

Abstract
Immunotherapy is amongst the most promising new treatment modalities to arise over the last two decades; antibody drugs are delivering immunotherapy to millions of patients with many different types of cancer. Initial success with antibody therapeutics came in the form of direct targeting or cytotoxic antibodies, such as rituximab and trastuzumab, which bind directly to tumor cells to elicit their destruction. These were followed by immunomodulatory antibodies, that elicit anti-tumor responses by either stimulating immune cells or relieving tumor-mediated suppression. By far the most successful approach in the clinic to date has been relieving immune suppression, with immune checkpoint blockade now a standard approach in the treatment of many cancer types. Despite equivalent and sometimes even more impressive effects in pre-clinical models, agonist antibodies designed to stimulate the immune system have lagged behind in their clinical translation. In this review we document the main receptors that have been targeted by agonist antibodies, consider the various approaches that have been evaluated to date, detail what we have learnt and consider how their anti-cancer potential can be unlocked.
Introduction
It is well recognised that the immune system is central to our health and well-being and that when its ability to maintain homeostasis is disturbed, pathology typically follows. Cancer is considered by many to be a direct consequence of immune-suppression and that for cancer to develop it must avoid elimination by the immune system, causing Hanahan and Weinberg to include it amongst their revised Hallmarks of Cancer.(1) Therefore, it is unsurprising that boosting anti-tumor immune responses and/or overcoming tumor immune-suppression are key goals of antibody immunotherapy for cancer. In order to achieve these goals and thereby benefit patients, detailed understanding of immune regulation is required. Immune cells have evolved to protect the host against pathogens and malignancy whilst avoiding destruction of self-tissues and commensals. To achieve this balance, immune cells like T cells only become fully functional after receiving a primary signal through their antigen receptor (T cell receptor; TCR) and a secondary antigen-independent signal known as costimulation.(2) As such, T cell costimulation is tightly regulated by both intrinsic factors including the expression of inhibitory receptors (CTLA-4 and PD-1) and through extrinsic mechanisms involving Toll-like receptor and regulatory T cell modulation of costimulatory ligands on antigen presenting cells.(3–5)
Costimulatory receptors belong to either the immunoglobulin superfamily (IgSF) or the TNF receptor superfamily (TNFRSF), with both contributing to the regulation of immunity. Considering T cells as an example, although the signal transduction mechanisms of these two costimulatory receptor families differ, both types of receptors transmit signals that combine with signalling pathways downstream of the TCR/CD3 complex to instigate quantitative and qualitative changes that culminate in increased T cell proliferation and survival, metabolic fitness, as well as differentiation into effector cells.(6–8) A similar set of co-stimulatory interactions serves to modulate the fate and function of many other immune cells including B cells and dendritic cells.
Given the essential role that costimulatory receptors play in the generation of successful immune responses their targeting has been proposed as an approach to stimulate anti-tumor responses.(9–11) Although non-antibody agonists, including fusion proteins, cyclic peptides and aptamers have been tested with variable success, the majority of agents destined for clinical development are antibodies or antibody-like molecules. Developing agonist antibodies that mimic the effects of natural membrane-bound costimulatory ligands, however, has proved considerably more challenging than developing checkpoint inhibitors directed against CTLA-4 and PD-1. This is in part due to differences in the valency and geometry of receptor binding between the natural ligand and antibody. For example, membrane anchoring of trimeric TNFRSF ligands such as CD27L, CD40L, 4-1BBL and OX40L is obligatory for optimal receptor signalling, likely due to the formation of higher order structures that facilitate assembly of the signalosome.(12) In contrast, trimeric GITRL, TL1A and lymphotoxin αβ are fully functional in solution.(12) Recent structural studies of the GITRL:GITR complex suggest that receptor homodimerization could enhance avidity for GITRL and also lead to formation of higher order structures upon receptor binding to soluble trimeric GITRL.(13) Thus, for TNFRSF ligands that function in solution this mechanism could provide an alternative way to assemble optimal receptor:ligand complexes on the cell surface.
Despite these challenges, agonist antibodies targeting the IgSF receptors CD28(14), ICOS(15) and CD96(16) as well as those targeting several members of the TNFRSF have been generated.(17) Interestingly, the agonistic activity of many of these antibodies requires Fc gamma receptor (FcγR) mediated hyper-crosslinking emphasising the importance of higher order receptor clustering for optimal signal transduction(17); see later sections.
Prediction of the therapeutic activity of immune agonists in human subjects is often complex and requires careful consideration of multiple factors including knowledge of immune cell subset specific expression as well as detailed analysis of receptor biology. In this regard, studies of inborn errors of immunity which are available for a subset of costimulatory receptors/ligands, including CD27/CD70(18), CD28(19), ICOS(20), 4-1BB/4-1BBL(21–23) and OX40(24) are highly informative and should be considered together with findings from studies in animal models. Meanwhile, detailed mechanistic studies in animal models are providing insights into the effects of combining agonists with checkpoint blockers to better understand how these treatments synergise for improved immunotherapy.(25–27) Despite equivalent and sometimes more profound effects in pre-clinical models(9,11) agonist antibodies have lagged behind in their clinical translation compared to checkpoint blockers. Below, we review the agents (focussing mainly on unmodified antibody isotypes and canonical antibody formats directed to CD40, 4-1BB, OX40, GITR, CD27 and DR5) that have been assessed clinically to date, detail areas where progress is being made and provide reasons for optimism for the future of this antibody class.
Mechanisms underpinning agonist activity
Whereas the “rules” for isotype selection for direct targeting or cytotoxic mAb are clear with hIgG1/3 in humans and mIgG2a/c in mice selected as having the highest activating:inhibitory FcγR engagement ratio and cytotoxic activity(28,29), they are not immediately evident for agonist antibodies. As described above, most agonist antibodies differ from their direct targeting/cytotoxic counterparts, by being optimally effective with isotypes that preferentially engage the inhibitory FcγR(30–34); or avoid FcγR binding entirely as summarised in Figure 1.
Figure 1. Current formats of monospecific IgG used to elicit direct targeting and/or receptor agonism.

Target cell killing (red) and/or receptor agonism (blue) can be evoked using various isotype and formats of mAb; clockwise from top centre illustrating wild-type hIgG1 and mIgG2a as powerful native isotypes capable of delivering direct cell killing. Afucosylation can augment this activity through enhanced affinity to FcγRIII or mFcγRIV, respectively as can amino-acid modifications in the Fc(61), where additional FcγRs can be engaged more effectively. Native hIgG2 can evoke FcγR-independent agonism, via IgG2B forms, which can also be generated through C-S hinge mutations resulting in “locked” IgG2B forms. Reduced affinity mAb in mIgG1 and hIgG2 isotypes can also elicit higher agonism, which can be similarly achieved through interaction with FcγRIIB, again through modifications to the Fc. Finally, Fc-null mutants can be generated through Fc mutations and/or deglycosylation which can allow mAb to deliver their activities (e.g. receptor blocking or agonism) independently of FcγR interaction.
In the murine system, the optimal isotype is mIgG1, which, following binding by mFcγRII enhances receptor cross-linking, mimicking the action of the natural ligand.(35) In all cases that have been reported, agonistic activity correlates with cell surface receptor clustering(36), with non-agonists or antagonists inert in this respect. Other means of providing such cross-linking provide scope for increasing the activity of agonist antibodies. As summarised in Figure 2, and described in part below, these strategies involve experimental methods such as secondary antibody cross-linking or antibody immobilisation/coating(37) in addition to means to increase receptor multimerization, exemplified by tetramerization(38) or hexamerisation(39,40) using antibodies, antibody-like molecules or ligand;antibody hybrids.
Figure 2. Means to evoke agonism.

