

Abstract
Desmin contributes to the stability of the myocardium and its amino-terminal domain influences intermediate filament formation and interacts with a variety of proteins and DNAs. Specific serine residues located in this domain are reversibly phosphorylated in a cell cycle and developmental stage-dependent manner as has been demonstrated also for other cytoplasmic type III intermediate filament proteins. Although absence of desmin apparently does not affect cardiomyogenesis, homozygous deletion of the amino-terminal domain of desmin severely inhibited in vitro cardiomyogenesis. To demonstrate the significance of phosphorylation of this domain in cardiomyogenic commitment and differentiation, we inhibited phosphorylation of serine residues 6, 7, and 8 by mutation to alanine, and investigated early cardiomyogenesis in heterozygous embryoid bodies. As control, serine residues 31 and 32, which are not phosphorylated by kinases mutating serine residues 6, 7, and 8, were mutated to alanine in a second set. DesminS6,7,8A interfered with cardiomyogenesis and myofibrillogenesis in a dominant negative fashion, whereas desminS31,32A produced only a mild phenotype. DesminS6,7,8A led to the down-regulation of the transcription factor genes brachyury, goosecoid, nkx2.5, and mef2C and increased apoptosis of presumptive mesoderm and differentiating cardiomyocytes. Surviving cardiomyocytes which were few in number had no myofibrils. Demonstration that some but not any mutant desmin interfered with the very beginning of cardiomyogenesis suggests an important function of temporarily phosphorylated serine residues 6, 7, and 8 in the amino-terminal domain of desmin in cardiomyogenic commitment and differentiation.
Keywords: cardiomyogenesis, desmin mutation, apoptosis, transcription factor, embryoid body
Introduction
Cardiomyopathies are often caused by mutations of cytoskeletal and sarcomeric proteins (Chien, 2003; Olson, 2004) responsible for function and intra- and inter-cellular integrity of cardiomyocytes. One of these proteins is desmin, the subunit of the muscle-specific type III intermediate filaments, for which an ever increasing number of mutations linked to various forms of cardiomyopathies and skeletal myopathies are described (OMIM 125660; Goldfarb et al., 2004). Most mutations described in humans are situated in the α-helical rod domains 1B and 2B of the desmin polypeptide chain (Paulin et al., 2004). The more complex phenotypes observed in patients with desmin mutations as compared with that of des −/− mice (Milner et al., 1996) suggest that mutant desmin may cause dominant negative phenotypes.
Substantial progress has been made in clarifying desmin’s contribution to function, stability, and longevity of the working myocardium (Weitzer et al., 1995; Capetanaki, 2002; Paulin and Li, 2004; Weisleder et al., 2004), of skeletal muscle (Balogh et al., 2003; Shah et al., 2004), and of smooth muscle containing tissues (Sjuve et al., 1998; Shardonofsky et al., 2006). However, less attention has been paid to another functional feature of desmin, common with other type III intermediate filament proteins, the reversible phosphorylation of free hydroxyl groups in their amino-terminal domain by many kinases in a cell cycle and developmental stage-dependent manner (Izawa and Inagaki, 2006).
In desmin, as in vimentin and glial fibrillary acidic protein, cell cycle-dependent disassembly and assembly of intermediate filaments is regulated by phosphorylation and dephosphorylation of some serine and threonine residues located in their amino-terminal domains. So far Rho kinase, p21-activated kinase, cdc2 kinase, protein kinases A and C, polo-like kinase, and aurora B kinase (Eriksson et al., 2004; Izawa and Inagaki, 2006) have been identified as enzymes phosphorylating type III intermediate filament proteins. All these kinases with the exception of MAPKAP kinase 2 (Cheng et al., 2003) contribute to the disassembly of intermediate filament proteins. Much less is known about their specific dephosphorylation. Protein phosphatase 2A is the only enzyme known to date specifically dephosphorylating vimentin (Cheng et al., 2000). Physiological significance of phosphorylation has been reported for Aurora-B and Rho-kinase which coordinatedly regulate desmin intermediate filament disassembly during cytokinesis (Kawajiri et al., 2003) and the cell cycle regulator cdc2 kinase phosphorylates desmin at serine residue number 6 (Kusubata et al., 1993). Unfortunately, so far not a single report demonstrates a molecular link between growth factors, signaling pathways utilizing these kinases, and the phosphorylation of amino-terminal domain of intermediate filaments.
The amino-terminal domain of desmin plays an important role in intermediate filament assembly (Nelson and Traub, 1983; Kaufmann et al., 1985; Bär et al., 2004), and is responsible for desmin’s muscle-specific behavior in xenopus (Cary and Klymkowsky, 1994). Amino acid residues 7–17 are necessary for hamster desmin intermediate filament assembly (Raats et al., 1990), the first 67 amino acid residues are required for the interaction with ankyrin (Georgatos and Blobel, 1987). Deletion of amino acid residues 1–48 of the amino-terminal domain severely hampered cardiomyogenesis in embryoid bodies (EBs) and prevents germline transmission of this allele (Höllrigl et al., 2002). In addition, desminΔ1 – 48 completely blocked smooth muscle development but rescued myoblast fusion in a null background in EBs. These results suggest different roles of desmin’s amino-terminal domain in cardiac, skeletal, and smooth muscle cell development and function. Introduction of several point mutations into both the amino-terminal domain and the rod domains of desmin (Haubold et al., 2003; Kaminska et al., 2004) demonstrated that many of them interfere with filament formation and affect the cytoarchitecture of cardiomyocytes. Finally, the amino-terminal domain protrudes from desmin filaments (Bär et al., 2004) and interacts with a variety of other cytoskeletal and regulatory proteins (Costa et al., 2004) such as plectin (Hijikata et al., 2003) and desmoplakin (Hofmann et al., 2000).
Cell cycle and developmental stage-dependent phosphorylation of the amino-terminal domain known to be involved in intermediate filament formation and interaction with other proteins led to the hypothesis that serine residues within this domain are indispensable for the proper function of desmin in developing and terminally differentiated cardiomyocytes.
