

Abstract
Aims
The molecular mechanisms underlying the association between type 2 diabetes (T2D) and gastrointestinal (GI) disease are unclear. To identify protein pathways, we conducted a two‐stage network Mendelian randomisation (MR) study.
Materials and Methods
Genetic instruments for T2D were obtained from a large‐scale summary‐level genome‐wide meta‐analysis. Genetic associations with blood protein levels were obtained from three genome‐wide association studies on plasma proteins (i.e. the deCODE study as the discovery and the UKB‐PPP and Fenland studies as the replication). Summary‐level data on 10 GI diseases were derived from genome‐wide meta‐analysis of the UK Biobank and FinnGen. MR and colocalisation analyses were performed. Pathways were constructed according to the directionality of total and indirect effects, and corresponding proportional mediation was estimated. Druggability assessments were conducted across four databases to prioritise protein mediators.
Results
Genetic liability to T2D was associated with 69 proteins in the discovery protein dataset after multiple testing corrections. All associations were replicated at the nominal significance level. Among T2D‐associated proteins, genetically predicted levels of nine proteins were associated with at least one of the GI diseases. Genetically predicted levels of SULT2A1 (odds ratio = 1.98, 95% CI 1.80–2.18), and ADH1B (odds ratio = 2.05, 95% CI 1.43–2.94) were associated with cholelithiasis and cirrhosis respectively. SULT2A1 and cholelithiasis (PH4 = 0.996) and ADH1B and cirrhosis (PH4 = 0.931) have strong colocalisation support, accounting for the mediation proportion of 72.8% (95% CI 45.7–99.9) and 42.9% (95% CI 15.5–70.4) respectively.
Conclusions
The study identified some proteins mediating T2D‐GI disease associations, which provided biological insights into the underlying pathways.
Keywords: drug target, gastrointestinal diseases, Mendelian randomisation, protein biomarker, type 2 diabetes
1. INTRODUCTION
Type 2 diabetes mellitus (T2D) affects around 480 million population globally. 1 As the fastest growing metabolic disorders, T2D imposes a significant disease burden worldwide. 2 Gastrointestinal (GI) disease is more prevalent in patients with T2D compared to the general population, and the presence of GI disease substantially deteriorates life quality. 3 Although accumulating evidence supports T2D as a risk factor for developing a wide range of GI diseases, 4 , 5 , 6 , 7 the underlying biological pathways remain unclear.
Circulating proteins play important roles in biological process and have high potentials as drug targets. 8 , 9 Unravelling protein links between T2D and GI disease may provide crucial insights into disease pathogenesis understanding, early disease prevention and target drug development. Blood protein alteration has been observed in patients with T2D 10 , 11 , 12 and implicated in GI diseases as well. 13 , 14 , 15 , 16
Mendelian randomisation (MR) using randomly allocated genetic variants as instrument variables has the advantages of minimising confounding and reversal causation. 17 MR approach has been widely used to understand disease pathophysiology. 18 Recent MR studies have identified several plasma proteins associated with T2D 19 , 20 and some GI diseases 21 , 22 , 23 ; however, the shared protein basis between T2D and GI diseases has been scarcely been studied. We here applied MR analysis under the two‐stage network framework to identify the intermediate proteins linking T2D and GI diseases.
2. MATERIALS AND METHODS
2.1. Study design
The study design is presented in Figure 1. Based on our previous explorations, 4 we included 10 GI diseases associated with T2D in this study. There are three steps of two‐stage MR analysis. First, we examined T2D‐GI disease associations. In the second step, we conducted a proteome‐wide MR to explore the proteins associated with genetic liability to T2D. To increase the reliability, only proteins survived after the multiple testing correction and consistently replicated in replication datasets were regarded as putative T2D‐associated proteins and retained for subsequent analyses. In the last step, we estimated the associations between T2D‐associated proteins and GI disease risk. Several extra analyses including genetic colocalisation analysis, mediation calculation and druggability assessment were conducted to prioritise protein mediators.