Multiple means through which receptor agonism can be evoked; clockwise from top left illustrating natural ligands, secondary “hyper” cross-linking often used in vitro, FcγR-mediated cross-linking, the related cross-linking achieved with another (non-FcγR) receptor such as a tumour associated antigen (TAA), low affinity mediated agonism, epitope-driven and hIgG2(B) mediated. Not shown are the multiple different multivalent methods through which cross-linking mediated agonism might be delivered including through tetra- and hexa-valent mAb and molecules, as well as IgM hexabody
For the canonical bivalent antibodies, based upon the experiences of targeting TNFRSF members in the mouse system, the most straightforward path to translation would have been to select a mIgG1-like human isotype. However, given the mouse:human differences in FcγR expression patterns and isotype binding profiles, there is no direct human equivalent to mIgG1.(41) Empirically, hIgG2 has been shown to elicit the greatest level of activity for several human agonist antibodies.(42) This was first demonstrated for anti-hCD40 agonists leveraging powerful B cell stimulation and antigen-specific CD8 T cell expansion, followed by similar evidence with mAb directed to 4-1BB, CD28(35), OX40(36) and CD27(43). The hIgG2 isotype was even able to convert anti-CD40 antagonists into maximally potent agonists.(44) Perhaps the most surprising property of the hIgG2 isotype was its ability to deliver agonism largely independently of FcγR.(35,44,45)
The agonistic capacity of hIgG2 relates to its unique hinge as evidenced by hinge-swaps with other isotypes.(42,44) The hIgG2 hinge contains additional cysteines which can undergo disulfide-shuffling to adopt different configurations, with IgG2A and IgG2B representing the extremes.(46,47) The IgG2B form is most agonistic (42,44), correlating with a more compact and less flexible mAb conformation; this is also true across IgG isotypes with hIgG2 the most compact and agonistically active and the highly flexible hIgG3 least agonistic.(48) A range of orthogonal biophysical techniques showed that agonism was linked to the presence of a disulphide cross-over between opposing heavy and light chains in engineered IgG2B forms(49), which imparted the more compact, less flexible agonist confirmation. The current model is that this more compact and less conformationally diverse molecular arrangement restricts receptor mobility in the plasma membrane, promoting more efficient receptor clustering and therein activation.
However, selection of hIgG2 alone is not sufficient to impart agonism for all antibodies and it is clear that epitope is also an important determinant. For example, although mAb binding to membrane distal domain CRD1 were shown to be agonistic for anti-CD40 mAb, and mAb directed to other CRDs less active, there was still a spectrum of activity for CRD1-targeting mAb. This was true for both mIgG1 and hIgG2 isotypes, with some mAb largely inert in any isotype, indicating that both epitope and isotype combine to mediate the net level of agonism for a given mAb.(45) These same studies also revealed that rare antibodies such as CP870,893 are agonistic independent of isotype.(35,44,45) Similar observations have been made for anti-4-1BB mAb where urelumab was demonstrated to be active in multiple isotypes (albeit more agonistic as hIgG2), whereas utomilumab was robustly inactive in all isotypes assessed.(45,50). This reveals that certain antibodies target rare epitopes that facilitate agonism without reliance on Fc or FcγR-dependent properties, although it has been shown that they may in certain contexts be augmented by FcγR engagement.(50–52)
A further determinant of agonist mAb activity is affinity. Following initial observations in a series of anti-CD95 mAb that demonstrated poorer apoptosis induction following affinity maturation(53), the impact of affinity on immunomodulatory antibodies was recently investigated.(54) Using antibody series targeting either CD40 or 4-1BB it was shown that reduced affinity increased receptor agonism, until a point where binding to the receptor was no longer detectable. Agonist activity still required bivalency, did not need FcγR interaction (but could be enhanced by it), was evoked in multiple isotypes (mIgG1 and hIgG2) and was correlated with increased receptor clustering on the cell surface. The hypothesis proposed is that lower affinity mAb bind bivalently, enabling them to bring 2 receptors together, before one F(ab) releases its receptor (due to high off-rate) and binds a third molecule and pulls it into the receptor complex before the released receptor has been able to migrate away; thereby nucleating larger receptor arrays that can better deliver agonistic signals. These findings add to our understanding of the various ways in which agonism can be achieved, as summarised in Figure 2.
Impact of the tumor micro-environment
There is a further complication to consider in delivering efficacy with agonist antibodies - the tumor microenvironment (TME). The TME comprises a wide array of host factors such as blood vessels, lymphatics, extracellular matrix, fibroblasts, innate and adaptive immune cells, and soluble factors in addition to tumor cells. Considering the context of this review, co-stimulatory receptors are largely expressed on myeloid (CD40) or T cell populations (OX40, 4-1BB, GITR and CD27). Consequently, these targets will be impacted differently dependent upon the varying TME and may require different mechanisms of action to induce therapeutic responses.
For example, CD40 expression is largely restricted to myeloid populations within the TME. Agonist mAb have been shown to act either directly through macrophages inducing T cell independent anti-tumor protection(55) or indirectly via dendritic cell cross-presentation leading to T cell mediated anti-tumor clearance.(56) Myeloid cells, particularly macrophages which can make up a significant proportion of the TME alter their phenotype and activation status in response to environmental cues. In the context of the TME there have been numerous studies observing that the inhibitory FcγRIIB becomes upregulated on monocytes/macrophages leading to a lowering of antibody-dependent effector capacity(57–60) whilst in contrast enhancing the potential for greater costimulatory agonism.(34) Therefore, the proportions of various myeloid cells in the TME and their relative activating to inhibitory FcγR levels will influence the mechanisms by which agonist mAb stimulate an anti-tumor response.
One approach seeking to take advantage of this phenomenon and overcome the relatively low affinity of human IgG isotypes for FcγRIIB is to use Fc engineering to selectively enhance mAb engagement with FcγRIIB using a number of established mutations. (51,61) This has been shown to work in vitro for several TNFR including CD40(40), OX40(40) and CD27(43). Although proof of concept has been demonstrated in pre-clinical models, intratumoral administration is required to overcome elevated toxicity.(62,63) Therefore, alternative strategies have attempted to overcome this, for example by replacing the FcγR-mediated cross-linking with an alternative receptor, such as a tumor associated antigen (TAA)(64–66) or avoiding activation of cell types associated with toxicity (macrophages and monocytes) by targeting receptors on dendritic cells (67,68). Such approaches are exciting with some showing clinical promise.(69,70) but it should be noted that their development also involves additional complexity in design, production and biological insight to elicit effective targeting and receptor activation.
In contrast, FcγR-independent solutions, such as through hIgG2, may provide a tractable approach for some agonist antibodies. They can function independently of hFcγR and so for some molecules and targets might better support systemic delivery, simplifying the route of administration and providing mAb access to sites outside the TME, for example where a TAA-bispecific may be restricted (e.g. draining lymph nodes for T cell priming etc.).
Also, as described above, Tregs may also be an important determinant of efficacy of agonist mAbs targeting T cell costimulatory receptors, such as 4-1BB, OX40 and GITR. Several studies demonstrate that these molecules are preferentially overexpressed on Treg in a range of human tumors.(71,72) In pre-clinical models, mAb targeting costimulatory receptors and engaging activating FcγR can deplete intratumoral Tregs and elicit anti-tumor CD8 T cell responses.(73) This TME effect clearly complicates the targeting of these molecules as should the mAb induce agonism of Tregs rather than depletion this could lead to their activation and expansion, thus restricting the anti-tumor response. Subsequent pre-clinical studies showed that for optimal activity in mice, mIgG1 can directly activate costimulatory receptor expressing CD8s when sufficient inhibitory FcγRIIB is present on tumor myeloid cells, whereas mIgG2a/c are most effective when target expression is high on intratumoral Treg and activating FcγR dominate the TME (73–75). Unfortunately, varying levels of both activating and inhibitory FcγR are commonly present in tumors and so the outcome is more difficult to predict, especially in humans. Moreover, often the potential stimulatory impact of the agonist mAb on the intratumoral Treg is often overlooked, and it is unclear whether it negatively contributes to outcome. Given that CD8 agonism and Treg depletion have opposing FcγR engaging requirements and show competition for FcγR availability, blunting their efficacy, the potential to specifically engage one of these mechanisms without inducing undesirable effects with the other are challenging. One means to do this and combine both mechanisms simultaneously is to disconnect the FcγR engaging requirement from one or both of these approaches.(74) This was previously achieved by grafting the agonistic hIgG2B hinge into a high activating:inhibitory FcγR, depleting mAb isotype (i.e. mIgG2a). This permitted preferential depletion of 4-1BB-high intratumoral Treg and FcγR-independent agonism of 4-1BB-low expressing CD8 T cells. Although this approach was successful pre-clinically whether it would be robust in clinical applications is an open question.
In summary, the TME can alter the expression of both costimulatory targets and FcγR that can be required to engage them through agonism or depletion in a cell-type specific manner. Understanding these factors and identifying patients with suitable expression profiles(76) and related effector mechanisms will be critical to effectively harness these agonist approaches. Furthermore, a recent study from Amit and colleagues (77) demonstrated that activating FcγR-mediated effector cell signaling could also reprogram the TME and enhance anti-tumor immune responses. Therefore, whether the pragmatic approach to avoid engaging FcγR and for example, utilize TAA-mediated clustering, means that potential beneficial FcγR-mediated signaling effects are lost remains to be determined. Ongoing efforts to further our understanding of FcγR biology may serve to clarify these issues. For now, the clinical promise being observed with TAA-bispecifics serve to keep the future of these preclinically powerful agents very much alive.
Clinical experience with agonists to date
To date, several agonist antibodies have been assessed clinically (Table 1). Here, we provide a summary of the clinical findings focussing on trials that include systemically administered mAb that target members of the TNFRSF as monotherapies; the latter to ensure the observed clinical effects are directly attributable to the agonists.