To test this hypothesis, we chose to mutate two sets of temporarily phosphorylated serine residues of desmin. Paralogous residues of the first set have significant functions in vimentin (Eriksson et al., 2004) and glial fibrillary acidic protein (GFAP) (Inagaki et al., 1996). Point mutations were introduced into one allele of exon 1 of the desmin gene, coding for the aminoterminal domain of desmin in embryonic stem cells (ESCs), resulting in the exchange of serine residues by alanine residues at position 6, 7, and 8, and 31 and 32, respectively, of the polypeptide chain (Höllrigl et al., 2001). Serine residues 6, 7, and/or 8 are potential targets of cdc2 kinase (Kusubata et al., 1993), protein kinases A and C (Kitamura et al., 1989), aurora-B kinase (Kawajiri et al., 2003), PAK (Ohtakara et al., 2000), whereas serine residues 31, 32 are so far not reported to be phosphorylated by any of these kinases.
Here we investigated early cardiomyogenesis in EBs and demonstrate that desminS6,7,8A interferes with the very beginning of cardiomyogenesis in a dominant negative fashion causing partial down-regulation of mesodermal and myocardial transcription factor genes, disruption of myofibrils, and finally causing apoptosis in differentiated cardiomyocytes, whereas desminS31,32A has a much less severe phenotype.
Materials and methods
Culture and in vitro differentiation of embryonic stem cells
Wild-type, AB2.2, and mutant des+/S31,32A and des+/S6,7,8A ESC lines (Höllrigl et al., 2001) were passaged on mitotically inactivated SNL76/7 fibroblasts (Bader et al., 2000). Each 800 ESCs were aggregated in 20 μl medium drops for 4 days, 80 ± 10 EBs were plated onto gelatinized 10 cm tissue culture dishes, and cardiomyogenesis was monitored essentially as described previously (Weitzer et al., 1995; Bader et al., 2000).
Analysis of cardiomyogenesis
Development of cardiomyocytes in EBs was compared by calculating the maximal daily increase in beating cardiomyocytes and the maximal percentage of EBs with beating cardiomyocytes at the day of maximum beating activity ± 2 days. Clusters of beating cardiomyocytes in EBs were counted and classified as small, when composed of less than 10, or as large, when composed of more than 10 cardiomyocytes. Number of clusters directly reflects commitment and size of clusters reflects proliferation of cardiomyocytes (Lauss et al., 2005). Experiments were performed with two independently isolated clonal des+/S31,32A and des+/S6,7,8A ESC lines.
Immunofluorescence microscopy
Immuno-staining of EBs was performed with desmin antibodies D8281 (Sigma, Vienna, Austria), Y20 (Santa Cruz Biotechnology, Heidelburg, Germany), and with cardiac troponin T (cTnT) anti-bodies (Neomarkers, Vienna, Austria), followed by FITC- and TRITC-conjugated secondary antibodies (Sigma F4018, T-5268). The annexin V binding assay was performed according to supplier’s protocol (1858777, Roche, Vienna, Austria). For terminal deoxynucleotidyl transferase-mediated dNTP-fluorescein nick end labeling (TUNEL) assays EBs were fixed in 4% para-formaldehyde solution, permeabilized and stained for cTnT, in this case using a Texas red-conjugated secondary antibody (59675, Jackson Immuno Research, Suffolk, UK). TUNEL assay was performed according to supplier’s protocol (1684809, Roche,) followed by DAPI staining. For the detection of dying cells 0.5 μg/ml propidium iodide (Calbiochem, Darmstadt, Germany) was added to living EBs in culture medium. Photomicrographs were taken from areas where typically cardiomyogenesis takes place in EBs (Weitzer, 2006) on a Leica TCS SP confocal microscope (Wetzlar, Germany) with × 40 and × 100 objectives and on a Zeiss Axiovert LSM 510 microscope (Vienna, Austria) with an × 63 objective. Images were prepared by Adobe Photoshop 7.0.
Fluorescence-activated cell sorting analysis
Until day 5 eighty EBs of each genotype were allowed to develop, then cells were separated by digestion with trypsin for 10 min at 37°C. 500,000 cells were resuspended in phosphate-buffered saline, dead cells were stained with propidium iodide (10 mg/ml) and sorted in a BD Becton&Dickinson (Schwechat, Austria) LSR1 fluorescence activated cell sorter.
Western blot analysis
Intermediate filament proteins were prepared from EBs essentially as described (Weitzer and Wiche, 1987). Western blot analysis was performed according to standard procedures using anti-desmin (Y20, Santa Cruz Biotechnology), anti-vimentin (ICN, Eschwepe, Germany), and anti-connexin 43 (Sigma-Aldrich, Vienna, Austria) antibodies. Bound primary antibodies were detected with alkaline phosphatase-conjugated secondary antibodies (Promega, Mannheim, Germany).
Semi-quantitative RT-PCR analysis
mRNA was prepared from ESCs and EBs with the RNeasy Mini Kit (Qiagen, Vienna, Austria) and reverse transcribed with Superscript II polymerase (Invitrogen, Merelbeke, Belgium) in at least three independent experiments. PCR was performed with primer pairs as indicated in Fig. 4. Oligonucleotide sequences and parameters of amplification are available on request. Number of cycles were carefully determined by several preparatory experiments and chosen so that none of the obtained signals were saturated. Statistical analysis of the brachyury, goosecoid, nkx2.5, and mef2c expression was performed by measuring the luminosity of ethidiumbromide stained RT-PCR products by Adobe Photoshop 7.0 tools of six independent experiments.
Fig. 4.

DesminS6,7,8A causes increased cell death in differentiating cardiomyocytes. Confocal micrographs of areas of cardiomyogenesis in embryoid bodies triple stained for nuclear DNA with DAPI (blue, left column), for damaged DNA by transferase-mediated dNTP-fluorescein nick end labeling assay (green, middle column), and with cTnT antibodies (red) at day 8 (A) and at day 12 (B) after embryonic stem cells aggregation. Genotypes as indicated in the left column. Scale bar, 25 µm.
Statistical analysis
All data are presented as the arithmetic mean ± standard deviation σx(n − 1). Statistical significance was evaluated using one sample and paired samples Student’s t-test, respectively, and values of p < 0.05 were considered to indicate statistical significance.
