

Abstract
Mycobacterium abscessus (Mab) has emerged as a challenging threat to individuals with cystic fibrosis. Infections caused by this pathogen are often impossible to treat due to the intrinsic antibiotic resistance leading to lung malfunction and eventually death. Therefore, there is an urgent need to develop new drugs against novel targets in Mab to overcome drug resistance and subsequent treatment failure. In this study, SAICAR synthetase (PurC) from Mab was identified as a promising target for novel antibiotics. An in-house fragment library screen and a high-throughput X-ray crystallographic screen of diverse fragment libraries were explored to provide crucial starting points for fragment elaboration. A series of compounds developed from fragment growing and merging strategies, guided by crystallographic information and careful hit-to-lead optimization, have achieved potent nanomolar binding affinity against the enzyme. Some compounds also show a promising inhibitory effect against Mab and Mtb. This work utilizes a fragment-based design and demonstrates for the first time the potential to develop inhibitors against PurC from Mab.
Keywords: structure-guided, fragment-based drug discovery, Mycobacterium abscessus, cystic fibrosis, PurC, SAICAR synthetase
Graphic abstract.

Mycobacterium abscessus (Mab) is a rapidly growing species of nontuberculous mycobacteria (NTM) which is responsible for a wide range of soft tissue infections which have emerged as a major threat to individuals with cystic fibrosis.1–4 These infections are often impossible to treat due to their intrinsic antibiotic resistance and result in extremely high treatment failure rates of around 55% despite years of combination chemotherapy and can often prove fatal.5–8The current drug regimen involves a combination of antibiotics, such as Amikacin, and this may take up to 2 years to be complete. Therefore, there is an urgent need to develop novel drugs targeting Mab, as an alternative antibacterial strategy.
Phosphoribosylaminoimidazole succinocarboxamide synthetase or PurC (also known as SAICAR synthetase) is an essential enzyme involved in the de novo purine biosynthesis in bacteria and fungi.9 The enzyme catalyzes the transformation of 5-aminoimidazole-4-carboxyribonucleotide (CAIR) and L-aspartate to phosphoribosylaminoimidazole-succinocarboxa-mide (SAICAR) in the presence of adenosine triphosphate (ATP) and Mg2+ cofactors (Scheme 1).10,11
Scheme 1. Purine Biosynthetic Pathway, the Pathway Transforms PRPP (Phosphoribosyl Pyrophosphate) into IMP (Inosine Monophosphate) in 10 Biosynthetic Stepsa.

aSAICAR synthetase (PurC) catalyzes seventh step in the biosynthetic pathway which transforms CAIR (carboxyaminoimidazole ribonucleotide) into SAICAIR (succinylaminoimidazole carboxyamide ribonucleotide) using ATP and L-aspartate.
It is known that bacteria rely on the de novo pathway for their survival, as the pathway plays a crucial role in the synthesis of nucleic acid and nucleotide phosphate precursors for energy metabolism. Several studies in the past have shown that purine biosynthesis is vital for bacterial growth and persistence in the gut, blood, and lungs.12–14
While most bacterial PurC enzymes exist as homodimers, Mab and Saccharomyces cerevisiae orthologs function as monomers.15–18 In contrast, the human bifunctional ortholog (PAICS) is an octameric enzyme combining a central C-terminal ring of the AIR carboxylase domain and an outer N-terminal SAICAR synthetase domain.19 A comparison of Mab PurC and the SAICAR synthetase domain of the human ortholog shows distinct structural and sequence differences, providing the basis for selective inhibition of this enzyme.20 The structural and functional differences between bacterial PurC and PAICS makes PurC an excellent target for antimicrobial drug discovery.15,19,21,22
Fragment-based drug discovery has emerged as a successful approach for the identification of new drugs. Recently, another drug derived from a fragment-based approach secured a breakthrough approval from the FDA and this methodology is seen as a reliable way forward in small-molecule drug design.23 Fragments are low-molecular-weight molecules with low structural complexity. The hits that come from a fragment-based screening usually exhibit lower potency than larger molecules identified from a standard high-throughput platform. However, these fragments bind by making high-quality interactions to a hotspot on the biomolecular target leading to highly ligand efficient (LE) molecules.24,25
In this work, the application of a fragment-based approach targeting Mab SAICAR synthetase (MabPurC) was used to discover a new class of 4-amino-6-(pyrazol-4-yl)pyrimidine based inhibitors. The fragment elaboration strategies described here utilized the hits identified from both our in-house fragment library and XChem fragment screening facilities as starting points for development.20 Through this study, for the first time, we were able to validate the essentiality of PurC in Mab and demonstrate the potential for developing inhibitors against this bacterial enzyme.
■. Results And Discussion
Generation of purC Gene Knockout Mutants
In order to assess the suitability of targeting PurC for antimicrobial drug development, essentiality studies of PurC in Mab were performed. This approach made use of a recombineering system to generate knockout mutants in M. abscessus subsp. massilliense. The resulting knockouts were analyzed by PCR and compared with that of the wild-type. The knockout strains did not grow in normal growth media, but colonies appeared when the media was supplemented with hypoxanthine, thus supporting the essentiality of the purC gene in bacterial growth and replication (Figure 1).
Figure 1.

(a) Schematic representation showing recombination of the allelic exchange substrate (AES) into the chromosome of M. abscessus subsp. massiliense followed by replacement of the purC gene with a Str cassette and PCR analysis of M. abscessus subsp. massiliense purC KO and wt confirming the insertion of the cassette. (b) Essentiality of the purC gene: Growth of M. abscessus subsp. massiliense purC KO strains on 7H11 ADC plates with or without added hypoxanthine (20 mg/mL).
Initial SAR Studies
Research within our group has identified several fragments that were found to bind into the adenine pocket of the ATP binding site of M. abscessus PurC. These were characterized using a range of biophysical techniques, including differential scanning fluorimetry (DSF), isothermal titration calorimetry (ITC), and X-ray crystallog-raphy.20 The fragment hits were identified using a dual strategy, first, the screening of an in-house fragment library of 960 fragments and, second, the screening of two diverse fragment libraries (125 and 768 fragments) at the XChem screening platform at the Diamond Light Source. The screening of the in-house fragment library led to the identification of eight fragments which were characterized using a range of biophysical techniques and X-ray crystallography. The screening of the two fragment libraries at XChem led to the identification of thirty five fragments, of which 60% of these were shown to bind at the ATP site by X-ray crystallography. However, no biophysical data was available for these fragments. One of the hits identified from the screening of the in-house fragment library was 4-amino-5-pyrimidine-carbonitrile, fragment 1. The X-ray crystal structure of MabPurC in complex with this fragment shows several key interactions which include (i) H-bonding of the amino moiety of 1 to the His69 side chain and to the main chain carbonyl of R91, (ii) H-bonding of the N3 of pyrimidine to the amide nitrogen of Leu93, and (iii) H-bonding of the nitrile nitrogen to the amide nitrogen of D213. These interactions are also made by the adenine ring of ATP when it binds to PurC. Along with its well-characterized interactions, this fragment also demonstrated a ΔTm of +3.6 °C and a Kd value of 340 μM (for ATP; Kd = 88 μM).
A further fragment screen was also performed on MabPurC with a more structurally diverse library using the high-throughput X-ray screening approach (XChem) available at the Diamond Light Source.26 With the access to the roboticized XChem facility,27–30 fragment 2 (XC1) was identified and its binding interactions to MabPurC were characterized (Figure 2).20 One of the distinct interactions that this fragment makes is the π-interaction between the pyridine ring of fragment 2 and the side chain of R17. The movement of the Arg side chain to accommodate the pyridine ring of fragment 2 was not previously observed from our in-house fragment screening and is not observed in the ATP-bound structure (PDB code 6YX3; Figure 2f). The flexibility of this R17 side chain was identified as beneficial towards further medicinal chemistry intervention to design chemical scaffolds that possess a slightly different mode of binding to that of ATP. This important aspect was therefore incorporated into a fragment-growing strategy from the ATP binding site.
Figure 2.