Table 1. Safety, efficacy and evidence of in vivo agonism in phase I-II agonist monotherapy trials in cancer.
| Target | mAb | Company | Isotype | Phase (Trial ID) | Disease population, N | Complete/ partial responses | Stable response rate (n, %) | Safety | MTD* or tolerable dose (mg/kg) | Evidence of in vivo agonism | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CD40 | SEA-CD40 | Seattle Genetics | Non-fucosylated hIgG1 | Phase I (NCT02376699) | Advanced cancers, 67 | 5 2(3.0%) | 9 (13.4%) | No DLT | 0.045-0.060 | B-cell depletion, transient T-cell and NK activation and depletion, transient cytokine activation (IP-10, MIG, MIP1β) in peripheral blood. | (88) |
| CP870,893 | Pfizer | hIgG2 | Phase I | Advanced solid malignancies, 29 | 4 (13.8%) | 7 (24%) | DLT (thromboembolism, headache) | 0.2* | B-cell depletion and activation (CD86) in peripheral blood | (78,89) | |
| Phase I (NCT02157831) | Advanced solid malignancies, 27 | 0 (0%) | 7 (26%) | DLT (grade 3 cytokine release and urticaria) | (78,89) | ||||||
| Mitazalimab | Alligator Bio-science | hIgG1 | Phase I (NCT02829099) | Advanced solid malignancies, 95 | 1 (1.1%) | 35 (36.8%) | 2 DLT (grade 3 headache and hepatotoxicity) | 0.9 | Transient increase in MCP-1, IP-10 and MIP-1β and transient reduction in B, NK and T cells in peripheral blood | (90) | |
| ChiLob7/4 | University of Southampton | hIgG1 | Phase I (NCT01561911) | Advanced cancers, 28 | 0 (0%) | 15 (53.6%) | DLT (liver transaminase elevation) | 2.1-3.3* | Transient increase in MIP1β in peripheral blood | (91) | |
| Dacetuzumab | Seattle Genetics | hIgG1 | Phase I (NCT00103779) | B-NHL, 50 | 6 (12%) | 13 (26%) | 2 DLT (grade 3 conjunctivitis and transient vision loss, grade 3 ALT elevation) | 4-8 | Transient increase in IL-1β, IL-6, IL-10, TNFα in peripheral blood | (92) | |
| Phase I (NCT00283101) | CLL, 12 | 0 (0%) | 5 (41.7%) | 1 DLT (grade 4 thrombocytopenia) | (93) | ||||||
| Phase II (NCT00435916) | DLBCL, 46 | 4 (9%) | 13 (37%) | Grade 3/4 events observed (ocular toxicity (2), transaminitis (1), cytokine release syndrome (1) | Not examined | (94) | |||||
| Phase I (NCT00079716) | Multiple myeloma, 44 | 0 (0%) | 9 (20%) | DLTs observed (cytokine release syndrome, ocular toxicity, transaminitis. | Not examined | (95) | |||||
| CD27 | Varlilumab | Celldex Therapeutics | hIgG1 | Phase I (NCT01460134) | Advanced solid tumors, 56; Lymphomas, 34 | Solid tumors: 1 (1.8%); Lymphomas: 1 (2.9%) | Solid tumors: 8 (14.3%); Lymphomas: 3 (8.8%) | 1 DLT (grade 3 transient hyponatremia) | 10 | Transient increase in proinflammatory cytokines, Treg depletion and augmentation of T-cell reactivity (in a subset of participants) in peripheral blood | (96,97) |
| 4-1BB | Urelumab | Bristol-Myers Squibb | hIgG4 (S228P hinge mutation) | Phase I (NCT00309023, NCT00612664, NCT01471210) | B-NHL (NCT01471210), 60; Advanced solid tumors, 347 | 6 (10%) (B-NHL) | 17 (28%) (B-NHL) | Hepatotoxicity; 2 deaths (n=347) | 0.1* | IFN-induced cytokines and IFN-response genes in peripheral blood | (98,99) |
| Utomilumab | Pfizer | hIgG2 | Phase I (NCT01307267) | Advanced solid tumors, 55 | 4 (7.3%) | 13 (24.5%) | No DLT | 10 | No consistent evidence in peripheral blood. | (100) | |
| GITR | GWN323 | Novartis | hIgG1 | Phase I/Ib (NCT02740270) | Advanced solid tumors, 39 | 0 (0%) | 7 (17.9%) | No DLT | ~21 (1500 mg flat dose)# | No evidence in peripheral blood or tumor | (101) |
| MK-4166 | MSD | hIgG1 | Phase I (NCT02132754) | Advanced solid tumors, 48 | 0 (0%) | 11 (22.9%) | DLT in 1 participant (grade 3 bladder perforation) | ~12.9 (900 mg flat dose) | Not examined | (102) | |
| TRX518 | Leap Therapeutics | Aglycosylated hIgG1 | Phase Ib (NCT02628574) | Advanced solid tumors, 43 (31 evaluable) | 1 (3.2%) | 22 (71.0%) | No DLT | 1 (4 mg/kg loading dose) | Treg reduction in periphery;Treg reduction and CD8 and Granzyme B increase in clinical responders. | (82) | |
| AMG 228 | Amgen | IgG1 | Phase I (NCT02437916) | Advanced solid tumors, 30 (27 evaluable) | 1 (0%) | 7 (23%) | No DLT | ~ 17.1 (1200 mg flat dose) | No evidence in peripheral blood | (103) | |
| BMS-986156 | Bristol-Myers Squibb | hIgG1 | Phase I (NCT02598960) | Advanced solid tumors, 34 | 0 (0%) | 11 (32.3%) | No DLT | ~11.4 (800 mg flat dose) | Trend to increased proinflam cytokines in peripheral blood; no evidence in tumor. | (104, 105) | |
| OX40 | MEDI6469 | Med-immune | murine IgG1 | Phase I (NCT01644968) | Advanced solid tumors, 30 | 1 (0%) | 12 (40%) | No DLT | 2 | Increased peripheral T and B cell vaccine reactivity and OX40 upregulation on Treg in tumor | (106) |
| Phase Ib (NCT02274155) | HNSCC, 17 | n/a Neoadjuvan t prior to surgical resection | n/a | No DLT | Increased CD4+ and CD8+ T cells in peripheral blood and tumor | (107) | |||||
| PF04518600 (Ivuxolimab) | Pfizer | hIgG2 | Phase I (NCT02315066) | Advanced cancers, 52 | 3 (5.6%) | 26 (56%) | No DLT | 10 | Increased profilferation of CD4 CM and EM cells in some participants in PB; Increased immune cell infiltration and immune activation/inflammation gene sets in tumor. | (108) | |
| BMS986178 | Bristol-Myers Squibb | hIgG1 | Phase I/IIa (NCT02737475) | Advanced cancers, 20 | 0 (0%) | 7 (35%) | No DLT | ~4.6 (320 mg flat dose) | Not examined | (109) | |
| MOXR0916 (Vonlerolizu-mab) | Genentech | hIgG1 | Phase I (NCT02219724) | Advanced solid tumors, 174 | 2 (1.1%) | 113 (66%) | No DLT | ~17 (1200 mg flat dose) | Immune activation in tumor | (110) | |
| MEDI0562 (Tavolimab) | AstraZeneca | hIgG1 | Phase I (NCT02318394) | Advanced solid tumors, 55 (50 evaluable) | 2 (4.0%) | 22 (44%) | 1 DLT (diarrhoea) | 10 | Increased peripheral Ki67+ CD4 and C8 memory T cells; decreased intratumoral OX40+ Treg | (111) | |
| INCAGN-01949 | Incyte Corporation | hIgG1 | Phase I (NCT02923349) | Advanced solid tumors, 87 | 1 (1.1%) | 23 (26.4%) | 1 DLT (grade 3 colitis) | ~20 (1400 mg flat dose) | No evidence in peripheral blood or tumor | (112) | |
| DR5 | PRO95780 (Drozitumab) | Genen-tech | hIgG1 | Phase I | Advanced cancers, 50 (41 evaluable) | 0 (0%) | 20 (49%) | 2 DLTs (grade 4 pulmonary embolism and grade 3 serum ALT elevation) | 20 | Not examined | (113) |
| INBRX-109 | Inhibrx, Inc. | Tetravalent hIgG1 | Phase I (NCT03715933) | Metastatic chondrosarcoma, 31 | 2 (6.5%) | 25 (80.6%) | No DLT; but 2 deaths due to hepatic failure | 3 | Not examined | (114) | |
| CS-1008 (Tigatuzumab) | Daiichi Sankyo, Inc. | hIgG1 | Phase I (NCT01220999) | Metastatic colorectal carcinoma, 19 | 1 (5.3%) | 8 (42.1%) | No DLT | 2 | No evidence in peripheral blood or tumor | (115) | |
| DS-8273a | Daiichi Sankyo, Inc. | hIgG1 | Phase I (NCT02076451) | Advanced cancers, 32 | 0 (0%) | 10 (31.3%) | No DLT | 24 | Decreased in peripheral myeloid derived suppressor cells | (116) | |
| Conatumumab | Amgen | hIgG1 | Phase I | Advanced solid tumors, 37 | 1 (2.7%) | 14 (37.8%) | No DLT | 20 | No evidence in tumor | (84) |
The flat dose is estimated based on a 70 kg participant weight
Complete/partial response rate(%)
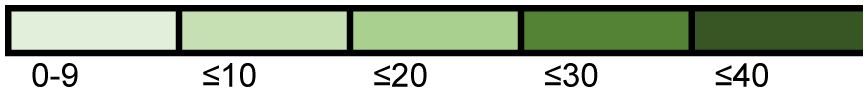
Stable response rate (%)
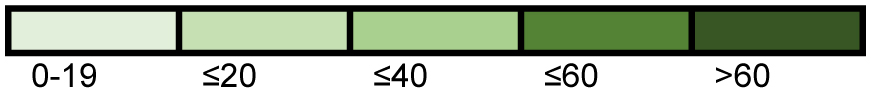
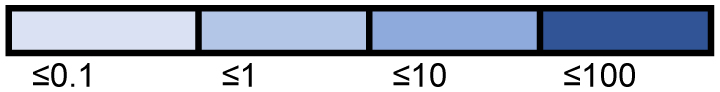
MTD/tolerable dose(mg/kg)
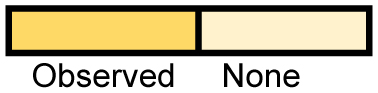
