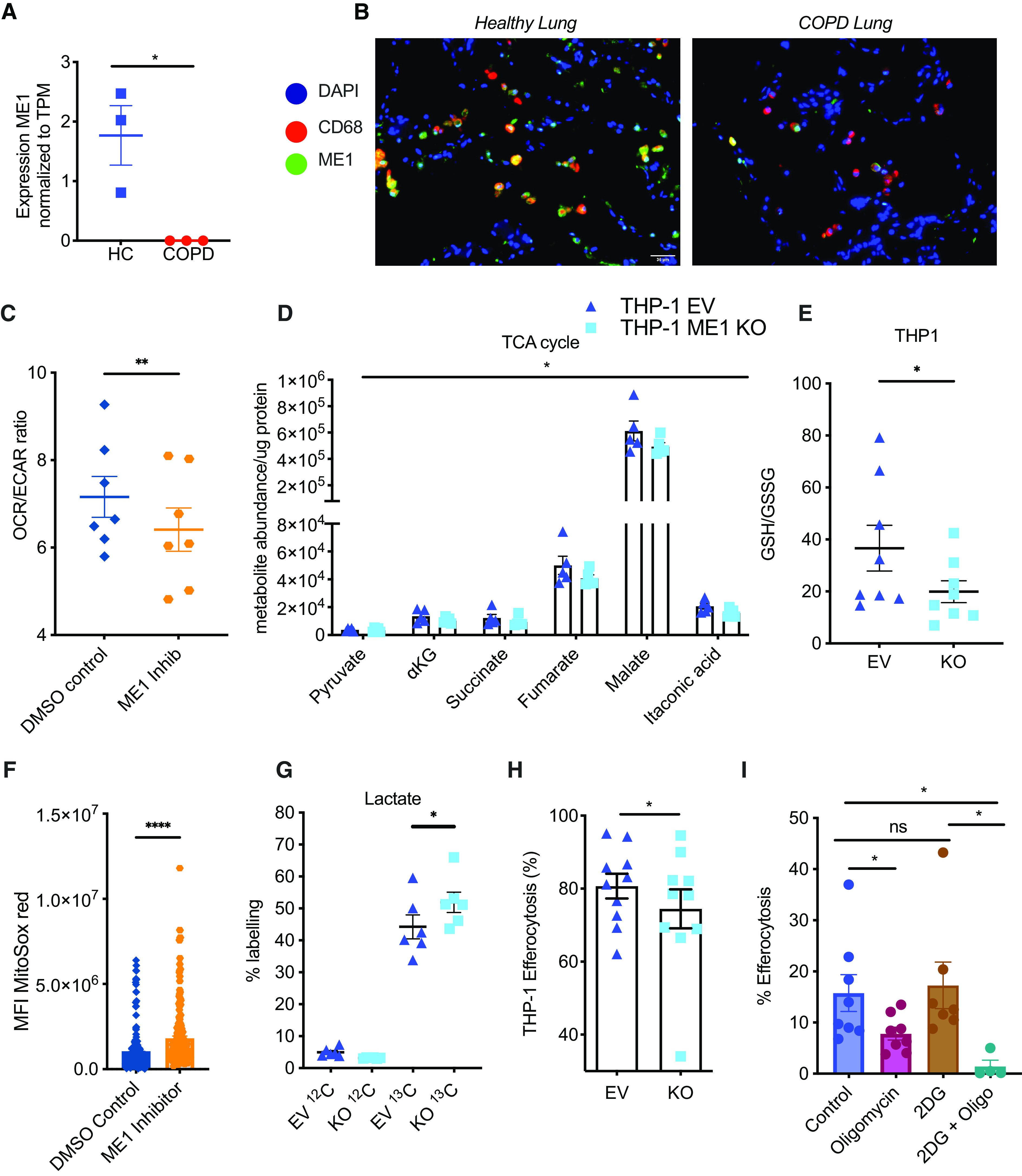
Malic enzyme 1 (ME1) plays a critical role in macrophage metabolism and efferocytosis. (A) Transcriptomic analysis of baseline ME1 expression in HC and chronic obstructive pulmonary disease (COPD) alveolar macrophages normalized to TPM (n = 3). (B) ME1 expression in healthy and COPD patient lung sections prepared from paraffin-embedded blocks; images taken at ×20 magnification. (C) Healthy monocyte-derived macrophages (MDMs) were treated ×16 hours with a chemical ME1 inhibitor or DMSO control before measuring the basal OCR:ECAR ratio on a Seahorse platform (n = 7). (D) High-performance liquid chromatography–mass spectrometry analysis of TCA cycle metabolite abundance in THP-1 empty vector control (EV) and ME1 knockout (ME1 KO) cells, normalized to protein content (n = 5). (E) GSH:GSSG ratio calculated in THP-1 EV and ME1 KO cells (n = 9). (F) Healthy MDMs were treated ×16 hours with a chemical ME1 inhibitor or DMSO control. Mean fluorescence intensity of MitoSox Red was then measured from more than 180 cells per condition for three donors. (G) U-13C glucose incorporation into lactate in THP-1 EV and ME1 KO cells after 6 hours of culture in U-13C glucose-containing media (n = 6). (H and I) THP-1 EV and ME1 knockout cells (H) and healthy MDMs pretreated with oligomycin 2 μM, 2DG 50 mM, or oligomycin and 2DG combined for 1 hour (I) before coincubation with PKH26-labeled 20-hour apoptotic neutrophils for 90 minutes and efferocytosis rate determined by flow cytometry (H: n = 8; I: n = 8, 8, 7, and 4, respectively). Data represent individual values and mean ± SEM. P values calculated via (A) adjusted t test values from total RNA-sequencing analysis as outlined in online supplement, (C, E, G, and H) paired t test, (D and I) two-way ANOVA with Dunnett’s multiple comparisons, or (F) Wilcoxon matched-pairs signed-rank test. *P ⩽ 0.05, **P ⩽ 0.01, and ****P ⩽ 0.0001. ECAR = extracellular acidification rate; HC = healthy control; MFI = mean fluorescence intensity; ns = not significant; OCR = oxygen consumption rate; TPM = transcripts per kilobase million.