Results
DesminS6,7,8A and desminS31,32A dominantly interfere with cardiomyogenesis in EBs Expression of mutant desmin alleles desminS6,7,8A and desminS31,32A under the control of the endogenous desmin promoter differentially affected commitment and differentiation of cardiomyocytes in a dominant-negative manner (Fig. 1A). In two independently generated desminS6,7,8A clones, in which serine residues 6, 7, and 8 no longer can be phosphorylated, cardiomyogenesis was delayed by 6 days, differentiation rate was drastically reduced (Fig. 1B), and differentiated cardiomyocytes finally developed only in very few EBs (Fig. 1C). Expression and synthesis of desminS31,32A, wherein serine residues 31 and 32 are not phosphorylated by kinases which phosphorylated serine residues 6, 7, and 8, had no significant effect on the onset of cardiomyogenesis in EBs (Fig. 1A), but dominantly affected differentiation rate of cardiomyocytes and the extent of cardiomyogenesis (Figs. 1B,1C). Thus serine residues 6, 7, and 8 are indispensable for desmin’s proper function during early development of cardiomyocytes, whereas serine residues 31 and 32 seem to be much less important. Synthesis of mutant desmin protein in EBs had no discernible effect on the protein levels of endogenous desmin and vimentin (Fig. 1D), and EB development was with the exception of cardiomyogenesis indistinguishable from that of wild-type EBs.
Fig. 1. DesminS6,7,8A and desminS31,32A affect cardiomyogenesis in embryoid bodies (EBs) in a dominant negative fashion.

(A) Development of beating cardiomyocytes in EBs generated from embryonic stem cells (ESCs) of genotypes as indicated. K1 and K2 are two different ESC clones. (B) Initial rate of differentiation of cardioblasts to beating cardiomyocytes. Starting 1 day after the first beating cardiomyocytes were observed, increase of beating, cardiomyocytes per day were measured for 3 consecutive days. (C) Influence of mutant desmin on the extent of cardiomyogenesis in EBs. Means were calculated from the day of maximum beating activity ± 2 days. (A–C) Data are means of at least four independent experiments, each performed in triplicate. Number of EBs analyzed in each case, N = 311; except for des+/+, N = 916. Error bars, standard deviation σx(n – 1). p-values relate to control (des+/+). (D) Western blot analysis of intermediate filament preparations from EBs at day 16. Immuno-detection of desmin, vimentin, and connexin 43. Ponceau S-stained gel as loading control.
DesminS6,7,8A affects morphogenesis and interferes with myofibrillogenesis and proliferation of cardiomyocytes Synthesis of 50% of desmin protein with the S6,7,8A mutation in differentiated cardiomyocytes at day 16 of EB development caused severe reduction in size and number of cardiomyocytes in des+/S6,7,8A EBs (Fig. 2A). Compared with des+/+ EBs, which have extended arrays of connected cardiomyocytes, only single cardiomyocytes with an aberrant morphology were found in des+/S6,7,8A EBs. In des+/S31,32A EBs the size of des+/S31,32A cardiomyocyte clusters was also significantly decreased as compared with those in des+/+ EBs.
Fig. 2. DesminS6,7,8A destroys the morphology of myofibrils in cardiomyocytes.

(A) Merged confocal double immunofluorescence micrographs of typical cardiomyocyte clusters in embryoid bodies (EBs) with desmin alleles as indicated. EBs were double stained with a polyclonal antibody to desmin and a secondary TRITC-conjugated antibody (red), a monoclonal antibody to cTnT and a secondary FITC-conjugated antibody (green) at day 16 after embryonic stem cells aggregation. Areas of colocalization are yellow. Scale bar: 25 µm. (B) High-resolution confocal double immunofluorescence micrographs of cardiomyocytes stained as in (A). Arrow-heads, irregularly assembled sarcomeres. Arrows, cytoplasmic aggregates of desmin. Right column, merged images. Scale bar, 10 µm.
DesminS6,7,8A severely affected myofibrillogenesis and intracellular morphology of differentiated cardiomyocytes, whereas in des+/S31,32A cardiomyocytes no obvious effect on the structure and organization of myofibrils was detected (Fig. 2B). In des+/S6,7,8A EBs, only single irregularly shaped cardiomyocytes with sarcomeres rarely assembled in abnormal myofibrils (arrowheads), and cytoplasmic aggregates of desmin (arrows) and cTnT were observed. Myofibrils with regularly aligned sarcomeres were completely absent in des+/S6,7,8A cardiomyocytes.
From extended statistical analysis, it became evident that the number of cardiomyocyte clusters per EB reflects commitment of mesodermal cells to the cardiomyogenic lineage, and that the size of cardiomyocyte clusters directly reflects proliferation of committed cardiomyocytes during early differentiation (Lauss et al., 2005). In des+/S6,7,8A EBs, the number of cardiomyocyte clusters per EB was significantly reduced but not in des+/S31,32A EBs (Fig. 3A), which is indicative of restricted commitment to the cardiomyogenic lineage or apoptosis. Indeed, fluorescence-activated cell sorting (FACS) analysis of dead or dying cells at day 5 with propidium iodide demonstrated an increased proportion of dead cell in des+/S6,7,8A EBs (25.77% ± 1.50% versus 17.97% ± 1.77% in wild-type EBs, N = 5, p = 0.0051). Additionally, 80% of des+/S6,7,8A clusters had a very small size (Fig. 3B) which indicates reduced proliferation. In accordance with the preponderance of irregularly organized sarcomeres, contraction rates of des+/S6,7,8A cardiomyocytes were significantly reduced (Fig. 3C). In des+/S31,32A EBs, the mean size of cardiomyocyte clusters was also reduced to some extent, which suggests a moderate negative influence on the proliferation of differentiating cardiomyocytes. However, desmin+/S31,32A seems not to affect desmin’s role in myofibrils because no effect on rhythmic contraction was observed. The negative influence of desminS6,7,8A on commitment of mesodermal precursors of cardiomyocytes and on proliferation of committed cardiomyocytes suggests that phosphorylation of serine residues 6, 7, and/or 8 is instrumental for commitment and proliferation of cardiomyocytes.
