

Abstract
We present chromosome-level genome assemblies from representative species of each of three independently evolved seagrass lineages, namely Posidonia oceanica, Cymodocea nodosa, Thalassia testudinum, and Zostera marina. We also include a draft genome of Potamogeton acutifolius, belonging to a freshwater sister lineage to Zosteraceae. All seagrass species share an ancient whole genome triplication, while additional whole genome duplications were uncovered for C. nodosa, Z. marina and P. acutifolius. Comparative analysis of selected gene families suggests that the transition from submerged-freshwater to submerged-marine environments mainly involved fine-tuning of multiple processes, e.g., osmoregulation, salinity, light capture, carbon acquisition and temperature, that all had to happen in parallel, likely explaining why adaptation to a marine lifestyle has been exceedingly rare. Major gene losses related to stomata, volatiles, defense, and lignification, are likely a consequence of the return to the sea rather than the cause of it. These new genomes will accelerate functional studies and solutions — as continuing losses of the ‘savannas of the sea’ are of major concern in times of climate change and loss of biodiversity.
Keywords: Alismatales, convergent evolution, Cymodocea nodosa, hexaploidy, Posidonia oceanica, Potamogeton acutifolium, seagrasses, Thalassia testudinum, whole genome duplication (WGD), whole genome triplication (WGT), Zostera marina
Introduction
Seagrasses are unique flowering plants, adapted to a fully submerged existence in the highly saline environment of the ocean, where they must root in reducing sediments, endure chronic light limitation, and withstand considerable hydrodynamic forces. In spite of these obstacles, the 80 or so species are among the most widely distributed flowering plants 1–3 with recently measured estimates of coverage ranging from 600,000 km2 4 to a modeled value of 1,6 million km2 5,6. Seagrasses fulfill many critical ecosystem functions and services including carbon sequestration, nutrient cycling, bacterial suppression, and coastal erosion protection 7–11. Along with mangroves, saltmarshes, and coral reefs, seagrass meadows are among the most biologically productive ecosystems on Earth. They act as breeding and nursery grounds for a huge variety of organisms including juvenile and adult fish, epiphytic and free-living algae, mollusks, bristle worms, nematodes, and other invertebrates such as scallops, crabs, and shrimp. Their importance for marine megafauna such as sea turtles, dugongs and manatees is unrivalled and their disappearance an important driver of the decline of these marine animals 12. Seagrasses also rank amongst the most efficient natural carbon sinks on Earth, sequestering CO2 through photosynthesis and storing organic carbon in sediments for millennia 13. While occupying only 0.1% of the ocean surface, seagrasses have been estimated to bury 27–44 Tg Corg per year globally, accounting for 10-18% of the total C burial in the oceans and being up to 40 times more efficient at capturing organic carbon than land-forests soils 14.
Previous work on Zostera marina 15,16 uncovered several unique gene family losses, as well as metabolic pathway losses and gains, that collectively underly novel structural and physiological traits, along with evidence for ancient polyploidy. Here, we expand on this work and present new chromosome-scale, high-quality reference genomes to understand the specific morphological and physiological adaptations that have enabled their worldwide distribution, except for Antarctica 1. These included Posidonia oceanica (L.) Delile (Posidoniaceae), Cymodocea nodosa (Ucria) Ascherson (Cymodoceaceae), and Thalassia testudinum K. D. Koenig (Hydrocharitaceae) to chromosome level assemblies, and a closely related freshwater-submerged alismatid, Potamogeton acutifolius Link (Potamogetonaceae), to draft level. Representative seagrass species within each family (Supplementary Figure 1.1) were chosen based on ecological importance, susceptibility to anthropogenic pressure, and availability of an extensive ecological literature. Briefly, Posidonia oceanica is the iconic Mediterranean seagrass and the largest in terms of plant size and physical biomass. It is a climax species characterized by extreme longevity and carbon storage capacity. Thalassia testudium (turtle grass) is a climax tropical species unique to the greater Caribbean region, with a single sister species endemic to the Indo-Pacific. Cymodocea nodosa is restricted mainly to the Mediterranean, Black and Caspian Seas, with an Atlantic extension along the Canary Island archipelago and along the subtropical Atlantic coast of Africa. It is the only temperate species of an otherwise disjunct tropical genus from the Indo-Pacific. The curly pondweed Potamogeton acutifolius belongs to the sister family of Zosteraceae and was chosen as its closest submerged freshwater sister taxon. We also included the recently upgraded genome of Zostera marina L. 17, which is found throughout the northern hemisphere and is arguably the most widespread species on the planet 18. To distinguish between adaptations to an aquatic lifestyle, and those unique to the ocean environment, our comparative analysis also included genomes of two recently sequenced emergent freshwater alismatids (which are rooted in underwater substrate, but have leaves and stems extending out of the water), along with the genomes of two distantly related salt-water tolerant mangrove species. In addition, representative transcriptomic data 16 of 89 Alismatales species was utilized to gain a more comprehensive view of shared and unique seagrass and freshwater adaptations within the order Alismatales (Supplementary Figure 1.1).
To better understand the extremely rare transition from a freshwater environment to a submerged saline environment, we compared gene family and pathway evolution across species, considering gene loss, as well as gene birth through small and large-scale gene duplication events, and investigated their effect on plant body structure (cell walls, stomata, hypolignification) and also investigated their relationship to physiological adaptations (hypoxia, plant defense, secondary metabolites, light perception, carbon acquisition, heat shock factors and especially salt tolerance mechanisms).
Results and Discussion
Genome assemblies and gene annotations
We assembled the genomes of T. testudinum, P. oceanica, and C. nodosa to chromosomal level using a combination of short sequence reads, PacBio HiFi, PacBio long reads, and Hi-C chromosome mapping. The novel seagrass genomes varied in haploid chromosome number from 6 to 18 and were very different in size, while containing approximately the same number of gene models (Supplementary Table 2.1.4). Further details of genome assembly and annotation, based on a combination of ab initio prediction, homology searches, RNA-aided evidence, and manual curation can be found in Methods, Supplementary Table 2.1.4., Supplementary Note 2.1, and Supplementary Table 2.1.3. BUSCO scores of >95% demonstrate the high level of completeness in the genomes. The prediction of non-protein coding RNA families (i.e., rRNAs, tRNAs, snoRNAs) for Z. marina, C. nodosa, P. oceanica, T. testudinum, and P. acutifolius can be found in Supplementary Note 3.1 and Supplementary Table 3.1.). Figure 1 shows the distribution of different genomic features along the reconstructed pseudochromosomes for the different seagrass species. Information on plastid and mitochondrial genomes can be found respectively in Supplementary Note 2.2 and Supplementary Note 2.3.
Figure 1. Distribution of the genomic features for the seagrass species T. testudinum, P. oceanica, Z. marina and C. nodosa.

Tracks from the inner to outer side correspond to gene density (blue); LTR/Gypsy density (green); LTR/Copia (orange); DNA transposable elements (pink) and chromosomes (with length in Mb). Curved lines through the center denote synteny between different genomic regions. Grey lines in A, B and C reflect synteny involving the WGD, whereas the three colored lines represent synteny with WGTs. Colored lines in D represent synteny and strong intragenomic conservation and should not be compared with colors in A, B and C (see text for further details). The distribution of the genomic features for the longest scaffolds of P. acutifolius, can be found in Supplementary Figure 2.1.2.
Information on Nuclear-mitochondria (NUMTs) and nuclear-chloroplast (NUPTs) integrants can be found in Supplementary Note 2.4 and Supplementary Table 2.4.
Genome Evolution
Transposable elements
Transposable elements (TE) comprise more than 85% of the genomes of T. testudinum and P. oceanica, as compared to only 65% for C. nodosa and Z. marina, and 40% for P. acutifolius (Supplementary Table 4.1). Long terminal-repeat retrotransposons (LTR-REs) are the major class of TEs and account for 72%, 66%, 46% and 42% in T. testudinum, P. oceanica, C. nodosa and Z. marina, respectively. LTR/Gypsy elements account for 63.18% in T. testudinum, 57.8% in P. oceanica and 32.11% in Z. marina, whereas the proportion of LTR/Copia elements was higher than that of LTR/Gypsy in C. nodosa and P. acutifolius. Bursts of TEs (especially LTRs) create new genetic variation that may be adaptive under conditions of stress. Over evolutionary time, different TE loads and distributions among species provide clues related to habitat differences and stress resistance 19,20. The insertion times of LTRs in the seagrass genomes (Methods) indicates a massive LTR/Gypsy burst around 200 thousand years ago (Kya) in T. testudinum (see y-axis), a moderate burst around 400 Kya in P. oceanica and Z. marina, but not in C. nodosa. By contrast, an expansion in Copia-elements happened around 2 Mya in C. nodosa but was weaker in P. oceanica, and nearly absent in T. testudinum and Z. marina. The recent TE gypsy burst (200 Kya) and older Copia burst (2 Mya median) coincide with drastic environmental fluctuations during Pleistocene ice ages (Supplementary Figure 4.1) and the timing of the trans-Arctic dispersal of Z. marina to the Atlantic from the Pacific 18. The Gypsy bursts at 400 and 200 Kya correspond to Marine Isotope Stage MIS12 and MIS6, two heavy glaciations that were followed by rapid warming 21.
Whole genome duplication, ancient (hexa)polyploidy and dating
Next, we revisited the established whole genome duplication (WGD) in Z. marina 15 and investigated whether evidence for ancient polyploidy could be found in the other seagrasses, which are all behaving as functional diploids 22. To this end, we used inferred age distributions of synonymous substitution rate (KS) for paralogs retained in collinear regions (anchor pairs), along with gene-tree/species-tree reconciliation methods (see Methods, Supplementary Note 4.2.1 and Supplementary Note 4.2.2). First, KS distributions of all seagrass species showed peaks indicative of ancient WGDs (Supplementary Figure 4.2.1)16. This was supported by intra- and intergenomic collinearity analysis (see Supplementary Note 4.2.1). Comparison of P. oceanica and T. testudinum with a reconstructed ancestral monocot karyotype (AMK 23) shows a clear 3:1 synteny relationship, while a comparison of Z. marina with the AMK exhibits a 1:6 synteny relationship (Supplementary Figure 4.2.2). Cymodocea nodosa was also found to show a 6:1 relationship compared to the AMK, while showing a 2:1 relationship with its sister species P. oceanica (Supplementary Figure 4.2.3), providing strong support for an additional WGD in C. nodosa after diverging from the P. oceanica lineage. Likewise, the freshwater species P. acutifolius was found to show a collinear relationship of 6:1 with the AMK and a 2:1 relationship with P. oceanica, and a 2:2 relationship with C. nodosa, while the colinearity relationship with its sister species Z. marina was more obscure (Supplementary Figure 4.2.4). However, these findings provide evidence that also P. acutifolius experienced an additional WGD event after its divergence with P. oceanica and C. nodosa. Of note, the overall 1:3 or 1:6 synteny relationships with the AMK suggested a hexaploid rather than a tetraploid ancestry for seagrasses and relatives.
Second, based on a KS analysis using ksrates 24, we were able to confirm that this paleohexaploidy is shared by P. oceanica, C. nodosa, Z. marina, and P. acutifolius, while the analysis was inconclusive for T. testudinum (Supplementary Figure 4.2.5). To resolve this issue, we applied a gene-tree/species-tree reconciliation approach using WHALE 25, which confirmed that the ancient whole genome triplication (WGT) event is shared by all seagrasses, and P. acutifolius. WHALE also supported the younger WGD in Z. marina is shared with P. acutifolius (Supplementary Note 4.2.2 and Supplementary Figure 4.2.6). Phylogenomic dating of the WGT (see Methods and Supplementary Note 4.2.3) further shows that most gene duplicates are reconciled on the branch leading to the most recent common ancestor (MRCA) of Potamogetonaceae, Zosteraceae, Posidoniaceae, Cymodoceaceae and Hydrocharitaceae, at approximately 86.96 (89.89 - 79.81) Mya (Figure 2 and Supplementary Figure 4.2.7a). Recently, Chen et al. 16 also reported a WGD shared by all core Alismatales (Supplementary Figure 1.1). However, these authors suggested a WGD rather than a WGT, which can be attributed to the lack of structural data, since their study was based solely on transcriptome data. Independent absolute dating of the shared WGD for P. acutifolius and Z. marina confirmed an earlier obtained date for the Zostera WGD of approximately 65 Mya (Supplementary Figure 4.2.7c-f), coinciding with the K/Pg boundary 15, which was also used to date a recent within-species phylogeographic study for Z. marina 18.
Figure 2. Time-calibrated phylogeny and WGT/WGD events across flowering plants that have chromosome-level genome assemblies.

The tree was inferred from 146 single-copy genes and show WGDs and WGTs based on inferences from the current study and previous analyses (Supplementary Table 4.2 and Supplementary Figure 4.2.8). For a more comprehensive tree showing the phylogenetic position of seagrasses within Alismatales, see Supplementary Figure 1.1. The dashed lines represent additional freshwater Alismatales species (phylogenetic position inferred using transcriptome data), mainly added for illustrative purposes to show non-monophyly of seagrass species. All branches have bootstrap support >98%. See text and Methods for details.
