

Abstract
The mechanisms underlying the rapid evolution of novel enzymatic activities from promiscuous side activities are poorly understood. Recently emerged enzymes catalyzing the catabolic degradation of xenobiotic substances that have been spread out into the environment during the last few decades provide an exquisite opportunity to study these mechanisms. A prominent example is the herbicide atrazine (2-chloro-4-ethylamino-6-isopropylamino-1,3,5-triazine), which is degraded through a number of enzymatic reactions, constituting the Atz pathway. Here, we analyzed the evolution of hydroxyatrazine ethylaminohydrolase AtzB, a Zn(II)-dependent metalloenzyme that adopts the amidohydrolase fold and catalyzes the second step of the Atz pathway. We searched for promiscuous side activities of AtzB, which might point to the identity of its progenitor. These investigations revealed that AtzB has a low promiscuous guanine deaminase activity. Furthermore, we found that the two closest AtzB homologues, which have not been functionally annotated until now, are guanine deaminases with modest promiscuous hydroxyatrazine hydrolase activity. Based on sequence comparisons with the closest AtzB homologues, the guanine deaminase activity of AtzB could be increased by three orders of magnitude through the introduction of only four active site mutations. Interestingly, introducing the four inverse mutations into the AtzB homologues significantly enhanced their hydroxyatrazine hydrolase activity and, in one case, is even equivalent to that of wild-type AtzB. Molecular dynamics simulations elucidated the structural and molecular basis for the mutation-induced activity changes. The example of AtzB highlights how novel enzymes with high catalytic proficiency can evolve from low promiscuous side activities by only a few mutational events within a short period of time.
Keywords: anthropogenic compound, atrazine, AtzB, enzyme evolution, herbicide, promiscuous activity, protein engineering, xenobiotics
Introduction
The advent of modern industrial chemistry has led to pervasive changes of the global ecosystem due to a multitude of anthropogenic substances being introduced into the environment at a large scale.1,2 Many of these compounds, including herbicides, insecticides, and synthetic antibiotics, do not occur in Nature and are thus referred to as xenobiotic. An important class of anthropogenic substances is derived from 1,3,5-triazine (s-triazine). This substance group includes a number of herbicidal compounds, which kill susceptible plants by coordinating to the quinone-binding protein in photosystem II, thereby inhibiting photosynthetic electron transfer.3,4 The most common s-triazine is atrazine (2-chloro-4-ethylamino-6-isopropylamino-1,3,5-triazine), which has been used since the 1950s as a herbicide for the selective control of grassy and broadleaf weeds in corn, sorghum, sugar cane, and pineapple.5,6
Remarkably, the introduction of large amounts of atrazine and related s-triazine herbicides into the environment led to the emergence of novel catabolic enzymes in soil bacteria within only a few decades, allowing for the utilization of these substances as a nutrient source.7 Various bacterial species possessing these metabolic capabilities have since been isolated all over the globe.8,9 The Atz pathway is the most abundant and best investigated pathway for the degradation of atrazine and other s-triazine herbicides. Its main evolutionary advantage is the utilization of the breakdown products as carbon and nitrogen sources.10,11 The relevant genes were first identified in Pseudomonas sp. strain ADP, which was isolated from an atrazine spillage site in Minnesota.12 The pathway is composed of eight proteins (AtzABCDEFGH) and can be divided into an upper pathway (Figure 1A) and a lower pathway (Figure 1B).
Figure 1.

The Atz pathway. (A) The herbicide atrazine (I) is converted by atrazine chlorohydrolase (AtzA) into hydroxyatrazine (II), which serves as a substrate for hydroxyatrazine ethylaminohydrolase (AtzB). AtzB releases ethylamine (NH2Et) from the s-triazine ring, yielding N-isopropylammelide (III), which is in turn hydrolyzed by N-isopropylammelide isopropylaminohydrolase (AtzC), giving isopropylamine (NH2iPr) and cyanuric acid (IV) as products. The sequential actions of AtzA, AtzB, and AtzC are considered to be the upper pathway. (B) Cyanuric acid hydrolase (AtzD) converts (IV) into 1-carboxybiuret (V), which is further hydrolyzed by 1-carboxybiuret hydrolase (AtzE) in complex with the small protein AtzG.13 The resulting 1,3-dicarboxyurea (VI) is decarboxylated through an unknown mechanism, possibly involving AtzH14 producing allophanate (VII), which is hydrolyzed by allophanate hydrolase (AtzF) to give two molecules each of NH3 and CO2. The reactions leading to the mineralization of cyanuric acid are considered the lower pathway.
In Pseudomonas sp. strain ADP, the genes encoding the enzymes required for atrazine catabolism are located on a single, self-transmissible plasmid (pADP-115). The lower pathway genes (atzDEFGH) are arranged in a regulated operon structure,16,17 which, together with the observation that in Nature cyanuric acid can also form nonenzymatically from isocyanic acid, points to an evolutionary origin of the involved enzymes that predates the introduction of atrazine.18 On the other hand, the genes for the upper pathway enzymes (atzABC) are expressed constitutively and without any known regulation and are spread over a wide area of the pADP-1 plasmid with long interjacent sequences. Moreover, those sequences are surrounded by inverted repeats that point to the recent assembly of the plasmid through transposase activity.19 Together, these findings indicate that AtzA, AtzB, and AtzC have evolved only in the last decades as a consequence of the presence of atrazine in the environment.20 The young evolutionary age of AtzA, AtzB, and AtzC suggests that their functions derived from low promiscuous side activities of evolutionarily old progenitor enzymes, with the old and the new activities being individually optimized in one of the two copies resulting from gene duplication events.21,22 AtzA, AtzB, and AtzC all belong to the amidohydrolase superfamily and catalyze their respective hydrolytic reactions with the assistance of a central metal ion.23−25 However, they share only limited sequence identity (AtzA–AtzB: 24%; AtzA–AtzC: 19%; AtzB–AtzC: 18%) pointing to the existence of three distinct progenitor enzymes.
Protein engineering starting from promiscuous side activities has previously been utilized to study enzyme evolution. Examples are the establishment of phosphoribosylanthranilate isomerase (TrpF) activity on the scaffolds of N′-[(5′-phosphoribosyl)formimino]-5-aminoimidazole-4-carboxamide ribonucleotide isomerase (HisA)26−28 and imidazole glycerol phosphate synthase (HisF),29 the establishment of o-succinylbenzoate synthase activity on the scaffolds of l-Ala-D/l-Glu epimerase and the muconate lactonizing enzyme II,30 and the conversion of anthranilate synthase into isochorismate synthase.31 Regarding enzymes degrading anthropogenic compounds, the evolution of organophosphate hydrolases from promiscuous lactonases has been analyzed through ancestral sequence reconstruction32,33 while the emergence of enzymes for the degradation of the herbicides molinate and phenylurea has been traced back to a transition from a binuclear to a mononuclear metal binding site.34 Notably, AtzA has previously been interconverted into the melamine deaminase TriA, which is another evolutionarily young xenobiotic-degrading enzyme with 98% sequence identity to AtzA.35 However, this work did not provide insights into the common “old” progenitor of these two enzymes.
Within the current work, we concentrated on elucidating the evolutionary history of AtzB. Using a broad substrate screen, we could identify a promiscuous guanine deaminase activity for this enzyme, which was then systematically increased by protein engineering. Vice versa, two hitherto uncharacterized AtzB homologues with about 60% sequence identity to AtzB were found to be guanine deaminases and their promiscuous hydroxyatrazine ethylaminohydrolase activities could be raised up to wild-type AtzB levels by only four amino acid substitutions. These experiments clearly indicated that AtzB descends from an evolutionarily old enzyme with guanine deaminase activity and allowed us to generate a plausible evolutionary trajectory between the two activities. Finally, the molecular mechanisms underlying this functional conversion were uncovered through molecular dynamics simulations.
Results
Substrate Screening of AtzB and Homologues
In the search for a likely progenitor of AtzB, we tested the enzyme for the promiscuous turnover of different substrates (adenine, ammeline, atrazine, cytosine, guanine, hydroxyatrazine, isoguanine, and melamine) accepted by various enzymes belonging to the amidohydrolase superfamily. Product analysis by HPLC showed that incubation with AtzB for 20 h resulted in complete conversion of hydroxyatrazine, as expected, as well as minor hydrolytic activities on guanine and ammeline (Figure S1). We performed a BLAST search to identify close homologues of AtzB. The two best matches were sequences from Haliea sp. SAOS-164 and Pleomorphomonas oryzae, which we termed AtzB_Hom_Hal (accession: WP_135441588.1; 64% global sequence identity to AtzB) and AtzB_Hom_Pleo (accession: WP_026793093.1; 56% global sequence identity to AtzB). Up until now, AtzB_Hom_Hal and AtzB_Hom_Pleo had no enzymatic functions assigned. Substrate screening analogous to that performed with AtzB showed complete conversion of guanine as well as minor activities on hydroxyatrazine and ammeline by these two proteins (Figure S1).