As ME1 loss has been associated with senescence (33), and senescence has been linked with skewing toward glycolysis in human fibroblasts (34), we next surveyed established markers of senescence (35) in the proteome of AMs recovered from healthy nonsmokers, healthy smokers, and patients with COPD. There was no consistent pattern of expression to support a senescent switch within the COPD AMs accounting for the change in metabolic and effector function (Figures E5A–E5F). We therefore propose that ME1 plays a key role in dictating redox buffering capacity and metabolic flux in macrophages and that loss of ME1 skews cells toward glycolysis and leads to impaired effector function. Metabolic and functional phenotyping of ME1 loss recapitulated core elements of COPD macrophage dysfunction, establishing ME1 to be an important regulator of macrophage function in both health and disease states.
Activation of NRF2 Rescues ME1 Expression and Reprograms Metabolism, Improving Cellular Energetics and Redox Balance, and Restores Function in COPD Macrophages
Previous GWAS (36), cellular (13, 37, 38), and murine (39) studies have identified a potential role for the NRF2-mediated antioxidant transcriptional response in the pathogenesis of COPD. More recently, work from our group has shown that augmentation of NRF2 activity can improve bacterial phagocytic capacity in COPD macrophages (7). Although the mechanisms by which NRF2 agonists recover COPD macrophage effector function remain to be fully elucidated, previous studies have suggested that NRF2 augmentation of nonopsonic bacterial phagocytosis in COPD occurs in part via upregulation of the scavenger receptor MARCO (13). We observed loss of expression of both MARCO and the NRF2 independent Class A scavenger receptors SRA1 and SRA4 in COPD AMs (Figures E6A–E6C). In MEFS isolated from mice lacking Nrf2, Nrf2 has also been shown to modulate mitochondrial respiration and ATP levels (40). Moreover, studies in human lymphoid and cancer cell lines have identified ME1 to be a target gene of NRF2 (41, 42). Given the inherent shared defect we observed in both efferocytosis and opsonic phagocytosis, we therefore questioned whether a specific NRF2 agonist, KI-696 (7, 43), could in part mediate its effect by overcoming the intrinsic metabolic defects observed in COPD AMs and MDMs.
Activation of NRF2 via KI-696 significantly increased ME1 expression in COPD macrophages, as anticipated (Figure 5A). In keeping with the established role for ME1 in generating NADPH, we also observed an improvement in GSH:GSSG ratios and an increase in total glutathione availability in COPD macrophages after treatment with KI-696 (Figures 5B and 5C). To more directly measure the effects of NRF2 agonists on the metabolic capacity of COPD AMs, we undertook LC-MS analysis of AMs from patients with COPD treated with KI-696 and compared these to untreated COPD and healthy control AMs. KI-696–mediated NRF2 activation did not significantly alter glycolytic metabolite abundance (Figure 5D). However, it did significantly increase the abundance of all TCA cycle intermediaries relative to baseline abundance in untreated COPD AMs (Figure 5E) and partially restored cellular energetics (Figure 5F).
Figure 5.