Adaptation to the Marine Environment
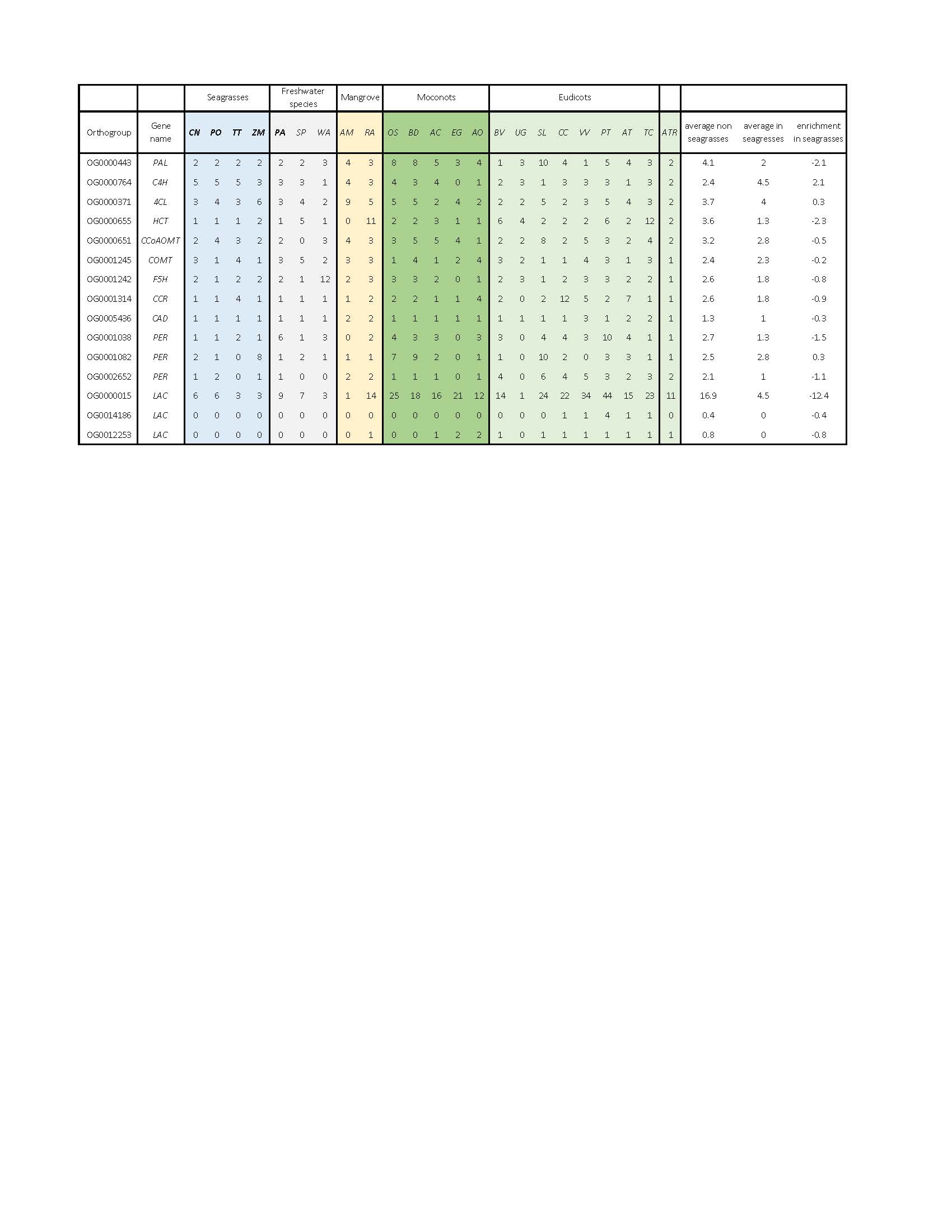
All three seagrass lineages characterized in this study share many specific morphological and physiological adaptations to their specific environment. Historically, a number of features were proposed as prerequisites for marine angiosperm life, such as tolerance to submergence, tolerance to salinity, hydrophilous pollination, and a capacity for vegetative anchorage 26,27. Previous studies have already reported genes potentially linked to the adaptation to the marine environment 15, while a recent study that conducted a broad transcriptome-based sampling of Alismatales uncovered some patterns of gene loss and gain also likely associated with aquatic and/or marine adaptation 16. Discrimination between aquatic (i.e., freshwater) and marine adaptations is not necessarily easy. To achieve greater insights into both adaptations, we used a common set of species for which full genome information is available (four seagrasses, three freshwater alismatids, and 16 other angiosperms, Figure 2 and Supplementary Note 4.3). We also utilized the extensive transcriptome dataset of Chen et al. (16) and broadly assessed commonalities and differences in gains and losses across gene families (further referred to as orthogroups, see Methods and Extended Data Table 1-10). The most important findings on adaptation to both aquatic-submerged, and marine conditions are summarized in Figure 3 (and Supplementary Figure 5.1).
Figure 3. The loss, contraction, and expansion of gene families involved in the adaption to a marine environment.

a) The normalized gene copy numbers for 4 seagrasses and 19 representative non-seagrass species. The normalization on the family dataset divides the gene count number of each species by the largest gene copy number within that family. The species order on the top of the heatmap is the same as that in Figure 1. The colors correspond to the different life-forms. The orange ones are terrestrial species; the green ones are emergent species (floating-leaved); the light blue ones are submerged species; the navy-blue ones are marine species (seagrasses) and the black ones are mangroves b) Salt stress signaling implies different ion channels. HAK5 encodes HIGH-AFFINITY POTASSIUM TRANSPORTER 5; CNGC, CYCLIC NUCLEOTIDE GATE CATION CHANNELs; AVP1 encodes Vacuolar H+-PPases c) Stomata differentiation from meristemoid mother cells (MMC) to guard mother cell (GMC), to guard cells. d) Ethylene synthesis and signaling. e) The hypoxia-responsive signaling in which the direct (ERF-VII) and indirect responsive (SnRK1) pathways are expanded. The rate-limiting enzyme (encoded by PFK4) in the glycolysis pathway, along with Lactate dehydrogenase (encoded by LDH), a rate-limiting enzyme in fermentation, are also expanded. F) Simplified schematic of the lignin and flavonoid biosynthesis pathways. Only steps that have significantly changed are shown. PAL encodes phenylalanine ammonialyase, which is the gateway enzyme of the general phenylpropanoid pathway; CHS encodes chalcone synthase, which is the first enzyme of flavonoid biosynthesis that directs the metabolic flux to flavonoid biosynthesis; GT1 encode flavonoid glycosyltransferases, which catalyze the final step of flavonoid biosynthesis to generate various flavonoid glycoside derivatives; GH1 encode flavonoid beta-glucosidase & myrosinase, which are responsible for the recycling of carbohydrate-based flavonoids; HCT encode Hydroxycinnamoyl‐CoA shikimate/quinate hydroxycinnamoyl transferase, channels phenylpropanoids via the “esters” pathway to monolignols”; LACCASEs encode the final enzymes in the pathway that oxidize monolignols to facilitate their polymerization into lignin. Panels d) e) f) genes in red are expanded; blue means contracted; The dashed line in the pathway means multiple metabolic steps.
Use it or lose it - convergence and specificity of gene losses
Under water, stomata are not required and may even be harmful for a submerged lifestyle because of the intrusion of water. Hence, seagrasses, and to a limited extent also freshwater alismatids, e.g., P. acutifolius, have reduced the number of genes involved in their development. Specifically, out of 30 orthogroups containing guard cell toolkit genes 28, eleven have been convergently and completely lost in seagrasses, while six others were significantly contracted compared to non-seagrass genomes (Figure 3a and Extended Data Table 1). Lost gene families include positive (SMF transcription factors), negative (EPIDERMAL PATTERNING FACTOR1 AND 2 (encoded by EPF1, EPF2), and TOO MANY MOUTHS (encoded by TMM)) regulators of stomatal development, as well as stomatal function (encoded by BLUS1, KAT1/2 and CHX20) (Figures 3a and 3c). Gene losses and contractions in the guard cell toolkit are also seen in the submerged freshwater alismatid P. acutifolius studied here, and to a less extreme degree in the floating alismatid S. polyrhiza (Figure 3a and Extended Data Table 1).
The aqueous habitat of seagrasses is also not conducive to emitting volatile substances as signals. Accordingly, we observed a convergent loss of orthogroups associated with volatile metabolites and signals. This includes the biosynthesis of triterpenes, and the volatile systemic acquired resistance signal, methyl salicylate 29 (Extended Data Table 2). Probably a more dramatic gene loss relates to ethylene biosynthesis and signaling (Extended Data Table 2). Two species, C. nodosa and Z. marina, do not contain ACS or ACO genes and hence, are not expected to produce ethylene or its precursor 1-aminocyclopropane 1-carboxylic acid (ACC). Moreover, they seem to have lost the ability to respond to ethylene, as indicated by a severe contraction of the early ethylene signal transduction components (Figure 3a and 3d) 15,16,30. In contrast, the downstream ethylene transcription factors (encoded by EIN3/EIL1/2) have been retained in all seagrasses, suggesting they can still exert ethyleneindependent functions. Remarkably, and unlike C. nodosa and Z. marina, T. testudinum and P. oceanica, as well as freshwater submerged species, retained some components for functional ethylene biosynthesis and signaling, as was also reported by Chen et al. 16. As diffusion of ethylene into water is extremely slow compared to diffusion in the air, ethylene rapidly accumulates in submerged organs. Such accumulation typically serves as a signal for submergence, and activating adaptive responses, such as formation of aerenchyma, adventitious rooting, shoot elongation, quiescence and priming the metabolism for efficient low-oxygen responses 31,32. However, while the accumulation of ethylene can be considered beneficial for the flooding tolerance of land plants, high levels and prolonged exposure to ethylene can have detrimental effects, such as stunted growth, senescence and abscission of leaves and flowers, root growth inhibition, and increased stress sensitivity 33. One possible mechanism that may prevent the accumulation of deleterious levels of ethylene, and thus explain its retention in T. testudinum and P. oceanica, is via epiphytic and endophytic bacteria that express ACC deaminases. This hypothesis is supported by the presence of multiple ACC deaminases in the metagenome of P. oceanica sediments 34, but needs further study.
Seagrasses increase their morphological flexibility to withstand hydrodynamic wave and current forces by a reduction in vascular tissues, the main site of lignification 35, consistent with the absence of vascular proliferation factor encoded by WOX4, and a contraction of the number of pericycle cell identity transcription factors (Figure 3a and Extended Data Table 3). This finding seems a more general adaption to aquatic lifestyles, as also suggested by analysis of the transcriptomes of different Alismatales (Supplementary Figure 5.1, this study, and ref (16)). The most severe reduction of the vascular bundle is seen in Z. marina which even lacks a pericycle36, a finding that correlates with the loss and divergence of the vascular proliferation regulators encoded by PXY and MONOPTEROS/ARF5 (Figure 3a and Extended Data Table 3). Notably, the lack of MONOPTEROS/ARF5 in Z. marina is further reflected in its inability to form an embryonic primary root 37. The general cellular hypolignification in seagrasses is reflected in the reduction in the number of LACCASEs encoding the final enzymes in the lignin pathway, which oxidize monolignols to facilitate their polymerization into lignin 38,39 (Figures 3a, 3e and Extended Data Table 4). The reduced need for the monolignol production is matched by a reduction of respectively PHENYLALANINE AMMONIA LYASE (encoded by PAL), and HYDROXYCINNAMOYL-COA SHIKIMATE/QUINATE HYDROXYCINNAMOYL TRANSFERASE (encoded by HCT) genes, which constitute entrance points into phenylpropanoid biosynthesis 40 (Figures 3a, 3f and Extended Data Table 4). Gene family contractions in lignin biosynthesis are also observed for the submerged freshwater species P. acutifolius and the freshwater floating species S. polyrhiza (Figure 3a).
Arbuscular mycorrhizal symbiosis (AMS) were and are critical for plant terretrialization 41,42 and are found in salt marsh plants, mangrove forests, and freshwater ecosystems 43–45. There is currently no evidence for any seagrass species to form mycorrhizal associations 46, which is reflected in the absence (secondary loss) of AMS-specific genes, with the sole exception of DMI3 in P. oceanica (Figure 3a). Gene loss of AMS-specific genes is also seen in freshwater submerged and floating species (Figure 3a). We also investigated so-called AMS-conserved genes, which have non-symbiotic roles 47 and discovered that seagrasses and P. acutifolius consistently retained a specific set of these conserved genes (DMI1, NUP85, NUP133, NENA, CCD7, CCD8 and MAX2) (Figure 3a). The absence of NSP1 and NSP2 is not unique to seagrasses but seems to be rather a common adaptation observed in aquatic environments (Supplementary Figure 5.1) and Proteales species 48.