Fig. 3. DesminS6,7,8A negatively affects commitment and early pro-liferation of cardiomyocytes and hampers rhythmic contraction.
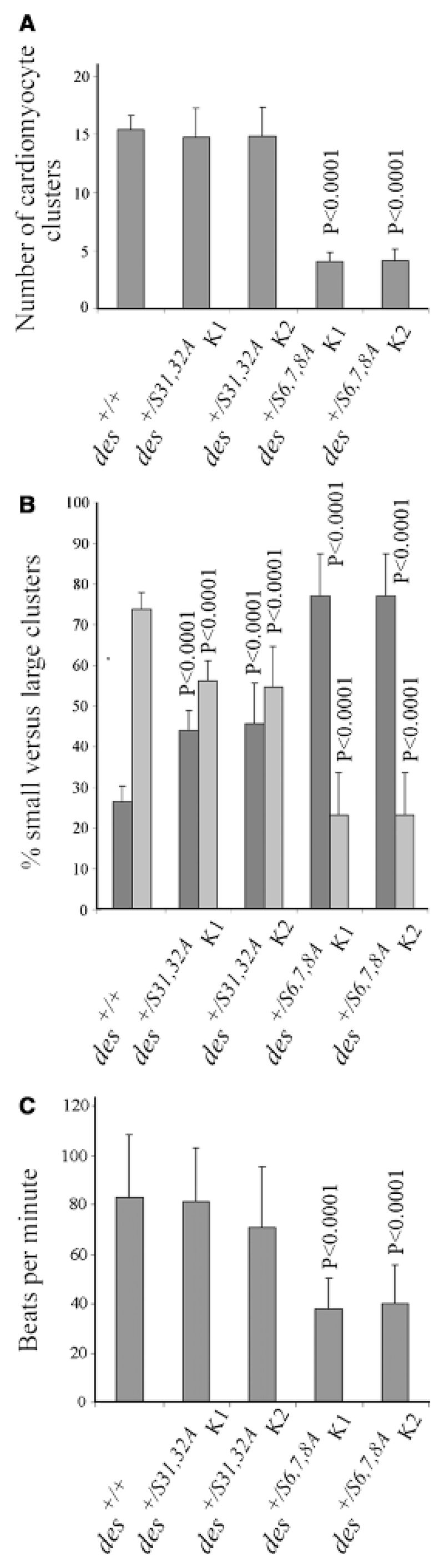
(A) Number of beating cardiomyocyte clusters per embryoid body (EB). Number of EBs analyzed in each case, N = 311; except for des+/+, N = 916. (B) Size distribution of cardiomyocyte clusters presented as the relative proportion of small clusters with less than 10 (dark gray bars) versus large clusters with up to several hundred cardiomyocytes (light gray bars). Number of EBs analyzed in each case: N = 386. (C) Beating rates of cardiomyocytes collected at day 16 ± 4 days. Number of cardiomyocyte clusters analyzed: des+/+, N = 85; all others N = 52. Error bars indicate standard deviation σx(n − 1). p-values relate to control (des+/+).
DesminS6,7,8A but not desminS31,32A causes death of differentiating cardiomyocytes Delayed cardiomyogenesis and reduced cluster size reflect either blocked molecular mechanisms instrumental for the differentiation of cardiomyocytes or might be a consequence of increased apoptosis of differentiating cardiomyocytes. Therefore, apoptosis was investigated by TUNEL and annexin V binding assays before beating cardiomyocytes develop in mutant EBs (day 8), during differentiation of cardiomyocytes (day 12) and in terminally differentiated cardiomyocytes (day 16). At day 8, TUNEL positive nuclei were prominent in small-defined areas of des+/S6,7,8A EBs but were extremely rarely observed in cTnT positive des+/+ or des+/S31,32A cardiomyocytes (Fig. 4A). Annexin V staining was never observed at the plasma membrane of cTnT positive cardiomyocytes of any stage but were seen sparsely on cells in areas of des+/S6,7,8A EBs where cardiomyocytes normally would form in des+/+ EBs (data not shown). At day 12, TUNEL-positive nuclei and fragmented nuclei were most abundant in des+/S6,7,8A cardioblast-like cells with a faint cTnT staining (Fig. 4B), and increased apoptosis was also evident in des+/S6,7,8A EBs at day 16 (data not shown). Thus cell death caused by desminS6,7,8A already starts during early steps of cardiomyocyte differentiation, consequently causing a reduction of cardiomyogenesis in EBs. The delay of cardiomyogenesis in des+/S6,7,8A EBs, however, also suggests a subtle involvement of desmin in regulatory mechanisms important for the commitment and differentiation of cardiomyocytes as has been demonstrated by overexpression of desmin in pre-cardiac mesoderm (Hofner et al., 2007).
DesminS6,7,8A already affects the expression of mesodermal and early myocardial transcription factors before the differentiation of cardiomyocytes Altered morphology and low contraction rates of differentiating cardiomyocytes in des+/S6,7,8A EBs suggest that mutant desmin also affects molecular decisions made during the commitment of primitive mesoderm to the cardiomyogenic lineages. To identify target genes by which desminS6,7,8A influences molecular mechanisms regulating early differentiation of cardiomyocytes timing and degree of expression of mesodermal and myocardial transcription factor genes were analyzed in differentiating EBs by semi-quantitative RT-PCR.