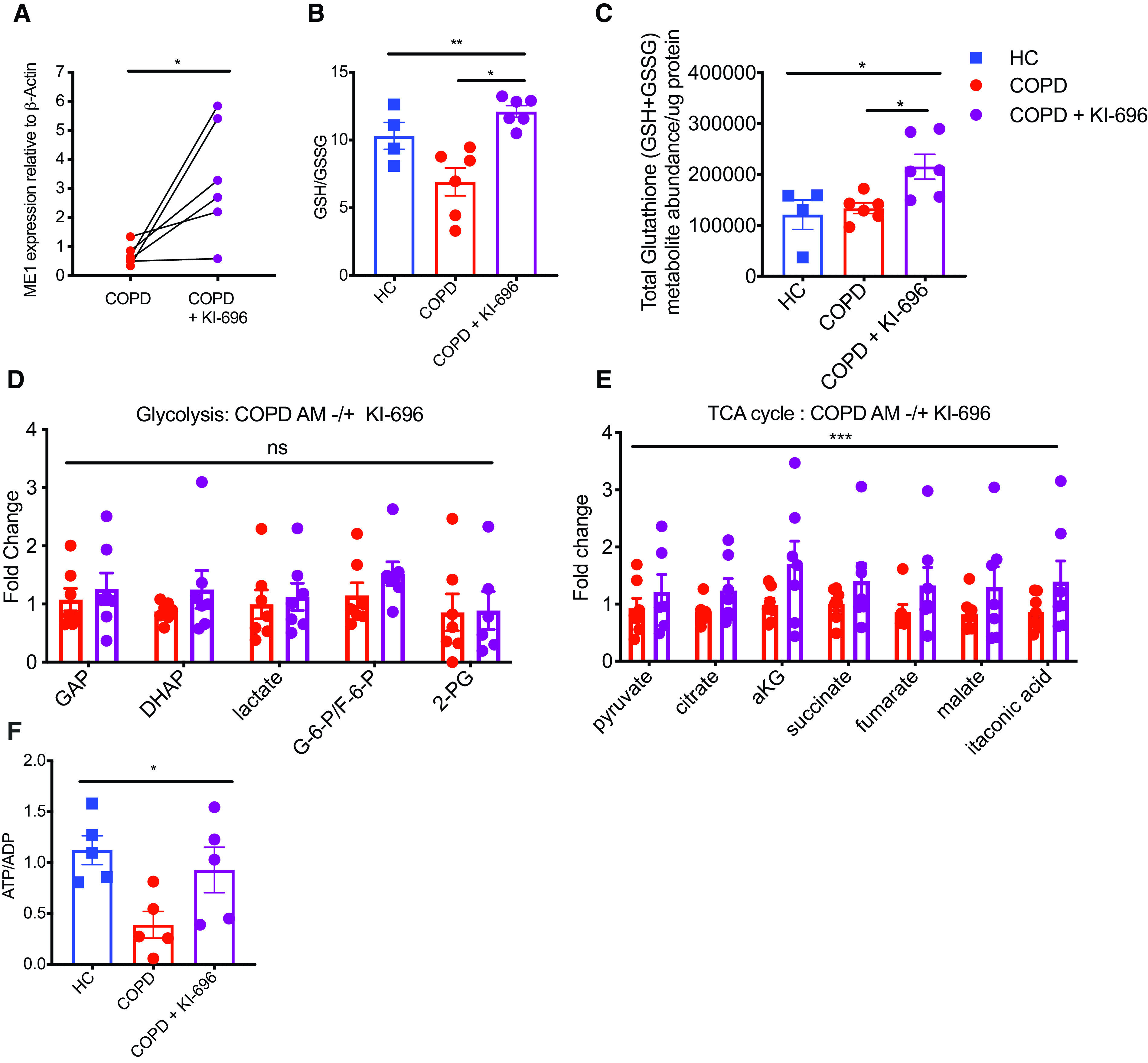
Cellular energetics and redox status are improved in chronic obstructive pulmonary disease (COPD) alveolar macrophages (AMs) after activation of malic enzyme 1 (ME1) via the NRF2 (nuclear factor erythroid 2–related factor 2) agonist KI-696. (A) Real-time PCR quantification of ME1 expression relative to β-actin in COPD AMs after culture in the absence or presence of KI-696 (n = 6). (B and C) Treatment with Ki-696 alters redox protection in COPD AMs as measured by (B) GSH:GSSG ratios and (C) absolute total glutathione abundance (GSH and GSSG) (HC, n = 4; COPD, n = 6). (D and E) Liquid chromatography–mass spectrometry analysis of healthy and COPD AMs (±KI-696). Metabolite abundance is plotted as relative to untreated COPD AMs (fold change) (n = 7). (F) Energy status expressed as an ATP-to-ADP ratio was calculated (n = 5). Data represent individual values and mean ± SEM. P values calculated via (A) paired t test, (B, C, and F) Kruskal-Wallis with Dunn’s multiple comparison tests, or (D and E) two-way ANOVA. *P ⩽ 0.05, **P ⩽ 0.01, and ***P ⩽ 0.001. HC = healthy control; ns = not significant.
To address the processes underlying this metabolic rescue, RNA-seq analysis was performed on AMs from healthy donors and patients with COPD at baseline and after treatment with KI-696. Correlation analysis across the 30,000-gene set revealed that COPD AMs were more highly correlated with healthy AMs after treatment with KI-696 (Figures 6A–6D). Regulation of metabolic processes was the most differentially upregulated biological process in COPD AMs after NRF2 activation (Figure 6E). Among ME1, other metabolic genes of interest altered by NRF2 activation of COPD AMs included G6PC3, MAPK14, TLK1, and OARD1 (Figures 6C and 6D). Finally, we questioned if the NRF2-mediated restoration of redox balance and correction of disturbances in oxidative metabolism and cellular energetics could rescue COPD macrophage efferocytosis. COPD MDMs and AMs both improved efferocytic capacity in the continued presence of the NRF2 agonist KI-696 (Figures 6F, 6G, and E6D). Failure of KI-696 to recover efferocytosis in ME1 knockout cells would suggest that augmentation of ME1 expression is required for NRF2-mediated enhancement of efferocytosis (Figure E6E). Figure 6H summarizes the functional outcomes associated with impaired metabolic plasticity in COPD macrophages and therapeutic rescue with NRF2 activation.