Increasing Promiscuous Activities by Site-Directed Mutagenesis
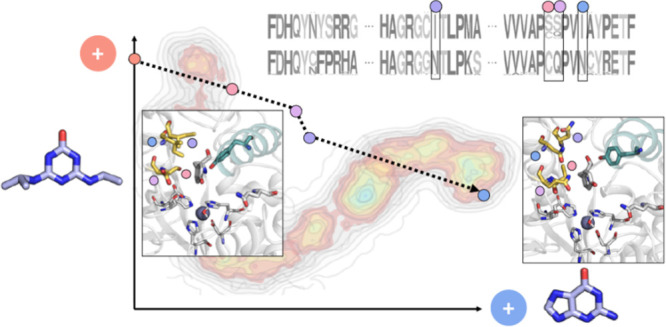
We attempted to increase the respective promiscuous activities of AtzB, AtzB_Hom_Hal, and AtzB_Hom_Pleo by rational protein engineering. For this purpose, we first generated a multiple sequence alignment (MSA) including the 10 known full-length (>300 amino acids) AtzB sequences and their 40 closest homologues (Table S1). Two sequence logos were generated based on this MSA, one for the AtzB group and the other one for the group comprising the AtzB homologues. Comparison of these logos allowed for the identification of seven group-specific amino acids, which were notably clustered at one region of the active site and subjected to site-directed mutagenesis (Figure 2).
Figure 2.

Selection of amino acid exchanges to increase promiscuous activities. (A) Sequence logos of AtzB proteins (top row) and their closest homologues (bottom row). Positions making up the active site and differing between the two groups are indicated by asterisks. These positions were chosen for mutual residue exchanges between AtzB and AtzB_Hom_Hal/AtzB_Hom_Pleo. (B) Model of the AtzB active site with residues selected for mutation (yellow sticks). The positions of the catalytic Zn(II) (gray sphere) and the substrate analogue xanthine (white sticks) are deduced from a superposition of the model with the crystal structure of a guanine deaminase (PDB Code 6OHA). (C) Schemes for AtzB and guanine deaminase (GuaD) reactions.
For AtzB, these residue exchanges are N139C, C169G, I170N, S218C, S219Q, I222N, and A223S. A223S was chosen instead of A223C, which is indicated by the sequence logos, as serine is present at the equivalent position in the two closest AtzB homologues. The corresponding recombinant proteins were produced and purified to homogeneity, and their catalytic activities for the conversion of guanine to xanthine were determined by a fast-screening method using a plate reader. AtzB-S218C (AtzB-C) was the most active single mutant, which was then combined with the remaining residue exchanges, leading to the identification of the most active double mutant AtzB-S218C S219Q (AtzB-CQ). The continuation of this procedure led to the most active triple mutant, AtzB-S218C S219Q I170N (AtzB-CQN), and the most active quadruple mutant, AtzB-S218C S219Q I170N I222N (AtzB-CQNN). Introduction of the remaining residue exchanges (N139C, C169G, and A223S) into this quadruple mutant did not further increase the activity for guanine and were therefore disregarded for the subsequent analyses. To cover the entire sequence space connecting AtzB with AtzB-CQNN, all possible permutations of I170N, S218C, S219Q, and I222N were produced and purified (Figure S2) and a spectrophotometer was used to precisely determine their steady-state guanine deaminase activities (Table S2). Moreover, the residual hydroxyatrazine hydrolase activities of the generated AtzB mutants were also monitored by steady-state kinetics (Table S3).
AtzB-CQNN displayed a catalytic efficiency for guanine (kcat/KM = 3.9e3 M–1 s–1) that represents an increase of about three orders of magnitude compared to AtzB. It even surpasses the guanine deaminase activities of AtzB_Hom_Hal (kcat/KM = 1.1e3 M–1 s–1) and AtzB_Hom_Pleo (kcat/KM = 2.2e3 M–1 s–1), mainly due to a slightly higher value for kcat (Table S2). Notably, along with the continuous increase of guanine deaminase activity from AtzB to AtzB-CQNN, the hydroxyatrazine hydrolase activity continuously decreased (Table S3). The corresponding trajectory is displayed as a solid line in Figure 3. Analysis of the AtzB mutants covering the remaining sequence space led to the identification of another path toward AtzB-CQNN, also with a continuous trade-off between guanine deaminase and hydroxyatrazine hydrolase activity. This alternative trajectory is displayed as a dashed line in Figure 3.
Figure 3.

Activity changes in the sequence space connecting AtzB and AtzB-S218C S219Q I170N I222N (AtzB-CQNN). Catalytic efficiency (kcat/KM) for hydroxyatrazine (HA) is plotted against kcat/KM for guanine (Gua) for all AtzB variants contained in the sequence space between wild-type AtzB and AtzB-CQNN. The mutations are listed in the order of their introduction. Values shown are the average of triplicate measurements and are explicitly listed in Tables S2 and S3. Activities that are too low to be determined are represented as kcat/KM = 1 M–1 s–1. Colors indicate the number of mutations relative to the wild type (0 = red; 1 = yellow; 2 = green; 3 = light blue; 4 = dark blue). The main trajectory that led from AtzB to AtzB-CQNN is indicated by a solid line. An alternative trajectory that features similar activity trade-offs is shown by a dashed line.
It was reasoned that the equivalents of the four functionally relevant residue exchanges that establish high guanine deaminase activity on AtzB might similarly increase the promiscuous hydroxyatrazine hydrolase activities of AtzB_Hom_Hal and AtzB_Hom_Pleo. To test this hypothesis, single, double, triple, and quadruple mutants were generated based on the best previous variant with respect to the hydroxyatrazine hydrolase activity. The iterative introduction of Q210S, N213I, N161I, and C209S into AtzB_Hom_Hal, finally leading to AtzB_Hom_Hal-SIIS, and the iterative introduction of N217I, N165I, Q214S, and C213S into AtzB_Hom_Pleo, finally resulting in AtzB_Hom_Pleo-IISS, produced mutants with successively increased catalytic efficiencies for the hydrolysis of hydroxyatrazine. This increase was mainly caused by a rise of kcat (Table S3). While AtzB_Hom_Hal-SIIS showed a hydroxyatrazine hydrolase activity with a kcat/KM = 2.3e4 M–1 s–1, the catalytic efficiency of AtzB_Hom_Pleo-IISS (kcat/KM = 2.7e5 M–1 s–1) corresponds to a 6400-fold increase over the wild-type enzyme and is equivalent to the native hydroxyatrazine hydrolase activity of wild-type AtzB (kcat/KM = 2.4e5 M–1 s–1). Intriguingly, both the KM and the kcat values of AtzB_Hom_Pleo-IISS and AtzB are almost identical (Table S3). In analogy to the observed activity trade-off when moving from AtzB to AtzB-CQNN, the conversion of AtzB_Hom_Hal and AtzB_Hom_Pleo into the highly active hydroxyatrazine hydrolases AtzB_Hom_Hal-SIIS and AtzB_Hom_Pleo-IISS comes at the cost of their guanine deaminase activities (Table S2), which is shown in Figure 4.
Figure 4.

Activity changes in the partial sequence space connecting the AtzB homologues with their respective quadruple mutants. Catalytic efficiency (kcat/KM) for hydroxyatrazine (HA) is plotted against kcat/KM for guanine (Gua) for all relevant variants of (A) AtzB_Hom_Hal and (B) AtzB_Hom_Pleo. The mutations are listed in the order of their introduction. Values shown are the average of triplicate measurements and are explicitly listed in Tables S2 and S3. Activities too low to be determined are represented as kcat/KM = 1 M–1 s–1. Colors indicate the number of mutations relative to the respective wild type (0 = red; 1 = yellow; 2 = green; 3 = light blue; 4 = dark blue). Trajectories leading to the most rapid continuous improvement of hydroxyatrazine hydrolase activity are indicated by lines.
Interestingly, the trajectories from AtzB_Hom_Hal and AtzB_Hom_Pleo to their corresponding quadruple mutants AtzB_Hom_Hal-SIIS and AtzB_Hom_Pleo-IISS (Figure 4A,B) mirror-image the ones from AtzB to AtzB-CQNN (Figure 3). In other words, when the trajectories in Figure 3 are read in the reverse direction (i.e., from AtzB-CQNN to wild-type AtzB), they are fully equivalent to the ones shown in Figure 4. For AtzB_Hom_Hal, the order of mutations is equivalent to the alternative trajectory of AtzB (dashed line in Figure 3) whereas AtzB_Hom_Pleo follows the main trajectory of AtzB (solid line in Figure 3). Hence, although the conversion of AtzB_Hom_Hal/AtzB_Hom_Pleo into AtzB_Hom_Hal-SIIS/AtzB_Hom_Pleo-IISS does not represent evolutionary history (the latter pair does not exist in Nature), it reflects the evolutionary potential of AtzB homologues to generate alternative AtzB enzymes and supports the plausibility of the AtzB trajectories.