(a and b) X-ray crystal structure of Mab PurC in complex with ATP (PDB code 6YX3). ATP is shown as a yellow stick model with the side chain and key interactions within the active sites highlighted, water molecules as red and Mg2+ as green spheres, respectively. (c and d) Overlay of fragment 1 (PDB code 6Z0R) and fragment 2 (PDB code 6Z0Q) in ATP bound PurC. (e) Interactions of fragment 1, shown as a green stick model, with PurC. (f) Interactions of fragment 2, shown as a gray stick model, with PurC. The amino acids are represented as a gray line model.
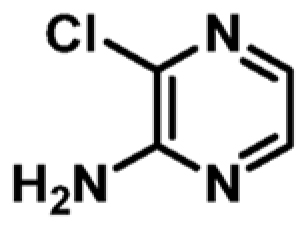
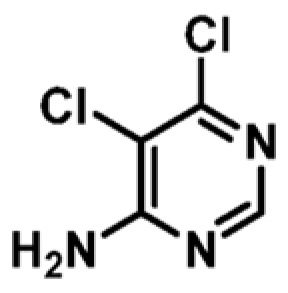
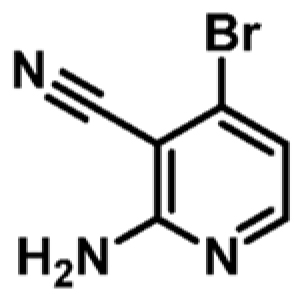
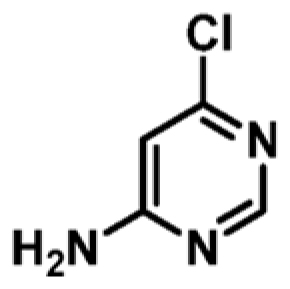
Prior to hit-to-lead elaboration, further DSF was performed on a number of close analogues of fragment 1 to examine preliminary SAR around the pyrimidine pharmacophore. As shown in Table 1, a comparison between fragment 1 (Kd = 341 μM, LE 0.53) and compound 4 (Kd = 1060 μM, LE 0.45) shows that the pyrimidine analogue exhibits a higher ΔTm and a 3-fold increase in the binding affinity compared to pyridine counterparts. The addition of a chlorine atom at the 6-position of the pyrimidine ring is tolerated as shown in compound 3 (Kd = 159 μM, LE 0.52) when compared to the original fragment 1. The replacement of a nitrile with a chlorine atom (compounds 3 vs 6) decreases ΔTm and shows a slight decrease in binding affinity (for compound 6; Kd = 442 μM, LE 0.51). This substitution could be beneficial for fine-tuning the physicochemical properties of a lead compound later in the optimization as compounds with a chlorine substituent show a higher clogP and lower PSA compared to compounds with a nitrile functional group.31 However, the removal of H-bond acceptor at this position (CN or Cl) diminishes the change in the melt-temperature (ΔTm) completely as seen in compound 8. This shows the significance of having a H-bonding acceptor present at the 5-position of pyrimidine. The change to the ring to a pyrazine ring 5 and the removal of N1 in pyrimidine 4 and 7 and removal of the cyano-group in the 5-position 8 all had a detrimental effect on the ΔTm.
Table 1. Biophysical Data for Selected Fragments Showing the Change in Protein Melting Temperatures (ΔTm), Binding Affinities (Kd), and Ligand Efficiencies (LE)a.
| Compound | Chemical structure | ΔTm (°C)b | MabPurC Kd (μM) | LEd |
|---|---|---|---|---|
| 1 |
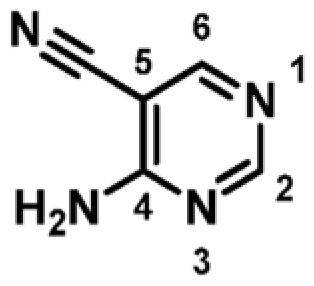
|
+3.6 | 341 ± 14 | 0.53 |
| 2 |
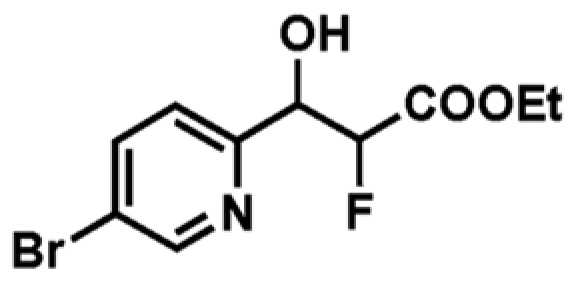
|
ND | ND | _ |
| 3 |
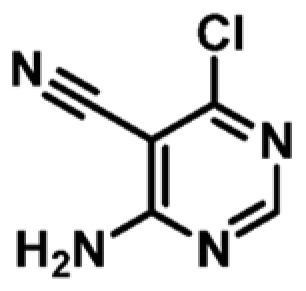
|
+4.0 | 159 ± 9 | 0.52 |
| 4 |
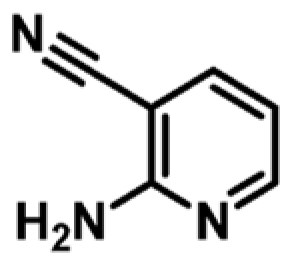
|
+1.7c | 1060 ± 252 | 0.45 |
| 5 |
|
+1.0 | 971 ± 29 | 0.51 |
| 6 |
|
+2.5 | 442 ± 22 | 0.51 |
| 7 |
|
+1.7 | ND | _ |
| 8 |
|
+0.7 | ND | _ |
ND not determined.
5 mM ligand and 100 μM MabPurC.
3 mM ligand and 70 μM MabPurC.
kcal mol–1 per heavy atom.
Hit-to-Lead Optimization
Fragment 1 was selected for further elaboration due to it having the highest ligand efficiency (LE) in the series. A closer examination of the X-ray crystal structure of fragment 1 with Mab PurC reveals a vector for elaboration, into the “ribose binding pocket”, at the 6-postion of the pyrimidine, which could be explored as an area for fragment growth as it is tolerated as seen in compound 3. Utilizing fragment elaboration at this position would allow the possibility of adding the feature of the pyridine moiety of fragment 2 into fragment 1. These two fragments are almost perpendicular to each other, and therefore it is necessary that a flexible linker be identified to join them together.
As shown in Table 2, the addition of a 4-pyrazolyl moiety effectively enhanced the binding affinity by an order of magnitude, resulting in the identification of compound 9 (Kd = 23 μM, LE 0.39). Even though the X-ray crystal structure of MabPurC in complex with compound 9 shows that the pyrazole does not make any significant polar interactions to MabPurC as demonstrated in Figure 3b, all the original polar contacts of the pyrimidine core have been retained. It is worth noting that the attached pyrazole 9 or pyridine (compounds 10 and 11) and the pyrimidine core align in a coplanar fashion (Figure 3).
Table 2. Exploration of SAR on the 6-Position of the Pyrimidine Ring against MabPurC 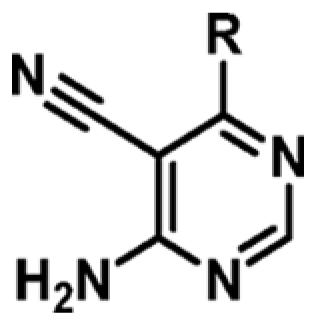 .
.
| Compound | R | ΔTm (°C)a | MabPurC Kd (μM) | LEb |
|---|---|---|---|---|
| 1 | H | +1.1 | 341 ± 14 | 0.53 |
| 3 | Cl | +2.1 | 159 ± 9 | 0.52 |
| 9 |
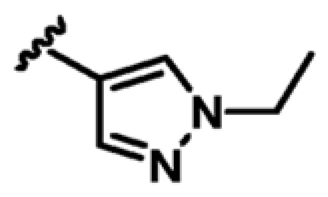
|
+4.4 | 23 ± 3 | 0.39 |
| 10 |
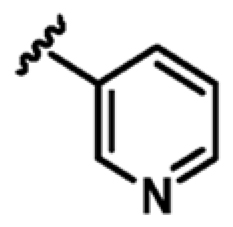
|
+2.0 | 588 ± 26 | 0.28 |
| 11 |
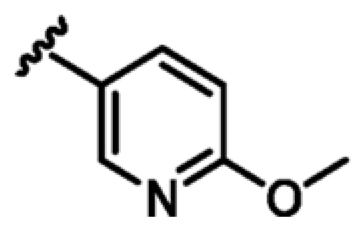
|
+2.6 | 84 ± 10 | 0.33 |
1 mM ligand and 100 μM Mab PurC.
kcal mol-1 per heavy atom.