With the onset of desmin expression in pre-cardiac mesoderm of EBs, desminS6,7,8A caused a significant temporal delay in the expression of mesodermal transcription factor genes brachyury and goosecoid (Fig. 5A). The mesoderm-specific gene of the Tgf-β family member nodal was temporally and partially down-regulated, but oct3/4 expression in the primitive ectoderm was not affected. This indicates that desminS6,7,8A affects definitive mesoderm once it formed from primitive ectoderm but not primitive ectoderm per se. Simultaneous within the limits of resolution desminS6,7,8A significantly inhibited and delayed expression of the early myocardial transcription factor nkx2.5 between days 4 and 8 and completely blocked expression of mef2c on day 6 and partially on day 8. Mlc1v expression was partially delayed but expression of structural genes mhcα and vimentin also expressed in other cell types that do not express desminS6,7,8A were not significantly affected. Timing and degree of expression of the myocardial transcription factors nkx2.5 and mef2c was much more affected than those of mesodermal transcription factors brachyury and goosecoid, suggesting that desminS6,7,8A specifically affects those mesodermal cells becoming cardiomyocytes (Fig. 5B). As expected, desminS31,32A did not cause any discernable effect on the expression of any transcription factor tested. Consequently, desminS6,7,8A but not desminS31,32A severely interferes with molecular mechanisms involved in the transcriptional control of transcription factor genes guiding the commitment of mesoderm to the cardiomyogenic lineages and the differentiation of committed cardiomyocytes.
Fig. 5.

DesminS6,7,8A negatively influences the onset of mesodermal and myocardial transcription factor expression. mRNA was isolated and reverse transcribed from embryonic stem cells (day 0) and embryoid bodies (EBs) of genotypes as indicated at days 4, 6, and 8. Semi-quantitative RT-PCR was performed with primer pairs as indicated. (A) Typical data from three independent experiments with each two clones are shown. GAPDH RT-PCR, loading control. (B) Time course of the expression of brachyury, goosecoid, nkx2.5, and mef2c in EBs between days 4 and 8. Statistical analysis of six independent experiments. Expression levels normalized to expression in wild-type EBs for each day. *, p-values were all smaller than 0.05 except for brachyury and goosecoid expression on day 8; p-values relate to control (des+/+) with an arbitrary expression level of 1.0.
Discussion
Desmin is an integral part of the molecular machinery maintaining the phenotype of differentiated muscle cells in the adult organism. Desmin’s very early expression (Kuisk et al., 1996) and its influence on the expression of skeletal muscle-specific transcription factor genes (Li et al., 1994; Weitzer et al., 1995) suggest that mutant desmin might interfere with the early differentiation of cardiomyocytes. Here we demonstrate that cardiomyogenesis was severely delayed in EBs with one mutant desS6,7,8A allele, and beating cardiomyocytes were reduced to near extinction. Viability of presumptive premyocardial mesoderm was reduced, number and size of cardiomyocyte clusters were drastically reduced in EBs and contraction of cardiomyocytes was reduced and often arrhythmic. Expression of mesodermal transcription factor genes brachyury and goosecoid were significantly attenuated, suggesting that desminS6,7,8A affects already cardioblasts, as has been observed for a mutant desmin lacking the amino-terminal domain (Höllrigl et al., 2002) and for overexpressed desmin (Hofner et al., 2007). Consequently, expression of early myocardial transcription factor genes nkx2.5 and mef2c were also delayed and remained low throughout differentiation of cardiomyocytes, suggesting that desminS6,7,8A affects to some extent the expression of mesodermal transcription factors and to a significantly increased degree the expression of cardiac-specific transcription factor genes nkx2.5 and mef2c. Changes in the expression pattern of cardiac muscle markers are specific for cardiomyogenesis because skeletal and smooth muscle cells start to differentiate much later in EB development (Weitzer et al., 1995).
These results demonstrate that desminS6,7,8A already affects mesodermal cells and cardioblasts during very early developmental processes. Because of the fact that the desS6,7,8A allele is under the control of the native desmin promoter, these results further imply that at least low levels of desmin protein are present in brachyury and goosecoid-positive pre-cardiac mesoderm before day 5 and that phosphorylation of one or more of serine residues 6, 7, and 8 contributes significantly to the function of desmin in the molecular regulation of early cardiomyogenesis. The influence of desmin and desminS6,7,8A on mesoderm and cardiac-specific transcription factors parallels the well-established influence of desmin on myogenic transcription factors myoD and myogenin in skeletal muscle cells (Li et al., 1994). In analogy to the model suggested by Yassemie Capetanaki for skeletal muscle cells, desmin may well influence brachyury or nkx2.5 expression in mesodermal precursors of cardiomyocytes.
Phosphorylation of the analog residues in other intermediate filament proteins regulated the disassembly of intermediate filaments during mitosis (Izawa and Inagaki, 2006), thus desminS6,7,8A might cause an irreversible aggregation of desmin intermediate filaments. Indeed, in cTnT-positive cardiomyocytes and surrounding desmin-positive presumptive cardiomyocytes, desmin was aggregated, although serine-to-alanine mutations remove only hydroxyl groups from an otherwise unaltered peptide chain backbone. In none of these aberrantly structured cardiomyocytes, myofibrils or typical intermediate filaments were found. Desmin containing irregularly shaped cytoplasmic material was reminiscent of aggresomes observed in desmin-related cardiomyopathies (Goldfarb et al., 2004).
As expected from the morphology of differentiated des+/S6,7,8A, cardiomyocytes and the low-overall contribution of des+/S6,7,8A ESCs to cardiomyogenesis in EBs, viability of cells was reduced at day 5, and apoptosis was significantly increased in des+/S6,7,8A EBs at day 8, the time when first cardiomyocytes start to contract rhythmically in control EBs. Apoptosis was most prominent in areas where primitive mesoderm forms (Bader et al., 2001; Weitzer, 2006) 1 day before first cardiomyocytes start to beat. The fact that apoptosis in cardiomyocytes starts very fast by proteolysis of Z-disk proteins (Maruyama et al., 2006) could explain why very few cardiomyocytes were simultaneously positive for cTnT and TUNEL assay or annexin V staining. Synthesis of desmin from the second wild-type allele and vimentin was not affected by desminS6,7,8A, suggesting that apoptosis was not merely caused by the absence of intermediate filament proteins as previously demonstrated for keratins 8 and 18 (Caulin et al., 2000). In line with this observation, a very small subpopulation of des+/S6,7,8A cardiomyocytes was resistant to the detrimental effects of desminS6,7,8A and continued to beat rhythmically as long as it was in wild-type EBs. Summarizing, at later developmental stages, desminS6,7,8A also affects the phenotype of fully differentiated cardiomyocytes which fits perfectly the well-described phenotype-maintaining role of desmin in muscle tissues of adult organisms.