Product Analysis of Hydroxyatrazine Hydrolases
Due to the near-symmetric nature of hydroxyatrazine, two different hydrolysis products (N-isopropylammelide resulting from ethylaminohydrolase activity and N-ethylammelide resulting from isopropylaminohydrolase activity) are conceivable, depending on the substrate orientation during catalysis. Wild-type AtzB almost exclusively produces N-isopropylammelide (cf. Figure 1A).24 As this preference could differ between the homologues and might be altered by the introduced mutations, we were interested in elucidating the product specificities of the mutants of AtzB, as well as AtzB_Hom_Hal and AtzB_Hom_Pleo, on the main trajectories between hydroxyatrazine hydrolase and guanine deaminase activities. For this purpose, the wild-type proteins and the mutants were incubated with hydroxyatrazine and the reaction mixtures were analyzed by HPLC. The resulting product ratios are shown in Figure 5.
Figure 5.

Product composition of hydroxyatrazine hydrolysis by wild-type AtzB, AtzB_Hom_Hal, and AtzB_Hom_Pleo, as well as relevant mutants. Mutants making up the main trajectories shown in Figures 3 and 4 were investigated for their product composition resulting from hydrolysis of 500 μM hydroxyatrazine by 2 μM enzyme over 20 h at 25 °C. The graph shows the resulting concentrations of the respective product as determined by HPLC analysis of three replicas. The raw data are shown in Figure S3.
The specific ethylaminohydrolase activity of AtzB is mostly maintained in the single S218C mutant but completely lost in the further AtzB mutants along the main trajectory. All mutants of the trajectory starting from AtzB_Hom_Hal show a preference for ethylaminohydrolase activity, which becomes nearly absolute in the quadruple mutant. In AtzB_Hom_Pleo, a strong preference for ethylaminohydrolase activity is first established in the triple mutant and also becomes nearly absolute in the quadruple mutant. Notably, neither N-ethylammelide nor N-isopropylammelide was further converted by any of the tested mutants, meaning that the observed product compositions are not distorted by any consecutive reaction. Conclusively, these findings demonstrate that the established trajectories of the homologues lead to highly active and specific hydroxyatrazine ethylaminohydrolases.
Rationalizing the Effect of Mutations on the Active Site Pocket and Their Impact on Functional Conversion
Intrigued by the question of how the introduced active site mutations convert AtzB into a guanine deaminase, we decided to perform nanosecond time-scale molecular dynamics (MD) simulations for each mutant along the trajectory from wild-type AtzB to AtzB-CQNN. For each mutant, three replicas of 500 ns of MD were simulated in the absence of any substrate. Principal component analysis (PCA) was applied, focusing on the distances between all heavy atoms located within 25 Å of the Zn(II) metal center, and conformational population analysis was performed (Figure 6).
Figure 6.

Principal component analysis (PCA) and representative structures for each AtzB mutant along the main trajectory. Conformational population analysis as described by PCA based on analyzing the distances between all heavy atoms located within 25 Å of the Zn(II) metal center along the MD simulations for each mutant along the trajectory from AtzB to AtzB-CQNN. PC1 mostly involves the change in conformation of E248 and Y138, whereas PC2 indicates the displacement of Y103 from the active site. A representative structure from each minimum is displayed. The introduced mutations at each step of the pathway are highlighted with a gold box. All distances between atoms connected by a dashed line are in Å.
The most populated minimum for wild-type AtzB contains in the active site two tyrosine residues Y103 and Y138, which establish a labile hydrogen bond (4.5 ± 1.6 Å, Figure 6 (AtzB panel), Figure S4) and are situated close to the Zn(II) metal center (the Y103-Zn(II) distance is 8.3 ± 1.9 Å, Figure 6 (AtzB panel)). Additionally, residue Q79 is also in the proximity of the metal site. These three residues could be potentially involved in substrate anchoring and in promoting catalysis similarly to what is found for Q87/Q105 and R209/R231 in Escherichia coli/Saccharomyces cerevisiae guanine deaminase (GuaD) and related enzymes.36 Y103 is located in an α-helix that additionally contains W98, which in AtzB positions its indole side chain in the active site pocket (Figure 6 (AtzB panel)).
Interestingly, the introduction of S218C into AtzB alters the position of the nearby Y138, which, in turn, affects the conformation of the α-helix containing Y103 and W98 (Figure S4). The distance between the hydroxyl groups of Y103 and Y138 is elongated to 8.5 ± 3.9 Å in AtzB-C, thus displacing Y103 farther from the Zn(II) metal center (14.0 ± 4.9 Å, Figure 6 (AtzB-C panel), Figure S4). Moreover, W98 is shifted from the active site in AtzB-C, and instead, W90, contained in a flexible loop, gets closer to the metal center. In AtzB-CQ, the newly introduced Q219 residue establishes a hydrogen bond with the backbone nitrogen of I222 (the mean distance along the MD trajectory between heavy atoms is 3.2 ± 0.2 Å), which brings back Y103 closer to the active site (distance of 7.3 ± 0.7 Å with respect to Zn(II)). A shorter distance between Y103 and Y138 (3.6 ± 0.5 Å) is also observed in this variant (Figure S4). Similarly, both tryptophan residues (W90 and W98) return to a conformation that is similar to the one found in AtzB (Figure 6 (AtzB-CQ and AtzB panels)). In AtzB-CQN, Y103 and Y138 are located at 3.4 ± 0.6 Å, and similarly to what occurs in AtzB-CQ, Y103 is situated at a long distance from the metal site (8.8 ± 0.7 Å). This also affects the conformation of W98 that is located far away from the metal center, thus providing additional space for W90 to fit in the active site pocket as observed in AtzB-C. The new asparagine N170 introduced in AtzB-CQN can potentially establish a labile hydrogen bond with the backbone of the nearby L172 residue (the distance between the carbonyl group of N170 and the amide backbone of L172 is 4.9 ± 1.5 Å, Figure 6 (AtzB-CQN panel), Figure S5A). In the quadruple variant AtzB-CQNN, Y103 is completely displaced from its original position mostly due to the S218C mutation as shown in AtzB-C (the distance between the hydroxyl groups of Y103 and Y138 is 4.8 ± 1.2 Å, and Y103 is located 11.8 ± 0.5 Å from Zn(II), Figure S4). Furthermore, N170 is in close proximity to form a potential hydrogen bond with the newly introduced N222 residue, which at the same time precisely positions Q219 close to Y138 through hydrogen bonding (the distance between N170 and N222 is 4.4 ± 0.9 Å, that between N222 and Q219 is 4.8 ± 1.5 Å, and that between Q219 and Y138 is 5.5 ± 1.2 Å, Figure 6 (AtzB-CQNN panel)). This reshapes the active site pocket and positions a new amide group (Q219) close to the reaction center, ready to assist guanine binding for catalysis (see discussion below and Figure 7D). As observed in the previous mutants, in AtzB-CQNN, W98 is displaced from its original position and, instead, W90 is located close to the Zn(II) metal site. This new positioning of the conserved W90 in the active site fits precisely with liganded crystal structures of GuaD (Figure S6), in which guanine is properly positioned on top of the Zn(II) metal center due to its interaction with W90 and Q79.
Figure 7.

Representative conformations explored in the 100 ns MD simulations with either hydroxyatrazine (HA) or guanine (Gua) bound in the AtzB and AtzB-CQNN active sites. The most populated conformation of the substrate is displayed in sticks. A volume containing the multiple conformations sampled by the substrates is shown with a transparent surface. Most stable substrate poses in the active site pocket are easily recognized as smaller deviations between the represented volume and the most visited substrate poses are found. The active site of wild-type AtzB binds HA in a productive conformation (A), while it cannot productively bind Gua (B). Inversely, AtzB-CQNN can no longer orient HA correctly for catalysis (C) while Gua is positioned in a way that a deamination reaction can occur (D). Distances between atoms connected by stippled lines are shown in Å and the catalytic distance between the oxygen of the hydroxide and the carbon of the substrate where deamination will take place is highlighted with a black box. The angle (a) for the deamination (Owat-CGUA/HA-NGUA/HA) is shown in degrees. The distance between the oxygen of the substrates and the hydroxyl group of Y138 is also represented. For AtzB-CQNN, the most relevant distances for productive binding of Gua (i.e., hydrogen bonds established between Gua and Q219, Y138, and Q79) in the active site pocket are averaged along the MD trajectory and shown in Å.
We further performed 100 ns MD simulations with both hydroxyatrazine and guanine bound in representative structures of AtzB and AtzB-CQNN to evaluate how the reshaping of the active site impacts substrate binding. A representative structure showing the overlay of the resulting conformations of both substrates in the active site pocket of AtzB and AtzB-CQNN is shown in Figure 7 and Figure S7.