Figure 3.

(a) Surface electrostatic representation of the X-ray crystal structures of Mab PurC in complex with fragment 1 (PDB code 6Z0R), occupying the ATP adenine pocket. (b–e) Mab PurC in complex with compounds 9 (PDB code 6YY6), 13 (PDB code 6YY8), 14 (PDB code 6YY7), and 16 (PDB code 6YY9) with the corresponding side chain and key interactions within the active sites illustrated beside each figure. The ligands are shown as a yellow stick model with the respective electron density map (|Fo| – |Fc| omit map) contoured at 2.0σ presented in gray mesh. The interacting amino acids are shown in gray line representation.
Following the identification of compound 9, guided by the structural information, various flexible groups and saturated side chains were examined in order to identify the best substituent to incorporate (Table 3). The morpholine ring was employed in a number of analogues, 12, 13, and 15, to assist with the solubility of the compounds. The compound 14 (Kd = 22 μM, LE 0.32) which has an ethyl ester side chain was chosen, and although an increase in its binding affinity was not observed, the added hydrophobic ethyl ester was identified by X-ray crystallography close to the region where the pyridine moiety of fragment 2 is located, Figure 3d. A similar binding mode was also observed in the X-ray crystal structure of 13 (Kd = 52 μM, LE 0.26), Figure 3c. Interestingly, the ethyl morpholine group in 13 occupies almost the same site as the pyridine in fragment 2. Even though these two compounds (14 and 13) did not show further improvement in binding affinity to MabPurC, the corresponding X-ray crystal structures have provided the critical information necessary for further fragment optimization (Figure 3c and d).
Table 3. Biophysical Data of 4-Amino-6-(pyrazol-4-yl)pyrimidine Derivatives against MabPurCa
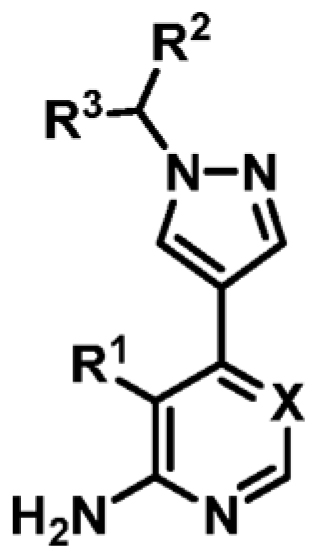 .
.
| Compound | R1 | R2 | R3 | X | ΔTm (°C)b | MabPurC Kd (μM) | LEd |
|---|---|---|---|---|---|---|---|
| 9 | CN | Me | H | N | +4.4 | 23 ± 3 | 0.39 |
| 12 | CN |
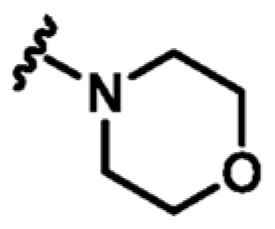
|
=o | N | +2.9 | 45 ± 5 | 0.27 |
| 13 | CN |
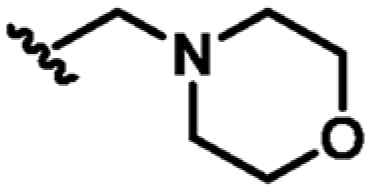
|
H | N | +3.5 | 52 ± 8 | 0.26 |
| 14 | CN |
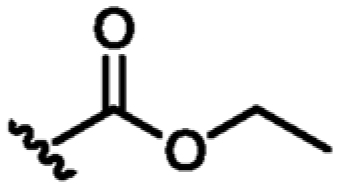
|
H | N | +4.5 | 22 ± 0 8 | 0.32 |
| 15 | CN |
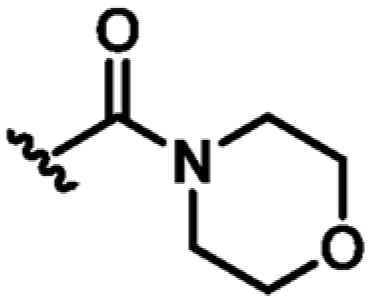
|
H | N | +2.6 | 47 ± 6 | 0.26 |
| 16 | CN | 3-pyridyl | H | N | +6.2 | 3.1 ± 0.3 | 0.36 |
| 17 | CN | 4-pyridyl | H | N | +6.0 | 7.5 ± 0.5 | 0.33 |
| 18 | CN | 5-methoxy-3-pyridyl | H | N | +6.5 | 2.1 ± 0.2 | 0.34 |
| 19 | CN | 3-fluorophenyl | H | N | +8.3 | 0.25 ± 0.03 | 0.41 |
| 20 | CN | 3-(trifluoromethyl)phenyl | H | N | +5.3 | ND | - |
| 21 | CN | 3-fluoro-5-methoxyphenyl | H | N | +8.8a | NDe | - |
| 22 | CN | 4-fluorophenyl | H | N | +9.4 | 0.22 ± 0.02 | 0.41 |
| 23 | CN | phenyl | H | N | +9.3 | 0.28 ± 0.03 | 0.43 |
| 24 | CN | phenyl | Me | N | +7.5 | 0.53 ± 0.03 | 0.38 |
| 25 | CN | benzyl | H | N | +5.3 | ND | - |
| 26 | CN | 3,5-difluorophenyl | H | N | +8.7e | 0.24 ± 0.05 | 0.37 |
| 27 | CN | 3,4-difluorophenyl | H | N | +9.0 | 0.15 ± 0.02 | 0.37 |
| 28 | Cl | 3-fluorophenyl | H | N | +7.1 | 0.35 ± 0.02 | 0.39 |
| 29 | CN | 3-fluorophenyl | H | CH | +5.1 | 1.4 ± 0.06 | 0.36 |
| 30 | Cl | 3-fluorophenyl | H | CH | +2.6c | ND | - |
| 31 | H | 3-fluorophenyl | H | N | +4.1 | ND | - |
ND not determined.
1 mM ligand and 100 μM Mab PurC
0.5 mM ligand and 100 μM Mab PurC
kcal mol–1 per heavy atom.
Due to poor solubility.
On the basis of the X-ray crystal structure of MabPurC in complex with compound 13, it was proposed that the incorporation of the 3-pyridylmethyl moiety at the N1 of the pyrazole ring would mimic the compound characteristics that engage the π-interaction seen with the pyridine moiety of fragment 2. The resulting compound 16 (Kd = 3.1 μM, LE 0.36) shows an order of magnitude increase in its binding affinity compared to compound 13. The alignment of this pyridine ring of compound 16 in the MabPurC binding site by forming π-stacking interaction to R17 side chain is crucial for the enhanced affinity observed (Figure 3e, Table 3).
Various substituted phenyl and pyridyl rings were examined to investigate the effect of the aromatic rings in forming the π interaction to R17. It is generally accepted that fluorobenzene is a good isostere of pyridine due to the similarity in dipole and electron density.32 There are many successful examples of an aryl C–F mimicking an azine nitrogen atom in drug discovery.33 This change is also deemed as a way to modulate the physicochemical properties of the compound as removing a nitrogen atom will reduce the compound’s basicity and increase its clogP at the same time. A number of analogues were synthesized, including pyridylmethyl and fluorobenzyl side chains. Compound 19 (Kd = 0.25 μM, LE 0.41) featuring a 3-fluorophenyl moiety shows another order of magnitude improvement in binding affinity while all key interactions to MabPurC are still observed in the corresponding X-ray crystal structure, Figure 4a.
Figure 4.