Figure 6.
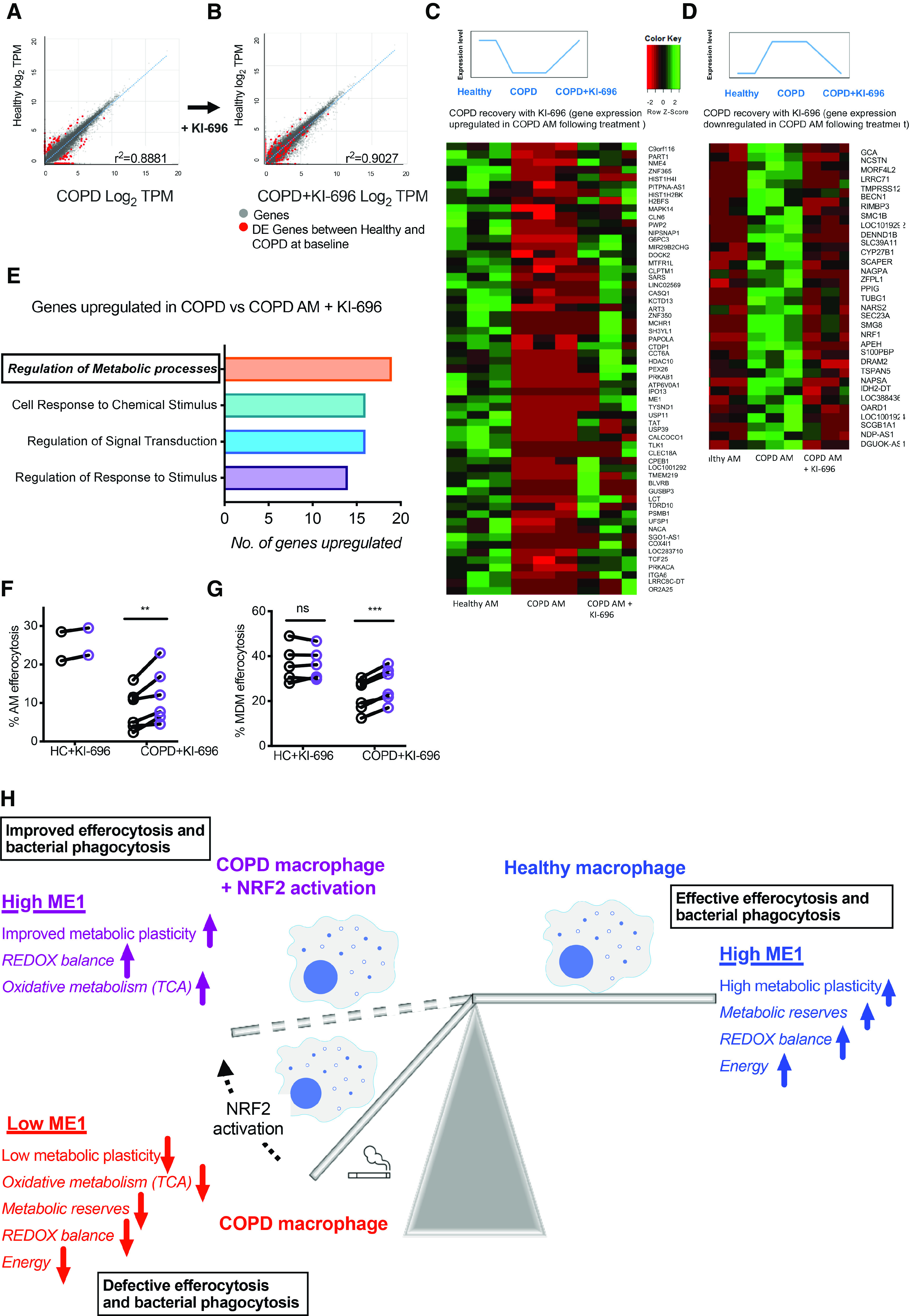
Activation of NRF2 (nuclear factor erythroid 2–related factor 2) with KI-696 reprograms alveolar macrophages (AMs) in chronic obstructive pulmonary disease (COPD) with consequences for effector function. (A–E) Healthy control (HC) and COPD AMs were cultured for 16 hours (±KI-696) before collection of RNA for total RNA-sequencing (n = 3). (A and B) Correlation analysis between healthy and COPD AMs before and after treatment with KI-696. Red dots represent significantly differentially expressed genes between COPD and healthy AMs at baseline, which are seen to move toward the trendline in B. (C and D) Heatmap of normalized z-scores showing genes that were initially comparatively downregulated (C) or upregulated (D) in COPD versus healthy AMs, which were transcriptionally reprogrammed by treatment with KI-696, in the direction of healthy AMs. (E) The lead Gene Ontology terms upregulated in COPD AMs after treatment with KI-696. (F and G) COPD AMs (F: HC, n = 2; COPD, n = 6) and MDMs (G: HC, n = 5; COPD, n = 6) were pretreated for 16 hours with the NRF2 activator KI-696, before coincubation with PKH26-labeled 20-hour apoptotic neutrophils and measurement of efferocytosis rates by flow cytometry. (H) Summary diagram of metabolic changes in COPD macrophages and the role of NRF2 augmentation. Data represent individual values and mean ± SEM. (A and B) Scatter plots were generated by plotting the average log2 tags per million (TPM) scores for healthy AMs versus the average log2 TPM scores for COPD AMs ±KI-696. R2 values were calculated from the slope of the correlation trendline. DE genes = FC > log21.5 and P value ⩽ 0.05. (F and G) P values calculated via (F) paired t test, or (G) donors with COPD paired t test, and healthy donors by Wilcoxon matched-pairs signed-rank test. **P ⩽ 0.01 and ***P ⩽ 0.001. DE = differentially expressed; FC = fold change; MDMs = monocyte-derived macrophages; ME1 = malic enzyme 1; ns = not significant; TCA = tricarboxylic acid cycle.