The MD simulations suggest that in wild-type AtzB, the hydroxyl group of hydroxyatrazine is positioned close to Y103, which is hydrogen bonded to Y138 (3.2 ± 0.4 Å, Figure S5B). While the distance between the hydroxyl group of hydroxyatrazine and Y138 is rather large (5.0 ± 1.5 Å), hydroxyatrazine is still properly maintained close to the Zn(II) metal center. The conformation of W98 contributes to this proper positioning, as it can establish a CH ··π interaction with the aromatic ring of the substrate (the distance between the center of mass of hydroxyatrazine and the closest carbon atom of W98 is 5.6 ± 0.9 Å, Figure 7A and Figures S5B and S8). This interaction maintains hydroxyatrazine on top of the Zn(II) metal site and allows the proper positioning of the carbon atom where deamination will take place close to the hydroxide coordinated to Zn(II) (with a distance of 5.8 ± 0.7 Å and an angle for the nucleophilic attack of 62.6 ± 18.3°, Figure 7A and Figure S7). The much bulkier guanine substrate does not fit the active site pocket of AtzB, and despite having a similar mean catalytic distance, guanine adopts multiple poses as shown with the large standard deviation found for the nucleophilic angle (Figure 7B and Figure S7).
As described above, W98 and Y103 are displaced from the active site in AtzB-CQNN, which provides further space for the bulkier guanine to fit in the pocket. In AtzB-CQNN, guanine can establish hydrogen bond interactions with Q79, Y138, and the newly introduced Q219 (Figure 7D and Figures S5C and S8). Interestingly, in AtzB-CQNN, W90 is situated close to the Zn(II) metal center and contributes to stabilize the guanine substrate in the active site through CH ··π interactions (the mean distance between the center of mass of both guanine and W90 is 5.5 ± 0.7 Å, Figure 7D and Figure S5C and S8). In AtzB-CQNN, guanine is well maintained in the active site pocket and displays a short distance between the hydroxide coordinated to the Zn(II) metal center and the carbon atom where deamination will take place (the mean distance is 3.9 ± 0.3 Å and the angle for the nucleophilic attack is 84.6 ± 6.9°, Figure 7D, Figure S5C). It should be noted that the predicted pose for guanine in AtzB-CQNN fits with the reported guanine-/xanthine-bound GuaD crystal structures (Figure S6). Inversely, the MD simulations with guanine and hydroxyatrazine bound in the active sites of AtzB and AtzB-CQNN, respectively, show an unstable and unproductive binding pose in line with the poor respective activities (Figure 7B,C). The overlay of the most populated conformations of liganded and unliganded AtzB and AtzB-CQNN structures indicates that the binding of the substrates does not substantially affect the active site conformation (Figure S9). The only exception is W90 in AtzB-CQNN that slightly changes the conformation of the indole ring in the presence of guanine for establishing and maximizing the CH ··π interaction with the substrate.
The results of the MD simulations further point to the mechanistic reason for the selectivity of AtzB for ethylaminohydrolysis. The conformation of W98 pointing toward the metal center in AtzB, and especially the position of W90, favors the binding of the smaller ethylamine group close to the Zn(II), thus dictating its preference for reaction over the isopropylamine substituent. Altogether, this analysis revealed a series of conformational changes triggered by the mutations that impact the active site shape and are responsible for the functional conversion. We observed that for enhancing guanine deamination activity in AtzB, Y103 and W98 must be displaced from the active site to allow the bulkier guanine to fit in the pocket and establish hydrogen bond interactions with Q79 and Y138. The newly introduced glutamine Q219, as well as W90 in its new conformation, contribute to stabilize guanine in the active site close to the Zn(II) metal center for deamination. These findings are in line with the structural features observed in guanine-bound crystal structures of guanine deaminases (Figure S6).
Finally, we decided to analyze the changes in the conformational dynamics induced by the mutations in the AtzB_Hom_Pleo trajectory. We computationally evaluated AtzB_Hom_Pleo and the AtzB_Hom_Pleo-IISS variant in the absence and presence of either hydroxyatrazine or guanine, following the same protocol used for AtzB. The overlay of the most populated conformation in the unliganded state shows a similar positioning for Y138 in AtzB-CQNN and Y133 in AtzB_Hom_Pleo as well as Q79 in AtzB-CQNN and Q74 in AtzB_Hom_Pleo but a somewhat different conformation for Y103/Y98, W98/W93, and W90/W85 (Figure 8A). The same changes are observed if the most populated conformations of AtzB and Atz_Hom_Pleo-IISS are compared (Figure 8B). As opposed to Y103 in AtzB-CQNN, Y98 in AtzB_Hom_Pleo is located in the active site pocket and actually together with Y133 helps position guanine close to the Zn(II) metal site for catalysis (the distance between the hydroxyl group of Y98/Y133 and N1 of Gua is 3.5 ± 0.6 Å, Figure 8C and Figure S10A). Y98 establishes a labile hydrogen bond with N217 (5.1 ± 1.6 Å), which at the same time is hydrogen bonded to the nearby Q214 (3.3 ± 0.4 Å, Figure 8C). This contrasts with what we observed in AtzB-CQNN, as Y103 (Y98 in AtzB_Hom_Pleo) is far from the active site, and instead Q219 (Q214 in AtzB_Hom_Pleo) contributes to the proper positioning of guanine in the pocket for catalysis (Figure 7D). Another interesting difference is that in AtzB_Hom_Pleo, W93 (W98 in AtzB-CQNN) instead of W85 (W90 in AtzB-CQNN) establishes a CH ··π interaction with guanine and contributes to its proper positioning (Figures 7D and 8C). W85 is displaced from the active site but can contribute to the retention of guanine in the pocket. As we observed in AtzB-CQNN, Q74 is hydrogen bonded to guanine (Figure S10A) and plays a key role in properly positioning the substrate close to the Zn(II) metal site for the nucleophilic attack of the hydroxide (the distance is 3.6 ± 0.2 Å and the angle 87.6 ± 12.8°).
Figure 8.

Comparison of the most populated conformations of AtzB and AtzB_Hom_Pleo as well as mutants, and representative conformations of AtzB_Hom_Pleo and AtzB_Hom_Pleo-IISS with bound hydroxyatrazine (HA) and guanine (Gua). (A) Representative conformation of AtzB-CQNN compared with AtzB_Hom_Pleo showing similar side-chain conformations of Q79/Q74, W98/W93, and Y138/Y133. (B) Representative conformation of AtzB compared with AtzB_Hom_Pleo-IISS showing a similar positioning to that of Y138/Y133. (C) AtzB_Hom_Pleo with bound Gua and (D) AtzB_Hom_Pleo-IISS with bound HA. All represented distances between atoms connected by stippled lines are averaged along the MD trajectories and shown in Å; the catalytic distance between the oxygen of the hydroxide and the carbon of the substrate where deamination will take place is highlighted with a black box. The angle (a) for the deamination (Owat-CGUA/HA-NGUA/HA) is shown in degrees.
Compared to AtzB, a slightly different binding pose of hydroxyatrazine is found in AtzB_Hom_Pleo-IISS (Figure 8D). In this case, Q74 is hydrogen bonded to the substrate (3.6 ± 0.8 Å) and contributes to its proper positioning close to the Zn(II) metal center. Both W93 and W85 establish CH ··π interactions with hydroxyatrazine (Figure S10B), and especially, the conformation of W85 favors the positioning of the smaller ethylamine group close to the metal center for hydrolysis. This results in a catalytic distance of 4.3 ± 1.3 Å and an angle for hydrolysis of 94.9 ± 20.5° (Figure 8D).
Discussion
Toward an AtzB Progenitor
In our effort to reconstruct the evolutionary history of the hydroxyatrazine ethylaminohydrolase AtzB, we performed substrate screening that identified a guanine deaminase side activity in AtzB. We succeeded in drastically increasing this activity by the stepwise introduction of only four residue exchanges, which were identified by comparison of AtzB with its closest homologues. Our results are in line with previous presumptions that the generation of a new activity by a low number of amino acid substitutions is evolutionary more plausible than the necessity of multiple mutations for functional innovation.30 We consider the end point of this trajectory, AtzB-CQNN, as a pseudoprogenitor of AtzB due to certain traits that would be expected for the true progenitor: Its guanine deaminase activity could have constituted its physiological role before the introduction of atrazine into the environment. Its promiscuous activity for hydroxyatrazine might then have provided an early selection advantage after this substance became available to soil organisms and was further optimized after a presumed gene duplication event, in a process called “subfunctionalization”.37 Alternatively, the AtzB activity may have become physiologically meaningful only after gene duplication and this is when the actual biologically relevant innovation happened, corresponding to a “neofunctionlization” process.38 It should be noted that both putative events presuppose the dechlorination of atrazine, either by an early atrazine chlorohydrolase or through photolytic reactions39,40 to provide the AtzB substrate hydroxyatrazine.