Surface electrostatic representation of the X-ray crystal structures of Mab PurC in complex with (a) compound 19 (PDB code 6YYA), (b) compound 24 (PDB code 6YYB), (c) compound 27 (PDB code 6YYD), and (d) compound 28 (PDB code 6YYC) with the side chain and key interactions within the active sites illustrated beside each figure. The ligands are shown as a yellow stick model with the respective electron density map (|Fo| – |Fc| omit map) contoured at 2.0σ presented in gray mesh. The interacting amino acids are shown in a gray line representation.
The modulation of the electronic nature of this aryl ring in compound 19 is crucial to the affinity jump observed, and various substituted phenyl analogues with electron-withdrawing and electron-donating groups were subsequently synthesized to test this hypothesis. The fluorine substituted compounds 22 (Kd = 0.22 μM, LE 0.41) and 27 (Kd = 0.15 μM, LE 0.37), Figure 4c, retain a strong binding affinity as observed in compound 19. Interestingly, compound 23, possessing an unsubstituted phenyl ring, also gave a Kd = 0.28 μM, with an LE of 0.43. Compound 24 (Kd = 0.53 μM, LE 0.38) where a methyl group was introduced on the methylene carbon, a derivative of compound 23, also displays a good affinity, suggesting that this modification is tolerated, and this could possibly offer a further vector for elaboration, Figure 4b.
The fragment SAR (Table 1) had shown that the interchange between the CN and Cl moieties is tolerated and the removal of the N1 atom in pyrimidine has a dramatic negative effect on binding affinity. As shown in Table 3, each compound (28, 29, 30, and 31) shares the same 3-fluorophenyl side chain as 19 but with a slightly different core structure. Compound 28 (Kd = 0.35 μM, LE 0.39) possessing a chlorine atom at the 5-position of pyrimidine rather than a nitrile group, exhibits a similar binding affinity as compared to 19 (Figure 4d), whereas compounds 29 and 30 led to a decrease in binding affinities. This observation corresponds to the results observed in early fragment SARs and could be important for further optimization, as the derivatives containing a Cl substituent would possess a higher lipophilicity in comparison to compounds having a CN moiety and therefore could play additional roles in determining compound permeability and retention in the mycobacteria
Phenotypic Screening of Compounds against Mab and Mtb
The minimal inhibitory concentrations (MIC) of the selected compounds with Kd < 50 μM were determined for both M. abscessus and M. tuberculosis in vitro (Table 4). The compounds were screened against Mtb as there is an overall percentage identity between the Mab and Mtb PurC orthologues of 75%. The important active site residues and those involved in the interactions with the compounds described above are 100% conserved between Mab and Mtb PurC (see Figure S7). Almost all of the compounds elaborated from the fragments exhibited complete growth inhibition at 50–200 μM against Mab. Whereas the compounds exhibited growth inhibition of 50 μM against Mtb, compounds 28 and 29 showed more promising inhibitory activity at 25 μM against Mtb (Table 3). The lack of significant MIC against Mab and Mtb shows the challenges of developing small molecules against mycobacteria. This lack of activity could be due to a number of factors. For example Mab has been shown to have a large number of efflux pumps, and any small molecules which get across the cell wall could be effluxed back out through these. In order to understand whether the compounds were getting across the Mab cell wall and engaging with PurC, hypoxanthine rescue experiments were carried out. Hypo-xanthine can be used as a salvage pathway of purine biosynthesis where this is converted to inosine monophosphate (IMP) by hypoxanthine-guanine phosphoribosyltransferase (HGPRT; Scheme 1). Compound 19 was examined with varying concentrations of hypoxanthine (Figure S8); however the results were inconclusive. While there seems to be rescue at a concentration of 200 μM, this is at the limit of the MIC measured. The chemotypes which have been identified that get into Mab tend to vary significantly. Many of the current antibiotics used to treat Mab infections have a log P less than zero (for example the aminoglycoside Amikacin) and many of these are highly hydrophilic. However, small molecule inhibitors (for example, those targeting Mmpl3 from Mab) which have been developed and show measurable effects on mycobacteria tend to have a log P > 4.34 The compounds that were developed in this work have log P values which range from −1.6 to 2.2, which could possibly explain the lack of activity on Mab.
Table 4. Antimycobacterial Profile of 4-Amino-6-(pyrazol-4-yl)pyrimidine Derivatives Screened against Mab and Mtb.
| compound | Mab PurC Kd (μM) | log Pa | Mab MIC (μM)b | Mtb MIC (μM)c |
|---|---|---|---|---|
| amikacin | NA | −8.9 | 3.1 | 12.5 |
| 12 | 45 ± 5 | −0.8 | 50–200 | NA |
| 13 | 52 ± 8 | −0.6 | 50–200 | NA |
| 14 | 22 ± 0.8 | −0.4 | 50–200 | NA |
| 15 | 47 ± 6 | −1.6 | 200–400 | 50 |
| 16 | 3.1 ± 0.3 | 0.2 | 50–200 | 50 |
| 17 | 7.5 ± 0.5 | 0.2 | 50–200 | 50 |
| 18 | 2.1 ± 0.2 | 0.03 | 50–200 | 50 |
| 19 | 0.25 ± 0.03 | 1.6 | 50–200 | 50 |
| 22 | 0.22 ± 0.02 | 1.6 | 50–200 | 50 |
| 23 | 0.28 ± 0.03 | 1.5 | 50–200 | 50 |
| 24 | 0.53 ± 0.03 | 1.8 | 50–200 | 50 |
| 26 | 0.24 ± 0.05 | 1.8 | 50–200 | 50 |
| 27 | 0.15 ± 0.02 | 1.8 | 50–200 | 50 |
| 28 | 0.35 ± 0.02 | 2.2 | 50–200 | 25 |
| 29 | 1.4 ± 0.06 | 1.8 | 50–200 | 25 |
log P was calculated using ChemDraw Professional Version 20.0
Mycobacterium abscessus subspecies abscessus (ATCC 19977) transformed with pmv310 plasmid expressing Lux ABDCE operon, grown in Middlebrook 7H9 broth supplemented with ADC.
Mycobacterium tuberculosis ΔleuD ΔpanCD (Bleupan) transformed with pSMT1 expressing Lux AB and GFP, grown in Middlebrook 7H9 broth supplemented with 0.5% glycerol, 0.05% Tween 80 (removed for 24 h prior to experiments), 10% OADC (BD), 0.05 mg/mL L-leucine, and 0.024 mg/mL calcium pantothenate, hygromycin, and zeocin (removed for 24 h prior to experiments).
Further work is currently ongoing first to further optimize the compounds developed and second to understand how these compounds engage with PurC within Mab.
Synthetic Chemistry
The early compounds in the series were synthesized following the routes shown in Scheme 2. Compounds 9, 10, and 11 were synthesized using a Suzuki coupling reaction between pyrazole boronic acid 32 or pyridine boronic acids 33 and 34 and 4-amino-6-chloropyr-imidine-5-carbonitrile 3 using Pd(t-Bu3P)2 as a catalyst in the presence of KF to afford the desired products. These were obtained in moderate to good yields (69% yield for 9, 25% for 10, and 24% for 11).35 Compound 12 was synthesized from its corresponding 4-iodopyrazole 35 and 4-morpholinecarbonyl chloride to afford compound 36. This then underwent a Miyaura borylation reaction using B2pin2 in the presence of a Pd(dppf)Cl2 catalyst to yield the boronate ester 37 in good yield (53%).36 The boronate ester 37 was then reacted with the 4-amino-6-chloropyrimidine-5-carbonitrile 3 heterocycle using Suzuki reaction conditions (Pd(t-Bu3P)2, KF, under MW irradiation, 150 °C over 40 min) to obtain 12 in 37% yield. Compounds 13, 14, and 15 were synthesized from the corresponding pyrazole boronate esters and 4-amino-6-chloropyrimidine-5-carbonitrile under the Suzuki reaction conditions to yield the products in moderate yields (32% yield for 13, 38% for 14, and 19% for 15).36
Scheme 2. Synthesis of 4-Aminopyrimidine and 2-Aminopyridine Derivativesa.