Discussion
COPD is characterized by persistent inflammation of the airways, destruction of lung tissue, and mucus hypersecretion resulting in airflow limitation and impaired gas exchange. Pathogen colonization and recurrent infective exacerbations are the largest contributors to morbidity and mortality within the disease (3). Failure of COPD macrophages to adequately phagocytose and kill bacteria and to instigate inflammation resolution via efferocytosis places macrophage dysfunction at the center of both disease pathology and progression in COPD (5, 12, 44). Study of AMs, directly influenced by the lung microenvironment, and peripherally derived MDMs, has enabled us to define an intrinsic metabolic defect in COPD macrophages that drives this functional impairment. Extending from previous descriptions of diminished baseline respiratory reserve capacity in COPD AMs (22), we describe a more global defect in COPD macrophage bioenergetics when these cells are engaged to undertake the highly energy-requiring process of efferocytosis. Moreover, we provide evidence that defective metabolism is not an exclusive consequence of an inflamed pulmonary niche by also characterizing this metabolic exhaustion, although to a lesser degree, in peripherally circulating MDMs. Within the limitations of the small number of healthy smoker control subjects included in this study, defective metabolism would also appear to be independent of current smoking status, with evidence of alterations in glycolytic enzyme abundance specific to disease state. This leads us to postulate that there is both central (bone marrow) reprogramming of newly formed monocytes that contribute to the replenishment of the AM compartment in the diseased lung and peripheral (lung tissue) reprogramming of the AM compartment after exposure to the local inflammatory milieu and metabolic intermediaries. This concept is supported by emerging evidence that reprogramming of myelopoiesis has consequence for myeloid cell tissue effector functions in acute (45) and chronic inflammatory disease states (6, 24), with overlapping transcriptional signatures previously reported in COPD MDM and AM populations (46) and evidence of changes in DNA accessibility and methylation marks in the AM compartment during trained immune responses (47, 48). Metabolic plasticity is a crucial feature of macrophage adaptability, with the capacity to generate ATP via multiple metabolic pathways, namely glycolysis, oxidative phosphorylation, and fatty acid oxidation (25, 28). Recent evidence has demonstrated that the dichotomy of glycolysis supporting acute inflammatory responses and oxidative metabolism fueling sustained energy production is an oversimplification of macrophage bioenergetics, with significant cross-talk required between these metabolic pathways (49–51). Importantly, we describe a refractory metabolism in COPD macrophages with both a significant depletion in glycolytic- and mitochondrial respiration–derived energy reserves and an overreliance on glycolysis as a source for ATP. Further work will be required to understand how these intrinsic metabolic adaptions influence and are themselves influenced by changes in the wider pulmonary niche, as exemplified by elevated BAL lactate levels in donors with COPD. This vulnerability of COPD macrophages to defects in reserve energy capacity has consequence for key effector functions, with glycolysis only partially supporting healthy macrophage efferocytosis in the absence of oxidative phosphorylation.