The limited number of residue exchanges required for functional conversion of AtzB into an enzyme with guanine deamination as its predominant activity allowed for the full analysis of the intermediate sequence space. This analysis revealed that two viable routes exist connecting the pseudoprogenitor AtzB-CQNN to wild-type AtzB across mutants with gradually increased hydroxyatrazine hydrolase activities. These two routes consist of the stepwise introduction of N222I, N170I, Q219S, and C218S or, alternatively, through the successive acquisition of Q219S, N222I, N170I, and C218S. For both potential trajectories, a negative trade-off between the original and the novel activity is observed (Figure 3), which is a general principle in the evolution of novel enzyme functions.41
Although one cannot know whether AtzB-CQNN ever existed in Nature, naturally occurring AtzB variants have been identified that carry the two amino acid exchanges S218C and S219Q (accession: AAY40323.2 and BAD69556.1). This could imply that these two exchanges were the most recent ones to be acquired, which would fit a scenario in which AtzB-CQNN is the true progenitor of AtzB. Furthermore, considering the nucleotide sequence encoding the AtzB protein from Pseudomonas sp. strain ADP (accession: AAC45138.1), two of the three types of residue exchanges required for the conversion from AtzB-CQNN to AtzB (C → S; N → I) are favored by the genetic code insofar as a single base exchange may be sufficient for them to arise (TGC → AGC; AAC/AAT → ATC/ATT). Only the exchange of glutamine with serine (Q → S) requires at least two base exchanges (CAA/CAG → TCA/TCG).
Evolutionary Potential of AtzB Homologues
The two closest known homologues of AtzB, AtzB_Hom_Hal and AtzB_Hom_Pleo, were investigated in this work for the first time. They were shown to be guanine deaminases and to exhibit promiscuous hydroxyatrazine hydrolase activities. Despite a global sequence identity between AtzB and AtzB_Hom_Hal/AtzB_Hom_Pleo of only 64%/56%, respectively, hydroxyatrazine hydrolase activities could be established in the background of the homologues. Remarkably, in the case of AtzB_Hom_Pleo-IISS, this activity even reached that of native AtzB. Importantly, this could be achieved by the introduction of the reverse residue exchanges that were required to establish high guanine deaminase activity on the scaffold of AtzB: Whereas AtzB_Hom_Pleo followed the main AtzB trajectory, AtzB_Hom_Hal followed the alternative AtzB trajectory (Figure 4). Moreover, in both trajectories, the stepwise increase of hydroxyatrazine hydrolase activity overall coincided with an increase of product specificities (Figure 5).
In summary, these findings underline that the evolutionary potential for the emergence of high and specific hydroxyatrazine ethylaminohydrolase activity exists more than once among members of the amidohydrolase superfamily. Hence, any enzyme with an active site sufficiently similar to that of AtzB_Hom_Hal or AtzB_Hom_Pleo could undergo or could have undergone the same evolutionary process, as described in this work.
Structure–Dynamics–Function Relationships of the Functional Conversion of AtzB and Its Homologues
We wanted to understand the structural basis of the functional conversion of the hydroxyatrazine ethylaminohydrolase AtzB into the guanine deaminase AtzB-CQNN. Enzymes belonging to subtype III of the amidohydrolase superfamily, such as AtzB, possess a highly conserved divalent metal binding site42 and a catalytic histidine (H280 in AtzB) that activates a metal-bound water molecule, which then initiates the hydrolytic reaction. Since this mechanism is most likely conserved between AtzB and AtzB-CQNN, it is plausible to assume that the different catalytic activities are based on different substrate affinities and/or orientations. Since hydroxyatrazine and guanine share similar core structures in the s-triazine ring and the pyrimidine portion of the purine moiety, substrate binding must be dominated by interactions of the isopropylamine group and the imidazole portion of the purine moiety with the respective active site. This conclusion is illustrated in Figures 7 and 8 by the comparison of the most populated conformation of both substrates obtained from the substrate-bound MD simulations for AtzB, AtzB_Hom_Pleo, AtzB-CQNN, and AtzB_Hom_Pleo-IISS. Hydroxyatrazine establishes a network of hydrogen bonds with Y138, Y103, and a CH ··π interaction with W98 in AtzB, whereas the ligand interacts with Q74 and W93 in AtzB_Hom_Pleo-IISS. This allows for the proper positioning of hydroxyatrazine on top of the Zn(II) metal center for efficient ethylaminohydrolysis. The bulkier guanine substrate is properly oriented for deamination in AtzB-CQNN and AtzB_Hom_Pleo. Thanks to the introduced mutations, Y103 and W98 are displaced from the active site in AtzB-CQNN, which maintain Q79 and allow the positioning of W90 close to the metal site. In addition to that, several hydrogen bonds between the side chains of the mutated residues are established, which ultimately favor the positioning of the new Q219 close to the reaction center, essential for productive guanine binding and catalysis. This is different in AtzB_Hom_Pleo as guanine establishes hydrogen bonds with Q74 and Y98, the latter of which is maintained in the active site pocket thanks to N217 and Q214. W93 in AtzB_Hom_Pleo contributes to the proper positioning of guanine close to the Zn(II) metal center.
By comparing the active site pocket of AtzB-CQNN and the different available guanine/xanthine-bound crystal structures of GuaD (Figure S6), we observed that indeed, Q79 and W90 are conserved and clearly involved in guanine binding. The position of the nonconserved R231 found in Saccharomyces cerevisiae GuaD that is hydrogen bonded to the product xanthine is instead occupied by Y138 and Q219 in AtzB-CQNN. R231 in ScGuaD establishes a salt bridge with E219, which occupies a space similar to that of Y103 found in AtzB-CQNN. Therefore, the analysis of the functional transition between AtzB and AtzB-CQNN with MD simulations identified some key conformational changes that reshape the active site pocket and convert the pocket of AtzB into a more GuaD-like active site.
The observed preference of AtzB, AtzB_Hom_Hal-SIIS, and AtzB_Hom_Pleo-IISS for ethylaminohydrolysis (Figure 5) requires the ethylamine group of hydroxyatrazine to be positioned toward the metal center. This preference might be attributed to the conformation of the conserved W98 (AtzB)/89 (AtzB_Hom_Hal)/93 (AtzB_Hom_Pleo) and W90/81/85 that impose some steric constraints in one side of the active site pocket and favor the positioning of the smaller ethylamine group close to the Zn(II) for efficient hydrolysis. W98/89/93 is clearly affected by the conformation of a conserved tyrosine residue Y103/94/98, situated close to another shared tyrosine Y138/129/133 that can establish a hydrogen bond with the hydroxyl group of hydroxyatrazine.
In AtzB-CQNN, AtzB_Hom_Hal, and AtzB_Hom_Pleo, which are predominantly guanine deaminases, Q219/210/214 is suited to direct guanine into a catalytically active conformation by interaction with the nitrogens of the imidazole portion of the molecule (N7, N9). The role of N170/161/165 and N222/213/217 is to rigidify and precisely position Q219/210/214 for its direct interaction with guanine (in AtzB-CQNN) or indirectly via Y98 (in AtzB_Hom_Pleo), which is achieved through a network of hydrogen bonds as shown by the MD simulations depicted in Figures 6, 7, and 8. Additionally, Q79/70/74 establishes hydrogen bonds with O6 of guanine for its proper positioning for catalysis. As opposed to hydroxyatrazine hydrolysis, the productive binding of guanine in the active site of AtzB-CQNN requires the displacement of both conserved Y103/94/98 and W98/89/93. Guanine is additionally stabilized in the active site pocket thanks to the dispersion interactions established with W90/81/85 and W98/89/93. Despite AtzB and AtzB_Hom_Pleo presenting different active sites (in line with the rather low sequence identity between them), the mutations introduced in both scaffolds reshape the pockets and induce a series of conformational changes in residues found to be involved in substrate binding in both trajectories: Q79/Q74, W90/W85, and W98/W93.
Conclusions
Using a combination of structure- and sequence-guided protein engineering, we have uncovered the mutations by which the evolutionarily young enzyme hydroxyatrazine hydrolase AtzB may have rapidly evolved from an evolutionarily old enzyme with guanine deaminase activity. Moreover, we could identify evolutionary trajectories between these two activities and uncover the structural and molecular basis of the functional transition. Hence, our findings highlight the power of protein engineering in combination with computational methods for gaining a detailed mechanistic understanding of enzyme evolution. Moreover, our study demonstrates how characterizing natural variants with unknown functions can advance the comprehension of both enzyme evolution and the principles of protein design.
Material and Methods
Bioinformatics
Sequences of AtzB proteins and their homologues were retrieved from NCBI43 utilizing protein BLAST44 with standard parameters. Pairwise sequence identities were determined by EMBOSS Needle with standard parameters.45 Multiple sequence alignments were generated with Clustal Omega46 and visualized as sequence logos with WebLogo.47
Gene Cloning and Site-Directed Mutagenesis
Genes coding for AtzB, AtzB_Hom_Hal, and AtzB_Hom_Pleo were codon optimized for expression in Escherichia coli (Table S4), ordered as gene strings (GeneArt, Regensburg), and subcloned into the pUR22 plasmid.48 A modified version of the QuickChange protocol49 was employed for site-directed mutagenesis. Primers containing specific mismatches (Table S5) were designed to amplify the entirety of the target plasmid. The resulting linear amplicon was treated with T4 polynucleotide kinase and T4 DNA ligase (5 U; 10 U; 30 min 37 °C; 30 min RT) in 1× T4 ligase buffer in a total volume of 50 μL. The resulting ligation solution, containing circular, mutated plasmids, was used for the transformation of E. coli cells without further purification. Integrity of the coding region and the presence of mutations were confirmed in each case by Sanger Sequencing (Microsynth Seqlab).