DLTs
CD40
CP870, 893 has a maximum tolerated dose (MTD) 10-40 fold lower than other anti-CD40 mAbs, ChiLob 7/4 and dacetuzumab (0.2 versus 3.3 versus 8 mg/kg), which are both hIgG1. The side effects observed with CD40 agonists were mostly mild-to-moderate but a few dose-limiting toxicities (DLTs) were noted, in particular, cytokine release syndrome, ocular toxicity and hepatotoxicity. These are hypothesized to be the result of on-target binding to CD40 on endothelium, conjunctiva and liver macrophages. Most events were transient and self-limiting. The anti-tumor efficacy with anti-CD40 agonists in these early-phase, dose-escalating monotherapy studies were modest, ranging from objective response rates (ORR) of 0-10% and stable response rates of 10-40%. Whether direct-targeting mechanisms or agonism were behind the observed anti-tumor efficacy are unclear. CD40 is expressed on B-cell lymphoid tumors and a proportion of solid tumors but the level of CD40 expression on tumor cells did not correlate with clinical responses. Tumor regression was also observed in metastatic melanoma lesions with CP870, 893 where CD40 is less likely to be expressed, altogether suggesting that agonism contributes to efficacy.(78) Transient peripheral blood B-cell depletion and upregulation of CD86 on residual B cells were observed in some participants, alongside elevation of inflammatory cytokines.
Whilst not reported within a monotherapy setting, the agonistic anti-CD40, sotigalimab, merits discussion as an example of ongoing clinical investigation. The PRINCE study randomized 99 participants with metastatic pancreatic adenocarcinoma to sotigalimab and chemotherapy (gemcitabine and nab-paclitaxel) versus nivolumab (anti-PD1) and chemotherapy vs sotigalimab, nivolumab and chemotherapy in first-line treatment.(79) Significant improvement in 1-year overall survival was observed with nivolumab and chemotherapy against a historical cohort (57.7% vs 35%) but not in the other two arms with the progression-free survival curves overlapping. Paired biopsies in two out of three participants showed increased tumor-infiltrating macrophages after sotigalimab/chemotherapy administration which was absent in other arms. Increased circulating numbers of Ki67+ non-naïve T-cells and IFNγ, CXCL9 and CXCL10 was observed across all arms, and interestingly, at an earlier time point for nivolumab-containing arms than sotigalimab (e.g. 2 weeks after treatment as opposed to 4-16 weeks).
4-1BB
Utomilumab is a hIgG2 mAb targeting 4-1BB. Unlike CP870, 893, it is a weak agonist, particularly in comparison to the hIgG4 urelumab (whose MTD is 100-fold lower). The clinical response rate for urelumab above 1 mg/kg has not been reported but it induced severe hepatotoxicity and resulted in two deaths at this level. Urelumab and utomilumab (administered <1 mg/kg and ≤10 mg/kg, respectively) had modest disease control rate (i.e. combined complete, partial and stable response rate). The distinct potency between the two mAbs is now understood to be due to differences in epitope and isotype. (36) Whilst 4-1BB is not highly expressed in the liver, FcγRIIb, which is preferentially bound by hIgG4 compared to hIgG2, is highly expressed by Kupffer cells. The combined effect of urelumab’s non-ligand blocking, membrane-distal binding epitope to 4-1BB on CD8+ T cells in the liver and hyper-crosslinking of 4-1BB by FcγRIIb on Kupffer cells has been shown by several groups to induce the observed hepatotoxicity.(80,81)
OX40
MEDI6469, the first OX40 agonist to enter clinical development, was deployed with as a murine IgG1. No objective responses or DLTs were observed in the phase I study in patients with advanced solid tumors, but the ability of MEDI16469 to elicit in vivo agonism was demonstrated by increased tumor-specific T- and B-cell responses in two patients with melanoma, and improved tetanus vaccine antibodies in a larger group of participants. Ivuxolimab, is an anti-OX40 hIgG2 agonist which displayed modest clinical efficacy in advanced stage cancers in the phase I study. Its dose was escalated to 10 mg/kg and no DLTs were observed. OX40 expression in the peripheral blood was heterogeneous and as expected, predominantly on CD4+ central and effector memory T cells. Some evidence of in vivo agonism was observed - paired tumor biopsies obtained at baseline and 6 weeks after treatment in participants dosed ≥ 1.5 mg/kg showed enrichment of inflammatory and immune activation gene signatures. The association between these signatures and clinical response is unclear. Only 1/29 participants experienced a partial response. Two other participants that experienced a partial response at lower dosing were not biopsied. The remaining published OX40 agonists are hIgG1 mAbs. This class of reagents have been largely well-tolerated and the MTD undefined but again, with modest clinical efficacy.
GITR
The clinically explored mAbs against GITR are all hIgG1 molecules. TRX518 is further aglycosylated for enhanced affinity for FcγRIII and augmented depleting ability. Similar to OX40 hIgG1 mAbs, these drugs have been well-tolerated with the MTD undefined. Low objective response rates are observed, ranging from 0-3%, but with higher stable disease rates (18-70%). TRX518-treated participants who achieved stable disease had reduced intratumoral Tregs compared to baseline biopsies, in contrast to participants with progressive disease, where an increase in intratumoral Tregs were observed. (82)
CD27
Varlilumab, a hIgG1 CD27 agonist was also selected with the aim of simultaneously mediating Treg depletion and agonism. Reduction in the peripheral CD4+ T-cell population including Tregs were observed in the phase I studies alongside a transient increase in peripheral inflammatory cytokines. A few participants with melanoma also showed increased CD8+ T-cell reactivity to melanoma antigens. Despite this, ORR was <5% and stable disease rate <20%. One DLT was observed (grade 3 hyponatremia) but otherwise varlilumab was well-tolerated. A further hIgG1 agonist (MK-5890) is currently undergoing clinical testing in advanced solid tumors but the mature data is yet to be published.(83)
DR5
DR5 is expressed on a wide range of haematopoietic and solid tumors. Therefore, distinct to the molecules above, DR5 agonists rely on the induction of direct tumor cell death. The ability of these agonists to induce objective responses has been low (<10%) although the tetravalent hIgG1 molecule, INBRX-109, induced stable disease in 80% of chondrosarcoma cases. In vivo evidence of DR5 agonism is lacking and often unreported. When conatumumab (an unmodified hIgG1 anti-DR5) was examined, intratumoral caspase-3 activation was observed in 2/7 cases, but not associated with clinical response(84). Hepatotoxicity was observed with both INBRX-109 and drozitumab (hIgG1). Two participants died of liver failure with INBRX-109 but this may have been accounted for, or aggravated by, other factors. A further 4/31 participants experienced grade 2/3 elevation of hepatic enzymes with drozitumab. The observed hepatotoxicity is likely to be due to on-target binding of the agonist to DR5-expressing hepatocytes.