FIGURE 1.

Study flow chart. GI, gastrointestinal; IVW, inverse variance weighted; SNP, single nucleotide polymorphisms; T2D, type 2 diabetes.
2.2. Study population and data sources
2.2.1. Genetic instruments for T2D
Genetic instruments for T2D were obtained from the Vujkovic et al GWAS incorporating 148 726 cases and 965 732 non‐cases of European ancestry. 24 We selected genetic variants (e.g. single‐nucleotide polymorphisms [SNPs]) associated with T2D at the genome‐wide significance level (p < 5× 10−8) and a low linkage disequilibrium (defined as r 2 < 0.001) as the instrumental variables. To minimise horizontal pleiotropy, we excluded variants in the FTO gene 4 , 25 via searching in PhenoScanner V2. 26
2.2.2. Blood protein data sources
We obtained GWAS data on blood protein levels from three large‐scale studies without sample overlap. The discovery analysis was based on the deCODE protein genetic database that assessed genetic variants' associations with 4719 unique plasma proteins measured in 35 559 Icelanders. 27 We further used two replication data sources from the UK Biobank Pharma Proteomics Project (UKB‐PPP) where 2923 unique proteins were profiled in 54 219 participants 28 as well as from the Fenland study where genetic associations were calculated for 4775 proteins in 10 708 Caucasian. 29 Both the deCODE and Fenland studies profiled blood protein data using the SomaScan version 4 assay (SomaLogic) while the UKB‐PPP used Olink Explore 3072 PEA. Regarding genetic instrumental variable selection for proteins, we obtained the index cis‐acting variant defined as the SNP located within 1 Mb upstream or downstream of the transcription start site of the protein encoding gene and with the smallest p value. 30
2.2.3. Data sources for GI diseases
The study included 10 GI diseases including four upper GI diseases (gastroesophageal reflux disease, gastric ulcer, acute gastritis, chronic gastritis), two lower GI diseases (irritable bowel syndrome, diverticular disease) and four hepato‐biliary and pancreatic diseases (cholelithiasis, cholecystitis, nonalcoholic fatty liver disease and cirrhosis). Summary‐level data on these outcomes were obtained from the UK Biobank and FinnGen R9 release. The UK Biobank is an ongoing cohort study, which recruited half a million of participant across the United Kingdom between 2006 and 2010 aged over 40. 31 Participants with GI diseases were ascertained by International Classification of Diseases (ICD)‐8, ICD‐9 and ICD‐10. Summary‐level data on the 10 GI diseases were based on individuals of European ancestry. GWAS analysis was performed by the Lee Lab (Seoul National University, Seoul, Republic of Korea; https://www.leelabsg.org/resources) with the adjustment of sex, birth year, genotyping batch and first four principal components. The FinnGen study included Finnish adults. GI diseases were ascertained by the ICD codes from nationwide health registers. GWAS analysis was adjusted for sex, age, genotyping batch and the first 10 genetic principal components. 32 Detailed information on two studies is presented in Additional File 1: Table S1. We performed the GWAS meta‐analysis of the UK Biobank and FinnGen R9 using the inverse‐variance fixed‐effects method by METAL software with genomic control correction. 33 To increase the statistical power, our MR analyses were based on these genome‐wide meta‐analysis data.