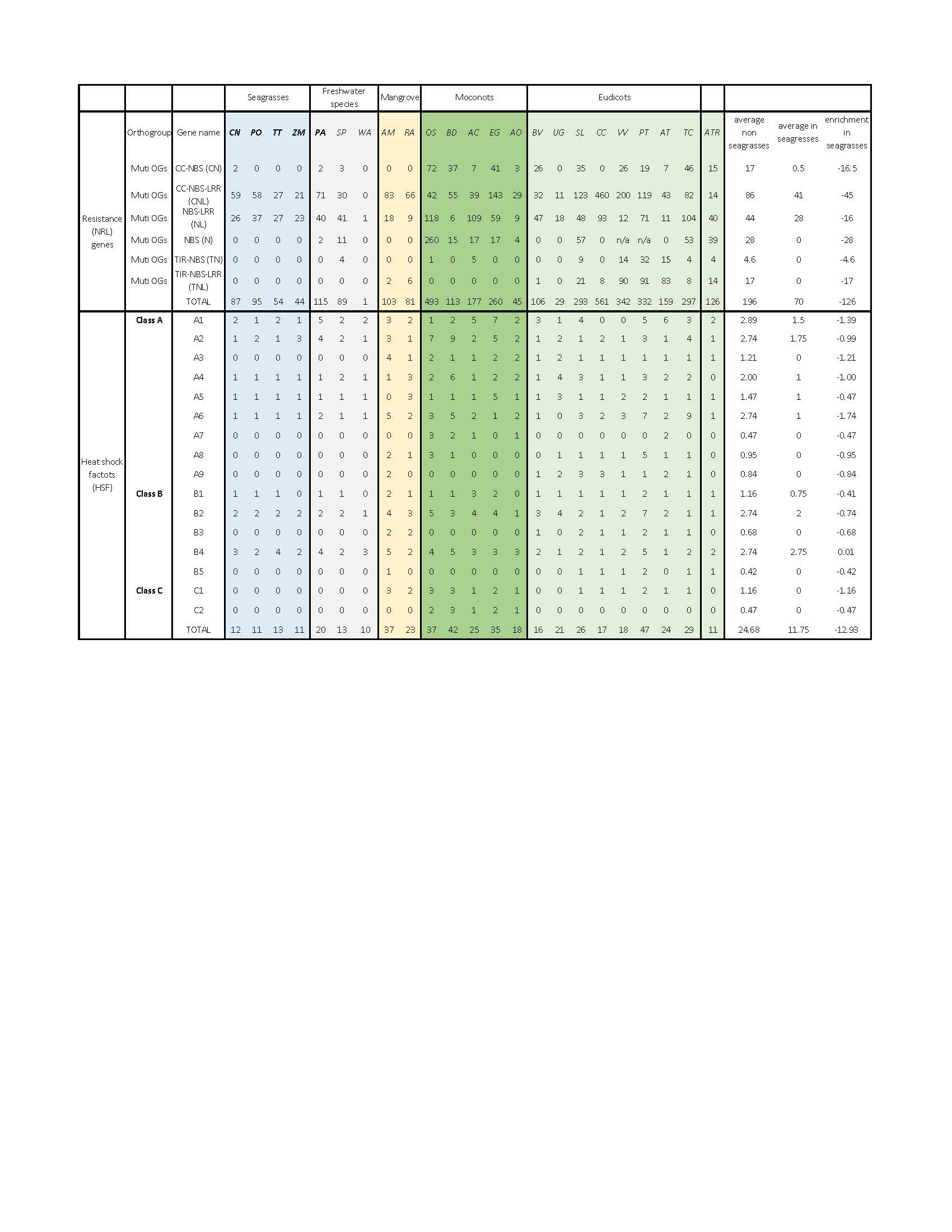
The pathogen landscape of the marine environment is associated with a different composition of plant resistance (R-genes) genes. In the seagrasses, there are fewer genes containing nucleotide-binding leucine-rich repeat receptors (NLRs) as compared to most other plants (Extended Data Table 5, Supplementary Note 5.2, Supplementary Table 5.2 and Supplementary Figure 5.2.1). As in many monocots, NLRs with a Toll/interleukin-1 receptor/resistance protein (TIR) domain are also completely absent in all seagrass lineages, as well as a few other NLR genes from the leucine rich repeat (LRR) domain. It is currently unclear what selective pressure was responsible for the unique R-gene composition of the seagrasses. Lower counts of disease resistance genes have also been observed for other aquatic plants 49.
Temperature fluctuations are much slower and show a lower amplitude in the marine compared to terrestrial environment 50. Accordingly, we observed a reduction in the number of plant heat shock transcription factors (HSFs) that are involved in the rapid activation of stress-responsive genes upon temperature changes, and which have been linked to the evolutionary adaptation of plants to the terrestrial environment 50. Seagrasses contain only about half the number of HSFs as compared with terrestrial plants (Extended Data Table 5, Supplementary Note 5.3 and Supplementary Table 5.3). Notably, only seagrasses belonging to the tropical genera retained some of the key heat stress-related HSFs from WGD and WGT events (Extended Data Table 5), which is consistent with their warmer native environment and higher heat stress tolerance compared to temperate seagrasses (P. oceanica and Z. marina).
Multi-level “tweaking” to adapt to the marine environment
Protective flavonoids and phenolics
Most seagrasses, except C. nodosa, seem to have greatly expanded the number of CHALCONE SYNTHASEs, which channel p-coumaroyl-CoA into flavonoid biosynthesis at the expense of monolignol biosynthesis (Figure 3a, 3f, and Extended Data Table 6). Flavonoids provide protection against UV and fungi, while enhancing recruitment of N-fixing bacteria 34,51,52. Flavonoids and other phenolics in seagrasses can be sulphated by the activity of cytosolic sulphotransferases to increase their water solubility and bioactivity in the marine environment 53,54. For example, the sulphated monolignol, zosteric acid (O-sulfonated p-coumaric acid) is an antifouling agent that prevents biofilm formation at the leaf surface 55. Cytosolic sulphotransferases are expanded in seagrasses, but significantly contracted in Potamogeton. However, flavonoid glycosyltransferases and flavonoid beta-glucosidases are contracted in both (Figure 3a, 3f, and Extended Data Table 6). Jointly, these data illustrate how rerouting precursors of the lignin biosynthesis pathway likely facilitated two traits, i.e., reduced rigidity, which appears to be a general aquatic adaptation, and sulphated protection, which contributes to the evolution of the marine lifestyle of seagrasses 34,54. In the case of P. oceanica, secreted phenolic compounds, together with anoxia, both inhibit microbial consumption of sucrose from root exudates 34.
Diverse mechanisms of cellular salt tolerance
Salt tolerance in flowering plants is a complex trait that involves multiple cellular processes 56. In the extreme case of invasion of highly-saline, marine environments, one might assume wholesale changes in salt tolerance mechanisms and/or the evolution of specialized features, such as salt glands in mangrove species. To date, no obvious specialized structures involved in salt tolerance have been identified in seagrasses. Instead, it seems that canonical salt tolerance mechanisms have been fine-tuned or “tweaked” towards higher efficiency on multiple levels. A major challenge associated with the marine environment is to prevent the accumulation of noxious levels of Na+ and Cl-, while allowing the efficient uptake of the essential ion K+. Angiosperms employ secondary Na+ transport mechanisms based on Na+/H+ antiporters fueled by a strong electrochemical H+ gradient. Surprisingly, no notable gene gains or losses were observed among the putative sodium transporting NHXs (NHX1 and SOS1/NHX7), except for C. nodosa, which contains a few extra copies of NHX1 and SOS1 orthologs (Extended Data Table 7). Instead of an increased number of genes, we observed similar amino-acid substitutions in regulatory domains of SOS1 orthologs in all four species (Supplementary Figure 5.4.1), indicating the possibility of altered regulation of SOS1/NHX7 in these species, a notion that is also supported by the loss of SOS3, a key regulator of SOS1 activity in C. nodosa (Extended Data Table 7). The electrochemical H+ gradients that fuel Na+ transport is established via H+ ATPases (encoded by AHA), V-ATPases and vacuolar H+-PPases (encoded by AVP1). Of these genes, only the AVP1 genes were obviously expanded in all the seagrasses, containing almost twice the number of AVP1 genes found on average in other angiosperms (Figure 3a and 3b). Interestingly, the expansion of AVP1-like genes can, at least partly, be linked to the ancient WGT followed by their specific retention, suggesting that these additional AVP copies were co-opted for adaptation to a marine lifestyle (Supplementary Figure 5.5.2). Indeed, overexpression of such PPases has been shown to improve salt tolerance in several angiosperms (e.g., Arabidopsis, poplar, sugar cane) 57–59, by enhancing Na+ sequestration in the vacuole 60. Analysis of the K+-channel repertoire in seagrasses reveals the loss of Shaker-type K+ channels (Supplementary Figure 5.4.2) 61, and a greatly reduced number of CYCLIC NUCLEOTIDE GATE CATION CHANNELs (Figure 3a, 3b and Extended Data Table 7). Moreover, the constant high K+ concentrations in seawater (9.7mM) renders high-affinity K+ transport systems superfluous, explaining the absence of AtHAK5 in all seagrass genomes (Figure 3a and 3b). Also, the Cl- transporter repertoire is reduced in seagrasses (Figure 3a and 3b), and seagrasses lack orthologs for NPF2.4 and ALMT12/QUAC1, CLC-A, B and CLC-E, likely reflecting their adaptation to a marine lifestyle (Figure 3a and 3b).
Maintaining the elasticity of the cell wall is another critical component of salt tolerance. The elasticity and structural strength of the cell wall are mainly dictated by components such as cellulose and pectins that cross-link the cellulose microfibrils. The bivalent cation Ca2+ stiffens the cell wall by establishing electrostatic bond between pectin strands. The excess of monovalent Na+ in seawater may displace the divalent calcium and hinder dimerization of homogalacturonan chains that are present in canonical pectin 62. In addition to the canonical pectin polysaccharides, seagrasses deposit apiogalacturonan in their cell walls 63. The borate-bridges that cross-link apiogalacturonan chains are less sensitive to sodium displacement, providing an advantage to plants grown under high salt condition 64. One of the few known key enzymes in the synthesis of apiogalaturonan is UDP-D-apiose/UDP-D-xylose synthase (encoded by Api), which converts UDP-D-glucuronate into UDP-D-apiose 65. Its expansion in seagrasses (in particular in Zostera and Cymodocea) is reflected in the cell-wall composition of seagrasses and therefore likely contributes to salt tolerance (Figure 3a). In addition, the apiogalacturonan could provide a way to incorporate boron into the cell wall, and protect seagrasses against its toxic effects.
Compared to terrestrial lineages, no major changes were observed for cellulose and hemi-cellulose biosynthesis (Extended Data Table 7). Notably, most of the salt related evolutionary changes in seagrasses are not reflected in the genomes of mangrove species (Avicinea marina and Rhizophora apiculate), which is consistent with the independent evolution of salt tolerance in mangrove species 66,67.
Coping with hypoxic sediments
The solubility of oxygen in seawater is limited (typically around 10 mL O2 L-1), while the sediments in which seagrasses grow are oxygen-free and reducing below a sediment depth of a few mm. This increases the O2 demand/draw-down by extensive belowground root-rhizome tissues that often comprise >50% of total plant biomass. Consistent with the increased risk of hypoxia, all seagrasses have expanded their repertoire of Plant Cysteine Oxidases (encoded by PCOs) and group VII Ethylene Responsive (ERF-VIIs) genes, for direct sensing and transcriptional adjustment to hypoxia (Figure 3a, 3e and Extended Data Table 8). As expected, most ERF-VIIs had higher expression in rhizomes and roots as compared to leaves (Supplementary Figure 5.5.1). Also, P. acutifolius contains an expanded hypoxia response machinery, reflecting its adaptation to submergence (Figure 3a). This is also supported by the transcriptome data of other Alismatales (Supplementary Figure 5.1) 16. Again, many, if not most, ERF-VII members reside within syntenic blocks retained from the WGT event in seagrasses, especially for P. oceanica and T. testudinum (Supplementary Figure 5.5.2). Such increases in the number of genes through whole genome duplication is also true for multiple hypoxia-related genes. Some examples are: (1) the PFK4 gene family, which encodes the rate-limiting enzyme in the glycolysis pathway (including enolases), expanded in both seagrasses and P. acutifolius, and derived from the WGT event (Supplementary Figure 5.5.2); (2) Lactate dehydrogenase, a rate-limiting enzyme in lactate fermentation, that is also expanded in seagrasses (Figure 3a and Extended Data Table 8) and has been shown to provide higher waterlogging tolerance in Arabidopsis upon overexpression 68; and (3) genes encoding the energy-sensing sucrose nonfermenting kinase SnRK1 69 and eIFiso4G1 (the dominant regulator in translational regulation by SnRK1 under hypoxia 70) (Extended Data Table 8) are increased as a result of the WGT (Supplementary Figure 5.5.2). In conclusion, we speculate that the increase and specific retention of many hypoxia responsive genes, subsequent to the WGT (dated at ~86 Mya), might have coincided with the Cenomanian-Turonian anoxic event (~91± 8.6 Mya, 71,72); if true, this low oxygen period may have helped to select for hypoxia tolerance in submerged species. In C. nodosa and P. acutifolius, additional recent lineage specific WGDs and tandem duplications may have also contributed to further expansion of the hypoxia responsive genes as a possible adaptation to submergence.
Light perception and photosynthetic carbon acquisition
Seagrass growth and zonation are constrained by light availability, as ocean waters rapidly attenuate photosynthetic active radiation with depth and modify its spectral quality, enriching blue while reducing red wavelengths 73. Most seagrass species grow in shallow water and even in the clearest waters, only a few species reach depths of 40 m or more. Dissolved inorganic carbon (DIC) is mainly available as bicarbonate (HCO3−) in seawater (nearly 90% DIC at normal pH) that needs to be exploited via special acquisition systems, as it cannot diffuse passively across the cell plasma membrane 74. The availability of dissolved CO2 for photosynthesis is instead limited to ~1% of the DIC pool, hence submerged plants and algae evolved CO2-concentration and convergent evolution of HCO3- to CO2 mechanisms (CCMs) to overcome this low availability. A recent report identified an evolutionary adaptation of RuBisCO kinetics across submerged angiosperms from marine, brackish-water and freshwater environments that correlates with the development and effectiveness of CCMs 75.