These early phase trials described above primarily aimed to assess the safety rather than clinical efficacy of the agonist antibodies. Regardless, the efficacy signal in in these monotherapy trials has been underwhelming with limitations in efficacy/activity or issues with toxicity. As such, attention has shifted towards combination with other agents or development of newer antibody formats. These modified antibodies have been readily adopted by the field and a second wave of ‘synthetic’ TNFRSF agonists have followed the classic bivalent mAb based approaches. These ‘second generation’ synthetic molecules are often non-FcγR dependent agonists which fall into two main classes; single valency TNFRSF bi/trispecifics that co-engage TAA or immune checkpoint molecules to deliver cross-linking and consequently higher-order clustering, and multivalent TNFRSF agents constituted of antibody-based molecules or ligand:antibody hybrids with intrinsic clustering potential. Interestingly, both these classes of synthetic agonist have, in general, shown less evidence of the cytokine release syndrome and increased liver enzymes associated with their bivalent antibody agonist predecessors. This may indicate that these non-FcγR dependent approaches have decoupled dose limiting toxicity from effective agonism as suggested in pre-clinical studies.(85) However, many of these molecules are still in the early stages of clinical investigation and are yet to report fully and so we must await definitive findings in this regard. Further detailed discussion of these synthetic agonist strategies is outside the scope of this review but they are evolving rapidly and have been reviewed recently conceptually(86) and with 4-1BB targeting as an exemplar.(69)
Conclusion
The earlier success of tumor-targeting antibodies like rituximab and the PD-1/PD-L1 antibodies drove academia and pharma to explore new antibody targets, including agonist antibodies. This led to a flurry of clinical trials involving these agonists, none of which have demonstrated significant clinical efficacy. However, the antibody formats chosen were perhaps not optimal, with understanding of epitope and isotype lacking at the time, coupled to under-appreciation of the impact of the TME. In addition, their kinetics of response compared to checkpoint blockade as indicated in the PRINCE study discussed above is an interesting aspect. That agonists might take longer to induce tumour reduction may require a reconsideration of conventional trial designs, even when pseudo progression is taken into account.
Despite the agonists’ failure to induce significant clinical responses, much has been learned from the early phase monotherapy trials and following pre-clinical work. Unlike immune checkpoint inhibitors, where diverse immune effects are observed, these have been less frequent with agonist antibodies. Instead, hepatotoxicity secondary to on-target toxicity has been the commonest safety concern. Hepatotoxicity is closely linked to agonistic potency, thus measures to broaden the therapeutic window will be important, including through bispecific and other approaches. One means of achieving this by localising the antibody in the tumor, such as exemplified by GEN1046, an antibody targeting 4-1BB and PD-L1, wherein the MTD was not reached at a dose >10 mg/kg and the safety profile was manageable.(87)
Another potentially important consideration for future clinical development is patient and/or cancer selection. Most of the early phase trials have recruited ‘all comers’ with advanced, multiply treated disease, and have not discriminated between cancer type on the basis that host immunity is being targeted instead of the tumor cells. Nevertheless, sufficient expression of the target in the tumor is likely to be important to enable the threshold for agonism to be reached; this is likely to differ between patients and tumor types and may necessitate targeting of multiple receptors to achieve the desired level of immune stimulation. Similarly, as described above, the nature of the target, FcγR expression pattern, TME composition and desired mechanism of action will all play their part. As we learn more about these aspects our ability to successfully leverage agonist antibodies in the clinic will surely follow.
Acknowledgements
We would like to thank the current and past members of the Antibody and Vaccine group for useful discussions and work on this topic. Funding was provided by Blood Cancer UK (Award number: 12050 and 14043) to MSC/SAB, Cancer Research UK (Award number: A18087 (all authors), A24721 (MSC/SAB), A25139 (AAS, SAB, MSC), A30681 (AAS), A27179 (SHL), A25778 (AAS) and DRCDDRPGMApr2020\100005 (AAS, SAB, MSC). We apologise to those authors whose work we have not cited for reasons of journal restrictions.
Footnotes
Conflicts:
M.S.C. has consulted for BioInvent, Boehringer Ingelheim, GSK, Radiant, iTeos Therapeutics, Surrozen, Hanall and Mestag and received research funding from BioInvent, GSK, UCB and iTeos. S.A.B. has consulted for Astex Pharmaceuticals, BioInvent, Epsilogen, ImCheck Therapeutics and LTZ Therapeutics and has received research funding from BioInvent and ImCheck Therapeutics. A.Al-S has received research funding and/or honoraria Auxeris Therapeuticx, Inc., Hoffman-La Roche Inc., UCB, F-star Therapeutics, Verastem, Celldex Therapeutics, Talix Therapeutics and AstraZeneca. S.H.L. has consulted for F-star Therapeutics, Celldex Therapeutics, AstraZeneca and Xcella and received research funding from Celldex Therapeutics and BioInvent. All authors hold patents related to TNFRSF agonists.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.:S0092-8674(11)00127-9 [pii] doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Jenkins MK. The ups and downs of T cell costimulation. Immunity. 1994;1(6):443–6. doi: 10.1016/1074-7613(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 3.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (New York, NY. 2017;355(6332):1428–33. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nature immunology. 2004;5(10):987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 5.Wing K, Yamaguchi T, Sakaguchi S. Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends in immunology. 2011;32(9):428–33. doi: 10.1016/j.it.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229(1):12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annual review of immunology. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wortzman ME, Clouthier DL, McPherson AJ, Lin GH, Watts TH. The contextual role of TNFR family members in CD8(+) T-cell control of viral infections. Immunol Rev. 2013;255(1):125–48. doi: 10.1111/imr.12086. [DOI] [PubMed] [Google Scholar]
- 9.Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nature medicine. 1997;3(6):682–5. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 10.Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, et al. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nature medicine. 1999;5(7):774–9. doi: 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- 11.French RR, Chan HTC, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nature medicine. 1999;5(5):548–53. doi: 10.1038/8426. [DOI] [PubMed] [Google Scholar]
- 12.Kucka K, Wajant H. Receptor Oligomerization and Its Relevance for Signaling by Receptors of the Tumor Necrosis Factor Receptor Superfamily. Front Cell Dev Biol. 2020;8:615141. doi: 10.3389/fcell.2020.615141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Chau B, West SM, Kimberlin CR, Cao F, Schwarz F, et al. Structures of mouse and human GITR-GITRL complexes reveal unique TNF superfamily interactions. Nature communications. 2021;12(1):1378. doi: 10.1038/s41467-021-21563-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luhder F, Huang Y, Dennehy KM, Guntermann C, Muller I, Winkler E, et al. Topological requirements and signaling properties of T cell-activating, anti-CD28 antibody superagonists. The Journal of experimental medicine. 2003;197(8):955–66. doi: 10.1084/jem.20021024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanson A, Elpek K, Duong E, Shallberg L, Fan M, Johnson C, et al. ICOS agonism by JTX-2011 (vopratelimab) requires initial T cell priming and Fc cross-linking for optimal T cell activation and anti-tumor immunity in preclinical models. PLoS One. 2020;15(9):e0239595. doi: 10.1371/journal.pone.0239595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogel A, Ibrahim FM, Thirdborough SM, Renart-Depontieu F, Birts CN, Buchan SL, et al. Fcgamma receptor-mediated cross-linking codefines the immunostimulatory activity of anti-human CD96 antibodies. JCI Insight. 2022;7(19) doi: 10.1172/jci.insight.158444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov. 2018;17(7):509–27. doi: 10.1038/nrd.2018.75. [DOI] [PubMed] [Google Scholar]