With high oxidative stress, mitochondrial dysfunction, and suppressed oxidative phosphorylation features of COPD (14, 23, 52–54), we were interested to note the basal suppression of transcript for the enzyme ME1 in COPD macrophages. ME1 is the rate-regulating enzyme for the cytosolic malate–pyruvate shunt. In catalyzing malate conversion to pyruvate, it generates NADPH, augmenting the replenishment of reduced glutathione for redox power (32) and in turn shuttling pyruvate into the TCA cycle (55). Replicating ME1 loss in vitro reveals its critical role in macrophage redox balance, with resulting changes in basal mitochondrial ROS levels, but also highlights that loss of ME1 skews macrophage metabolism away from oxidative metabolism, with consequence for effector function such as efferocytosis. The specific mechanisms by which ME1 loss drives increased glycolytic flux remain to be fully explored and may relate not to only redox capacity but also a need to replenish intracellular pyruvate levels. To date, there is no evidence linking ME1 activity directly to macrophage polarization states. Specifically, a malic enzyme signature was not detected in the seminal paper on macrophage phenotype metabolism by Jha and colleagues (28). The specific mechanisms driving the depletion of ME1 in COPD are a subject for future work. Of note, however, we observe changes in ME1 expression to occur at both the protein and the transcript level. Coupled with evidence of altered activity of the ME1 transcriptional regulator NRF2 in the small airways of patients with COPD (56), global changes in DNA methylation states, and correlation between HDAC2 and NRF2 expression in COPD MDMs (57), this supports the concept of intrinsic rewiring of macrophage responses. We speculate that epigenetic changes within the bone marrow compartment would enable this reprogramming of the mononuclear phagocyte system with further modification by local cues within the airways of patients with COPD. We propose that it is the interplay between these central and peripheral epigenetic programs that regulates ME1 expression, with consequent changes in AM and MDM core effector functions.
Although ME1 was the only gene directly regulated by NRF2 that showed differential regulation at baseline in RNA-seq data sets comparing healthy control AMs with COPD AMs, previous work by our group has demonstrated a relative increase in protein expression of the NRF2 targets GLCL, NQO1, and HO-1 after NRF2 activation of COPD macrophages (7). It is likely, therefore, that ME1 is one of a number of factors contributing to the metabolic defects observed in COPD and responsive to NRF2 augmentation. This is further supported by our observation that other metabolic genes, including G6PC3, MAPK14, TLK1, and OARD1, are altered by activation of NRF2 activation in COPD AMs and the incomplete rescue of COPD macrophage effector function by KI-696. Understanding how regulators of transcriptional programs and metabolic plasticity in COPD macrophages interact with processes that are responsive to NRF2 augmentation to rescue effector function will be essential for the development of new therapeutic strategies. Equally, study of patients with early disease and smokers who have airway inflammation but are yet to develop airflow obstruction will also be of importance in defining the temporal relationship between defective macrophage metabolism and disease progression.
Acknowledgments
Acknowledgment
The authors thank Thomson Bioinformatics, Edinburgh, United Kingdom, for analyzing the transcriptomics data and K. Survana for procuring and L. Boswell (SUrF Facility, Edinburgh University) for processing histological samples. Flow cytometry data were generated with support from the QMRI Flow Cytometry facility, University of Edinburgh.
Footnotes
Supported by Wellcome Trust Senior Clinical Fellowship awards 098516 and 209220 (S.R.W.), Wellcome Trust Clinical Research Training Fellowship R43999 (E.M.R.), and an GlaxoSmithKline (investigator-led grant; D.H.D. and M.K.B.W.).
Author Contributions: E.M.R., P.S., P. Coelho, A.Z., A.J.M.H., J.C., W.V., B.G., G.R.B., B.J.M., and G. Cooper performed the research. E.M.R., J.M., G. Choudhury, and D.S. recruited patient cohorts and performed bronchoscopy. E.R.W., M.A.S.-G., M.B., P. Carmeliet, and D.A.C. contributed technical expertise. Y.S., W.R., and J.F.C. provided the GSK drug compound and contributed to experimental design. E.M.R., P.S., A.V.K., N.P., K.B., D.H.D., M.K.B.W., and S.R.W. provided scientific interpretation of the data. E.M.R., M.K.B.W., and S.R.W. designed the research and wrote the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202203-0482OC on February 1, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Willemse BW, ten Hacken NH, Rutgers B, Lesman-Leegte IG, Postma DS, Timens W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J . 2005;26:835–845. doi: 10.1183/09031936.05.00108904. [DOI] [PubMed] [Google Scholar]
- 2. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med . 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 3. Patel IS, Seemungal TAR, Wilks M, Lloyd-Owen SJ, Donaldson GC, Wedzicha JA. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax . 2002;57:759–764. doi: 10.1136/thorax.57.9.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oltmanns U, Sukkar MB, Xie S, John M, Chung KF. Induction of human airway smooth muscle apoptosis by neutrophils and neutrophil elastase. Am J Respir Cell Mol Biol . 2005;32:334–341. doi: 10.1165/rcmb.2004-0321OC. [DOI] [PubMed] [Google Scholar]