aReagents and conditions: (a) Pd(t-Bu3P)2, KF, 1,4-dioxane, water, MW irradiation, 150 °C, 40 min; (b) 4-morpholinecarbonyl chloride, Et3N, DCM, overnight; (c) B2pin2, Pd(dppf)Cl2.DCM, KOAc, DMSO, MW irradiation, 80 °C, 3 h.
For most of the final products in the series (16–31), as depicted in Scheme 3, the synthesis began with the substitution reaction between pyrazole-4-boronic acid pinacol ester and an alkyl halide of choice (39–50) in the presence of a carbonate base (Cs2CO3 or K2CO3) to obtain the corresponding N-alkylated pyrazole boronate esters (51–62) in moderate to good yields (31% to quantitative yield).37 The Suzuki coupling reactions under the same conditions as described previously using Pd(t-Bu3P)2 as a catalyst were employed in the preparation of the final compounds, resulting in 20–81% yield of the desired products.
Scheme 3. Synthesis of 4-Aminopyrimidine and 2-Aminopyridine Derivativesa.

aReagents and conditions: (a) K2CO3 or Cs2CO3, MeCN, overnight. (b) Pd(t-Bu3P)2, KF, 1,4-dioxane, water, MW irradiation, 150 °C, 40 min. (c) (i) Pd2(dba)3, BINAP, t-BuONa, benzophenone imine, toluene, 80 °C, overnight; (ii) 1 M HCl, THF, 5 h.
Compound 7 was prepared according to the reported literature procedure starting from the bromination reaction of 4-methoxy-2-pyridone using phosphorus oxybromide (POBr3) followed by nucleophilic amination using aqueous ammonia to replace the bromine at the 2-position of pyridine.38 Compound 29 was synthesized using the same conditions as those for the Suzuki coupling reaction between previously prepared compound 54 and compound 7 in 61% yield. The synthesis of 30 began with the Suzuki coupling reaction between 54 and 2,3-dichloro-4-iodopyridine 65. The reaction proceeded in a 56% yield of 66 with the 4-iodo moiety being replaced with a pyrazole side chain. Compound 66 underwent a palladium-catalyzed Buchwald–Hartwig reaction with benzophenone imine, and the imine product obtained was then hydrolyzed in situ under acidic conditions using 1 M HCl to afford the desired 2-aminopyridine 30 (63% yields over two steps).39
■. Conclusion
The application of a fragment-based approach towards the discovery of novel inhibitors of M. abscessus PurC is described. A combination of in-house fragment screening and high-throughput X-ray crystallographic screening provided crucial starting points for the fragment elaboration process. The fragment growing and merging strategies combined the structural characteristics of the two parent fragments, fragment 1 and fragment 2. The hit-to-lead elaboration, guided by X-ray crystallographic information and binding affinity data, resulted in the identification of compound 19 (Kd = 50 nM, LE 0.41) and compound 27 (Kd =150 nM, LE 0.39) both possessing promising in vitro binding affinities. The fragment growing and merging approaches described further demonstrate the strength of fragment-based design in accelerating the overall hit-to-lead progression. These results are encouraging and this study, as a first example of designing inhibitors against bacterial PurC, shows the potential for developing novel antimicrobials targeting this enzyme. However, this study also highlights the challenges in developing small molecules against targets in M. abscessus. While small molecules with high affinities can be developed using the fragment-based approach, the challenge is ensuring that these inhibitors get across the cell wall of M. abscessus and engage with the target in vivo. Work is currently underway to improve the overall physicochemical profiles of the lead compounds described herein in order to enhance their affinity against M. abscessus.
■. Methods
The characterization data for all of the compounds described can be found in the Supporting Information.
Mycobacterium abscessus purC gene Knockout Studies
Deletion of purC gene from M. abscessus subsp massilliense CIP108297 was carried out by recombineering as described previously. Briefly, primers were designed to amplify 1000 bp flanking regions upstream and downstream of the gene. A streptomycin cassette obtained from pHP45W was cloned between these fragments to create an allelic exchange substrate (AES). M. abscessus subsp massilliense containing a modified version of pJV53 with the xylE gene (pJV53-xylE) was induced for 4 h with 0.2% acetamide and electroporated with 500 ng of AES. Cells were incubated at 37 °C overnight and plated on 7H11 ADC containing streptomycin 200 mg/mL for selection and hypoxanthine 20 mg/mL to complement growth of the mutant. After 5 days colonies were picked on plates with or without hypoxanthine and checked by PCR to confirm deletion of the gene.
Amplification of purC Gene from Mycobacterium abscessus Genomic DNA
Genomic DNA sample was obtained from Mycobacterium abscessus (ATCC 19977). The stock of genomic DNA (6 ng/mL) was diluted using sterile water to make a working concentration of 0.6 ng/mL. The purC gene (MAB_0689) was amplified using the following primers (Sigma):
Forward Primer: 5’-ATTCCATGGTGCGTCCTTCG-CTGTCCGATTAC-3’
Reverse Primer: 5’-TATCTCGAGTCACGCCGACG-GGCCAATCC-3’
The following thermocycle program was used: stage 1 × 1 cycle, activation of polymerase at 95 °C for 2 min; stage 2 × 35 cycles, denaturation at 95 °C for 20 s, annealing at 70 °C for 20 s, extension at 70 °C for 45 s. This was followed by stage 3, final extension at 70 °C for 10 min.
Molecular Cloning
The purified PCR product and a PHAT2 E. coli expression vector containing Ampr and noncleavable N-terminal His-tag were then subjected to restriction digestion with Nco1 and XhoI restriction endonucleases (ThermoScientific). The ligation of the digested insert and vector was performed using T4 DNA ligase (New England Biolabs), by incubation at room temperature for 10 min. The ligation product was transformed into E. coli DH5a competent cells by the heat-shock method and plated on LB agar-kanamycin plates and incubated at 37 °C. Single colonies were randomly picked on the following day and inoculated in LB media with kanamycin (30 mg/mL) and grown overnight at 37 °C. Plasmids from the resulting cultures were isolated and purified (ThermoScientific GeneJet Plasmid Miniprep Kit). The integrity of the clones was confirmed by sequencing (DNA Sequencing Facility, Department of Biochemistry, University of Cambridge, UK).
Expression and Purification of PurC Protein
E. coli BL21 (DE3) strains containing N-His-PurC pHAT2 plasmids were grown overnight at 37 °C in LB-media containing Ampicillin (100 μg/mL). This seed-stage culture was used to inoculate six shake flasks containing 1 L each of 2XYT media with Ampicillin (100 μg/mL) until the optical density (A600nm) reached 0.6. Expression of the recombinant construct was induced by the addition of isopropyl β-D-1-thiogalactopyrano-side (IPTG) to a final concentration of 0.5 mM and further allowed to grow at 18 °C for 16 h. Cells were harvested by centrifugation at 4 °C for 20 min at 5000g, and the pellet was resuspended in buffer A (50 mM Tris-HCl pH 7.5, 350 mM NaCl, 20 mM Imidazole). Then, 10 μg/mL of DNaseI, 5 mM MgCl2, and protease inhibitor cocktail tablets (New England Biolabs) were added to the cell suspension. The cells were lysed by sonication (Branson), and the lysate was clarified by centrifugation at 4 °C for 40 min at 25 568 g. The clarified lysate was filtered using a 0.45 μm syringe filter and passed through a pre-equilibrated (with buffer A) 10 mL prepacked nickel-sepharose column (HiTrap IMAC FF, GE Healthcare). The column was washed with five column volumes of buffer A, and the bound protein was eluted as 2 × 12 mL elutes using buffer B (50 mM Tris-HCl pH 7.5, 350 mM NaCl, and 500 mM imidazole). The protein was analyzed on a 15% SDS-PAGE gel, and elutes from the HiTrap IMAC column containing the sample protein were pooled and subjected to dialysis against 2 L of buffer C (50 mM Tris-HCl pH 7.5, 350 mM NaCl) overnight at 4 °C. After overnight dialysis, the protein was concentrated using a 10 kDa centrifugal concentrator (Sartorius Stedim) and loaded onto a pre-equilibrated (with buffer D: 50 mM Tris-HCl, pH 7.5, 150 mM NaCl) 120 mL Superdex200 16/600 column (GE Healthcare). Two milliliter fractions were collected and analyzed on a 15% SDS-PAGE gel, followed by MALDI-Mass fingerprinting. Fractions corresponding to pure PurC protein were pooled, concentrated to 26 mg/mL, flash frozen in liquid nitrogen, and stored at −80 °C.