To emphasize that the phenotype observed in des+/S6,7,8A EBs was specific to mutation of serine residues 6, 7, and 8, we analyzed cardiomyogenesis in EBs with a different mutant allele of desmin resulting in the synthesis of desminS31,32A. Serine residues 31 and 32 have no exact functional counterparts in vimentin (Izawa and Inagaki, 2006) and GFAP (Inagaki et al., 1990), and are so far not reported to be phosphorylated by any of the kinases which target serine residues 6, 7, or 8 in a cell cycle-specific manner, and thus should cause a dissimilar if any phenotype in cardiomyocytes.
Indeed, cardiomyogenesis in des+/S31,32A EBs was not delayed as in des+/S6,7,8A EBs, and overall development of cardiomyocytes was only weakly affected, demonstrating a clearly distinct effect of this mutation. Differentiation of cardiomyocytes, although starting at the same time as in wild-type EBs, resulted in a lower number of EBs with beating cardiomyocytes. The number of cardiomyocyte clusters was unchanged but the mean size of clusters was significantly reduced. Myofibril formation, sarcomere length, and rhythmic contraction of cardiomyocytes was apparently not affected, and apoptosis was much lower than in des+/S6,7,8A EBs. Timely onset of cardiomyogenesis suggests that desminS31,32A does not affect commitment of mesodermal precursors to the cardiomyogenic lineages, and this was corroborated by the total absence of any obvious effect on the expression of mesodermal and early myocardial transcription factor genes. Reduction of cardiomyocyte cluster size in consideration of constant numbers of clusters per EB, however, suggests that desminS31,32A affects proliferation of differentiating cardiomyocytes.
An ever increasing number of mutations within the desmin gene are linked to human cardiomyopathies (Goldfarb et al., 2004; Paulin et al., 2004). The majority of them manifests at adult age and are located in the conserved α-helical domains 1B and 2B of the desmin protein. Here we demonstrate that mutations of amino-terminally located serine residues known to be reversibly phosphorylated during the cell cycle, but not those residues which are not phosphorylated, affect in vitro cardiomyogenesis in a dominant negative manner. Point mutations were introduced by homologous recombination avoiding introduction of any transgenic DNA into the desmin locus. This perfectly mimics the situation in potential patients with a heterozygous mutation in one desmin allele, because mutant alleles are under the strict control of the endogenous desmin promoter throughout commitment and differentiation of cardiomyocytes. The dramatic defects occurring at the beginning of cardiomyogenesis in des+/S6,7,8A EBs suggest that mutation of serine residues 6, 7, and/or 8 would cause an embryonic lethal phenotype in humans, and thus, so far have not been found in any patient with a cardiomyopathy of juvenile or adult onset. We have sequenced exon 1 coding for the amino-terminal domain of desmin in both desmin alleles of 13 elderly patients with HYMA-stages I–IV cardiomyopathies, but did not find any mutation in this part of the desmin gene (J. Falkowski and G. Weitzer, unpublished results).
The dominant negative effects observed in des+/S6,7,8A EBs suggest a vital function for desmin in general and, in particular, for its amino-terminal domain containing serine residues 6, 7, and 8 in commitment of mesoderm to the cardiomyogenic lineage or in early differentiation of cardiomyocytes and myofibrillogenesis. Most importantly, these effects seem to be specific for cardiomyogenesis because later on in development when skeletal and smooth muscle cells develop in EBs, no significant changes in the expression pattern of muscle-specific genes or in the morphology and contraction of skeletal and smooth muscle cells were detected in EBs.
These data support the current working model that desmin-containing intermediate filaments can only disassemble before mitosis if desmin is properly phosphorylated (Izawa and Inagaki, 2006). Incorporation of desminS6,7,8A but not of desminS31,32A in intermediate filaments thus may result in reduced disassembly negatively affecting cytokinesis and causing premature death of cardiomyocytes.
In summary, EBs provide a powerful tool to demonstrate desmin’s early influence on commitment of mesoderm to the cardiomyogenic lineage which was impossible to assess in vivo so far due to the experimental inaccessibility of implanting and gastrulating embryo. By these means we could provide evidence to the hypothesis that serine residues located within the amino-terminal domain of desmin are required for the proper function of desmin and in addition to its well established role in maintenance of the muscle cell phenotype, we demonstrated an influence of desmin on the regulation of expression of transcription factors during commitment and early differentiation of cardiomyocytes.
Acknowledgments
We thank Karin Habegger and Sabine Enzinger for technical assistance, Thomas Sauer for FACS analysis, and Allan Bradley for the AB2.2 ESCs. This work was supported by funds from the Austrian Federal Ministry of Education, Science and Culture (GZ70.078/0002-Pr/472002), the Austrian Fonds zur Förderung der wissenschaftlichen Forschung, Grants P11189, P15303, and P18659 and the Hochschuljubiläumsstiftung der Stadt Wien, Grant H-933/2003.