2.3. Statistical analyses
2.3.1. MR analysis
Data harmonisation was performed based on both effect and other alleles. Due to low levels of missingness, we did not replace SNPs that were not available in one of the datasets with proxies. The inverse variance weighted (IVW) and the Wald ratio methods were used as the primary analysis depending on the number of SNPs included in the analysis. To test the robustness of the results, MR‐Egger, 34 weighted median 35 and weighted mode 36 analyses were performed as the sensitivity analyses. Cochran's Q statistic was calculated to measure the heterogeneity. The IVW analysis was guided by the heterogeneity test. Specifically, the fixed‐effects IVW method was applied when the p‐value for Cochran's Q statistic was ≥0.05; otherwise, the random‐effects IVW method was used. 37 Horizontal pleiotropy was assessed by MR–Egger intercept test. 34 The false discovery rate (FDR) based on Benjamini‐Hochberg approach was used for multiple testing correction. A significance level of FDR‐corrected p < 0.05 was considered significant. All tests were two‐sided, and the TwoSampleMR package in R software were used to perform analyses.
2.3.2. Colocalisation analysis
To investigate whether the protein and GI disease share a causal variant, 38 we conducted genetic Bayesian colocalisation analysis using the coloc R package. For each protein, we included SNPs within the 1 Mb region around the lead cis‐pQTL in the primary analysis and included SNPs within 500 kb in the supplementary analysis. Five exclusive hypotheses are as follows: (1) no association with either protein or GI outcomes (PH0); (2) one causal variant for protein only (PH1); (3) one causal variant for GI disease only (PH2); (4) two distinct causal variants for protein and GI disease were associated (PH3); (5) a shared causal variant for both protein and GI disease (PH4). The prior probability for the causal variant associated with trait 1 only (P1), trait 2 only (P2), both traits 1 and 2 (P12) were set as 1 × 10−4, 1 × 10−4 and 1 × 10−5. We defined the PH4 ≥0.8 as strong evidence of colocalisation, 0.5≤PH4<0.8 as medium evidence and PH4 <0.5 as low evidence.
2.3.3. Mediation analysis
We performed the mediation analysis according to directional consistency of between total effect (β T2D‐GI disease) and indirect effect (β T2D‐protein × β Protein‐GI disease) among the associations reaching the FDR significance level in both step 2 (β T2D‐protein) and step 3 (β Protein‐GI disease) MR analyses. We then estimated the proportion mediated of protein in the T2D‐GI disease association. In detail, the product of coefficient method was applied to estimate the indirect effect. 39 The proportion was calculated by multiplying the estimate of T2D‐protein association and the estimate of protein‐GI disease association (indirect effect = β T2D‐protein × β protein‐GI disease) then dividing by the estimate of T2D‐GI disease association (β T2D‐GI disease). The propagation method was used to calculate the CI. 39
2.3.4. Appraisal of druggability
We assessed the druggability of potential protein mediators with the aim of prioritising therapeutic targets. This assessment was conducted with data from the DrugBank, 40 ChEMBL, 41 Dependency Map and the Connectivity Map (https://clue.io/repurposing-app). Treating protein as the drug target, we documented the information on the drug name, drug indication and development process in the pipeline were documented. We characterised the therapeutic development process into four categories: (1) approved, (2) in clinical trials, (3) preclinical and (4) druggable. 21
3. RESULTS
3.1. Genetic liability to T2D in relation to GI disease
Genetic liability to T2D was associated with all 10 GI diseases after FDR correction, with ORs ranging from 1.04 (95% CI 1.00, 1.07, p = 0.035) for diverticular disease to 1.36 (95% CI 1.21, 1.53, p = 2.94 × 10−7) for nonalcoholic fatty liver disease (Figure 2). The associations remained overall consistent in the sensitivity analyses (Additional File 1: Table S2).
FIGURE 2.