Crystallization of M. abscessus PurC
Screening of commercial sparse matrix libraries for the identification of appropriate crystallization conditions for M. abscessus PurC protein was performed as described previously.20 Drops containing 18 mg/mL of the protein in storage buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl) and reservoir were set up at drop ratios of 1:1 and 2:1 (of protein/reservoir, respectively) using a Mosquito crystallization robot (TTP labtech), in 96-well sitting drop plates. The drops were equilibrated against 80 μL of the corresponding reservoir solution at 19 °C. The best diffracting crystals were observed in the crystal condition containing 0.2 M LiSO4, 0.1 M Bis-Tris pH 5.5, and 25% PEG 3350.
Soaking of Fragments and Compounds on PurC Native Crystals
Crystals for soaking were grown at 19 °C in 48-well sitting drop plates in a grid consisting of 0.2 M lithium sulfate, 21–28% PEG 3350, and 0.1 M Bis-Tris pH 5.5–6.5, set up at drop ratios of 1:1 μL (of protein and reservoir, respectively). The crystals appeared in 3–5 days and were then moved into 24-well hanging drop plates to allow soaking in 4 μL drops containing the respective reservoir solutions and 10 mM fragments/compounds. The drops were then equilibrated against 800 μL of the respective reservoir solution for 16 h at 19 °C in 24-well hanging drop plates.
Co-crystallization of PurC Protein with ATP and Lead Compounds
For cocrystallization of PurC, 18 mg/mL of the protein was mixed with a 2–5 mM final concentration of the ligand/compound and incubated for 2 h on ice. The crystallization drops were set up using the incubated samples at a protein to reservoir drop ratio of 0.3:0.3 μL, in 96-well (MRC2) sitting drop plates in 0.2 M lithium sulfate, 21–28% PEG 3350, 0.1 M Bis-Tris at a pH of 5.5–6.5. The drops were equilibrated against 70 μL of reservoir at 19 °C.
X-ray Data Collection and Processing
The crystals were flash-cooled in cryo-solution consisting of 25% ethylene glycol in the corresponding reservoir. X-ray data sets were collected at the Diamond Light Source in the UK, using the rotation method at a wavelength of 0.979 Å; omega start, 0°; omega oscillation, 0.15°; total images, 2100–2400; exposure time, 0.05–0.08 s, and at the French National Synchrotron facility (Soleil), at a wavelength of 0.979 Å; omega start, 0°; omega oscillation, 1°; total oscillation, 210–240°; exposure time, 0.5 s. The diffraction images were processed using AutoPROC,40 utilizing XDS41 for indexing and integration, and POINTLESS,42 AIMLESS,43 and TRUNCATE44 programs from the CCP4 Suite45 for data reduction, scaling, and calculation of structure factor amplitudes and intensity statistics. PurC ligand bound structures were solved by molecular replacement using PHASER46 with the atomic coordinates of the Mycobacterium abscessus PurC apo structure as a search model, as described previously.20 Structures were refined using REFMAC47 and PHENIX.48 Model building was performed using the COOT49 interactive graphics program, and electron density maps were calculated with 2|Fo| – |Fc| and |Fo| – |Fc| coefficients. OMIT difference maps |mFo – DFc|50 were calculated and analyzed to verify positions of modelled ligands.
Thermal Shift Assay
Reactions were carried out in triplicate in a 96-well plate. Each reaction contained a final concentration of 20 μM PurC, 5 × SYPRO Orange, 100 mM HEPES at pH 7.5, 150 mM NaCl, 1 mM MgCl2, 5% DMSO, and 5 mM compounds 1–3 and 5–8. Compound 4 was screened at a concentration of 3 mM, and all other compounds were screened at a concentration of 1 mM. The plate was centrifuged briefly to remove bubbles and analyzed in a Bio-Rad CFX96 Touch or CFX Connect RT-qPCR machine. Samples were incubated at 25 °C for 10 min, and the temperature was then raised to 96 °C at a rate of 1 °C min–1 while fluorescence was recorded with excitation/emission wavelengths of 490/575 nm. Data were analyzed in Microsoft Excel to calculate the melting temperature (Tm) and ΔTm for each condition relative to the protein and DMSO-only control.
Isothermal Titration Calorimetry
ITC experiments were carried out in a buffer of 100 mM HEPES at pH 7.5, 150 mM NaCl, and 1 mM MgCl2. Protein was dialyzed against this buffer overnight before experiments and subsequent dilutions of protein and ligands used the remaining dialysis buffer. Generally, ligands at a concentration of 4 mM (ATP and CAIR), 2–5 mM (fragments and weakly binding compounds), or 0.2–1.5 mM (strongly binding compounds) were titrated against 20–100 μM PurC in a Malvern MicroCal iTC200 machine. To correct for enthalpy of dilution, ligands were also titrated against a buffer and the peak values of this run subtracted from the experimental run as a reference. When ligands were dissolved in DMSO, an appropriate concentration of DMSO (5–10% v/v) was added to other solutions. Data were analyzed using the Origin software (OriginLab, Northampton, MA, USA). The titration of an initial injection (0.2 μL) was discarded during data processing.
Phenotypic Screening against Mab
M. abscessus subspecies abscessus (ATCC19977) was transformed with pmv310 plasmid expressing Lux ABDCE operon, grown in Middlebrook 7H9 broth supplemented with ADC (Sigma, UK). Minimum inhibitory concentrations (MIC) were determined according to the Clinical and Laboratory Standards Institute (CLSI) method M07-A9. Mycobacteria were grown to an OD600 of 0.2–0.3 (Jenway 6300 spectrophotometer) in liquid culture, and 1 × 105 bacteria were added to each well of 96-well plates containing serial dilutions of compounds (400, 200, 100, 50, 25, 12.5, 6.3, 3.1, 1.6, 0.8, 0.4, 0 μM), in triplicate wells per condition, and incubated at 37 °C until growth was seen in the control wells. The MIC value was determined as the last well which showed no bacterial growth.
Supplementary Material
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.1c00432.
Compound synthesis and X-ray crystallography tables. LCMS of screened compounds (PDF)
■. Acknowledgments
This paper is dedicated to the memory of Professor Chris Abell, who passed away on October 26th, 2020. The authors would like to thank the Diamond Light Source for beam-time (proposals mx9537, mx14043, mx18548) and the staff of beamlines I03, I02, I04, I04-1, and I24 and the French National Synchrotron facility (Soleil) for assistance with data collection. The authors are grateful to Prof. Frank von Delft, Dr. Patrick Collins, and the XChem team at the DLS I04-1 beamline for access to fragment libraries and the high-throughput fragment screening facility. We thank Prof. Adam Nelson and his group at the University of Leeds for the threedimensional fragment library at the DLS XChem facility. S.E.T. was funded by the Cystic Fibrosis Trust and Fondation Botnar (Grant No. 6063). S.C. and S.G.G. were funded by the Fondation Botnar (Grant No. 6063). A.W. was funded by the EPSRC. R.A.F. is funded by the Cystic Fibrosis Trust Strategic Research Centre (SRC010) and Innovation Hub (IH01) awards, Wellcome Trust (I07032AIA), and Royal Papworth Hospital Research Award. V.M. was funded by the Bill and Melinda Gates Foundation SHORTEN-TB (OPP1158806). T.L.B. is funded by the Wellcome Trust (Investigator Award 200814_Z_16_Z). M.J. and J.M.B. are funded by the Cystic Fibrosis Strategic Research Centre (SRC010), M.A.G.d.E. is funded by the American Leprosy Mission (G88726).