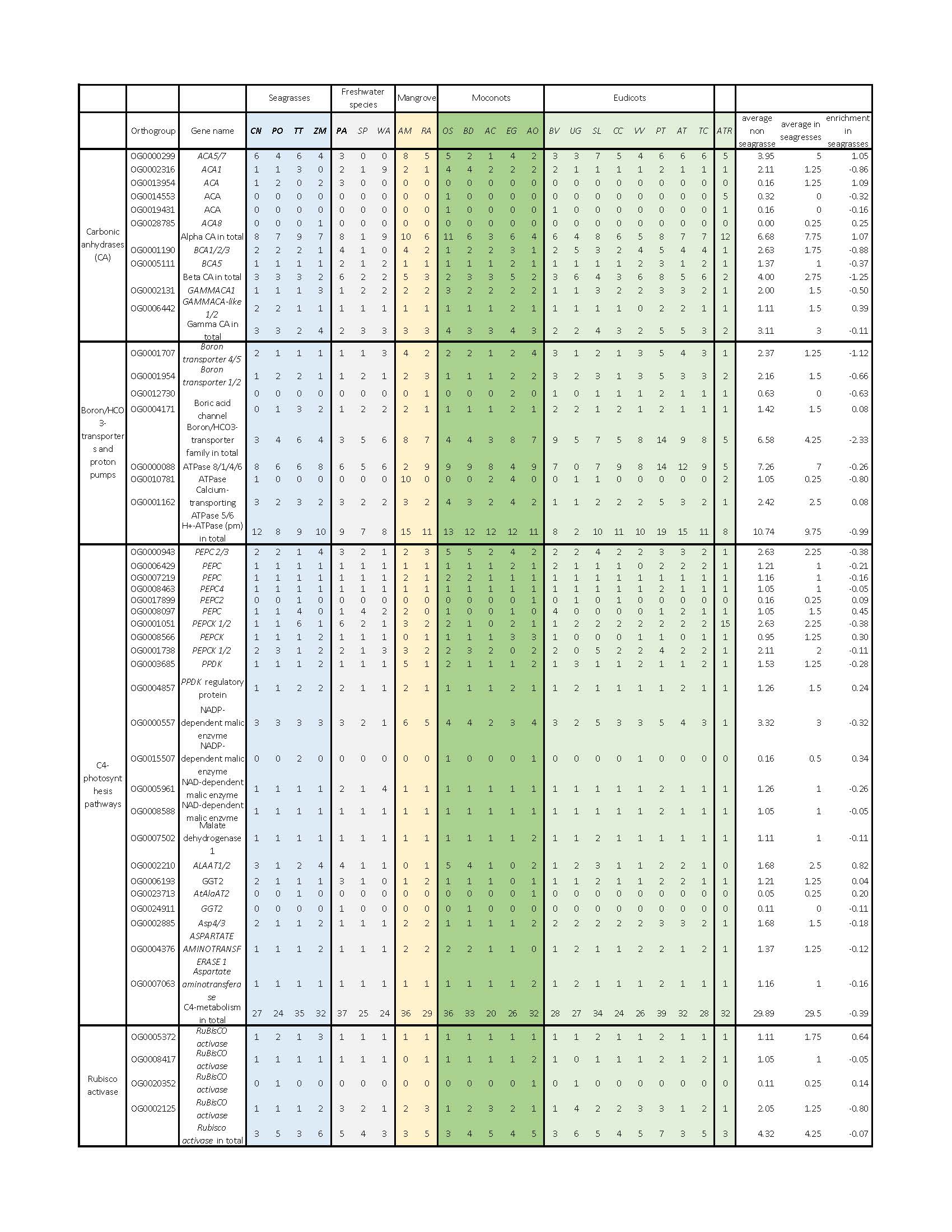
The analysis of genes related to inorganic carbon (Ci) acquisition revealed a slight increase in extracellular α-CA (encoding Carbonic Anhydrase α-type) copy number across the studied species (Supplementary Note 5.6.1). In P. oceanica and P. acutifolius, extra genes again have been specifically retained following the WGT event, although some copies also evolved local tandem duplications. α-CA OG0013954 was found to be specific to seagrasses (except for T. testudinum) and P. acutifolius (Extended Data Table 9 and Supplementary Table 5.6), and most of the corresponding genes are highly expressed in leaves (Supplementary Figure 5.6.1). This supports their involvement in Ci acquisition and possibly CCMs, as the presence of external CAs catalyzing the apoplastic dehydration of HCO3− to the RuBisCO substrate CO2, together with a higher activity of the extrusion proton pumps 76, likely evolved to alleviate dissolved inorganic carbon limitation in most seagrass species 77.
Our findings of a retention of 15 C4-related genes after WGT or WGD events (of which two encode PEPC) support the hypothesis that C. nodosa could be a C4 species 78, similar to what has been observed in P. acutifolius (Extended Data Table 9). Notably, none of the studied seagrass species possesses the Serine-residue characteristic of C4 Phosphoenolpyruvate carboxylase (PEPC), thus likely ruling out that a terrestrial-like C4-based (biochemical) CCM system is operating in seagrasses. This would suggest the presence of some kind of C3-C4 intermediate metabolism. Alternatively, homologs to C4 genes could have a role in the resistance of seagrasses to a variety of abiotic stresses, including salt stress 79.
Consistent with an augmented need for light capture, seagrasses show an expansion of LHCB (encoding light-harvesting complex B) as compared to freshwater plants that occur close to the water surface (Supplementary Figure 5.6.2 and Supplementary Note 5.6.2). Only C. nodosa had a number of LHCB genes comparable to the freshwater P. acutifolius and Spirodela spp. Other components of the photosynthetic machinery, including Photosystems I and II, are similar in gene number to other species, either freshwater or terrestrial (Supplementary Figure 5.6.2). Seagrasses have conserved the full repertoire of orthologous genes encoding photosensory proteins and components of the light signaling systems (Supplementary Figure 5.6.4 and Supplementary Note 5.6.3) that evolved in the green lineages during the different stages of plant terrestrialization 80.
Species-specific adaptation to UV tolerance and downstream regulation, and its relation to light habitat features during the invasion of the marine environment, appear to have differed among seagrass lineages (Supplementary Note 5.6.3). Those living at lower latitudes with intense UV-B radiation throughout the year (T. testudinum and C. nodosa) have kept the typical UVR8 of land plants along with their main regulatory proteins (encoded by RUP1,2). In contrast, Z. marina, as a higher latitude species, has lost the genes for both photoreceptors and their main negative regulatory proteins (Supplementary Figure 5.6.4), consistent with its lower exposure to UV-B radiation. In P. oceanica, a species restricted to the Mediterranean, the orthologous gene for UVR8 lacks the sequence region C27 engaged in the regulation of UVR8 reversion state from the activated to the inactivated state. The species-specific adaptation in the UV-signaling and its negative feedback regulation (Supplementary Figure 5.6.5), further reinforce the idea that ‘tweaking’ and not massive change of key traits and their regulatory mechanisms facilitated the invasion of the marine environment.
Perception of surrounding light cues is also critical for the entrainment of the circadian clock system which in turn is essential for regulation of basic physiology and the life cycle, e.g., daily water and carbon availability, and hormone signaling pathways 81. All seagrass species, except T. testudinum have lost the TIMING OF CAB1 (encoded by TOC1) gene (Supplementary Figure 5.6.4). The general reduction of clock genes in aquatic species suggests that the “absence of drought”, has led to a reduction of the regulatory daily-timing constraints for some metabolic and developmental plant processes. We find it interesting that all seagrasses have retained some genes related to the phytochromes light-signaling pathway. These include PIFs and LAF1 (Supplementary Figure 5.6.4) following WGT and WGD events, as well as genes related to the circadian clock and photoperiodism such as GI and ZTL (Supplementary Figure 5.6.4).
No Apical Meristem (NAC) Transcription Factors (TF)
NAC transcription factors (TF) are among the largest plant-specific-transcription factor (TF) families involved in signaling crosstalk events. They mediate development and aging programs and environmental stress signals. While a comparable number of sequences are found in seagrasses as compared to land plants, freshwater and mongrove species, specific orthogroups were restricted to seagrasses. One of them is annotated as Transcription factor JUNGBRUNNEN 1 (encoded by JUB1), a central longevity regulator that is also involved in (salt) stress tolerance. A detailed screening of sequences annotated as JUB1 across other plant genomes reveals sequence similarities and functional reorganizations among JUB1 found in C. nodosa and P. oceanica. Besides the sequence similarity between the two species, only C. nodosa sequences are expressed (Supplementary Note 5.7 and Supplementary Figure 5.7). This difference in functional regulation could potentially be linked to the different ecological tolerance of the two species to environmental factors. Although the two species can coexist, C. nodosa can colonize enclosed and shallow environments, which have higher fluctuation range and speed of salinity, light and temperature.
Nitrogen Metabolism
Key genes linked to nitrogen uptake/transport and assimilation have been retained in all seagrasses examined, although nitrate transporters (encoded by NRTs) are strongly contracted (Extended Data Table 10 and Supplementary Note 5.8). This implies that seagrasses may have evolved alternative mechanisms for nitrogen uptake and utilization. Although our results are not particularly revealing in this regard, recent work on seagrass microbiomes has shown that nitrogen acquisition involves nitrogen-fixing bacteria in the roots 82 and that epiphytic micro-organisms on the leaves mineralize amino acids via their heterotrophic metabolism 83. Gaining a more mechanistic understanding of the plant role in these interactions, is now possible for future investigations, given these new genomes.
Flower Development
Sexual reproduction in seagrasses occurs underwater (hydrophilous) by completely submerged male and female (unisexual) flowers. Their floral structures are simplified, often having reduced, or no, sepals and petals, which may represent an adaption to hydrophilous, and mostly abiotic, pollination 84. However, this striking morphological adaption is not reflected by a striking loss of genes defining the well-known ABC(D)E model for floral organ-specification 85,86 (Supplementary Table 5.9 and Figure 4a). In Z. marina, the B-function (encoding PISTILATA) homolog seems to be mainly expressed in the staminate (“male”) flower, while two C-function homologs (AGAMOUS; AGa and AGb) were mainly expressed in the pistillate (“female”) flower (Figure 4d), suggesting involvement of the B-function only in stamen development, and C-function in carpel development. In P. oceanica, the expression patterns differ from those in Z. marina, but largely agree with previously ascribed roles in floral organ patterning: B-function PI and C-function AG homologs are highly expressed in both staminate and pistillate flowers (Figure 4e). However, in both seagrasses, one A-function homolog, AGL6, is highly expressed in pistillate flowers, indicating the possibility of A-function neofunctionalization, transitioning from a role in sepals and petals to one being associated with pistillate flower development. The two SEP E-function homologs of three seagrasses are highly expressed in pistillate and staminate flowers, indicating an essential role of these flower-specific co-factors in organ specification. The discrepancy between the floral simplification and the presence of all types of floral organ identity genes in the seagrass genomes may reflect the instability of the floral ground plan between alismatid lineages 87, and is possibly affected by neofunctionalisation and shifts in expression domains of floral identity genes.
Figure 4. Flower development (like MADS-box genes) and pollen toolkit genes.

a) Phylogenetic tree of type II MADS-box genes in seagrasses and P. acutifolius, including Arabidopsis thaliana (AT) and Oryza sativa (Os) for reference. b) Gene expression patterns for type II MADS-box genes from various organs of Z. marina. Expression values were scaled by log2(TPM+ 1). c) Gene expression patterns for type II MADS-box genes from various organs of P. oceanica. Expression values were scaled by log2(TPM+ 1). d) The flowering ABCE model in Z. marina specifying female and male organs as proposed based on gene expression values (bar heights) from b. e) The flowering ABCE model in P. oceanica specifying female and male organs as proposed based on the gene expression values (bar heights) from c. f) Normalized gene copy numbers for MADS-box and pollen toolkit genes for 4 seagrasses and 19 representative non-seagrass species. Normalization for each gene family was obtained by dividing the number of genes in that gene family for a particular species by the largest gene copy number within that family (considering all species). Genes in black are absent. Taxa are arranged phylogenetically and colored by life form.
Hydrophilous pollination is extremely rare outside the seagrasses, leading to the proposal that it is one of the defining features of seagrasses 26. The majority of seagrasses have flexible, filiform pollen in which a rigid exine layer is structurally reduced or absent 88, likely facilitating hydrophilous pollination. Consistent with the loss or severe reduction of the exine layer, many genes involved in the biosynthesis and secretion of the exine layer (Supplementary Note 5.9) are absent in Z. marina 15, while C. nodosa, P. oceanica, and T. testudinum show partial gene loss (Figure 4f). It will be of interest to also investigate the role of pollen-specific genes, such as an orthologs of RESTORER OF FERTILITY 1 (encoded by RF-1), in the evolution of hydrophylous pollination. Supplementary Figure 5.9 shows flower and pollen development toolkit gene family expansion and contraction values for 96 species, including the 90 species-transcriptome data set of Chen et al. 16.