Protein Production and Purification
The main strain for gene expression was E. coli BW25113 guaD::Kan (JW5466-350) to avoid contamination with E. coli guanine deaminase. AtzB_Hom_Hal and variants thereof gave poor yields in BW25113 guaD::Kan and were instead produced in E. coli BL21 (DE3) Gold (Agilent Technologies). All proteins were produced as C-terminal His6-tagged fusion constructs. BW25113 guaD::Kan or E. coli BL21 (DE3) Gold cells were transformed with expression vectors and grown in LB medium (37 °C; 140 rpm) supplemented with applicable antibiotics to an OD600 of 0.6–0.8 before the addition of 0.5 mM of isopropyl β-d-1-thiogalactopyranoside to induce gene expression. Cells were cultivated overnight (20 °C; 16 h), harvested (JFA 8.1; 16,000g; 20 min; 4 °C), and resuspended in 100 mM Tris/HCl (pH 7.5), 300 mM KCl, 10 mM imidazole. Cells were disrupted by sonication, debris was removed by centrifugation (16,000g; 45 min; 4 °C), and proteins were purified from the supernatant by immobilized Ni2+ ion affinity chromatography in 100 mM Tris/HCl (pH 7.5) and 300 mM KCl, by applying a linear imidazole gradient (10 mM → 1 M). Further purification was achieved by preparative size exclusion chromatography on a HiLoad 26/60 Superdex 75 or a HiLoad 26/60 Superdex 200 column equilibrated with SEC buffer (50 mM Tris/HCl (pH 7.5), 50 mM KCl). Protein elution was monitored by a continuous measurement of OD280. Relevant fractions were further analyzed by SDS-PAGE before selected fractions were pooled, frozen in liquid nitrogen, and stored at −80 °C. Protein concentrations were determined by absorption spectroscopy using a molar extinction coefficient at 280 nm that was calculated from the amino acid sequence.51 Yields for variants of AtzB ranged from 2.4 mg of protein per liter of culture medium to 12 mg/L. Variants of AtzB_Hom_Pleo gave yields of around 3.0 mg/L, while AtzB_Hom_Hal could only be produced in small quantities with yields ranging from 0.1 to 0.4 mg/L.
Compound Synthesis
N-Ethylammelide and N-isopropylammelide were synthesized by reacting 6 mg of 6-chloro-1,3,5-triazine-2,4(1H,3H)-dione in 300 μL of KOH (0.2 M) with either 2.6 μL of ethylamine solution (70%) or 3.5 μL of isopropylamine. The reaction proceeded for 5 min at 90 °C. The reaction mixtures were analyzed by HPLC (Eclipse XDB-C18 (4.6 × 150); gradient 5–100% acetonitrile in water) and mass spectrometry (ESI-QTOF) to confirm the identity of the respective substance. To quantify N-ethylammelide and N-isopropylammelide, selected volumes of each compound were incubated with 10 nM AtzC. After total turnover, the absorbance change was used to determine the concentrations by applying Lambert–Beer’s law and previously published molar extinction coefficients.25
Qualitative Enzyme Assays
To confirm the activity of wild-type and mutant enzymes toward a certain substrate or to identify product composition, qualitative assays based on HPLC analysis were established. All enzymatic assays contained 500 μM of the respective substrate in a total volume of 150 μL of potassium phosphate buffer (50 mM; pH 7.0) as well as 10 μM of the enzyme variant to be tested. Genuine substance standards were set up in the same way without enzyme present. Assays were incubated for 20 h at RT, at which point all enzymes were removed by ultrafiltration and the filtrate was analyzed by HPLC (Eclipse XDB-C18 (4.6 × 150); gradient 5–100% acetonitrile in water).
Steady-State Kinetic Enzyme Assays
Quantification of the hydrolysis of hydroxyatrazine and guanine was achieved under steady-state conditions through direct photometric assays. The conversion of hydroxyatrazine into ethylamine and N-isopropylammelide produces a photometric signal with a differential extinction coefficient of Δε242 = 10,957 M–1 cm–1 as deduced by the differential spectrum of (a) genuine hydroxyatrazine and (b) the reaction mixture of complete conversion of hydroxyatrazine by AtzB (adjusted for the absorption of the AtzB enzyme). Hydrolysis of guanine, resulting in xanthine and ammonia, is accompanied by a photometric signal with a differential extinction coefficient of Δε242 = 5023 M–1 cm–1. This value was derived from the differential spectra of genuine guanine and xanthine. Standard assays contained 5–50 μM hydroxyatrazine or 10–300 μM guanine in a total volume of 300 μL of potassium phosphate buffer (50 mM; pH 7.5). The reactions were performed at 25 °C and initiated with 1 nM–10 μM of enzyme. In all cases, it was assured that the enzyme concentration was at least 10 times lower than the substrate concentration. Fast screening of catalytic activities was performed using a plate reader (Tecan Infinite M200 PRO), and subsequently selected variants were assayed in a spectrophotometer (JASCO V-650). Initial rates of change of A242 or A253 were determined by linear regression and converted into the rate constant (v/E0). Rate constants were then plotted against substrate concentration, and the resulting graph was fitted either with a hyperbolic function when possible, revealing both kcat and KM, or with a linear function, revealing kcat/KM.
Computational Methods
Structural models of AtzB and AtzB_Hom_Pleo were generated using AlphaFold2.52 The position of Zn(II) and the catalytic water molecule were generated using the AlphaFill program.53 Correct protonation states of the structures were generated using the H++ server.54 The metal ion binding site parameters were generated using the MCPB.py program in the Amber18 software package.55 The partial charges and bonded parameters for metal binding site residues, Zn(II), and water molecules were generated in the Gaussian09 software package by utilizing the B3LYP/6-31G* level of theory. The structures for the simulation were prepared using the tleap program.56 The AtzB mutants resulting from the sequential introduction of S218C, S219Q, I170N, and I222N and for AtzB_Hom_Pleo, mutations N217I, N165I, Q214S, C213S were generated with PyMoL and further validated by AlphaFold2 prediction. These apo structures were simulated for 500 ns (in triplicate) in the Amber18 program using the ff14SB force field and the TIP3P water model.57 The accumulated simulation time (i.e., 1500 ns for each variant) was sufficient for capturing the key conformational changes between variants involved in the functional transition. The data were then analyzed by applying PCA considering all heavy atoms located around 25 Å from the Zn(II) metal center. A conformational population analysis (as shown in Figure 6) was then performed. Different minima structures from the PCA were taken and used to dock the ligands (guanine and hydroxyatrazine) using AutoDock4.2.58 The keto form of guanine59 and the enol form of hydroxyatrazine were procured from the PubChem database and optimized with Gaussian09 at the B3LYP/6-31G* level of theory. The ligands and the AtzB, AtzB-CQNN, AtzB_Hom_Pleo, and AtzB_Hom_Pleo-IISS enzymes were prepared separately for the docking, and a grid box of 20 Å was created around the Zn metal ion binding site containing all mutant residues. Furthermore, the parameters for ligand molecules were generated using Gaussian09. These parameters were utilized to prepare the input files for ligand-bound holo structures. The best-ranked docked poses were then further studied and refined by means of shorter 100 ns MD simulations (in triplicate). In this case, we observed that 100 ns MD simulations were enough to properly discriminate between variants and to elucidate the molecular basis of the large difference in activity between AtzB and AtzB-CQNN toward hydroxyatrazine and guanine. The mean value and standard deviation of all distances reported were computed using the MDTraj program60 considering the entire MD trajectories. Structures were analyzed and rendered by using PyMoL.
Acknowledgments
We thank Dr. Sandra Schlee for critical reading of the manuscript. This work was supported by a grant of the Deutsche Forschungsgemeinschaft to R.S. (STE 891/16-1) and a fellowship from the Fonds der Chemischen Industrie to L.D. We thank the Generalitat de Catalunya for the consolidated group TCBioSys (SGR 2021 00487) and grant projects PID2021-129034NB-I00 and PDC2022-133950-I00 funded by Spanish MICIN. S.O. is grateful to the funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (ERC-2015-StG-679001,ERC-2022-POC-101112805, and ERC-2022-CoG-101088032), and the Human Frontier Science Program (HFSP) for project grant RGP0054/2020. D.R.M. was supported by a research grant from ERC-StG (ERC-2015-StG-679001) and an HFSP grant (RGP0054/2020).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c04010.