■. Abbreviations
- SAICAR
phosphoribosylaminoimidazole-succinocarboxamide
- Mab
Mycobacterium abscessus
- Mtb
Mycobacterium tuberculosis
- DSF
differential scanning fluorimetry
- ITC
isothermal titration calorimetry
- SAR
structure activity relationship
- PSA
polar surface area
- LE
ligand efficiency
Footnotes
Author Contributions
C.A., A.G.C., T.L.B., R.A.F., V.M., and M.J. designed and supervised the project. S.C., A.C., A.J.W., and S.G.G. synthesized the compounds used in this study. S.E.T. and M.A.G.d.E. carried out the structural biology and biochemical studies. K.B. and J.S. devised and carried out the screening of the compounds against Mab and Mtb. J.M.B. and M.J. generated the knockout mutants. S.C., S.E.T., and A.G.C. wrote the manuscript, and all authors commented on the final version of the manuscript.
Notes
The authors declare no competing financial interest.
Contributor Information
Sitthivut Charoensutthivarakul, Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, United Kingdom; School of Bioinnovation and Biobased Product Intelligence, Faculty of Science, Mahidol University, Bangkok 10400, Thailand.
Sherine E. Thomas, Department of Biochemistry, University of Cambridge, Cambridge CB2 1GA, United Kingdom
Amy Curran, Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, United Kingdom.
Karen P. Brown, Molecular Immunity Unit, Department of Medicine, MRC Laboratory of Molecular Biology, University of Cambridge, Cambridge CB2 0QH, United Kingdom; Cambridge Centre for Lung Infection, Royal Papworth Hospital, Cambridge CB23 3RE, United Kingdom
Juan M. Belardinelli, Mycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, Colorado 80523-1682, United States
Andrew J. Whitehouse, Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, United Kingdom
Marta Acebrón-García-de-Eulate, Department of Biochemistry, University of Cambridge, Cambridge CB2 1GA, United Kingdom.
Jaspar Sangan, Molecular Immunity Unit, Department of Medicine, MRC Laboratory of Molecular Biology, University of Cambridge, Cambridge CB2 0QH, United Kingdom; Cambridge Centre for Lung Infection, Royal Papworth Hospital, Cambridge CB23 3RE, United Kingdom.
Subramanian G. Gramani, Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, United Kingdom
Mary Jackson, Mycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, Colorado 80523-1682, United States.
Vitor Mendes, Department of Biochemistry, University of Cambridge, Cambridge CB2 1GA, United Kingdom.
R. Andres Floto, Molecular Immunity Unit, Department of Medicine, MRC Laboratory of Molecular Biology, University of Cambridge, Cambridge CB2 0QH, United Kingdom; Cambridge Centre for Lung Infection, Royal Papworth Hospital, Cambridge CB23 3RE, United Kingdom
Chris Abell, Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, United Kingdom.
References
- (1).Liu H, Dong F, Liu J, Liu J, Pang Y, Zhao S, Lu J, Li H. Successful management of Mycobacterium abscessus complex lung disease in an otherwise healthy infant. Infect Drug Resist. 2019;12:1277–1283. doi: 10.2147/IDR.S198461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Esther CR, Jr, Esserman DA, Gilligan P, Kerr A, Noone PG. Chronic Mycobacterium abscessus infection and lung function decline in cystic fibrosis. J Cyst Fibros. 2010;9(2):117–23. doi: 10.1016/j.jcf.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bar-On O, Mussaffi H, Mei-Zahav M, Prais D, Steuer G, Stafler P, Hananya S, Blau H. Increasing nontuberculous mycobacteria infection in cystic fibrosis. J Cyst Fibros. 2015;14(1):53–62. doi: 10.1016/j.jcf.2014.05.008. [DOI] [PubMed] [Google Scholar]
- (4).Qvist T, Taylor-Robinson D, Waldmann E, Olesen HV, Hansen CR, Mathiesen IH, Hoiby N, Katzenstein TL, Smyth RL, Diggle PJ, Pressler T. Comparing the harmful effects of nontuberculous mycobacteria and Gram negative bacteria on lung function in patients with cystic fibrosis. J Cyst Fibros. 2016;15:380–5. doi: 10.1016/j.jcf.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chen J, Zhao L, Mao Y, Ye M, Guo Q, Zhang Y, Xu L, Zhang Z, Li B, Chu H. Clinical Efficacy and Adverse Effects of Antibiotics Used to Treat Mycobacterium abscessus Pulmonary Disease. Front Microbiol. 2019;10:1977. doi: 10.3389/fmicb.2019.01977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sfeir M, Walsh M, Rosa R, Aragon L, Liu SY, Cleary T, Worley M, Frederick C, Abbo LM. Mycobacterium abscessus Complex Infections: A Retrospective Cohort Study. Open Forum Infect Dis. 2018;5(2):ofy022. doi: 10.1093/ofid/ofy022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Raats D, Lorent N, Saegeman V, Vos R, van Ingen J, Verleden G, Van Raemdonck D, Dupont L. Successful lung transplantation for chronic Mycobacterium abscessus infection in advanced cystic fibrosis, a case series. Transpl Infect Dis. 2019;21(2):e13046. doi: 10.1111/tid.13046. [DOI] [PubMed] [Google Scholar]
- (8).Osmani M, Sotello D, Alvarez S, Odell JA, Thomas M. Mycobacterium abscessus infections in lung transplant recipients: 15-year experience from a single institution. Transpl Infect Dis. 2018;20(2):e12835. doi: 10.1111/tid.12835. [DOI] [PubMed] [Google Scholar]
- (9).Moffatt BA, Ashihara H. Purine and pyrimidine nucleotide synthesis and metabolism. Arabidopsis Book. 2002;1:e0018. doi: 10.1199/tab.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Manjunath K, Jeyakanthan J, Sekar K. Catalytic pathway, substrate binding and stability in SAICAR synthetase: A structure and molecular dynamics study. J Struct Biol. 2015;191(1):22–31. doi: 10.1016/j.jsb.2015.06.006. [DOI] [PubMed] [Google Scholar]
- (11).Tuntland ML, Fung LW. Substrate independent ATPase activity may complicate high throughput screening. Anal Biochem. 2016;510:18–20. doi: 10.1016/j.ab.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Samant S, Lee H, Ghassemi M, Chen J, Cook JL, Mankin AS, Neyfakh AA. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 2008;4(2):e37. doi: 10.1371/journal.ppat.0040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vogel-Scheel J, Alpert C, Engst W, Loh G, Blaut M. Requirement of purine and pyrimidine synthesis for colonization of the mouse intestine by Escherichia coli. Appl Environ Microbiol. 2010;76(15):5181–7. doi: 10.1128/AEM.00242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang N, Ozer EA, Mandel MJ, Hauser AR. Genomewide identification of Acinetobacter baumannii genes necessary for persistence in the lung. MBio. 2014;5(3):e01163-14. doi: 10.1128/mBio.01163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ginder ND, Binkowski DJ, Fromm HJ, Honzatko RB. Nucleotide complexes of Escherichia coli phosphoribosylaminoimidazole succinocarboxamide synthetase. J Biol Chem. 2006;281(30):20680–8. doi: 10.1074/jbc.M602109200. [DOI] [PubMed] [Google Scholar]
- (16).Levdikov VM, Barynin VV, Grebenko AI, Melik-Adamyan WR, Lamzin VS, Wilson KS. The structure of SAICAR synthase: an enzyme in the de novo pathway of purine nucleotide biosynthesis. Structure. 1998;6(3):363–76. doi: 10.1016/s0969-2126(98)00038-0. [DOI] [PubMed] [Google Scholar]
- (17).Wolf NM, Abad-Zapatero C, Johnson ME, Fung LW. Structures of SAICAR synthetase (PurC) from Streptococcus pneumoniae with ADP, Mg2+, AIR and Asp. Acta crystallographica. Section D, Biological crystallography. 2014;70(3):841–50. doi: 10.1107/S139900471303366X. [DOI] [PubMed] [Google Scholar]