Conclusion
Seagrasses are now recognized as foundational species for invaluable ecosystems that provide multiple functions and services 9. They prevent erosion and hence preserve coastal seascapes, serve as biodiversity hotspots for associated animals, algae and plants, and have recently been proposed as a nature-based solution for climate mitigation owing to their carbon storage capacity in belowground biomass 89. Seagrasses also represent an extremely rare adaptation in the world of flowering plants, unlike (re-)adaptation to freshwater environments, which occurred at least 222 times in embryo-bearing plants 90. As far as is known, in part due to an extremely poor fossil record, seagrasses have evolved only on three different occasions from freshwater ancestors to (a group of) species that lives continuously submerged in a highly saline environment, including subaqueous pollination (except in Enhalus acoroides 91). Why only 84 species, spread across the three lineages, emerged in a time interval of 100 Mya, remains unresolved, but it may be related to high ocean connectivity on one hand 92, while within-species, ecological tolerance and phenotypic plasticity is high 93.
Comparative genome analysis has unveiled considerable convergence in seagrasses, but mainly for processes and pathways that have become redundant or even detrimental in a submerged marine environment. These include genes for stomata development, ethylene biosynthesis and signaling, pollen-coat formation, disease resistance, and heat shock transcription factors (HSFs). Jointly, these results illustrate that the invasion of the marine environment is associated with a significant loss of genes in multiple pathways that are no longer needed, a compelling example of “use it or lose it.”
Clear evidence of convergent positive (or gain of function) adaptation among the different lineages of seagrasses is harder to establish. Rather than unveiling major biological innovations including the rewiring of biological networks, adaptation to the marine environment seems mainly to involve the fine-tuning of many different/supportive processes that likely all had to happen in parallel, possibly explaining why the transitioning to a marine lifestyle has been exceedingly rare. For instance, adaptation of seagrasses to a marine (saline) environment was not accompanied by massive changes to individual salt tolerance traits, but rather involved more subtle changes in gene copy number and regulatory mechanisms, along with structural adaptations of the cell walls. This gradual modulation of preexisting mechanisms is consistent with the presence of multiple less extreme halophytes within alismatid families 94. The fine-tuning of many biological processes may also have facilitated the considerable phenotypic plasticity displayed by seagrass populations allowing their colonization from the tropics to the poles.
Many of the genes co-opted in different pathways in seagrasses seem to have been specifically retained following WGDs and WGTs that occurred long ago, suggesting important interdependencies of large-scale (or major) genome evolution events and evolutionary adaptation. Prime examples identified here are hypoxia-responsive genes, genes involved in salt tolerance, flavonoid metabolism, carbon acquisition, and C4-like photosynthesis. Therefore, the co-option of extra genes specifically retained following ancient whole genome duplications likely played a crucial role in facilitating survival in a marine environment.
We expect that the new, high-quality, seagrass genomes presented here will accelerate experimental and functional studies and contribute to transformative solutions in the management and conservation of seagrass ecosystems, which is an urgent concern in times of climate change and marine biodiversity crisis given the continuing worldwide loss of seagrass meadows.
Methods
Sampling metadata, DNA and RNA preparation
Whole plants from each species were collected from the field, transported to the lab in a cool box, cleaned, frozen in LN2 and then stored at -80°C. Collection and processing information are summarized in Supplementary Table 1.1. All samples were made with collection permits and followed the CBD-Nagoya Protocol. Care was taken to use tissue harvested from the basal area of young, clean leaves (10-cm pieces) to minimize epiphytic diatoms and bacteria If necessary. The seagrass tissues were then sent by overnight courier on dry ice to the Arizona Genomics Institute, Tucson, AZ, USA for extraction of nucleic acids (https://www.genome.arizona.edu). Quality controlled nucleic acid samples were then shipped on dry ice to the Joint Genome Institute (JGI) in Berkeley, CA, USA (https://jgi.doe.gov/) for further diagnostics and sequencing library preparation. For P. acutifolius, nucleic acids were extracted, QC’d and sequenced at the Max Planck-Genome-Centre Cologne, Germany (https://mpgc.mpipz.mpg.de/home/).
High Molecular Weight (HWM) DNA was extracted from young leaves of T. testudinium, P. oceanica, and C. nodosa, using the protocol of Doyle and Doyle (1987)95 with minor modifications. Young leaves, that had been flash frozen in LN2 and kept frozen at -80C, were ground to a fine powder in a frozen pestle and mortar with LN2 followed by very gentle extraction in CTAB buffer (that included proteinase K, PVP-40 and β-mercaptoethanol) for 20 mins at 37°C, followed by 20 mins at 50°C. Following centrifugation, the supernatant was gently extracted twice with 24:1 chloroform: iso-amyl alcohol. The upper phase was adjusted to 1/10th volume with 3M Sodium acetate (pH=5.2), gently mixed, and DNA precipitated with iso-propanol. DNA was collected by centrifugation, washed with 70% EtOH, air dried for few minutes and dissolved thoroughly in 1x TE at room temperature. Size was validated by pulsed field electrophoresis. HMW DNA for P. acutifolius was extracted from 2 g of young leaves with the NucleoBond HMW DNA kit (Macherey Nagel). Quality was assessed with a FEMTOpulse device (Agilent) and the quantity was measured by a Quantus fluorometer (Promega).
RNA was extracted from seagrass leaves, rhizomes, roots, and flowers (Supplementary Table 1.1) with the NucleoSpin RNA Plant and Fungi Kit (Macherey-Nagel, USA), and checked for integrity by capillary electrophoresis using an Agilent (Santa Clara, CA, USA) 2100 Bioanalyzer with the Agilent RNA 6000 Nano Kit following manufacturer’s instructions. RNA was extracted from leaves and roots of P. acutifolius with the RNAeasys Plant Kit (Qiagen), including an on-column DNase I treatment. Quality was assessed with an Agilent Bioanalyser and the quantity was calculated by an RNA-specific kit from Quantus (Promega).
Genome Sequencing
The genomes of T. testudinium, P. oceanica, and C. nodosa were determined following a whole genome shotgun sequencing strategy and standard sequencing protocols. Sequencing reads were produced using the Illumina NovaSeq platform and the PacBio SEQUEL II platform at the Department of Energy (DOE) Joint Genome Institute (JGI) in Walnut Creek, California, and the Hudson Alpha Institute in Huntsville, Alabama. One 400bp insert 2x150 Illumina fragment library and one HiC library was sequenced for each organism. Technical sequencing statistics are summarized in Supplementary Table 2.1.1. Prior to assembly, Illumina fragment reads were screened for PhiX contamination and reads composed of >95% simple sequences were removed. Furthermore, Illumina reads <50bp, after trimming for adapter and checking for quality (q<20), were also removed. For the Illumina sequencing, the final combined read set consisted of 4,284,278,120 high-quality reads with 161x coverage for T. testudinium, 6,543,657,580 high-quality reads with 327x coverage for P. oceanica, and 693,903,610 high-quality reads with 208x coverage for C. nodosa. For the PacBio sequencing, a total of 18 PB chemistry 3.1 chips (30-hour movie time) were sequenced with a HiFi read yield of 231.8 Gb with 51.53x coverage, 238.3 Gb with 79.44x coverage and 39.6 Gb with 79.24x coverage for T. testudinium, P. oceanica and C. nodosa, respectively.
For P. acutifolius, all libraries (PacBio, RNA and Tell-seq) and PacBio HiFi sequencing were performed at the Max Planck-Genome-Centre Cologne, Germany (https://mpgc.mpipz.mpg.de/home/). Short-read libraries and sequencing (RNA-seq and Tell-seq) were performed at Novogene Ltd (UK), using a NovaSeq 6000 S4 flowcell Illumina system. An Illumina-compatible was prepared with the NEBNext® Ultra™ II RNA Library Prep Kit for Illumina. PacBio-HiFi libraries were prepared according to the manual “Procedure & Checklist - Preparing HiFi SMRTbell® Libraries using SMRTbell Express Template Prep Kit 2.0” with an initial DNA fragmentation by g-Tubes (Covaris) and final library size selection on BluePippin (Sage Science). Size distribution was again controlled by FEMTOpulse (Agilent). Size-selected libraries were sequenced on a Sequel II with Binding Kit 2.0 and Sequel II Sequencing Kit 2.0 for 30 h (Pacific Biosciences). The same genomic DNA was used for TELL-seq but without fragmentation. Library preparation was done as outlined in the manual “TELL-Seq™ WGS Library Prep User Guide” (ver. November 2020). Illumina “sequencing-by-synthesis” was performed on a HiSeq 2500, 2 x 250 bp with additional index sequencing cycles to read out the unique fragment barcodes. Sequences were analyzed as recommended by Universal Sequencing Technology (UST, Canton, U.S.A). The final combined read set consisted of 54,401,190 Illumina high-quality reads with 13.4 coverage and 1,900,000 PacBio HiFi reads with 43.5 coverage (Supplementary Table 2.1.1)
Genome assembly
For T. testudinium, P. oceanica and C. nodosa, the following assembly strategy was used: the PacBio HiFi data was assembled using HiFiAsm and subsequently polished using RACON (https://github.com/lbcb-sci/racon). Due to the high heterozygosity of our sequenced seagrasses, both haplotypes were nearly complete resulting in a genome assembly composed of a highly contiguous primary set of chromosomes and a more fragmented alternative set of chromosomes (Supplementary Figure 2.1.1). For T. testudinium, the initial primary assembly consisted of 1,987 contigs with a contig N50 of 483.4 Mb, and a total assembled size of 4,866.1 Mb. For P. oceanica, the initial primary assembly consisted of 3,470 contigs, with a contig N50 of 355.8 Mb, and a total assembled size of 3,192.0 Mb (Supplementary Table 2.1.2). For C. nodosa, we produced an initial primary assembly of 1,362 contigs, with a contig N50 of 18.5 Mb, and a total assembled size of 466.0 Mb (Supplementary Table 2.1.2). Misjoins in the assemblies were identified using HiC data as part of the JUICER/JuiceBox pipeline96 for each of the three seagrass genomes. After resolving the misjoins, the broken contigs were then oriented, ordered, and joined together with HiC data using the JUICER/JuiceBox pipeline. In T. testudinum, there were 5 misjoins identified in the polished primary assembly, and a total of 15 joins were applied to the primary assembly to form the final assembly consisting of 9 chromosomes. In both the P. oceanica and C. nodosa polished primary genomes, there were no misjoins identified. A total of 6 joins were applied to the primary assemblies of P. oceanica and C. nodosa to form the final assembly consisting of 10 chromosomes and 18 chromosomes, respectively. Each chromosome join is padded with 10,000 Ns. Significant telomeric sequence was identified using the (TTTAGGG)n repeat, and care was taken to make sure that contigs terminating in telomere were properly oriented in the production assembly. The remaining scaffolds were screened against bacterial proteins, organelle sequences, GenBank nr and removed if found to be a contaminant. Heterozygous SNP/indel phasing errors were corrected using the HiFi data (51.53x for T. testudinum, 79.44x for P. oceanica and 79.24x for C. nodosa). Finally, homozygous SNPs and indels were corrected in the releases using Illumina reads (2x150, 400bp insert). A total of 2,613 homozygous SNPs and 82,421 homozygous indels were corrected in T. testudinum. A total of 1,643 homozygous SNPs and 100,570 homozygous indels were corrected in P. oceanica and total of 1,426 homozygous SNPs and 12,492 homozygous indels were corrected in the C. nodosa. Due to the high heterozygosity of the three genomes, both haplotypes of each chromosome were well represented in the assemblies. The primary set of chromosomes were constructed from the primary assembly, while an alternative set of chromosomes were constructed from the alternate assembly. Chromosomes for the alternate haplotype were then oriented, ordered, and joined together using synteny from the primary chromosomes (Supplementary Table 2.1.3).
For Potamogeton acutifolius, we used HiFiAsm 97 to assemble a draft genome assembly of a total length of 611 Mb with N50 = 3.09 Mb and scaffolded it further with Tell-seq data (linked reads; bioRxiv 2019, 852947) using the ARCS software 98 and reaching final N50 = 4.45 Mb (6,705 scaffolds in total, the length of the largest scaffold = 31.2 Mb).
Genome annotation
Structural and functional annotation of genes
Our annotation pipeline integrated three independent approaches, the first one based on transcriptome data, the second one being an ab initio prediction and the third based on protein homology. Both RNA-seq and Iso-seq data from different tissues (Supplementary Table 3.2.1 – Supplementary Table 3.2.4) were used to aid the structural annotation and RNA-seq datasets were first mapped using Hisat2 (v2.1.0, arguments dta) 99 and subsequently assembled into transcript sequences by Stringtie2 100, whereas Iso-seq sequences were aligned to the seagrass genome using GMAP 101. All transcripts from RNA-seq and Iso-seq were combined using Cuffcompare (v2.2.1) and subsequently merged with Stringtie2 (arguments merge -m 150) to remove fragments and redundant structures 100. Transdecoder v5.0.2 (github.com/TransDecoder) was then used to predict protein sequences with diamond v2.0.14 results (evalue 1e-5 max-target-seqs 1 -f 6). BARKER v2.1.2 102 was used for ab initio gene prediction using model training based on RNA-seq data. Homology-based annotation was based on the protein sequences from related species (Z. marina v1.0, Spirodela polyrhiza, Oryza sativa and Arabidopsis thaliana) as query sequences to search the reference genome using TBLASTN with e-value ≤1e–5, then regions mapped by these query sequences were subjected to Exonerate to generate putative transcripts. Additionally, an independent, homology-based gene annotation was performed using GeMoMa 103 using the same species with TBLASTN.