Substrate screening of AtzB, AtzB_Hom_Hal, and AtzB_Hom_Pleo; assessment of protein purity by SDS-PAGE; analysis of product mixtures of hydroxyatrazine hydrolysis; plot of key active site distances for AtzB, AtzB-C, AtzB-CQ, AtzB-CQN, AtzB-CQNN; histogram and distribution plot of key active site distances in apo structures, and AtzB liganded with hydroxyatrazine (HA) and guanine (Gua); structural and sequence comparison of liganded guanine deaminases (GuaD) and a representative conformation of AtzB-CQNN with guanine bound from 100 ns MD simulation; overlay of conformations explored in the 100 ns MD simulations with either hydroxyatrazine (HA) or guanine (Gua) bound in the AtzB and AtzB-CQNN active sites; plot of key active site distances for AtzB and AtzB-CQNN in the presence of either hydroxyatrazine (HA) or guanine (Gua); overlay of the most populated representative conformation of AtzB and AtzB-CQNN in the unliganded and liganded states; distance distribution plots for AtzB_Hom_Pleo bound with guanine and hydroxyatrazine substrates; AtzB proteins and AtzB homologues used to construct sequence logos; catalytic parameters of mutants of AtzB, AtzB_Hom_Hal, and AtzB_Hom_Pleo for the deamination of guanine; catalytic parameters of mutants of AtzB, AtzB_Hom_Hal, and AtzB_Hom_Pleo for the hydrolysis of hydroxyatrazine; synthesized genes used in this work; and nucleotide primers used in this work (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Copley S. D. Evolution of a Metabolic Pathway for Degradation of a Toxic Xenobiotic: The Patchwork Approach. Trends Biochem. Sci. 2000, 25 (6), 261–265. 10.1016/S0968-0004(00)01562-0. [DOI] [PubMed] [Google Scholar]
- Russell R. J.; Scott C.; Jackson C. J.; Pandey R.; Pandey G.; Taylor M. C.; Coppin C. W.; Liu J.-W.; Oakeshott J. G. The Evolution of New Enzyme Function: Lessons from Xenobiotic Metabolizing Bacteria versus Insecticide-Resistant Insects. Evol Appl. 2011, 4 (2), 225–248. 10.1111/j.1752-4571.2010.00175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullet J. E.; Arntzen C. J. Identification of a 32–34-Kilodalton Polypeptide as a Herbicide Receptor Protein in photosystem II. BBA-Bioenergetics 1981, 635 (2), 236–248. 10.1016/0005-2728(81)90023-2. [DOI] [PubMed] [Google Scholar]
- Wackett L. P.; Sadowsky M. J.; Martinez B.; Shapir N. Biodegradation of atrazine and Related S-Triazine Compounds: From Enzymes to Field Studies. Appl. Microbiol. Biotechnol. 2002, 58 (1), 39–45. 10.1007/s00253-001-0862-y. [DOI] [PubMed] [Google Scholar]
- Belluck D. A.; Benjamin S. L.; Dawson T.. Groundwater Contamination by atrazine and Its Metabolites. In Pesticide Transformation Products; ACS Symposium Series; American Chemical Society, 1991; Vol. 459, pp 254–273.
- Fan X.; Song F. Bioremediation of atrazine: Recent Advances and Promises. J. Soils Sediments 2014, 14 (10), 1727–1737. 10.1007/s11368-014-0921-5. [DOI] [Google Scholar]
- Udiković-Kolić N.; Scott C.; Martin-Laurent F. Evolution of atrazine-Degrading Capabilities in the Environment. Appl. Microbiol. Biotechnol. 2012, 96 (5), 1175–1189. 10.1007/s00253-012-4495-0. [DOI] [PubMed] [Google Scholar]
- de Souza M. L.; Seffernick J.; Martinez B.; Sadowsky M. J.; Wackett L. P. The atrazine Catabolism Genes atzABC Are Widespread and Highly Conserved. J. Bacteriol. 1998, 180 (7), 1951–1954. 10.1128/JB.180.7.1951-1954.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutz L. J.; Shaner D. L.; Weaver M. A.; Webb R. M.; Zablotowicz R. M.; Reddy K. N.; Huang Y.; Thomson S. J. Agronomic and Environmental Implications of Enhanced S-Triazine Degradation. Pest. Manage. Sci. 2010, 66 (5), 461–481. 10.1002/ps.1909. [DOI] [PubMed] [Google Scholar]
- Strong L. C.; Rosendahl C.; Johnson G.; Sadowsky M. J.; Wackett L. P. Arthrobacter Aurescens TC1Metabolizes Diverse S-Triazine Ring Compounds. Appl. Environ. Microbiol. 2002, 68 (12), 5973–5980. 10.1128/AEM.68.12.5973-5980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govantes F.; Porrúa O.; García-González V.; Santero E. atrazine Biodegradation in the Lab and in the Field: Enzymatic Activities and Gene Regulation. Microb Biotechnol 2009, 2 (2), 178–185. 10.1111/j.1751-7915.2008.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelbaum R. T.; Allan D. L.; Wackett L. P. Isolation and Characterization of a Pseudomonas Sp. That Mineralizes the s-Triazine Herbicide atrazine. Appl. Environ. Microbiol. 1995, 61 (4), 1451–1457. 10.1128/aem.61.4.1451-1457.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquirol L.; Peat T. S.; Wilding M.; Liu J.-W.; French N. G.; Hartley C. J.; Onagi H.; Nebl T.; Easton C. J.; Newman J.; Scott C. An Unexpected Vestigial Protein Complex Reveals the Evolutionary Origins of an S-Triazine Catabolic Enzyme. J. Biol. Chem. 2018, 293 (20), 7880–7891. 10.1074/jbc.RA118.001996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquirol L.; Peat T. S.; Wilding M.; Hartley C. J.; Newman J.; Scott C. A Novel Decarboxylating Amidohydrolase Involved in Avoiding Metabolic Dead Ends during cyanuric Acid Catabolism in Pseudomonas Sp. Strain ADP. PLoS One 2018, 13 (11), e0206949 10.1371/journal.pone.0206949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza M. L.; Wackett L. P.; Sadowsky M. J. The atzABC Genes Encoding atrazine Catabolism Are Located on a Self-Transmissible Plasmid in Pseudomonas Sp. Strain ADP. Appl. Environ. Microbiol. 1998, 64 (6), 2323–2326. 10.1128/AEM.64.6.2323-2326.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-González V.; Govantes F.; Porrúa O.; Santero E. Regulation of the Pseudomonas Sp. Strain ADP cyanuric Acid Degradation Operon. J. Bacteriol. 2005, 187 (1), 155–167. 10.1128/JB.187.1.155-167.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govantes F.; García-González V.; Porrúa O.; Platero A. I.; Jiménez-Fernández A.; Santero E. Regulation of the atrazine-Degradative Genes in Pseudomonas Sp. Strain ADP. FEMS Microbiol Lett. 2010, 310 (1), 1–8. 10.1111/j.1574-6968.2010.01991.x. [DOI] [PubMed] [Google Scholar]
- Seffernick J. L.; Wackett L. P. Ancient Evolution and Recent Evolution Converge for the Biodegradation of cyanuric Acid and Related Triazines. Appl. Environ. Microbiol. 2016, 82 (6), 1638–1645. 10.1128/AEM.03594-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez B.; Tomkins J.; Wackett L. P.; Wing R.; Sadowsky M. J. Complete Nucleotide Sequence and Organization of the atrazine Catabolic Plasmid pADP-1 from Pseudomonas Sp. Strain ADP. J. Bacteriol. 2001, 183 (19), 5684–5697. 10.1128/JB.183.19.5684-5697.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seffernick J. L.; Wackett L. P. Rapid Evolution of Bacterial Catabolic Enzymes: A Case Study with atrazine Chlorohydrolase. Biochemistry 2001, 40 (43), 12747–12753. 10.1021/bi011293r. [DOI] [PubMed] [Google Scholar]
- Copley S. D. Shining a Light on Enzyme Promiscuity. Curr. Opin Struct Biol. 2017, 47, 167–175. 10.1016/j.sbi.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Tawfik D. S. Enzyme Promiscuity and Evolution in Light of Cellular Metabolism. FEBS J. 2020, 287 (7), 1260–1261. 10.1111/febs.15296. [DOI] [PubMed] [Google Scholar]
- Seffernick J. L.; McTavish H.; Osborne J. P.; de Souza M. L.; Sadowsky M. J.; Wackett L. P. atrazine Chlorohydrolase from Pseudomonas Sp. Strain ADP Is a Metalloenzyme. Biochemistry 2002, 41 (48), 14430–14437. 10.1021/bi020415s. [DOI] [PubMed] [Google Scholar]