References
- Bader A, Al-Dubai H, Weitzer G. Leukemia inhibitory factor modulates cardiogenesis in embryoid bodies in opposite fashions. Circ Res. 2000;86:787–794. doi: 10.1161/01.res.86.7.787. [DOI] [PubMed] [Google Scholar]
- Bader A, Gruss A, Höllrigl A, Al-Dubai H, Capetanaki Y, Weitzer G. Paracrine promotion of cardiomyogenesis in embryoid bodies by LIF modulated endoderm. Differentiation. 2001;68:31–43. doi: 10.1046/j.1432-0436.2001.068001031.x. [DOI] [PubMed] [Google Scholar]
- Balogh J, Li Z, Paulin D, Arner A. Lower active force generation and improved fatigue resistance in skeletal muscle from desmin deficient mice. J Muscle Res Cell Motil. 2003;24:453–459. doi: 10.1023/a:1027353930229. [DOI] [PubMed] [Google Scholar]
- Bär H, Strelkov SV, Sjoberg G, Aebi U, Herrmann H. The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol. 2004;148:137–152. doi: 10.1016/j.jsb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Capetanaki Y. Desmin cytoskeleton: a potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc Med. 2002;12:339–348. doi: 10.1016/s1050-1738(02)00184-6. [DOI] [PubMed] [Google Scholar]
- Cary RB, Klymkowsky MW. Differential organization of desmin and vimentin in muscle is due to differences in their head domains. J Cell Biol. 1994;126:445–456. doi: 10.1083/jcb.126.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulin C, Ware CF, Magin TM, Oshima RG. Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J Cell Biol. 2000;149:17–22. doi: 10.1083/jcb.149.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TJ, Lin YL, Chiang AS, Lai YK. Association of protein phosphatase 2A with its substrate vimentin intermediate filaments in 9L rat brain tumor cells. J Cell Biochem. 2000;79:126–138. [PubMed] [Google Scholar]
- Cheng TJ, Tseng YF, Chang WM, Chang MD, Lai YK. Retaining of the assembly capability of vimentin phosphorylated by mitogen-activated protein kinase-activated protein kinase-2. J Cell Biochem. 2003;89:589–602. doi: 10.1002/jcb.10511. [DOI] [PubMed] [Google Scholar]
- Chien KR. Genotype, phenotype: upstairs, downstairs in the family of cardiomyopathies. J Clin Invest. 2003;111:175–178. doi: 10.1172/JCI17612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa ML, Escaleira R, Cataldo A, Oliveira F, Mermelstein CS. Desmin: molecular interactions and putative functions of the muscle intermediate filament protein. Braz J Med Biol Res. 2004;37:1819–1830. doi: 10.1590/s0100-879x2004001200007. [DOI] [PubMed] [Google Scholar]
- Eriksson JE, He T, Trejo-Skalli AV, Harmala-Brasken AS, Hellman J, Chou YH, Goldman RD. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J Cell Sci. 2004;117:919–932. doi: 10.1242/jcs.00906. [DOI] [PubMed] [Google Scholar]
- Georgatos SD, Blobel G. Lamin B constitutes an intermediate filament attachment site at the nuclear envelope. J Cell Biol. 1987;105:117–125. doi: 10.1083/jcb.105.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG, Vicart P, Goebel HH, Dalakas MC. Desmin myopathy. Brain. 2004;127:723–734. doi: 10.1093/brain/awh033. [DOI] [PubMed] [Google Scholar]
- Haubold K, Herrmann H, Langer SJ, Evans RM, Leinwand LA, Klymkowsky MW. Acute effects of desmin mutations on cytoskeletal and cellular integrity in cardiac myocytes. Cell Motil Cytoskeleton. 2003;54:105–121. doi: 10.1002/cm.10090. [DOI] [PubMed] [Google Scholar]
- Hijikata T, Murakami T, Ishikawa H, Yorifuji H. Plectin tethers desmin intermediate filaments onto subsarcolemmal dense plaques containing dystrophin and vinculin. Histochem Cell Biol. 2003;119:109–123. doi: 10.1007/s00418-003-0496-5. [DOI] [PubMed] [Google Scholar]
- Hofmann I, Mertens C, Brettel M, Nimmrich V, Schnolzer M, Herrmann H. Interaction of plakophilins with desmoplakin and intermediate filament proteins: an in vitro analysis. J Cell Sci. 2000;113:2471–2483. doi: 10.1242/jcs.113.13.2471. [DOI] [PubMed] [Google Scholar]
- Hofner M, Höllrigl A, Puz S, Stary M, Weitzer G. Desmin stimulates differentiation of cardiomyocytes and upregulation of brachyury and nkx2.5. Differentiation, same issue as this publication. 2007 doi: 10.1111/j.1432-0436.2007.00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höllrigl A, Hergovich A, Görzer I, Bader A, Ellersdorfer G, Habegger K, Hammer E, Enzinger S, Capetanaki Y, Weitzer G. High-throughput site-directed mutagenesis in ES cells. Biochem Biophys Res Commun. 2001;289:329–336. doi: 10.1006/bbrc.2001.5980. [DOI] [PubMed] [Google Scholar]
- Höllrigl A, Puz S, Al-Dubai H, Kim JU, Capetanaki Y, Weitzer G. Amino-terminally truncated desmin rescues fusion of des(– / –) myoblasts but negatively affects cardiomyogenesis and smooth muscle development. FEBS Lett. 2002;523:229–233. doi: 10.1016/s0014-5793(02)02995-2. [DOI] [PubMed] [Google Scholar]
- Inagaki M, Gonda Y, Nishizawa K, Kitamura S, Sato C, Ando S, Tanabe K, Kikuchi K, Tsuiki S, Nishi Y. Phosphorylation sites linked to glial filament disassembly in vitro locate in a non-alpha-helical head domain. J Biol Chem. 1990;265:4722–4729. [PubMed] [Google Scholar]
- Inagaki N, Tsujimura K, Tanaka J, Sekimata M, Kamei Y, Inagaki M. Visualization of protein kinase activities in single cells by antibodies against phosphorylated vimentin and GFAP. Neurochem Res. 1996;21:795–800. doi: 10.1007/BF02532302. [DOI] [PubMed] [Google Scholar]
- Izawa I, Inagaki M. Regulatory mechanisms and functions of intermediate filaments: a study using site- and phosphorylation state-specific antibodies. Cancer Sci. 2006;97:167–174. doi: 10.1111/j.1349-7006.2006.00161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminska A, Strelkov SV, Goudeau B, Olive M, Dagvadorj A, Fidzianska A, Simon Casteras M, Shatunov A, Dalakas MC, Ferrer I, Kwiecinski H, et al. Small deletions disturb desmin architecture leading to breakdown of muscle cells and development of skeletal or cardioskeletal myopathy. Hum Genet. 2004;114:306–313. doi: 10.1007/s00439-003-1057-7. [DOI] [PubMed] [Google Scholar]