All structural gene annotations were joined with EvidenceModeller 104 v1.1.1, and BUSCO v4.0.4 (Benchmarking Universal Single-Copy Orthologs) 105 was used to assess the quality of the annotation results. Finally, we used GenomeView 106 to do the gene curations manually based on the RNA-seq and Iso-seq data. Putative gene functions were identified using InterProScan 107 with different databases, including PFAM, Gene3D, PANTHER, CDD, SUPERFAMILY, ProSite and GO. Meanwhile, functional annotation of these predicted genes was obtained by aligning the protein sequences of these genes against the sequences in public protein databases and the UniProt database using BLASTP with the e-value ≤1e − 5.
Annotation of non-protein coding RNA families
Finished genome assemblies and annotations (genome.fasta and genome.gff files for Z. marina, C. nodosa, P. oceanica, T. testudinum and P. acutifolius) were uploaded to, and later downloaded from, JGI Phytozome 108. Infernal v1.1.4 (Dec 2020) 109 was used to perform sequence similarity searches of each genome sequence versus the RFAM database (RNA families database, Dec2021) 110. The output from Infernal was filtered, keeping only the hits with an E-value threshold E<0.01. A second filtering step was performed to remove redundant information, i.e., overlapping matches with similar hits. A third filtering step was performed by retaining all the hits matching with a coverage of at least 95% and removing all partial/fragmented matches with incomplete hits from the reference collection. rRNA, tRNA, snoRNA and miRNA regions were selected and annotated in the annotation.jff files for each species. An updated functional annotation including the identified loci in the genomes was performed by scanning the Uniprot database 111 with BLASTp 3. Introns and the corresponding sequence regions were extracted by GenomeTools 112 and Bedtools 113 programs. The functional annotation of the long introns (>= 20kb) was performed by similarity searches in the NCBI nucleotide 114 database with the BLASTn tool 3.
Annotation of repeats and transposable elements (TEs)
Two complementary approaches were used to identify repetitive DNA sequences. First, a de novo repeat identification was carried out with RepeatModeler v2.0.1 (https://www.repeatmasker.org/RepeatModeler/) based on the default TE Rfam database, followed by RepeatMasker v4.1 (https://www.repeatmasker.org/) to discover and classify repeats based on the custom repeat libraries from RepeatModeler v2.0.1. Second, LTR_Finder 115 (v1.0.7), LTR_harvest 116 from genometools (v1.5.9) and LTR_retriever 117 (v2.9.0) were used to identify and trace the LTR elements, which were subsequently characterized at clade/lineage level by searching coding domains within the sequences, using the tool Domain based ANnotation of Transposable Elements (DANTE) (https://github.com/kavonrtep/dante). Transposable elements not classified by RepeatModeler were analyzed using DeepTE 118. We merged the libraries from RepeatModeler, LTR_retriever and DeepTE using USEARCH 119 with 80% identity as the minimum threshold for combining similar sequences into the final non-redundant de novo repeat library. Finally, we used RepeatMasker v4.1.0 (-e rmblast -gff -xsmall -s -norna -no_is - lib) to identify and classify repeats in the genome assemblies of seagrasses and Potamogeton.
Dating bursts of repeats in seagrass genomes
The identification of high-quality intact LTR-RTs and the calculation of insertion age for intact LTR-RTs were carried out using LTR_retriever (v2.9.0), using the formula T=K/2r. The nucleotide substitution rate “r” was set to 1.3e-8 substitutions per site per year 120.
Identifying Whole Genome Duplications
KS age distributions and gene tree-species tree reconciliation
Ks age distribution analysis was performed using the wgd package 121. Anchor pairs (i.e., paralogous genes lying in collinear or syntenic regions of the genome) were obtained using i-ADHoRe 122. Ks distribution analysis was also performed using the KSRATES software 123, which locates ancient polyploidization events with respect to speciation events within a phylogeny, comparing paralog and ortholog KS distributions, while correcting for substitution rate differences across the involved lineages (see Supplementary Note 4.2.1).
OrthoFinder 124 was used to build orthologous gene families. For each orthogroup, a multiple sequence alignment (MSA) based on amino acid sequences was obtained using PRANK 125 and then used as input for Markov Chain Monte Carlo (MCMC) analysis in MrBayes 126. A time-calibrated species tree was inferred by MCMCtree from the PAML package 127, using reference speciation times of 42–52 million years ago (MYA) for the divergence between Oryzae sativa and Brachypodium distachyon, 118-129 MYA for that between Spirodela polyrhiza and Z. marina, and 130-140 for that between Spirodela and other terrestrial monocots 128. A gene duplication-loss (DL)+WGD model, under critical and relaxed branch-specific rates, was implemented for the inference of the significance and corresponding retention rates of the assumed WGD events under Bayesian inference 25. (see Supplementary Note 4.2.2)
Absolute dating of WGDs
Absolute dating of WGD events followed an approach previously described for Zostera marina 15. Paralogous gene pairs located in duplicated segments (so-called anchors) and duplicated pairs lying under the WGD peak (so-called peak-based duplicates) were collected for phylogenetic dating. Anchors, which are assumed to correspond to the most recent WGD, were detected using i-ADHoRe 3.0 122. For each WGD paralogous pair, an orthogroup was created that included the two paralogues plus several orthologues from other plant species, as identified by InParanoid (v. 4.1) 129, using a broad taxonomic sampling. Gene duplicates were then dated using the BEAST v. 1.7 package 130 under an uncorrelated relaxed clock model with the LG+G (four rate categories) evolutionary model. A starting tree with branch lengths satisfying all fossil-prior-constraints was created according to the consensus APGIII phylogeny. Fossil calibrations were implemented using log-normal calibration priors (see Supplementary Note 4.2.3).
Time-calibrated tree construction
Protein sets were collected for 23 species (see Supplementary Note 4.3). These species were selected as representatives for monocots and eudicots, and representing different habitats from terrestrial, freshwater-floating, freshwater-submerged, to marine-submerged. Orthofinder v2.3 131 was used to delineate gene families with mcl inflation factor 3.0. All-versus-all Diamond blast with an E-value cutoff of 1e−05 was performed and orthologous genes were clustered using OrthoFinder. Single-copy orthologous genes were extracted from the clustering results. MAFFT 132) with default parameters was used to perform multiple sequence alignment of protein sequences for each set of single-copy orthologous genes, and to transform the protein sequence alignments into codon alignments after removing the poorly aligned or divergent regions using trimAl 133. The resulting codon alignments from all single copy orthologs were then concatenated into one supergene for species phylogenetic analysis. A maximum-likelihood phylogenetic tree of single-copy protein alignments and codon alignments was constructed using IQ-TREE 134 with the GTR+G model and 1,000 bootstrap replicates. Divergence times between species were estimated using MCMCtree from the PAML package under the GTR+G model (see Supplementary Note 4.3).
Gene family comparisons
Gene families analyzed in the paper were searched in the output from Orthofinder and a master table was compiled to show the detailed information for each orthogroup, which is defined as the group of genes from multiple species descended from a single gene in the last common ancestor. For the superfamilies, we used the phylogenetic tree to further classify them into subfamilies. We adopted a custom criterion to assess the expansion and contraction of gene families. If the average gene number in seagrasses increased or reduced by >40% compared to non-seagrass species, we called it expansion or contraction. Syntenic analysis of genes are performed using MCScanX 135 and i-ADHoRe 122. Lastly, circos plots were drawn using Circos 136.
Extended Data










Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Y.VdP., J.L.O., T.B.H.R. and G.P. acknowledge funding from the US-Dept. of Energy, Joint Genome Institute, Berkeley, California, USA, under the Community Sequencing Program 2018, Project Number 504341 (Marine Angiosperm Genomes Initiative-MAGI). The CSP award also included support sequencing and plant bioinformatics from HudsonAlpha Institute for Biotechnology, Huntsville, AL; and DNA/RNA extraction and processing from the Arizona Genomics Institute, Tuscon, AZ. Y.VdP. acknowledges funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (No. 833522) and from Ghent University (Methusalem funding, BOF.MET.2021.0005.01). P.N. acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 497665889, 1606/3-1 for research on Potamogeton. M.K. acknowledge funding through the Helmholtz School for Marine Data Science (MarDATA), Grant No. HIDSS-0005. The work of G.P., E.D., J.P., and M.R. was partially supported by the project Marine Hazard, PON03PE_00203_1 (MUR, Italian Ministry of University and Research) and by the National Biodiversity Future Centre (NBFC) Program, Italian Ministry of University and Research, PNRR, Missione 4 Componente 2 Investimento 1.4 (Project: CN00000033). M.D.D., L.L.W., M.P.T. and Y.Y.S. acknowledge funding from Universiti Malaysia Terengganu (SRG Vot55317). The work (proposal: 10.46936/10.25585/60001196) conducted by the U.S. Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy operated under Contract No. DE-AC02-05CH11231. The work of A.B. was performed within the Papanin Institute for Biology of Inland Waters RAS state assignment (theme 121051100099-5).
Footnotes
Author contributions table
X.M., V.S. and J.Ch. contributed equally to the work and are joint first authors. Y.VdP., J.L.O., T.B.H.R. and G.P. conceived the project, provided the overall evolutionary context, and wrote the proposal. Y.VdP., J.L.O., T.B.H.R. and G.P. and S.V. wrote and edited the main manuscript, and organized and further edited the individual contributions for the Supplementary Notes (as listed for the Supplementary Information below). All co-authors then provided specific feedback in forming the final version.
J.L.O., J.Ca., G.P., L.M.G., T.B.H.R., A.B., A.M., and P.N. contributed to sample tissue collection, preparation, and shipping for DNA extraction. S.Raj., L.B., G.H., J.W., M.Y performed the HMW DNA extractions and QC, as well as RNA extractions and QC for annotation assistance. J.S. and J.G. coordinated genome sequencing management steps for the seagrasses. A.M. and P.N. coordinated genome sequencing management steps for Potamogeton. K.B. was responsible for overall JGI technical coordination, liaison with principal investigators and project manager.
J.J., C.P., J.S., Y.VdP., S.R., A.S., J.vV. and T.B. performed analysis activities surrounding genome assembly (PacBio, HiC), supporting transcriptomics for annotation. J.J., Y.VdP., S.R., X.M. were responsible for deposition and maintenance of the species on the ORCAE site, and deposition of the new genomes to NCBI and Phytozome. J.Ch., X.M., S.M., J.Ch. were responsible for manuscript graphics.
Analysis of architectural features of genome evolution and annotation of specific gene families, including the written contributions to the main paper and Supplementary Information sections as follows: M.L.C., L.A. for the Orthogroups Master Extended Data; M.L.C., A.S., X.M., JCh. for overview of gene families; H.C., X.M., J.Ch., Y.VdP. for Whole Genome Duplications/Triplications and dating; M.L.C., X.M. for Transposable Elements and repeat elements; M.K., T.B.H.R. for Organellar genomes; M.L.C., L.A. for Non-protein coding RNA families; S.V., X.M. for Stomata; S.V., X.M. for Volatile metabolites and signaling, ethylene; X.M., S.V. for Plant body development, lignification, vascular tissue; T.B. for Plant defense, R-genes; L.M.G. for Heat shock factors; S.V., X.M. for Flavenoids and phenolics; S.V. for Cellular salt tolerance; S.V., B.V. for Cell wall plasticity; S.V., X.M. for Hypoxia; G.P. for Light perception, photosynthesis, light harvesting, transcription factors; G.P., M.R. Carbon acquisition, CCMs; G.P. for UVB tolerance; G.P., E.D. for Clock genes; J.P. for NAC genes; D.M., L.W., M.P.T., Y.Y.S. for Nitrogen metabolism.
X.M., J.Ch., S.R. were responsible for data deposit on ORCAE platform. K.B., J.G. were in charge of data deposit on NCBI.
Competing Interests
The authors declare no competing interests
Contributor Information
Thorsten B.H. Reusch, Email: treusch@geomar.de.
Gabriele Procaccini, Email: gpro@szn.it.
Jeanine L. Olsen, Email: j.l.olsen@rug.nl.
Yves Van de Peer, Email: yves.vandepeer@psb.ugent.be.