- Seffernick J. L.; Aleem A.; Osborne J. P.; Johnson G.; Sadowsky M. J.; Wackett L. P. Hydroxyatrazine N-Ethylaminohydrolase (AtzB): An Amidohydrolase Superfamily Enzyme Catalyzing Deamination and dechlorination. J. Bacteriol. 2007, 189 (19), 6989–6997. 10.1128/JB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapir N.; Osborne J. P.; Johnson G.; Sadowsky M. J.; Wackett L. P. Purification, Substrate Range, and Metal Center of AtzC: The N-Isopropylammelide Aminohydrolase Involved in Bacterial atrazine Metabolism. J. Bacteriol. 2002, 184 (19), 5376–5384. 10.1128/JB.184.19.5376-5384.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgens C.; Strom A.; Wegener D.; Hettwer S.; Wilmanns M.; Sterner R. Directed Evolution of a (βα)8-Barrel Enzyme to Catalyze Related Reactions in Two Different Metabolic Pathways. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (18), 9925–9930. 10.1073/pnas.160255397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claren J.; Malisi C.; Höcker B.; Sterner R. Establishing Wild-Type Levels of Catalytic Activity on Natural and Artificial (βα)8-Barrel Protein Scaffolds. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (10), 3704–3709. 10.1073/pnas.0810342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton M. S.; Guo X.; Söderholm A.; Näsvall J.; Lundström P.; Andersson D. I.; Selmer M.; Patrick W. M. Structural and Functional Innovations in the Real-Time Evolution of New (βα)8-Barrel Enzymes. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (18), 4727–4732. 10.1073/pnas.1618552114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopoldseder S.; Claren J.; Jürgens C.; Sterner R. Interconverting the Catalytic Activities of (βα)8-Barrel Enzymes from Different Metabolic Pathways: Sequence Requirements and Molecular Analysis. J. Mol. Biol. 2004, 337 (4), 871–879. 10.1016/j.jmb.2004.01.062. [DOI] [PubMed] [Google Scholar]
- Schmidt D. M. Z.; Mundorff E. C.; Dojka M.; Bermudez E.; Ness J. E.; Govindarajan S.; Babbitt P. C.; Minshull J.; Gerlt J. A. Evolutionary Potential of (β/α)8-Barrels: Functional Promiscuity Produced by Single Substitutions in the Enolase Superfamily. Biochemistry 2003, 42 (28), 8387–8393. 10.1021/bi034769a. [DOI] [PubMed] [Google Scholar]
- Plach M. G.; Löffler P.; Merkl R.; Sterner R. Conversion of anthranilate Synthase into Isochorismate Synthase: Implications for the Evolution of Chorismate-Utilizing Enzymes. Angew. Chem., Int. Ed. Engl. 2015, 54 (38), 11270–11274. 10.1002/anie.201505063. [DOI] [PubMed] [Google Scholar]
- Afriat-Jurnou L.; Jackson C. J.; Tawfik D. S. Reconstructing a Missing Link in the Evolution of a Recently Diverged Phosphotriesterase by Active-Site Loop Remodeling. Biochemistry 2012, 51 (31), 6047–6055. 10.1021/bi300694t. [DOI] [PubMed] [Google Scholar]
- Yang G.; Anderson D. W.; Baier F.; Dohmen E.; Hong N.; Carr P. D.; Kamerlin S. C. L.; Jackson C. J.; Bornberg-Bauer E.; Tokuriki N. Higher-Order Epistasis Shapes the Fitness Landscape of a Xenobiotic-Degrading Enzyme. Nat. Chem. Biol. 2019, 15 (11), 1120–1128. 10.1038/s41589-019-0386-3. [DOI] [PubMed] [Google Scholar]
- Sugrue E.; Fraser N. J.; Hopkins D. H.; Carr P. D.; Khurana J. L.; Oakeshott J. G.; Scott C.; Jackson C. J. Evolutionary Expansion of the Amidohydrolase Superfamily in Bacteria in Response to the Synthetic Compounds Molinate and Diuron. Appl. Environ. Microbiol. 2015, 81 (7), 2612–2624. 10.1128/AEM.04016-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor S.; Taylor M. C.; Russell R. J.; Jermiin L. S.; Jackson C. J.; Oakeshott J. G.; Scott C. Intramolecular Epistasis and the Evolution of a New Enzymatic Function. PLoS One 2012, 7 (6), e39822 10.1371/journal.pone.0039822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shek R.; Hilaire T.; Sim J.; French J. B. Structural Determinants for Substrate Selectivity in Guanine Deaminase Enzymes of the Amidohydrolase Superfamily. Biochemistry 2019, 58 (30), 3280–3292. 10.1021/acs.biochem.9b00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Cantú A.; Ascencio D.; Barona-Gómez F.; DeLuna A. Gene Duplication and the Evolution of Moonlighting Proteins. Front. Genet. 2015, 6, 227. 10.3389/fgene.2015.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley S. D. Evolution of New Enzymes by Gene Duplication and Divergence. FEBS J. 2020, 287 (7), 1262–1283. 10.1111/febs.15299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiaozhen F.; Bo L.; Aijun G. Dynamics of Solar Light Photodegradation Behavior of atrazine on Soil Surface. J. Hazard Mater. 2005, 117 (1), 75–79. 10.1016/j.jhazmat.2004.09.011. [DOI] [Google Scholar]
- Chen C.; Yang S.; Guo Y.; Sun C.; Gu C.; Xu B. photolytic Destruction of Endocrine Disruptor atrazine in Aqueous Solution under UV Irradiation: Products and Pathways. J. Hazard Mater. 2009, 172 (2–3), 675–684. 10.1016/j.jhazmat.2009.07.050. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. S. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem. 2010, 79, 471–505. 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Seibert C. M.; Raushel F. M. Structural and Catalytic Diversity within the Amidohydrolase Superfamily. Biochemistry 2005, 44 (17), 6383–6391. 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- Sayers E. W.; Bolton E. E.; Brister J. R.; Canese K.; Chan J.; Comeau D. C.; Connor R.; Funk K.; Kelly C.; Kim S.; Madej T.; Marchler-Bauer A.; Lanczycki C.; Lathrop S.; Lu Z.; Thibaud-Nissen F.; Murphy T.; Phan L.; Skripchenko Y.; Tse T.; Wang J.; Williams R.; Trawick B. W.; Pruitt K. D.; Sherry S. T. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50 (D1), D20–D26. 10.1093/nar/gkab1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215 (3), 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Madeira F.; Pearce M.; Tivey A. R. N.; Basutkar P.; Lee J.; Edbali O.; Madhusoodanan N.; Kolesnikov A.; Lopez R. Search and Sequence Analysis Tools Services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50 (W1), W276–W279. 10.1093/nar/gkac240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Higgins D. G. The Clustal Omega Multiple Alignment Package. Methods Mol. Biol. 2021, 2231, 3–16. 10.1007/978-1-0716-1036-7_1. [DOI] [PubMed] [Google Scholar]
- Crooks G. E.; Hon G.; Chandonia J.-M.; Brenner S. E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14 (6), 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohweder B.; Semmelmann F.; Endres C.; Sterner R. Standardized Cloning Vectors for Protein Production and Generation of Large Gene Libraries in Escherichia Coli. BioTechniques 2018, 64 (1), 24–26. 10.2144/000114628. [DOI] [PubMed] [Google Scholar]
- Wang W.; Malcolm B. A. Two-Stage PCR Protocol Allowing Introduction of Multiple Mutations, Deletions and Insertions Using QuikChange Site-Directed Mutagenesis. Biotechniques 1999, 26 (4), 680–682. 10.2144/99264st03. [DOI] [PubMed] [Google Scholar]
- Baba T.; Ara T.; Hasegawa M.; Takai Y.; Okumura Y.; Baba M.; Datsenko K. A.; Tomita M.; Wanner B. L.; Mori H. Construction of Escherichia Coli K-12 in-Frame, Single-Gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol. 2006, 2, 2006.0008. 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31 (13), 3784–3788. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekkelman M. L.; de Vries I.; Joosten R. P.; Perrakis A. AlphaFill: Enriching AlphaFold Models with Ligands and Cofactors. Nat. Methods 2023, 20 (2), 205–213. 10.1038/s41592-022-01685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon J. C.; Myers J. B.; Folta T.; Shoja V.; Heath L. S.; Onufriev A. H++: A Server for Estimating PKas and Adding Missing Hydrogens to Macromolecules. Nucleic Acids Res. 2005, 33 (Web Server issue), W368–W371. 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Merz K. M. Jr. MCPB.Py: A Python Based Metal Center Parameter Builder. J. Chem. Inf Model 2016, 56 (4), 599–604. 10.1021/acs.jcim.5b00674. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E.; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Gilson M. K.; Gohlke H.; Goetz A. W.; Greene D.; Harris R.; Homeyer N.; Huang Y.; Izadi S.; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Mermelstein D. J.; Merz K. M.; Miao Y.; Monard G.; Nguyen C.; Nguyen H.; Omelyan I.; Onufriev A.; Pan F.; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Smith J.; Salomon-Ferrer R.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER 2018. University of California: San Francisco, 2018.
- Lee T.-S.; Cerutti D. S.; Mermelstein D.; Lin C.; LeGrand S.; Giese T. J.; Roitberg A.; Case D. A.; Walker R. C.; York D. M. GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf Model 2018, 58 (10), 2043–2050. 10.1021/acs.jcim.8b00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colominas C.; Luque F. J.; Orozco M. Tautomerism and Protonation of Guanine and Cytosine. Implications in the Formation of Hydrogen-Bonded Complexes. J. Am. Chem. Soc. 1996, 118 (29), 6811–6821. 10.1021/ja954293l. [DOI] [Google Scholar]
- McGibbon R. T.; Beauchamp K. A.; Harrigan M. P.; Klein C.; Swails J. M.; Hernández C. X.; Schwantes C. R.; Wang L.-P.; Lane T. J.; Pande V. S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109 (8), 1528–1532. 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.