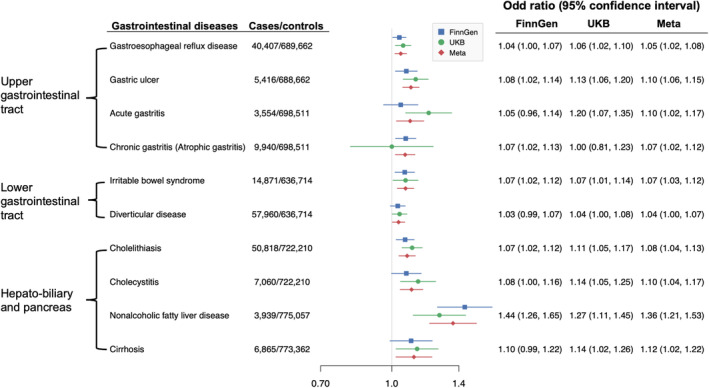
Associations of genetically predicted type 2 diabetes with gastrointestinal diseases. The ORs were scaled to 1‐unit increase in log‐transformed OR of type 2 diabetes. CI, confidence interval; OR, odd ratio.
3.2. Genetic liability to T2D in relation to protein levels
The association between genetic liability to T2D and levels of blood proteins was examined in three protein genome‐wide association datasets. In the discovery dataset, a total of 464 of 4907 proteins was associated with genetic liability to T2D after FDR correction (Figure 3A, and Additional File 1: Table S3). Among these, 69 associations were directionally replicated in UKB‐PPP and Fenland (p < 0.05; Figure 3B, Additional File 1: Tables S4 and S5).
FIGURE 3.
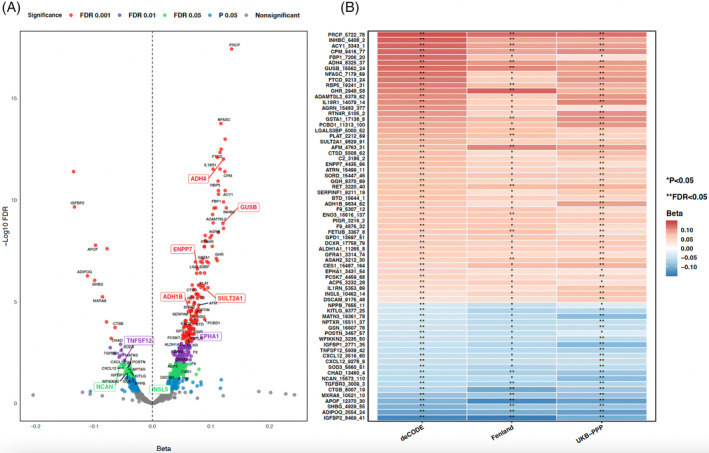
(A) Volcano plots of MR association between genetically predicted T2D and circulating protein levels in the discovery dataset (deCODE). The labelled proteins are 69 out of 464 discovered proteins with FDR <0.05 that directionally consistent and maintained nominal significant (p < 0.05) in the UKB‐PPP and Fenland datasets. (B) Heatmap of associations replicated using protein data from UKB‐PPP and Fenland. The association with a p value <0.05, but FDR‐corrected p value ≥0.05 was labelled with *, while FDR‐corrected p value <0.05 was labelled as ** in the heatmap. FDR performed among all proteins in each dataset. ADH1B, alcohol dehydrogenase 1B; ADH4, alcohol dehydrogenase 4; ENPP7, ectonucleotide pyrophosphatase/phosphodiesterase family member 7; EPHA1, ephrin type‐A receptor 1; FDR, false discovery rate; GI, gastrointestinal; GUSB, beta‐glucuronidase; INSL5, insulin‐like peptide INSL5; NCAN, neurocan core protein; SULT2A1, bile salt sulfotransferase; T2D, type 2 diabetes; TNFSF12, tumour necrosis factor ligand superfamily member 12.