Data availability
The DNA sequencing data for C. nodosa genome assembly has been deposited in the NCBI databases under the BioProject PRJNA1041560 via the link: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1041560 All assemblies and annotations for all seagrass species discussed in the current paper can be found at https://bioinformatics.psb.ugent.be/gdb/seagrasses/. Transcriptome data (including raw data and clean data) and sequencing QC Reports for C. nodosa can be found at https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Cymnodnscriptome_2; transcriptome data and sequencing QC Reports for P. oceanica can be found at https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Posocenscriptome_2; transcriptome data and sequencing QC Reports for T. testudinum can be found at https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Thatesnscriptome_4; transcriptome data for Z. marina is from Jeanine et al. (2016). For the public databases, RFAM database v14.7 can be downloaded at https://ftp.ebi.ac.uk/pub/databases/Rfam/14.7/; UniProt database can be accessed from the web at http://www.uniprot.org and downloaded from http://www.uniprot.org/downloads; NCBI nucleotide database can be accessed via https://www.ncbi.nlm.nih.gov/
References
- 1.Green EP, Short FT. World Atlas of Seagrasses Prepared by the UNEP World Conservation Monitoring Centre. Berkeley, USA: Univ. of California Press; 2003. pp. 48–58. [Google Scholar]
- 2.Short F, Carruthers T, Dennison W, Waycott M. Global seagrass distribution and diversity: A bioregional model. Journal of Experimental Marine Biology and Ecology. 2007;350:3–20. doi: 10.1016/j.jembe.2007.06.012. [DOI] [Google Scholar]
- 3.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenzie LJ, et al. The global distribution of seagrass meadows. Environmental Research Letters. 2020;15:074041. doi: 10.1088/1748-9326/ab7d06. [DOI] [Google Scholar]
- 5.Duffy JE, et al. Toward a Coordinated Global Observing System for Seagrasses and Marine Macroalgae. Frontiers in Marine Science. 2019;6 [Google Scholar]
- 6.Gallagher AJ, et al. Tiger sharks support the characterization of the world’s largest seagrass ecosystem. Nature Communications. 2022;13:6328. doi: 10.1038/s41467-022-33926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertelli CM, Unsworth RKF. Protecting the hand that feeds us: Seagrass (Zostera marina) serves as commercial juvenile fish habitat. Marine Pollution Bulletin. 2014;83:425–429. doi: 10.1016/j.marpolbul.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Nordlund L, Koch E, Barbier E, Creed J. Seagrass Ecosystem Services and Their Variability across Genera and Geographical Regions. PLOS ONE. 2016;11:e0163091. doi: 10.1371/journal.pone.0163091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unsworth RKF, Cullen-Unsworth LC, Jones BLH, Lilley RJ. The planetary role of seagrass conservation. Science. 2022;377:609–613. doi: 10.1126/science.abq6923. [DOI] [PubMed] [Google Scholar]
- 10.Waycott M, et al. Accelerating loss of seagrasses across the globe threatens coastal ecosystems. Proc Natl Acad Sci U S A. 2009;106:12377–12381. doi: 10.1073/pnas.0905620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reusch TBH, et al. Lower Vibrio spp. abundances in Zostera marina leaf canopies suggest a novel ecosystem function for temperate seagrass beds. Marine Biology. 2021;168:149. doi: 10.1007/s00227-021-03963-3. [DOI] [Google Scholar]
- 12.Sievers M, et al. The Role of Vegetated Coastal Wetlands for Marine Megafauna Conservation. Trends in Ecology & Evolution. 2019;34:807–817. doi: 10.1016/j.tree.2019.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Duarte CM, Sintes T, Marbà N. Assessing the CO2 capture potential of seagrass restoration projects. Journal of Applied Ecology. 2013;50:1341–1349. doi: 10.1111/1365-2664.12155. [DOI] [Google Scholar]
- 14.Macreadie PI, et al. Blue carbon as a natural climate solution. Nature Reviews Earth & Environment. 2021;2:826–839. doi: 10.1038/s43017-021-00224-1. [DOI] [Google Scholar]
- 15.Olsen JL, et al. The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature. 2016;530:331–335. doi: 10.1038/nature16548. [DOI] [PubMed] [Google Scholar]
- 16.Chen LY, et al. Phylogenomic Analyses of Alismatales Shed Light into Adaptations to Aquatic Environments. Molecular Biology and Evolution. 2022;39:msac079. doi: 10.1093/molbev/msac079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma X, et al. Improved chromosome-level genome assembly and annotation of the seagrass, Zostera marina (eelgrass. F1000Res. 2021;10:289. doi: 10.12688/f1000research.38156.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu L, et al. Ocean current patterns drive the worldwide colonization of eelgrass (Zostera marina. Nature Plants. 2023;9:1207–1220. doi: 10.1038/s41477-023-01464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubin MJ, Mittelsten Scheid O, Becker C. Transposons: a blessing curse. Curr Opin Plant Biol. 2018;42:23–29. doi: 10.1016/j.pbi.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Vicient CM, Casacuberta JM. Impact of transposable elements on polyploid plant genomes. Annals of Botany. 2017;120:195–207. doi: 10.1093/aob/mcx078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Böse M, Lüthgens C, Lee JR, Rose J. Quaternary glaciations of northern Europe. Quaternary Science Reviews. 2012;44:1–25. doi: 10.1016/j.quascirev.2012.04.017. [DOI] [Google Scholar]
- 22.Van de Peer Y, Mizrachi E, Marchal K. The evolutionary significance of polyploidy. Nat Rev Genet. 2017;18:411–424. doi: 10.1038/nrg.2017.26. [DOI] [PubMed] [Google Scholar]
- 23.Murat F, Armero A, Pont C, Klopp C, Salse J. Reconstructing the genome of the most recent common ancestor of flowering plants. Nature Genetics. 2017;49:490–496. doi: 10.1038/ng.3813. [DOI] [PubMed] [Google Scholar]
- 24.Sensalari C, Maere S, Lohaus R. ksrates: positioning whole-genome duplications relative to speciation events in KS distributions. Bioinformatics. 2022;38:530–532. doi: 10.1093/bioinformatics/btab602. [DOI] [PubMed] [Google Scholar]
- 25.Zwaenepoel A, Van de Peer Y. Inference of Ancient Whole-Genome Duplications and the Evolution of Gene Duplication and Loss Rates. Mol Biol Evol. 2019;36:1384–1404. doi: 10.1093/molbev/msz088. [DOI] [PubMed] [Google Scholar]
- 26.Arber A. Water plants: a study of aquatic angiosperms. Cambridge University Press; 1920. [Google Scholar]
- 27.Den Hartog C. The seagrasses of the world. North Holland Publishing Co; 1970. [Google Scholar]
- 28.Harris BJ, Harrison CJ, Hetherington AM, Williams TA. Phylogenomic Evidence for the Monophyly of Bryophytes and the Reductive Evolution of Stomata. Current Biology. 2020;30:2001–2012.:e2002. doi: 10.1016/j.cub.2020.03.048. [DOI] [PubMed] [Google Scholar]
- 29.Shulaev V, Silverman P, Raskin I. Airborne signalling by methyl salicylate in plant pathogen resistance. Nature. 1997;385:718–721. doi: 10.1038/385718a0. [DOI] [Google Scholar]
- 30.Golicz AA, et al. Genome-wide survey of the seagrass Zostera muelleri suggests modification of the ethylene signalling network. J Exp Bot. 2015;66:1489–1498. doi: 10.1093/jxb/eru510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasidharan R, Voesenek LACJ. Ethylene-Mediated Acclimations to Flooding Stress. Plant Physiology. 2015;169:3–12. doi: 10.1104/pp.15.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartman S, et al. Ethylene-mediated nitric oxide depletion pre-adapts plants to hypoxia stress. Nat Commun. 2019;10:4020. doi: 10.1038/s41467-019-12045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van de Poel B, Smet D, Van Der Straeten D. Ethylene and Hormonal Cross Talk in Vegetative Growth and Development. Plant Physiol. 2015;169:61–72. doi: 10.1104/pp.15.00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sogin EM, et al. Sugars dominate the seagrass rhizosphere. Nat Ecol Evol. 2022 doi: 10.1038/s41559-022-01740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuo J, Cambridge ML, Kirkman H. In: Seagrasses of Australia: Structure, Ecology and Conservation. Larkum Anthony WD, Kendrick Gary A, Ralph Peter J., editors. Springer International Publishing; 2018. pp. 93–125. [Google Scholar]
- 36.Barnabas AD, Arnott HJ. Zostera capensis Setchell: root structure in relation to function. Aquatic Botany. 1987;27:309–322. [Google Scholar]
- 37.Taylor ARA. Studies of the development of Zostera marina L.: II. Germination and seedling development. Can J Botany. 1957;35:477–499. [Google Scholar]
- 38.Zhuo C, et al. Developmental changes in lignin composition are driven by both monolignol supply and laccase specificity. Sci Adv. 2022;8:eabm8145. doi: 10.1126/sciadv.abm8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Q, et al. Laccase is necessary and nonredundant with peroxidase for lignin polymerization during vascular development in Arabidopsis. Plant Cell. 2013;25:3976–3987. doi: 10.1105/tpc.113.117770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barros J, Dixon RA. Plant Phenylalanine/Tyrosine Ammonia-lyases. Trends Plant Sci. 2020;25:66–79. doi: 10.1016/j.tplants.2019.09.011. [DOI] [PubMed] [Google Scholar]
- 41.Wang B, et al. Presence of three mycorrhizal genes in the common ancestor of land plants suggests a key role of mycorrhizas in the colonization of land by plants. New Phytol. 2010;186:514–525. doi: 10.1111/j.1469-8137.2009.03137.x. [DOI] [PubMed] [Google Scholar]
- 42.Strullu-Derrien C, Selosse MA, Kenrick P, Martin FM. The origin and evolution of mycorrhizal symbioses: from palaeomycology to phylogenomics. New Phytologist. 2018;220:1012–1030. doi: 10.1111/nph.15076. [DOI] [PubMed] [Google Scholar]
- 43.Kohout P, et al. Surprising spectra of root-associated fungi in submerged aquatic plants. FEMS Microbiology Ecology. 2012;80:216–235. doi: 10.1111/j.1574-6941.2011.01291.x. [DOI] [PubMed] [Google Scholar]
- 44.Moora M, et al. AM fungal communities inhabiting the roots of submerged aquatic plant Lobelia dortmanna are diverse and include a high proportion of novel taxa. Mycorrhiza. 2016;26:735–745. doi: 10.1007/s00572-016-0709-0. [DOI] [PubMed] [Google Scholar]
- 45.Bohrer KE, Friese CF, Amon JP. Seasonal dynamics of arbuscular mycorrhizal fungi in differing wetland habitats. Mycorrhiza. 2004;14:329–337. doi: 10.1007/s00572-004-0292-7. [DOI] [PubMed] [Google Scholar]
- 46.Nielsen SL, Thingstrup I, Wigand C. Apparent lack of vesicular–arbuscular mycorrhiza (VAM) in the seagrasses Zostera marina L. and Thalassia testudinum Banks ex König. Aquatic Botany. 1999;63:261–266. doi: 10.1016/S0304-3770(98)00123-5. [DOI] [Google Scholar]
- 47.Gomez-Roldan V, et al. Strigolactone inhibition of shoot branching. Nature. 2008;455:189–194. doi: 10.1038/nature07271. [DOI] [PubMed] [Google Scholar]
- 48.Chang J, et al. The genome of the king protea, Protea cynaroides. Plant J. 2023;113:262–276. doi: 10.1111/tpj.16044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, et al. An angiosperm NLR Atlas reveals that NLR gene reduction is associated with ecological specialization and signal transduction component deletion. Mol Plant. 2021;14:2015–2031. doi: 10.1016/j.molp.2021.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Scharf KD, Berberich T, Ebersberger I, Nover L. The plant heat stress transcription factor (Hsf) family: structure, function and evolution. Biochim Biophys Acta. 2012;1819:104–119. doi: 10.1016/j.bbagrm.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Papazian S, Parrot D, Buryskova B, Weinberger F, Tasdemir D. Surface chemical defence of the eelgrass Zostera marina against microbial foulers. Sci Rep. 2019;9:3323. doi: 10.1038/s41598-019-39212-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lamb JB, et al. Seagrass ecosystems reduce exposure to bacterial pathogens of humans, fishes, and invertebrates. Science. 2017;355:731–733. doi: 10.1126/science.aal1956. [DOI] [PubMed] [Google Scholar]
- 53.Teles YCF, Souza MSR, Souza MFV. Sulphated Flavonoids: Biosynthesis, Structures, and Biological Activities. Molecules. 2018;23 doi: 10.3390/molecules23020480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grignon-Dubois M, Rezzonico B. Phenolic chemistry of the seagrass Zostera noltei Hornem. Part 1: First evidence of three infraspecific flavonoid chemotypes in three distinctive geographical regions. Phytochemistry. 2018;146:91–101. doi: 10.1016/j.phytochem.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 55.Vilas-Boas C, Sousa E, Pinto M, Correia-da-Silva M. An antifouling model from the sea: a review of 25 years of zosteric acid studies. Biofouling. 2017;33:927–942. doi: 10.1080/08927014.2017.1391951. [DOI] [PubMed] [Google Scholar]
- 56.van Zelm E, Zhang Y, Testerink C. Salt Tolerance Mechanisms of Plants. Annu Rev Plant Biol. 2020;71:403–433. doi: 10.1146/annurev-arplant-050718-100005. [DOI] [PubMed] [Google Scholar]
- 57.Gaxiola RA, et al. Drought-and salt-tolerant plants result from overexpression of the AVP1 H+-pump. Proc Natl Acad Sci U S A. 2001;98:11444–11449. doi: 10.1073/pnas.191389398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar T, Khan Uzma MR, Abbas Z, Ali GM. Genetic improvement of sugarcane for drought and salinity stress tolerance using Arabidopsis vacuolar pyrophosphatase (AVP1) gene. Mol Biotechnol. 2014;56:199–209. doi: 10.1007/s12033-013-9695-z. [DOI] [PubMed] [Google Scholar]
- 59.Yang Y, et al. Overexpression of a Populus trichocarpa H+-pyrophosphatase gene PtVP1.1 confers salt tolerance on transgenic poplar. Tree Physiol. 2015;35:663–677. doi: 10.1093/treephys/tpv027. [DOI] [PubMed] [Google Scholar]
- 60.Duan XG, Yang AF, Gao F, Zhang SL, Zhang JR. Heterologous expression of vacuolar H(+)-PPase enhances the electrochemical gradient across the vacuolar membrane and improves tobacco cell salt tolerance. Protoplasma. 2007;232:87–95. doi: 10.1007/s00709-007-0268-5. [DOI] [PubMed] [Google Scholar]