- 18.Gennery AR. CD27-CD70 defects: a wolf in wolf’s clothing? Blood. 2020;136(23):2600–2. doi: 10.1182/blood.2020007763. [DOI] [PubMed] [Google Scholar]
- 19.Beziat V, Rapaport F, Hu J, Titeux M, Bonnet des Claustres M, Bourgey M, et al. Humans with inherited T cell CD28 deficiency are susceptible to skin papillomaviruses but are otherwise healthy. Cell. 2021;184(14):3812–28.:e30. doi: 10.1016/j.cell.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warnatz K, Bossaller L, Salzer U, Skrabl-Baumgartner A, Schwinger W, van der Burg M, et al. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood. 2006;107(8):3045–52. doi: 10.1182/blood-2005-07-2955. [DOI] [PubMed] [Google Scholar]
- 21.Somekh I, Thian M, Medgyesi D, Gulez N, Magg T, Gallon Duque A, et al. CD137 deficiency causes immune dysregulation with predisposition to lymphomagenesis. Blood. 2019;134(18):1510–6. doi: 10.1182/blood.2019000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen K, Wang J, Zhou K, Mu W, Zhang M, Deng X, et al. CD137 deficiency because of two novel biallelic TNFRSF9 mutations in a patient presenting with severe EBV-associated lymphoproliferative disease. Clin Transl Immunology. 2023;12(5):e1448. doi: 10.1002/cti2.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fournier B, Hoshino A, Bruneau J, Bachelet C, Fusaro M, Klifa R, et al. Inherited TNFSF9 deficiency causes broad Epstein-Barr virus infection with EBV+ smooth muscle tumors. The Journal of experimental medicine. 2022;219(7) doi: 10.1084/jem.20211682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byun M, Ma CS, Akcay A, Pedergnana V, Palendira U, Myoung J, et al. Inherited human OX40 deficiency underlying classic Kaposi sarcoma of childhood. The Journal of experimental medicine. 2013;210(9):1743–59. doi: 10.1084/jem.20130592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vezys V, Penaloza-MacMaster P, Barber DL, Ha SJ, Konieczny B, Freeman GJ, et al. 4-1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. J Immunol. 2011;187(4):1634–42. doi: 10.4049/jimmunol.1100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buchan SL, Fallatah M, Thirdborough SM, Taraban VY, Rogel A, Thomas LJ, et al. PD-1 Blockade and CD27 Stimulation Activate Distinct Transcriptional Programs That Synergize for CD8(+) T-Cell-Driven Antitumor Immunity. Clin Cancer Res. 2018;24(10):2383–94. doi: 10.1158/1078-0432.CCR-17-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pichler AC, Carrie N, Cuisinier M, Ghazali S, Voisin A, Axisa PP, et al. TCR-independent CD137 (4-1BB) signaling promotes CD8(+)-exhausted T cell proliferation and terminal differentiation. Immunity. 2023;56(7):1631–48.:e10. doi: 10.1016/j.immuni.2023.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nimmerjahn F, Vidarsson G, Cragg MS. Effect of posttranslational modifications and subclass on IgG activity: from immunity to immunotherapy. Nature immunology. 2023;24(8):1244–55. doi: 10.1038/s41590-023-01544-8. [DOI] [PubMed] [Google Scholar]
- 29.Nimmerjahn F, Ravetch JV. FcgammaRs in health and disease. Curr Top Microbiol Immunol. 2011;350:105–25. doi: 10.1007/82_2010_86. [DOI] [PubMed] [Google Scholar]
- 30.White AL, Chan HTC, Roghanian A, French RR, Mockridge CI, Tutt AL, et al. Interaction with FcγRIIB Is Critical for the Agonistic Activity of Anti-CD40 Monoclonal Antibody. The Journal of Immunology. 2011;187(4):1754–63. doi: 10.4049/jimmunol.1101135. [DOI] [PubMed] [Google Scholar]
- 31.Li F, Ravetch JV. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science (New York, NY. 2011;333(6045):1030–4.:333/6045/1030 [pii] doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, et al. An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19(1):101–13. doi: 10.1016/j.ccr.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Li F, Ravetch JV. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcgamma receptor engagement. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(27):10966–71.:1208698109 [pii] doi: 10.1073/pnas.1208698109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hussain K, Hargreaves CE, Roghanian A, Oldham RJ, Chan HT, Mockridge CI, et al. Upregulation of FcgammaRIIb on monocytes is necessary to promote the superagonist activity of TGN1412. Blood. 2015;125(1):102–10. doi: 10.1182/blood-2014-08-593061. [DOI] [PubMed] [Google Scholar]
- 35.White AL, Dou L, Chan HTC, Field VL, Mockridge CI, Moss K, et al. Fcγ Receptor Dependency of Agonistic CD40 Antibody in Lymphoma Therapy Can Be Overcome through Antibody Multimerization. The Journal of Immunology. 2014;193(4):1828–35. doi: 10.4049/jimmunol.1303204. [DOI] [PubMed] [Google Scholar]
- 36.Yu X, James S, Felce JH, Kellermayer B, Johnston DA, Chan HTC, et al. TNF receptor agonists induce distinct receptor clusters to mediate differential agonistic activity. Commun Biol. 2021;4(1):772. doi: 10.1038/s42003-021-02309-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ball C, Fox B, Hufton S, Sharp G, Poole S, Stebbings R, et al. Antibody C region influences TGN1412-like functional activity in vitro. J Immunol. 2012;189(12):5831–40. doi: 10.4049/jimmunol.1201795. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y, Yeh SH, Madireddi S, Matochko WL, Gu C, Pacheco Sanchez P, et al. Tetravalent biepitopic targeting enables intrinsic antibody agonism of tumor necrosis factor receptor superfamily members. MAbs. 2019;11(6):996–1011. doi: 10.1080/19420862.2019.1625662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Horst HJ, Gelderloos AT, Chamuleau MED, Breij ECW, Zweegman S, Nijhof IS, et al. Potent preclinical activity of HexaBody-DR5/DR5 in relapsed and/or refractory multiple myeloma. Blood Adv. 2021;5(8):2165–72. doi: 10.1182/bloodadvances.2020003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang D, Goldberg MV, Chiu ML. Fc Engineering Approaches to Enhance the Agonism and Effector Functions of an Anti-OX40 Antibody. The Journal of biological chemistry. 2016;291(53):27134–46. doi: 10.1074/jbc.M116.757773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–43. doi: 10.1158/1078-0432.CCR-12-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White AL, Chan HTC, French RR, Willoughby J, Mockridge CI, Roghanian A, et al. Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer cell. 2015;27(1):138–48. doi: 10.1016/j.ccell.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heckel F, Turaj AH, Fisher H, Chan HTC, Marshall MJE, Dadas O, et al. Agonistic CD27 antibody potency is determined by epitope-dependent receptor clustering augmented through Fc-engineering. Commun Biol. 2022;5(1):229. doi: 10.1038/s42003-022-03182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu X, Chan HTC, Fisher H, Penfold CA, Kim J, Inzhelevskaya T, et al. Isotype Switching Converts Anti-CD40 Antagonism to Agonism to Elicit Potent Antitumor Activity. Cancer Cell. 2020;37(6):850–66.:e7. doi: 10.1016/j.ccell.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu X, Chan HTC, Orr CM, Dadas O, Booth SG, Dahal LN, et al. Complex Interplay between Epitope Specificity and Isotype Dictates the Biological Activity of Anti-human CD40 Antibodies. Cancer Cell. 2018;33(4):664–75.:e4. doi: 10.1016/j.ccell.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. The Journal of biological chemistry. 2008;283(23):16194–205. doi: 10.1074/jbc.M709987200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. The Journal of biological chemistry. 2008;283(23):16206–15. doi: 10.1074/jbc.M709988200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X, Zhao Y, Shi H, Zhang Y, Yin X, Liu M, et al. Human immunoglobulin G hinge regulates agonistic anti-CD40 immunostimulatory and antitumour activities through biophysical flexibility. Nature communications. 2019;10(1):4206. doi: 10.1038/s41467-019-12097-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Orr CM, Fisher H, Yu X, Chan CH, Gao Y, Duriez PJ, et al. Hinge disulfides in human IgG2 CD40 antibodies modulate receptor signaling by regulation of conformation and flexibility. Sci Immunol. 2022;7(73):eabm3723. doi: 10.1126/sciimmunol.abm3723. [DOI] [PubMed] [Google Scholar]