3.3. T2D‐associated proteins in relation to GI diseases
We then performed two‐sample MR analysis to estimate the associations between 69 T2D‐driven proteins and GI diseases. Given some proteins had no suitable instruments, 64 proteins with cis‐acting (protein quantitative trait loci) pQTL (p < 5 × 10−8, r 2 < 0.01) were included. Eleven pairs of putative T2D‐driven protein associated with GI diseases were identified using the deCODE database. This included three proteins (ADH4 [alcohol dehydrogenase 1B], ENPP7 [ectonucleotide pyrophosphatase/phosphodiesterase family member 7] and SULT2A1 [bile salt sulfotransferase]) associated with cholelithiasis, two proteins (ADH1B [alcohol dehydrogenase 1B] and NCAN [neurocan core protein]) associated with cirrhosis, two proteins (GUSB [beta‐glucuronidase] and NCAN) associated with NAFLD, two proteins (EPHA1 [ephrin type‐A receptor 1] and SULT2A1 [bile salt sulfotransferase]) with cholecystitis, one protein (TNFSF12 [tumour necrosis factor ligand superfamily member 12]) associated with diverticular disease and one protein (INSL5 [insulin‐like peptide 5]) associated with gastric ulcer (Table 1 and Additional File 1: Table S6). All these pairs were directionally replicated in the UKB‐PPP database, while ADH1B‐cirrhosis and EPHA1‐cholecystitis did not reach the significant level (Additional File 1: Table S7). In the Fenland study, all identified pairs reached nominal significant level (p < 0.05) except for ENPPT (the analysis could not be performed due to lack of available variants) (Additional File 1: Table S8).
TABLE 1.
Associations of putative type 2 diabetes–associated proteins with gastrointestinal diseases.
| Gene a | GI disease | Discovery (deCODE) | Replication (UKB‐PPP) | Replication (Fenland) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | p | PH4 b | OR (95% CI) | p | PH4 b | OR (95% CI) | p | PH4 b | ||
| ADH1B | Cirrhosis | 2.05 (1.43, 2.94) | 8.96E−05 | 0.931 | 1.81 (0.82, 4.03) | 0.145 | 0.181 | 3.49 (1.87, 6.51) | 8.96E−05 | 0.933 |
| ADH4 | Cholelithiasis | 1.26 (1.10, 1.43) | 0.001 | 0.363 | NA | NA | NA | 1.20 (1.05, 1.38) | 8.54E−03 | 0.107 |
| ENPP7 | Cholelithiasis | 1.04 (1.02, 1.06) | 4.19E−04 | 0.028 | NA | NA | NA | NA | NA | NA |
| EPHA1 | Cholecystitis | 0.87 (0.80, 0.95) | 0.001 | 0.013 | 1.05 (0.87, 1.25) | 0.629 | 0.009 | 0.99 (0.94, 1.03) | 0.542 | 0.016 |
| GUSB | NAFLD | 1.92 (1.39, 2.67) | 8.61E−05 | 0.017 | 2.10 (1.45, 3.04) | 8.61E‐05 | 0.723 | 1.79 (1.31, 2.44) | 2.51E−04 | 0.562 |
| INSL5 | Gastric ulcer | 0.39 (0.23, 0.65) | 3.18E−04 | 0.774 | 0.74 (0.63, 0.87) | 3.18E‐04 | 0.800 | 0.60 (0.46, 0.79) | 3.18E−04 | 0.817 |
| NCAN | Cirrhosis | 0.54 (0.44, 0.65) | 1.85E−10 | <0.001 | 0.48 (0.38, 0.61) | 1.09E‐09 | <0.001 | 0.48 (0.39, 0.61) | 1.85E−10 | <0.001 |
| NCAN | NAFLD | 0.34 (0.26, 0.43) | 1.34E−18 | 0.009 | 0.30 (0.22, 0.41) | 1.07E‐14 | 0.001 | 0.28 (0.21, 0.37) | 1.34E−18 | 0.008 |
| SULT2A1 | Cholecystitis | 1.45 (1.17, 1.81) | 0.001 | 0.589 | 1.23 (1.09, 1.39) | 8.03E‐04 | 0.638 | 1.35 (1.13, 1.62) | 1.03E−03 | 0.593 |
| SULT2A1 | Cholelithiasis | 1.98 (1.80, 2.18) | 1.28E−45 | 0.996 | 1.47 (1.39, 1.55) | 1.28E‐45 | 0.998 | 1.64 (1.52, 1.77) | 1.28E−36 | 0.961 |
| TNFSF12 | Diverticular disease | 1.11 (1.05, 1.17) | 2.78E−04 | 0.001 | 1.15 (1.08, 1.22) | 1.08E‐05 | 0.001 | 1.05 (1.01, 1.08) | 8.55E−03 | <0.001 |
Abbreviations: ADH1B, alcohol dehydrogenase 1B; ADH4, alcohol dehydrogenase 4; CI, confidence interval; ENPP7, ectonucleotide pyrophosphatase/phosphodiesterase family member 7; EPHA1, ephrin type‐A receptor 1; FDR, false discovery rate; GI, gastrointestinal; GUSB, beta‐glucuronidase; INSL5, insulin‐like peptide INSL5; NA, absence of cis‐pQTL; NCAN, neurocan core protein; NAFLD; nonalcoholic fatty liver disease; OR, odd ratio; SULT2A1, bile salt sulfotransferase; TNFSF12, Tumour necrosis factor ligand superfamily member 12.