- 61.Nakamura RL, Gaber RF. Ion selectivity of the Kat1 K+ channel pore. Mol Membr Biol. 2009;26:293–308. doi: 10.1080/09687680903188332. [DOI] [PubMed] [Google Scholar]
- 62.Morris ER, Powell DA, Gidley MJ, Rees DA. Conformations and interactions of pectins. I. Polymorphism between gel and solid states of calcium polygalacturonate. J Mol Biol. 1982;155:507–516. doi: 10.1016/0022-2836(82)90484-3. [DOI] [PubMed] [Google Scholar]
- 63.Gloaguen V, et al. Structural characterization and cytotoxic properties of an apiose-rich pectic polysaccharide obtained from the cell wall of the marine phanerogam Zostera marina. J Nat Prod. 2010;73:1087–1092. doi: 10.1021/np100092c. [DOI] [PubMed] [Google Scholar]
- 64.Byrt CS, Munns R, Burton RA, Gilliham M, Wege S. Root cell wall solutions for crop plants in saline soils. Plant Science. 2018;269:47–55. doi: 10.1016/j.plantsci.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 65.Mølhøj M, Verma R, Reiter WD. The biosynthesis of the branched-chain sugar d-apiose in plants: functional cloning and characterization of a UDP-d-apiose/UDP-d-xylose synthase from Arabidopsis. Plant J. 2003;35:693–703. doi: 10.1046/j.1365-313x.2003.01841.x. [DOI] [PubMed] [Google Scholar]
- 66.Xu S, et al. The origin, diversification and adaptation of a major mangrove clade (Rhizophoreae) revealed by whole-genome sequencing. National Science Review. 2017;4:721–734. doi: 10.1093/nsr/nwx065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Natarajan P, et al. A reference-grade genome identifies salt-tolerance genes from the salt-secreting mangrove species Avicennia marina. Communications Biology. 2021;4:851. doi: 10.1038/s42003-021-02384-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dolferus R, et al. Functional analysis of lactate dehydrogenase during hypoxic stress in Arabidopsis. Functional Plant Biology. 2008;35:131–140. doi: 10.1071/FP07228. [DOI] [PubMed] [Google Scholar]
- 69.Baena-González E, Rolland F, Thevelein JM, Sheen J. A central integrator of transcription networks in plant stress and energy signalling. Nature. 2007;448:938–942. doi: 10.1038/nature06069. [DOI] [PubMed] [Google Scholar]
- 70.Cho HY, Lu MYJ, Shih MC. The SnRK1-eIFiso4G1 signaling relay regulates the translation of specific mRNAs in Arabidopsis under submergence. New Phytologist. 2019;222:366–381. doi: 10.1111/nph.15589. [DOI] [PubMed] [Google Scholar]
- 71.Monteiro FM, Pancost RD, Ridgwell A, Donnadieu Y. Nutrients as the dominant control on the spread of anoxia and euxinia across the Cenomanian-Turonian oceanic anoxic event (OAE2): Model-data comparison. Paleoceanography. 2012;27 doi: 10.1029/2012PA002351. [DOI] [Google Scholar]
- 72.Selby D, Mutterlose J, Condon DJ. U–Pb and Re–Os geochronology of the Aptian/Albian and Cenomanian/Turonian stage boundaries: Implications for timescale calibration, osmium isotope seawater composition and Re–Os systematics in organic-rich sediments. Chemical Geology. 2009;265:394–409. doi: 10.1016/j.chemgeo.2009.05.005. [DOI] [Google Scholar]
- 73.Kirk J. Light and Photosynthesis in Aquatic Systems. Light and Photosynthesis in Aquatic Ecosystems. (Third edition) 2010;VI:1–651. doi: 10.1017/CBO9781139168212. [DOI] [Google Scholar]
- 74.Campbell JE, Fourqurean JW. Mechanisms of bicarbonate use influence the photosynthetic carbon dioxide sensitivity of tropical seagrasses. Limnology and Oceanography. 2013;58:839–848. doi: 10.4319/lo.2013.58.3.0839. [DOI] [Google Scholar]
- 75.Capó-Bauçà S, Iñiguez C, Aguiló-Nicolau P, Galmés J. Correlative adaptation between Rubisco and CO2-concentrating mechanisms in seagrasses. Nature Plants. 2022;8:706–716. doi: 10.1038/s41477-022-01171-5. [DOI] [PubMed] [Google Scholar]
- 76.Rubio L, et al. Direct uptake of HCO3− in the marine angiosperm Posidonia oceanica (L.) Delile driven by a plasma membrane H+ economy. Plant, Cell & Environment. 2017;40:2820–2830. doi: 10.1111/pce.13057. [DOI] [PubMed] [Google Scholar]
- 77.Larkum AWD, Davey PA, Kuo J, Ralph PJ, Raven JA. Carbon-concentrating mechanisms in seagrasses. Journal of Experimental Botany. 2017;68:3773–3784. doi: 10.1093/jxb/erx206. [DOI] [PubMed] [Google Scholar]
- 78.Koch M, Bowes G, Ross C, Zhang XH. Climate change and ocean acidification effects on seagrasses and marine macroalgae. Global Change Biology. 2013;19:103–132. doi: 10.1111/j.1365-2486.2012.02791.x. [DOI] [PubMed] [Google Scholar]
- 79.Chen S, Peng W, Ansah EO, Xiong F, Wu Y. Encoded C4 homologue enzymes genes function under abiotic stresses in C3 plant. Plant Signal Behav. 2022;17:2115634. doi: 10.1080/15592324.2022.2115634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han X, et al. Origin and Evolution of Core Components Responsible for Monitoring Light Environment Changes during Plant Terrestrialization. Mol Plant. 2019;12:847–862. doi: 10.1016/j.molp.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 81.McClung CR. The Plant Circadian Oscillator. Biology. 2019;8 doi: 10.3390/biology8010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mohr W, et al. Terrestrial-type nitrogen-fixing symbiosis between seagrass and a marine bacterium. Nature. 2021;600:105–109. doi: 10.1038/s41586-021-04063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tarquinio F, et al. Microorganisms facilitate uptake of dissolved organic nitrogen by seagrass leaves. ISME J. 2018;12:2796–2800. doi: 10.1038/s41396-018-0218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kuo J, Hartog Cd. In: SEAGRASSES: BIOLOGY, ECOLOGYAND CONSERVATION. Larkum Anthony WD, Orth Robert J, Duarte Carlos M., editors. Springer Netherlands; 2006. pp. 51–87. [Google Scholar]
- 85.Krizek BA, Fletcher JC. Molecular mechanisms of flower development: an armchair guide. Nature Reviews Genetics. 2005;6:688–698. doi: 10.1038/nrg1675. [DOI] [PubMed] [Google Scholar]
- 86.Lohmann JU, Weigel D. Building beauty: the genetic control of floral patterning. Dev Cell. 2002;2:135–142. doi: 10.1016/s1534-5807(02)00122-3. [DOI] [PubMed] [Google Scholar]
- 87.Remizowa MV, Sokoloff DD, Rudall PJ. Evolutionary History of the Monocot Flower. Annals of the Missouri Botanical Garden. 2010;97:617–645. [Google Scholar]
- 88.Ackerman JD. In: Seagrasses: Biology, Ecologyand Conservation. Larkum Anthony WD, Orth Robert J, Duarte Carlos M., editors. Springer Netherlands; 2006. pp. 89–109. [Google Scholar]
- 89.Orth RJ, et al. Restoration of seagrass habitat leads to rapid recovery of coastal ecosystem services. Science Advances. 6:eabc6434. doi: 10.1126/sciadv.abc6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cook CDK. The number and kinds of embryo-bearing plants which have become aquatic: a survey. Perspectives in Plant Ecology, Evolution and Systematics. 1999;2:79–102. doi: 10.1078/1433-8319-00066. [DOI] [Google Scholar]
- 91.Ackerman JD. In: Seagrasses: Biology, Ecology and Conservation. Larkum WD, Orth RJ, Duarte CM, editors. Springer, NL; 2006. pp. 89–109. [Google Scholar]
- 92.Waycott M, Biffin E, Les DH. In: Seagrasses of Australia: Structure, Ecology and Conservation. Larkum Anthony WD, Kendrick Gary A, Ralph Peter J., editors. Springer International Publishing; 2018. pp. 129–154. [Google Scholar]
- 93.Pazzaglia J, Reusch TBH, Terlizzi A, Marín-Guirao L, Procaccini G. Phenotypic plasticity under rapid global changes: The intrinsic force for future seagrasses survival. Evolutionary Applications. 2021;14:1181–1201. doi: 10.1111/eva.13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flowers TJ, Galal HK, Bromham L. Evolution of halophytes: multiple origins of salt tolerance in land plants. Functional Plant Biology. 2010;37:604–612. [Google Scholar]
- 95.Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical bulletin. 1987 [Google Scholar]
- 96.Dudchenko O, et al. The Juicebox Assembly Tools module facilitates <em>de novo</em> assembly of mammalian genomes with chromosome-length scaffolds for under $1000. bioRxiv. 2018:254797. doi: 10.1101/254797. [DOI] [Google Scholar]
- 97.Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods. 2021;18:170–175. doi: 10.1038/s41592-020-01056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yeo S, Coombe L, Warren RL, Chu J, Birol I. ARCS: scaffolding genome drafts with linked reads. Bioinformatics. 2018;34:725–731. doi: 10.1093/bioinformatics/btx675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kovaka S, et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biology. 2019;20:278. doi: 10.1186/s13059-019-1910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 102.Bruna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform. 2021;3:qaa108. doi: 10.1093/nargab/lqaa108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Keilwagen J, Hartung F, Grau J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol Biol. 2019;1962:161–177. doi: 10.1007/978-1-4939-9173-0_9. [DOI] [PubMed] [Google Scholar]
- 104.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Seppey M, Manni M, Zdobnov EM. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol. 2019;1962:227–245. doi: 10.1007/978-1-4939-9173-0_14. [DOI] [PubMed] [Google Scholar]
- 106.Abeel T, Van Parys T, Saeys Y, Galagan J, Van de Peer Y. GenomeView: a next-generation genome browser. Nucleic Acids Research. 2012;40 doi: 10.1093/nar/gkr995. e12-e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Quevillon E, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nordberg H, et al. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014;42:D26–31. doi: 10.1093/nar/gkt1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kalvari I, et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021;49:D192–d200. doi: 10.1093/nar/gkaa1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Consortium U. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–d489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gremme G, Steinbiss S, Kurtz S. GenomeTools: A Comprehensive Software Library for Efficient Processing of Structured Genome Annotations. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2013;10:645–656. doi: 10.1109/TCBB.2013.68. [DOI] [PubMed] [Google Scholar]
- 113.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Benson DA, et al. GenBank. Nucleic Acids Res. 2013;41:D36–42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 2008;9:18. doi: 10.1186/1471-2105-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ou S, Jiang N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiology. 2017;176:1410–1422. doi: 10.1104/pp.17.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yan H, Bombarely A, Li S. DeepTE: a computational method for de novo classification of transposons with convolutional neural network. Bioinformatics. 2020;36:4269–4275. doi: 10.1093/bioinformatics/btaa519. [DOI] [PubMed] [Google Scholar]
- 119.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 120.Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004;101:12404–12410. doi: 10.1073/pnas.0403715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zwaenepoel A, Van de Peer Y. wgd—simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics. 2019;35:2153–2155. doi: 10.1093/bioinformatics/bty915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Proost S, et al. i-ADHoRe 3.0 fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 2012;40:e11. doi: 10.1093/nar/gkr955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sensalari C, Maere S, Lohaus R. ksrates: positioning whole-genome duplications relative to speciation events in KS distributions. Bioinformatics. 2021 doi: 10.1093/bioinformatics/btab602. [DOI] [PubMed] [Google Scholar]
- 124.Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology. 2019;20:238. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Löytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. P Natl Acad Sci USA. 2005;102:10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 127.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 128.An D, et al. Plant evolution and environmental adaptation unveiled by long-read whole-genome sequencing of Spirodela. Proc Natl Acad Sci U S A. 2019;116:18893–18899. doi: 10.1073/pnas.1910401116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.O’Brien KP, Remm M, Sonnhammer EL. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005;33:D476–480. doi: 10.1093/nar/gki107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Research. 2019;47:W5–W10. doi: 10.1093/nar/gkz342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Minh BQ, et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Molecular Biology and Evolution. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang Y, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DNA sequencing data for C. nodosa genome assembly has been deposited in the NCBI databases under the BioProject PRJNA1041560 via the link: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1041560 All assemblies and annotations for all seagrass species discussed in the current paper can be found at https://bioinformatics.psb.ugent.be/gdb/seagrasses/. Transcriptome data (including raw data and clean data) and sequencing QC Reports for C. nodosa can be found at https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Cymnodnscriptome_2; transcriptome data and sequencing QC Reports for P. oceanica can be found at https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Posocenscriptome_2; transcriptome data and sequencing QC Reports for T. testudinum can be found at https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Thatesnscriptome_4; transcriptome data for Z. marina is from Jeanine et al. (2016). For the public databases, RFAM database v14.7 can be downloaded at https://ftp.ebi.ac.uk/pub/databases/Rfam/14.7/; UniProt database can be accessed from the web at http://www.uniprot.org and downloaded from http://www.uniprot.org/downloads; NCBI nucleotide database can be accessed via https://www.ncbi.nlm.nih.gov/