- 50.Leitner J, Egerer R, Waidhofer-Söllner P, Grabmeier-Pfistershammer K, Steinberger P. FcγR requirements and costimulatory capacity of Urelumab, Utomilumab, and Varlilumab. Frontiers in Immunology. 2023;14 doi: 10.3389/fimmu.2023.1208631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV. Therapeutic Activity of Agonistic, Human Anti-CD40 Monoclonal Antibodies Requires Selective FcgammaR Engagement. Cancer Cell. 2016;29(6):820–31. doi: 10.1016/j.ccell.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richman LP, Vonderheide RH. Anti-human CD40 monoclonal antibody therapy is potent without FcR crosslinking. Oncoimmunology. 2014;3:e28610. doi: 10.4161/onci.28610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chodorge M, Zuger S, Stirnimann C, Briand C, Jermutus L, Grutter MG, et al. A series of Fas receptor agonist antibodies that demonstrate an inverse correlation between affinity and potency. Cell Death Differ. 2012;19(7):1187–95. doi: 10.1038/cdd.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu X, Orr CM, Chan HTC, James S, Penfold CA, Kim J, et al. Reducing affinity as a strategy to boost immunomodulatory antibody agonism. Nature. 2023;614(7948):539–47. doi: 10.1038/s41586-022-05673-2. [DOI] [PubMed] [Google Scholar]
- 55.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (New York, NY. 2011;331(6024):1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.French RR, Taraban VY, Crowther GR, Rowley TF, Gray JC, Johnson PW, et al. Eradication of lymphoma by CD8 T cells following anti-CD40 monoclonal antibody therapy is critically dependent on CD27 costimulation. Blood. 2007;109(11):4810–5. doi: 10.1182/blood-2006-11-057216. [DOI] [PubMed] [Google Scholar]
- 57.Dahal LN, Dou L, Hussain K, Liu R, Earley A, Cox KL, et al. STING activation reverses lymphoma-mediated resistance to antibody immunotherapy. Cancer research. 2017 doi: 10.1158/0008-5472.CAN-16-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell. 2018;33(4):649–63.:e4. doi: 10.1016/j.ccell.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hussain K, Liu R, Smith RCG, Muller KTJ, Ghorbani M, Macari S, et al. HIF activation enhances FcgammaRIIb expression on mononuclear phagocytes impeding tumor targeting antibody immunotherapy. J Exp Clin Cancer Res. 2022;41(1):131. doi: 10.1186/s13046-022-02294-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simpson AP, Roghanian A, Oldham RJ, Chan HTC, Penfold CA, Kim HJ, et al. FcgammaRIIB controls antibody-mediated target cell depletion by ITIM-independent mechanisms. Cell reports. 2022;40(3):111099. doi: 10.1016/j.celrep.2022.111099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu R, Oldham RJ, Teal E, Beers SA, Cragg MS. Fc-Engineering for Modulated Effector Functions-Improving Antibodies for Cancer Treatment. Antibodies (Basel) 2020;9(4) doi: 10.3390/antib9040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garris CS, Wong JL, Ravetch JV, Knorr DA. Dendritic cell targeting with Fc-enhanced CD40 antibody agonists induces durable antitumor immunity in humanized mouse models of bladder cancer. Science translational medicine. 2021;13(594) doi: 10.1126/scitranslmed.abd1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knorr DA, Dahan R, Ravetch JV. Toxicity of an Fc-engineered anti-CD40 antibody is abrogated by intratumoral injection and results in durable antitumor immunity. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(43):11048–53. doi: 10.1073/pnas.1810566115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sum E, Rapp M, Durr H, Mazumdar A, Romero PJ, Trumpfheller C, et al. The tumor-targeted CD40 agonist CEA-CD40 promotes T cell priming via a dual mode of action by increasing antigen delivery to dendritic cells and enhancing their activation. J Immunother Cancer. 2022;10(3) doi: 10.1136/jitc-2021-003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trub M, Uhlenbrock F, Claus C, Herzig P, Thelen M, Karanikas V, et al. Fibroblast activation protein-targeted-4-1BB ligand agonist amplifies effector functions of intratumoral T cells in human cancer. J Immunother Cancer. 2020;8(2) doi: 10.1136/jitc-2019-000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, et al. Tumor-Targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-The-shelf therapy. Science translational medicine. 2019;11(496) doi: 10.1126/scitranslmed.aav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salomon R, Rotem H, Katzenelenbogen Y, Weiner A, Cohen Saban N, Feferman T, et al. Bispecific antibodies increase the therapeutic window of CD40 agonists through selective dendritic cell targeting. Nat Cancer. 2022;3(3):287–302. doi: 10.1038/s43018-022-00329-6. [DOI] [PubMed] [Google Scholar]
- 68.Salomon R, Dahan R. Next Generation CD40 Agonistic Antibodies for Cancer Immunotherapy. Front Immunol. 2022;13:940674. doi: 10.3389/fimmu.2022.940674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Claus C, Ferrara-Koller C, Klein C. The emerging landscape of novel 4-1BB (CD137) agonistic drugs for cancer immunotherapy. MAbs. 2023;15(1):2167189. doi: 10.1080/19420862.2023.2167189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Melero I, Tanos T, Bustamante M, Sanmamed MF, Calvo E, Moreno I, et al. A first-in-human study of the fibroblast activation protein-targeted, 4-1BB agonist RO7122290 in patients with advanced solid tumors. Science translational medicine. 2023;15(695):eabp9229. doi: 10.1126/scitranslmed.abp9229. [DOI] [PubMed] [Google Scholar]
- 71.De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity. 2016;45(5):1135–47. doi: 10.1016/j.immuni.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Magnuson AM, Kiner E, Ergun A, Park JS, Asinovski N, Ortiz-Lopez A, et al. Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(45):E10672–E81. doi: 10.1073/pnas.1810580115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. The Journal of experimental medicine. 2013;210(9):1685–93. doi: 10.1084/jem.20130573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, et al. Antibodies to Costimulatory Receptor 4-1BB Enhance Anti-tumor Immunity via T Regulatory Cell Depletion and Promotion of CD8 T Cell Effector Function. Immunity. 2018;49(5):958–70.:e7. doi: 10.1016/j.immuni.2018.09.014. [DOI] [PubMed] [Google Scholar]
- 75.Griffiths J, Hussain K, Smith HL, Sanders T, Cox KL, Semmrich M, et al. Domain binding and isotype dictate the activity of anti-human OX40 antibodies. J Immunother Cancer. 2020;8(2) doi: 10.1136/jitc-2020-001557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Melake MJ, Smith HG, Mansfield D, Davies E, Dillon MT, Wilkins AC, et al. OX40 and 4-1BB delineate distinct immune profiles in sarcoma. Oncoimmunology. 2022;11(1):2066050. doi: 10.1080/2162402X.2022.2066050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yofe I, Landsberger T, Yalin A, Solomon I, Costoya C, Demane DF, et al. Anti-CTLA-4 antibodies drive myeloid activation and reprogram the tumor microenvironment through FcgammaR engagement and type I interferon signaling. Nat Cancer. 2022;3(11):1336–50. doi: 10.1038/s43018-022-00447-1. [DOI] [PubMed] [Google Scholar]
- 78.Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25(7):876–83. doi: 10.1200/JCO.2006.08.3311. doi 25/7/876 [pii] [DOI] [PubMed] [Google Scholar]
- 79.Padron LJ, Maurer DM, O’Hara MH, O’Reilly EM, Wolff RA, Wainberg ZA, et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nature medicine. 2022;28(6):1167–77. doi: 10.1038/s41591-022-01829-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bartkowiak T, Jaiswal AR, Ager CR, Chin R, Chen CH, Budhani P, et al. Activation of 4-1BB on Liver Myeloid Cells Triggers Hepatitis via an Interleukin-27-Dependent Pathway. Clin Cancer Res. 2018;24(5):1138–51. doi: 10.1158/1078-0432.CCR-17-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qi X, Li F, Wu Y, Cheng C, Han P, Wang J, et al. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcgammaR affinity. Nature communications. 2019;10(1):2141. doi: 10.1038/s41467-019-10088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Davar D, Zappasodi R, Wang H, Naik GS, Sato T, Bauer T, et al. Phase IB Study of GITR Agonist Antibody TRX518 Singly and in Combination with Gemcitabine, Pembrolizumab, or Nivolumab in Patients with Advanced Solid Tumors. Clin Cancer Res. 2022;28(18):3990–4002. doi: 10.1158/1078-0432.CCR-22-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guelen L, Fischmann TO, Wong J, Mauze S, Guadagnoli M, Babala N, et al. Preclinical characterization and clinical translation of pharmacodynamic markers for MK-5890: a human CD27 activating antibody for cancer immunotherapy. J Immunother Cancer. 2022;10(9) doi: 10.1136/jitc-2022-005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Herbst RS, Kurzrock R, Hong DS, Valdivieso M, Hsu CP, Goyal L, et al. A first-in-human study of conatumumab in adult patients with advanced solid tumors. Clin Cancer Res. 2010;16(23):5883–91. doi: 10.1158/1078-0432.CCR-10-0631. [DOI] [PubMed] [Google Scholar]