Presented results reached FDR <0.05 in MR analysis based on deCODE database.
PH4 values were based on colocalisation analysis under ±1000 kb window.
3.4. Colocalisation analysis
SULT2A1 (PH4 = 0.996) and ADH1B (PH4 = 0.931) had high support colocalisation with cholelithiasis and cirrhosis in the deCODE dataset respectively. SULT2A1 maintained strong colocalisation using other two data sources (PH4 = 0.998 in UKB‐PPP and PH4 = 0.961 in Fenland). While strong colocalisation for ADH1B was replicated in Fenland study (PH4 = 0.933), evidence was weaker in UKB‐PPP (PH4 = 0.181). Moderate colocalisation were observed between GUSB and NAFLD, and INSL5 and gastric ulcer (0.8 > PH4 > 0.7) (Table 1, and Additional File 1: Table S9).
3.5. Mediation analysis
Among 11 protein‐GI disease pairs, mediation effect was not found between EPHA1 and cholecystitis, INSL5 and gastric ulcer, TNFSF12 and diverticular disease due to directionality inconsistency. SULT2A1 mediated around 72.8% (95% CI, 45.7%, 99.9%) of the association between T2D and cholelithiasis. This mediation was observed in UKB‐PPP and Fenland albeit attenuated. ADH1B mediated 42.9% (95% CI, 15.5%, 70.4%) of the association between T2D and cirrhosis using deCODE protein data and 45.9% (95% CI 0%, 92.6%) using the Fenland protein study. (Figure 4, and Additional File 1: Table S10). There were some other mediations with moderate effects including ADH4‐cholelithiasis (36.2%, 95% CI, 13.6%, 58.8%), ENPP7‐cholithiasis (3.8%, 95% CI, 1.3%, 6.3%), GUSB‐NAFLD (25.7%, 95% CI, 10.9%, 40.4%), NCAN‐cirrhosis (31.8%, 95% CI, 10.5%, 53.1%), NCAN‐NAFLD (20.1%, 95% CI, 7.4%, 32.9%) and SULT2A1‐cholecystitis (30.9%, 95% CI, 10.0%, 51.8%) (Additional File 1: Table S10).
FIGURE 4.