- Kaufmann E, Weber K, Geisler N. Intermediate filament forming ability of desmin derivatives lacking either the amino-terminal 67 or the carboxy-terminal 27 residues. J Mol Biol. 1985;185:733–742. doi: 10.1016/0022-2836(85)90058-0. [DOI] [PubMed] [Google Scholar]
- Kawajiri A, Yasui Y, Goto H, Tatsuka M, Takahashi M, Nagata K, Inagaki M. Functional significance of the specific sites phosphorylated in desmin at cleavage furrow: aurora-B may phosphorylate and regulate type III intermediate filaments during cytokinesis coordinatedly with Rho-kinase. Mol Biol Cell. 2003;14:1489–1500. doi: 10.1091/mbc.E02-09-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S, Ando S, Shibata M, Tanabe K, Sato C, Inagaki M. Protein kinase C phosphorylation of desmin at four serine residues within the non-alpha-helical head domain. J Biol Chem. 1989;264:5674–5678. [PubMed] [Google Scholar]
- Kuisk IR, Li H, Tran D, Capetanaki Y. A single MEF2 site governs desmin transcription in both heart and skeletal muscle during mouse embryogenesis. Dev Biol. 1996;174:1–13. doi: 10.1006/dbio.1996.0046. [DOI] [PubMed] [Google Scholar]
- Kusubata M, Matsuoka Y, Tsujimura K, Ito H, Ando S, Kamijo M, Yasuda H, Ohba Y, Okumura E, Kishimoto T, Inagaki M. cdc2 kinase phosphorylation of desmin at three serine/threonine residues in the amino-terminal head domain. Biochem Biophys Res Commun. 1993;190:927–934. doi: 10.1006/bbrc.1993.1138. [DOI] [PubMed] [Google Scholar]
- Lauss M, Stary M, Tischler J, Egger G, Puz S, Bader-Allmer A, Seiser C, Weitzer G. Single inner cell masses yield embryonic stem cell lines differing in lifr expression and their developmental potential. Biochem Biophys Res Commun. 2005;331:1577–1586. doi: 10.1016/j.bbrc.2005.04.068. [DOI] [PubMed] [Google Scholar]
- Li H, Choudhary SK, Milner DJ, Munir MI, Kuisk IR, Capetanaki Y. Inhibition of desmin expression blocks myoblast fusion and interferes with the myogenic regulators MyoD and myogenin. J Cell Biol. 1994;124:827–841. doi: 10.1083/jcb.124.5.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama R, Takemura G, Tohse N, Ohkusa T, Ikeda Y, Tsuchiya K, Minatoguchi S, Matsuzaki M, Fujiwara T, Fujiwara H. Synchronous progression of calcium transient-dependent beating and sarcomere destruction in apoptotic adult cardiomyocytes. Am J Physiol Heart Circ Physiol. 2006;290:H1493–H1502. doi: 10.1152/ajpheart.00669.2005. [DOI] [PubMed] [Google Scholar]
- Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol. 1996;134:1255–1270. doi: 10.1083/jcb.134.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ, Traub P. Proteolysis of vimentin and desmin by the Ca2+-activated proteinase specific for these intermediate filament proteins. Mol Cell Biol. 1983;3:1146–1156. doi: 10.1128/mcb.3.6.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtakara K, Inada H, Goto H, Taki W, Manser E, Lim L, Izawa I, Inagaki M. p21-activated kinase PAK phosphorylates desmin at sites different from those for Rho-associated kinase. Biochem Biophys Res Commun. 2000;272:712–716. doi: 10.1006/bbrc.2000.2854. [DOI] [PubMed] [Google Scholar]
- Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10:467–474. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- Paulin D, Huet A, Khanamyrian L, Xue Z. Desminopathies in muscle disease. J Pathol. 2004;204:418–427. doi: 10.1002/path.1639. [DOI] [PubMed] [Google Scholar]
- Paulin D, Li Z. Desmin: a major intermediate filament protein essential for the structural integrity and function of muscle. Exp Cell Res. 2004;301:1–7. doi: 10.1016/j.yexcr.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Raats JM, Pieper FR, Vree Egberts WT, Verrijp KN, Ramaekers FC, Bloemendal H. Assembly of aminoterminally deleted desmin in vimentin-free cells. J Cell Biol. 1990;111:1971–1985. doi: 10.1083/jcb.111.5.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SB, Davis J, Weisleder N, Kostavassili I, McCulloch AD, Ralston E, Capetanaki Y, Lieber RL. Structural and functional roles of desmin in mouse skeletal muscle during passive deformation. Biophys J. 2004;86:2993–3008. doi: 10.1016/S0006-3495(04)74349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shardonofsky FR, Capetanaki Y, Boriek AM. Desmin modulates lung elastic recoil and airway responsiveness. Am J Physiol Lung Cell Mol Physiol. 2006;290:L890–L896. doi: 10.1152/ajplung.00397.2005. [DOI] [PubMed] [Google Scholar]
- Sjuve R, Arner A, Li Z, Mies B, Paulin D, Schmittner M, Small JV. Mechanical alterations in smooth muscle from mice lacking desmin. J Muscle Res Cell Motil. 1998;19:415–429. doi: 10.1023/a:1005353805699. [DOI] [PubMed] [Google Scholar]
- Weisleder N, Soumaka E, Abbasi S, Taegtmeyer H, Capetanaki Y. Cardiomyocyte-specific desmin rescue of desmin null cardiomyopathy excludes vascular involvement. J Mol Cell Cardiol. 2004;36:121–128. doi: 10.1016/j.yjmcc.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Weitzer G. Embryonic stem cell-derived embryoid bodies: an in vitro model of eutherian pregastrulation development and early gastrulation. Handb Exp Pharmacol. 2006;174:21–51. [PubMed] [Google Scholar]
- Weitzer G, Milner DJ, Kim JU, Bradley A, Capetanaki Y. Cytoskeletal control of myogenesis: a desmin null mutation blocks the myogenic pathway during embryonic stem cell differentiation. Dev Biol. 1995;172:422–439. doi: 10.1006/dbio.1995.8070. [DOI] [PubMed] [Google Scholar]
- Weitzer G, Wiche G. Plectin from bovine lenses: chemical properties, structural analysis and initial identification of interaction partners. Eur J Biochem. 1987;169:41–52. doi: 10.1111/j.1432-1033.1987.tb13578.x. [DOI] [PubMed] [Google Scholar]