- 85.Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Science translational medicine. 2019;11(496) doi: 10.1126/scitranslmed.aav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fromm G, de Silva S, Schreiber TH. Reconciling intrinsic properties of activating TNF receptors by native ligands versus synthetic agonists. Front Immunol. 2023;14:1236332. doi: 10.3389/fimmu.2023.1236332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muik A, Garralda E, Altintas I, Gieseke F, Geva R, Ben-Ami E, et al. Preclinical Characterization and Phase I Trial Results of a Bispecific Antibody Targeting PD-L1 and 4-1BB (GEN1046) in Patients with Advanced Refractory Solid Tumors. Cancer Discov. 2022;12(5):1248–65. doi: 10.1158/2159-8290.CD-21-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coveler AL, Smith DC, Phillips T, Curti BD, Goel S, Mehta AN, et al. Phase 1 dose-escalation study of SEA-CD40: a non-fucosylated CD40 agonist, in advanced solid tumors and lymphomas. J Immunother Cancer. 2023;11(6) doi: 10.1136/jitc-2022-005584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ruter J, Antonia SJ, Burris HA, Huhn RD, Vonderheide RH. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol Ther. 2010;10(10):983–93. doi: 10.4161/cbt.10.10.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moreno V, Perets R, Peretz-Yablonski T, Fourneau N, Girgis S, Guo Y, et al. A phase 1 study of intravenous mitazalimab, a CD40 agonistic monoclonal antibody, in patients with advanced solid tumors. Invest New Drugs. 2023;41(1):93–104. doi: 10.1007/s10637-022-01319-2. [DOI] [PubMed] [Google Scholar]
- 91.Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, et al. Clinical and Biological Effects of an Agonist Anti-CD40 Antibody: A Cancer Research UK Phase I Study. Clin Cancer Res. 2015;21(6):1321–8. doi: 10.1158/1078-0432.CCR-14-2355. [DOI] [PubMed] [Google Scholar]
- 92.Advani R, Forero-Torres A, Furman RR, Rosenblatt JD, Younes A, Ren H, et al. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. J Clin Oncol. 2009;27(26):4371–7. doi: 10.1200/JCO.2008.21.3017. doi JCO.2008.21.3017 [pii] [DOI] [PubMed] [Google Scholar]
- 93.Furman RR, Forero-Torres A, Shustov A, Drachman JG. A phase I study of dacetuzumab (SGN-40, a humanized anti-CD40 monoclonal antibody) in patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2010;51(2):228–35. doi: 10.3109/10428190903440946. [DOI] [PubMed] [Google Scholar]
- 94.de Vos S, Forero-Torres A, Ansell SM, Kahl B, Cheson BD, Bartlett NL, et al. A phase II study of dacetuzumab (SGN-40) in patients with relapsed diffuse large B-cell lymphoma (DLBCL) and correlative analyses of patient-specific factors. Journal of hematology & oncology. 2014;7:44. doi: 10.1186/1756-8722-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hussein M, Berenson JR, Niesvizky R, Munshi N, Matous J, Sobecks R, et al. A phase I multidose study of dacetuzumab (SGN-40; humanized anti-CD40 monoclonal antibody) in patients with multiple myeloma. Haematologica. 2010;95(5):845–8. doi: 10.3324/haematol.2009.008003. doi haematol.2009.008003 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, et al. Safety and Activity of Varlilumab, a Novel and First-in-Class Agonist Anti-CD27 Antibody, in Patients With Advanced Solid Tumors. J Clin Oncol. 2017;35(18):2028–36. doi: 10.1200/JCO.2016.70.1508. [DOI] [PubMed] [Google Scholar]
- 97.Ansell SM, Flinn I, Taylor MH, Sikic BI, Brody J, Nemunaitis J, et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, for hematologic malignancies. Blood Adv. 2020;4(9):1917–26. doi: 10.1182/bloodadvances.2019001079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin Cancer Res. 2017;23(8):1929–36. doi: 10.1158/1078-0432.CCR-16-1272. [DOI] [PubMed] [Google Scholar]
- 99.Timmerman J, Herbaux C, Ribrag V, Zelenetz AD, Houot R, Neelapu SS, et al. Urelumab alone or in combination with rituximab in patients with relapsed or refractory B-cell lymphoma. Am J Hematol. 2020;95(5):510–20. doi: 10.1002/ajh.25757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin Cancer Res. 2018;24(8):1816–23. doi: 10.1158/1078-0432.CCR-17-1922. [DOI] [PubMed] [Google Scholar]
- 101.Piha-Paul SA, Geva R, Tan TJ, Lim DW, Hierro C, Doi T, et al. First-in-human phase I/Ib open-label dose-escalation study of GWN323 (anti-GITR) as a single agent and in combination with spartalizumab (anti-PD-1) in patients with advanced solid tumors and lymphomas. J Immunother Cancer. 2021;9(8) doi: 10.1136/jitc-2021-002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Papadopoulos KP, Autio K, Golan T, Dobrenkov K, Chartash E, Chen Q, et al. Phase I Study of MK-4166, an Anti-human Glucocorticoid-Induced TNF Receptor Antibody, Alone or with Pembrolizumab in Advanced Solid Tumors. Clin Cancer Res. 2021;27(7):1904–11. doi: 10.1158/1078-0432.CCR-20-2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tran B, Carvajal RD, Marabelle A, Patel SP, LoRusso PM, Rasmussen E, et al. Dose escalation results from a first-in-human, phase 1 study of glucocorticoid-induced TNF receptor-related protein agonist AMG 228 in patients with advanced solid tumors. J Immunother Cancer. 2018;6(1):93. doi: 10.1186/s40425-018-0407-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang R, Baxi V, Li Z, Locke D, Hedvat C, Sun Y, et al. Pharmacodynamic activity of BMS-986156, a glucocorticoid-induced TNF receptor-related protein agonist, alone or in combination with nivolumab in patients with advanced solid tumors. ESMO Open. 2023;8(2):100784. doi: 10.1016/j.esmoop.2023.100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Heinhuis KM, Carlino M, Joerger M, Di Nicola M, Meniawy T, Rottey S, et al. Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor-Related Protein Agonist Alone or in Combination With Nivolumab for Patients With Advanced Solid Tumors: A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 2020;6(1):100–7. doi: 10.1001/jamaoncol.2019.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer research. 2013;73(24):7189–98. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Duhen R, Ballesteros-Merino C, Frye AK, Tran E, Rajamanickam V, Chang SC, et al. Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells. Nature communications. 2021;12(1):1047. doi: 10.1038/s41467-021-21383-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Diab A, Hamid O, Thompson JA, Ros W, Eskens F, Doi T, et al. A Phase I Open-Label, Dose-Escalation Study of the OX40 Agonist Ivuxolimab in Patients with Locally Advanced or Metastatic Cancers. Clin Cancer Res. 2022;28(1):71–83. doi: 10.1158/1078-0432.CCR-21-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gutierrez M, Moreno V, Heinhuis KM, Olszanski AJ, Spreafico A, Ong M, et al. OX40 Agonist BMS-986178 Alone or in Combination With Nivolumab and/or Ipilimumab in Patients With Advanced Solid Tumors. Clin Cancer Res. 2021;27(2):460–72. doi: 10.1158/1078-0432.CCR-20-1830. [DOI] [PubMed] [Google Scholar]
- 110.Kim TW, Burris HA, de Miguel Luken MJ, Pishvaian MJ, Bang YJ, Gordon M, et al. First-In-Human Phase I Study of the OX40 Agonist MOXR0916 in Patients with Advanced Solid Tumors. Clin Cancer Res. 2022;28(16):3452–63. doi: 10.1158/1078-0432.CCR-21-4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Glisson BS, Leidner RS, Ferris RL, Powderly J, Rizvi NA, Keam B, et al. Safety and Clinical Activity of MEDI0562, a Humanized OX40 Agonist Monoclonal Antibody, in Adult Patients with Advanced Solid Tumors. Clin Cancer Res. 2020;26(20):5358–67. doi: 10.1158/1078-0432.CCR-19-3070. [DOI] [PubMed] [Google Scholar]
- 112.Davis EJ, Martin-Liberal J, Kristeleit R, Cho DC, Blagden SP, Berthold D, et al. First-in-human phase I/II, open-label study of the anti-OX40 agonist INCAGN01949 in patients with advanced solid tumors. J Immunother Cancer. 2022;10(10) doi: 10.1136/jitc-2021-004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Camidge DR, Herbst RS, Gordon MS, Eckhardt SG, Kurzrock R, Durbin B, et al. A phase I safety and pharmacokinetic study of the death receptor 5 agonistic antibody PRO95780 in patients with advanced malignancies. Clin Cancer Res. 2010;16(4):1256–63. doi: 10.1158/1078-0432.CCR-09-1267. [DOI] [PubMed] [Google Scholar]
- 114.Subbiah V, Chawla SP, Conley AP, Wilky BA, Tolcher A, Lakhani NJ, et al. Preclinical Characterization and Phase 1 Trial Results of INBRX-109, a Third-Generation, Recombinant, Humanized, Death Receptor 5 Agonist Antibody, in Chondrosarcoma. Clin Cancer Res. 2023 doi: 10.1158/1078-0432.CCR-23-0974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ciprotti M, Tebbutt NC, Lee FT, Lee ST, Gan HK, McKee DC, et al. Phase I Imaging and Pharmacodynamic Trial of CS-1008 in Patients With Metastatic Colorectal Cancer. J Clin Oncol. 2015;33(24):2609–16. doi: 10.1200/JCO.2014.60.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Forero A, Bendell JC, Kumar P, Janisch L, Rosen M, Wang Q, et al. First-in-human study of the antibody DR5 agonist DS-8273a in patients with advanced solid tumors. Invest New Drugs. 2017;35(3):298–306. doi: 10.1007/s10637-016-0420-1. [DOI] [PubMed] [Google Scholar]