- (18).Zhang R, Skarina T, Evdokimova E, Edwards A, Savchenko A, Laskowski R, Cuff ME, Joachimiak A. Structure of SAICAR synthase from Thermotoga maritima at 2.2 angstroms reveals an unusual covalent dimer. Acta crystallographica Section F, Structural biology and crystallization communications. 2006;62(4):335–339. doi: 10.1107/S1744309106009651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li SX, Tong YP, Xie XC, Wang QH, Zhou HN, Han Y, Zhang ZY, Gao W, Li SG, Zhang XC, Bi RC. Octameric structure of the human bifunctional enzyme PAICS in purine biosynthesis. J Mol Biol. 2007;366(5):1603–14. doi: 10.1016/j.jmb.2006.12.027. [DOI] [PubMed] [Google Scholar]
- (20).Thomas SE, Collins P, James RH, Mendes V, Charoensutthivarakul S, Radoux C, Abell C, Coyne AG, Floto RA, von Delft F, Blundell TL. Structure-guided fragment-based drug discovery at the synchrotron: screening binding sites and correlations with hotspot mapping. Philos Trans A Math Phys Eng Sci. 2019;377(2147):20180422. doi: 10.1098/rsta.2018.0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ducati RG, Breda A, Basso LA, Santos DS. Purine Salvage Pathway in Mycobacterium tuberculosis. Curr Med Chem. 2011;18(9):1258–75. doi: 10.2174/092986711795029627. [DOI] [PubMed] [Google Scholar]
- (22).Wolf NM, Abad-Zapatero C, Johnson ME, Fung LWM. Structures of SAICAR synthetase (PurC) from Streptococcus pneumoniae with ADP, Mg2+, AIR and Asp. Acta Crystallographica Section D Biological Crystallography. 2014;70(3):841–850. doi: 10.1107/S139900471303366X. [DOI] [PubMed] [Google Scholar]
- (23).Murray CW, Newell DR, Angibaud P. A successful collaboration between academia, biotech and pharma led to discovery of erdafitinib, a selective FGFR inhibitor recently approved by the FDA. Med Chem Comm. 2019;10(9):1509–1511. [Google Scholar]
- (24).Mendes V, Blundell TL. Targeting tuberculosis using structure-guided fragment-based drug design. Drug Discovery Today. 2017;22(3):546–554. doi: 10.1016/j.drudis.2016.10.003. [DOI] [PubMed] [Google Scholar]
- (25).Scott DE, Coyne AG, Hudson SA, Abell C. Fragmentbased approaches in drug discovery and chemical biology. Biochemistry. 2012;51(25):4990–5003. doi: 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- (26).Collins PM, Douangamath A, Talon R, Dias A, Brandao-Neto J, Krojer T, von Delft F. Achieving a good crystal system for crystallographic X-Ray fragment screening. Methods Enzymol. 2018;610:251–264. doi: 10.1016/bs.mie.2018.09.027. [DOI] [PubMed] [Google Scholar]
- (27).Pearce NM, Krojer T, Bradley AR, Collins P, Nowak RP, Talon R, Marsden BD, Kelm S, Shi J, Deane CM, von Delft F. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat Commun. 2017;8:15123. doi: 10.1038/ncomms15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Krojer T, Talon R, Pearce N, Collins P, Douangamath A, Brandao-Neto J, Dias A, Marsden B, von Delft F. The XChemExplorer graphical workflow tool for routine or large-scale protein-ligand structure determination. Acta Crystallogr D Struct Biol. 2017;73(3):267–278. doi: 10.1107/S2059798316020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Aimon A, Karageorgis G, Masters J, Dow M, Craven PGE, Ohsten M, Willaume A, Morgentin R, Ruiz-Llamas N, Lemoine H, Kalliokoski T, et al. Realisation of small molecule libraries based on frameworks distantly related to natural products. Org Biomol Chem. 2018;16(17):3160–3167. doi: 10.1039/c8ob00688a. [DOI] [PubMed] [Google Scholar]
- (30).Foley DJ, Craven PGE, Collins PM, Doveston RG, Aimon A, Talon R, Churcher I, von Delft F, Marsden SP, Nelson A. Synthesis and Demonstration of the Biological Relevance of sp(3)-rich Scaffolds Distantly Related to Natural Product Frameworks. Chem Eur J. 2017;23(60):15227–15232. doi: 10.1002/chem.201704169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Jones LH, Summerhill NW, Swain NA, Mills JE. Aromatic chloride to nitrile transformation: medicinal and synthetic chemistry. Med Chem Comm. 2010;1(5):309–318. [Google Scholar]
- (32).Meanwell NA. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J Med Chem. 2018;61(14):5822–5880. doi: 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- (33).Gillis EP, Eastman KJ, Hill MJ, Donnelly DJ, Meanwell NA. Applications of Fluorine in Medicinal Chemistry. J Med Chem. 2015;58(21):8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- (34).Li W, Yazidi A, Pandya AN, Hegde P, Tong W, Calado Nogueira de Moura V, North EJ, Sygusch J, Jackson M. Mmpl3 as a Target for the Treatment of Drug Resistant Nontuberculosis Mycobacterial Infections. Front Microbiol. 2018;9:1547. doi: 10.3389/fmicb.2018.01547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Williamson DS, Smith GP, Acheson-Dossang P, Bedford ST, Chell V, Chen IJ, Daechsel JCA, Daniels Z, David L, Dokurno P, Hentzer M, et al. Design of Leucine-Rich Repeat Kinase 2 (LRRK2) Inhibitors Using a Crystallographic Surrogate Derived from Checkpoint Kinase 1 (CHK1) J Med Chem. 2017;60(21):8945–8962. doi: 10.1021/acs.jmedchem.7b01186. [DOI] [PubMed] [Google Scholar]
- (36).Katz JD, Jewell JP, Guerin DJ, Lim J, Dinsmore CJ, Deshmukh SV, Pan B-S, Marshall CG, Lu W, Altman MD, Dahlberg WK, et al. Discovery of a 5HBenzo[4,5]cyclohepta[1,2-b]pyridin-5-one (MK-2461) Inhibitor of c-Met Kinase for the Treatment of Cancer. J Med Chem. 2011;54(12):4092–4108. doi: 10.1021/jm200112k. [DOI] [PubMed] [Google Scholar]
- (37).Westaway SM, Preston AGS, Barker MD, Brown F, Brown JA, Campbell M, Chung C-W, Drewes G, Eagle R, Garton N, Gordon L, et al. Cell Penetrant Inhibitors of the KDM4 and KDM5 Families of Histone Lysine Demethylases. 2. Pyrido[3,4-d]pyrimidin-4(3H)-one Derivatives. J Med Chem. 2016;59(4):1370–1387. doi: 10.1021/acs.jmedchem.5b01538. [DOI] [PubMed] [Google Scholar]
- (38).Trabanco AA, Tresadern G, Macdonald GJ, Vega JA, de Lucas AI, Matesanz E, García A, Linares ML, Alonso de Diego SA, Alonso JM, Oehlrich D, et al. Imidazo[1,2-a]pyridines: Orally Active Positive Allosteric Modulators of the Metabotropic Glutamate 2 Receptor. J Med Chem. 2012;55(6):2688–2701. doi: 10.1021/jm201561r. [DOI] [PubMed] [Google Scholar]
- (39).Herr RJ, Cioffi CL, Berlin ML. Convenient Palladium-Catalyzed Preparation of Primary Anilines Using a Fluorous Benzophenone Imine Reagent. Synlett. 2004;5:0841–0845. [Google Scholar]
- (40).Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne T. Data processing and analysis with the AutoPROC toolbox. Acta crystallographica Section D, Biological crystallography. 2011;67(4):293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kabsch W XDS. Acta crystallographica Section D, Biological crystallography. 2010;66(2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta crystallographica Section D, Biological crystallography. 2011;67(4):282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta crystallographica. Section D, Biological crystallography. 2013;69(7):1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).French SWK, Wilson K. On the treatmet of negatice intensity observations. Acta Crystallogr. 1978;A34:517–525. [Google Scholar]
- (45).Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, et al. Overview of the CCP4 suite and current developments. Acta crystallographica Section D, Biological crystallography. 2011;67(4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser Crystallographic Software. J Appl Crystallogr. 2007;40(4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta crystallographica Section D, Biological crystallography. 2011;67(4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66(2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60(12):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (50).Hodel A, Kim S-H, Brünger AT. Model Bias in Macromolecular Crystal Structures. Acta Cryst A. 1992;48:851–858. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.