- 5. Berenson CS, Garlipp MA, Grove LJ, Maloney J, Sethi S. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis . 2006;194:1375–1384. doi: 10.1086/508428. [DOI] [PubMed] [Google Scholar]
- 6. Taylor AE, Finney-Hayward TK, Quint JK, Thomas CM, Tudhope SJ, Wedzicha JA, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J . 2010;35:1039–1047. doi: 10.1183/09031936.00036709. [DOI] [PubMed] [Google Scholar]
- 7. Bewley MA, Budd RC, Ryan E, Cole J, Collini P, Marshall J, et al. COPDMAP Opsonic phagocytosis in chronic obstructive pulmonary disease is enhanced by Nrf2 agonists. Am J Respir Crit Care Med . 2018;198:739–750. doi: 10.1164/rccm.201705-0903OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol . 2007;37:748–755. doi: 10.1165/rcmb.2007-0025OC. [DOI] [PubMed] [Google Scholar]
- 9. Russell REK, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol . 2002;26:602–609. doi: 10.1165/ajrcmb.26.5.4685. [DOI] [PubMed] [Google Scholar]
- 10. Molet S, Belleguic C, Lena H, Germain N, Bertrand CP, Shapiro SD, et al. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res . 2005;54:31–36. doi: 10.1007/s00011-004-1319-4. [DOI] [PubMed] [Google Scholar]
- 11. Hiemstra PS. Altered macrophage function in chronic obstructive pulmonary disease. Ann Am Thorac Soc . 2013;10:S180–S185. doi: 10.1513/AnnalsATS.201305-123AW. [DOI] [PubMed] [Google Scholar]
- 12. Bewley MA, Preston JA, Mohasin M, Marriott HM, Budd RC, Swales J, et al. Impaired mitochondrial microbicidal responses in chronic obstructive pulmonary disease macrophages. Am J Respir Crit Care Med . 2017;196:845–855. doi: 10.1164/rccm.201608-1714OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med . 2011;3:78ra32. doi: 10.1126/scitranslmed.3002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabón MA, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med . 2016;22:163–174. doi: 10.1038/nm.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ryan EM, Coelho P, Cole J, Bewley MA, Budd R, Callahan J, et al. T1 defective metabolism drives macrophage dysfunction in COPD [abstract] Thorax . 2021;76:A1. [Google Scholar]
- 16. Ryan EM, Budd R, Bewley MA, Coelho P, Rumsey W, Sanchez Y, et al. Impaired bioenergetic profile of COPD macrophages [abstract] Am J Respir Crit Care Med . 2018:A7443. [Google Scholar]
- 17. Ryan EM, Budd R, Bewley MA, Coelho P, Rumsey W, Sanchez Y, et al. S115 mechanisms to reverse impaired macrophage efferocytosis in COPD [abstract] Thorax . 2017;72:A70. [Google Scholar]
- 18. Cacciottolo TM, Perikari A, van der Klaauw A, Henning E, Stadler LKJ, Keogh J, et al. DDD Study, CHANGE Study Collaborators, CheartED Study Collaborators Scientific business abstracts of the 113th annual meeting of the Association of Physicians of Great Britain and Ireland. QJM . 2019;112:724–729. doi: 10.1093/qjmed/hcz175. [DOI] [PubMed] [Google Scholar]
- 19. Gordon SB, Irving GR, Lawson RA, Lee ME, Read RC. Intracellular trafficking and killing of Streptococcus pneumoniae by human alveolar macrophages are influenced by opsonins. Infect Immun . 2000;68:2286–2293. doi: 10.1128/iai.68.4.2286-2293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bewley MA, Belchamber KBR, Chana KK, Budd RC, Donaldson G, Wedzicha JA, et al. Differential effects of p38, MAPK, PI3K or Rho kinase inhibitors on bacterial phagocytosis and efferocytosis by macrophages in COPD. PLoS One . 2016;11:e0163139. doi: 10.1371/journal.pone.0163139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hodge S, Matthews G, Mukaro V, Ahern J, Shivam A, Hodge G, et al. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am J Respir Cell Mol Biol . 2011;44:673–681. doi: 10.1165/rcmb.2009-0459OC. [DOI] [PubMed] [Google Scholar]
- 22. O’Beirne SL, Kikkers SA, Oromendia C, Salit J, Rostmai MR, Ballman KV, et al. Alveolar macrophage immunometabolism and lung function impairment in smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2020;201:735–739. doi: 10.1164/rccm.201908-1683LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim WJ, Lim JH, Lee JS, Lee S-D, Kim JH, Oh Y-M. Comprehensive analysis of transcriptome sequencing data in the lung tissues of COPD subjects. Int J Genomics . 2015;2015:206937–206939. doi: 10.1155/2015/206937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Belchamber KBR, Singh R, Batista CM, Whyte MK, Dockrell DH, Kilty I, et al. COPD-MAP consortium Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J . 2019;54:1802244. doi: 10.1183/13993003.02244-2018. [DOI] [PubMed] [Google Scholar]
- 25. Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol . 2010;185:605–614. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 26. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem . 2014;289:7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab . 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jha AK, Huang SC-C, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity . 2015;42:419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 29. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab . 2019;29:443–456.e5. doi: 10.1016/j.cmet.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Becker L, Liu N-C, Averill MM, Yuan W, Pamir N, Peng Y, et al. Unique proteomic signatures distinguish macrophages and dendritic cells. PLoS One . 2012;7:e33297. doi: 10.1371/journal.pone.0033297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Court M, Petre G, Atifi ME, Millet A. Proteomic signature reveals modulation of human macrophage polarization and functions under differing environmental oxygen conditions. Mol Cell Proteomics . 2017;16:2153–2168. doi: 10.1074/mcp.RA117.000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng F-J, Ye H-B, Wu M-S, Lian Y-F, Qian C-N, Zeng Y-X. Repressing malic enzyme 1 redirects glucose metabolism, unbalances the redox state, and attenuates migratory and invasive abilities in nasopharyngeal carcinoma cell lines. Chin J Cancer . 2012;31:519–531. doi: 10.5732/cjc.012.10088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murai S, Ando A, Ebara S, Hirayama M, Satomi Y, Hara T. Inhibition of malic enzyme 1 disrupts cellular metabolism and leads to vulnerability in cancer cells in glucose-restricted conditions. Oncogenesis . 2017;6:e329. doi: 10.1038/oncsis.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol . 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hassibi S, Baker J, Barnes P, Donnelly L. COPD monocyte-derived macrophages display hallmarks of senescence [abstract] 2022;60:LSC-0131. [Google Scholar]
- 36. Korytina GF, Akhmadishina LZ, Aznabaeva YG, Kochetova OV, Zagidullin NS, Kzhyshkowska JG, et al. Associations of the NRF2/KEAP1 pathway and antioxidant defense gene polymorphisms with chronic obstructive pulmonary disease. Gene . 2019;692:102–112. doi: 10.1016/j.gene.2018.12.061. [DOI] [PubMed] [Google Scholar]
- 37. Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, Kaga K, et al. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol . 2008;39:673–682. doi: 10.1165/rcmb.2007-0424OC. [DOI] [PubMed] [Google Scholar]
- 38. Goven D, Boutten A, Leçon-Malas V, Marchal-Sommé J, Amara N, Crestani B, et al. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax . 2008;63:916–924. doi: 10.1136/thx.2007.091181. [DOI] [PubMed] [Google Scholar]
- 39. Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest . 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Holmström KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open . 2013;2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res . 2002;62:5196–5203. [PubMed] [Google Scholar]
- 42. Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res . 2012;40:7416–7429. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, et al. Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein-protein interaction with high cell potency identified by fragment-based discovery. J Med Chem . 2016;59:3991–4006. doi: 10.1021/acs.jmedchem.6b00228. [DOI] [PubMed] [Google Scholar]
- 44. Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol . 2003;81:289–296. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x. [DOI] [PubMed] [Google Scholar]
- 45. Mirchandani AS, Jenkins SJ, Bain CC, Sanchez-Garcia MA, Lawson H, Coelho P, et al. Hypoxia shapes the immune landscape in lung injury and promotes the persistence of inflammation. Nat Immunol . 2022;23:927–939. doi: 10.1038/s41590-022-01216-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poliska S, Csanky E, Szanto A, Szatmari I, Mesko B, Szeles L, et al. Chronic obstructive pulmonary disease-specific gene expression signatures of alveolar macrophages as well as peripheral blood monocytes overlap and correlate with lung function. Respiration . 2011;81:499–510. doi: 10.1159/000324297. [DOI] [PubMed] [Google Scholar]
- 47. Schultze JL, Mass E, Schlitzer A. Emerging principles in myelopoiesis at homeostasis and during infection and inflammation. Immunity . 2019;50:288–301. doi: 10.1016/j.immuni.2019.01.019. [DOI] [PubMed] [Google Scholar]
- 48. Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, et al. Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell . 2018;175:1634–1650.e17. doi: 10.1016/j.cell.2018.09.042. [DOI] [PubMed] [Google Scholar]
- 49. Namgaladze D, Brüne B. Fatty acid oxidation is dispensable for human macrophage IL-4-induced polarization. Biochim Biophys Acta . 2014;1841:1329–1335. doi: 10.1016/j.bbalip.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 50. Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabón MA, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med . 2016;22:1002–1012. doi: 10.1038/nm.4153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep . 2016;17:684–696. doi: 10.1016/j.celrep.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 52. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, et al. COPDMAP Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol . 2015;136:769–780. doi: 10.1016/j.jaci.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H, et al. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J . 2015;29:2912–2929. doi: 10.1096/fj.14-268276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gleeson LE, O’Leary SM, Ryan D, McLaughlin AM, Sheedy FJ, Keane J. Cigarette smoking impairs the bioenergetic immune response to Mycobacterium tuberculosis infection. Am J Respir Cell Mol Biol . 2018;59:572–579. doi: 10.1165/rcmb.2018-0162OC. [DOI] [PubMed] [Google Scholar]
- 55. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA . 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vucic EA, Chari R, Thu KL, Wilson IM, Cotton AM, Kennett JY, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol . 2014;50:912–922. doi: 10.1165/rcmb.2013-0304OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mercado N, Thimmulappa R, Thomas CMR, Fenwick PS, Chana KK, Donnelly LE, et al. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem Biophys Res Commun . 2011;406:292–298. doi: 10.1016/j.bbrc.2011.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]