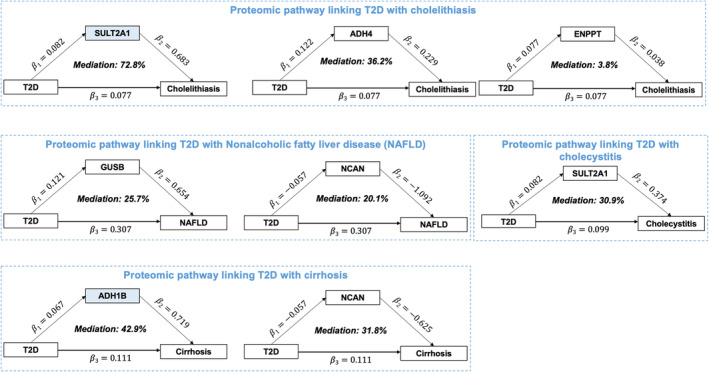
Proteomic mediators of the associations between T2D and GI diseases. The association was based on the deCODE protein dataset. Blue coloured protein encoding genes (SULT2A1 and ADH1B) demonstrating high evidence of colocalisation with associated GI diseases (PH4 ≥0.8). ADH1B, alcohol dehydrogenase 1B; GI, gastrointestinal; SULT2A1, bile salt sulfotransferase; T2D, type 2 diabetes.
3.6. Druggability analysis
We assessed druggability for nine protein mediators in T2D‐GI disease association. As summarised in Additional File 1: Table S11, SULT2A1 and ADH1B were listed as druggable target with wide implications in cancer and chronic pain treatment. However, neither of them has been recognised to treat GI diseases.
4. DISCUSSION
4.1. Principal findings
The study employed a two‐stage network MR analysis and studied the potential roles of blood proteins mediating association between T2D and a wide range of GI diseases. Putative causal plasma proteins associated withT2D were screened among more than 4000 proteins. We identified 69 proteins associated with genetically predicted T2D. We then established the MR associations between these proteins and GI disease risk. Along with genetic colocalisation and mediation construction, we highlighted two potential protein pathways linking T2D and GI diseases. SULT2A1 mediated around 72.8% of T2D–cholelithiasis association and ADH1B mediated 42.9% of T2D‐cirrhosis association. These findings provided biological insights into the underlying pathways for T2D and GI disease associations.
4.2. Comparison with previous studies
The results from the present study are in line with previous findings. Some identified T2D‐associated proteins were reported including well‐studied proteins like SHBG (sex hormone‐binding globulin), 42 C2 (complement C2) 43 and MXRA8 (matrix remodelling associated 8). 44 A prior MR‐based association study identified T2D was associated with SULT2A1 (bile salt sulfotransferase). 44 Supported by earlier observations, reduced level of plasma TNFSF12 (TWEAK) 45 and elevated level of GUSB (β‐glucuronidase) 46 were reported in prevalent T2D. ENPP7 has been identified as novel biomarkers associated with glycaemic deterioration in T2D. 47 Two alcohol dehydrogenase (ADH) enzymes, ADH1B and ADH4, were identified as the putative T2D‐associated proteins. ADH protein regulates alcohol metabolism and may influence the development of T2D. The association between ADH4 and incident T2D was identified in a previous cohort study, 46 but the effect of T2D on the ADH1B and ADH4 is less explored. However, considering the complex biological pathways due to possible pleiotropy, further study is warranted to disclose the underlying chain of T2D‐to‐protein effect.
A recent study revealed shared genetic mechanisms between T2D and gastroesophageal reflux disease, irritable bowel syndrome, peptic ulcer disease (i.e. gastric ulcer) and gastritis‐duodenitis (i.e. acute gastritis). 48 The study found multiple genes and biological pathways are shared between T2D and various GI disorders, involving autoimmune, viral and proinflammatory‐mediated mechanisms. 48 Our study reveals underlying connections between T2D and GI diseases from a protein perspective by integrating human genetic and proteomics data. These findings offer valuable insights for clinical translation and potential opportunities for drug target repurposing and development.
Our findings offer protein insights into the association between the protein links to T2D and GI diseases. Among 11 identified protein‐GI disease pairs, the evidence of colocalisation for most associations were weak, indicating the possible confounding from linkage disequilibrium. The strongest signal was SULT2A1 that may play a role in the development of cholelithiasis among T2D. SULT2A1, known as bile salt sulfotransferase, belongs to hydroxysteroid sulfotransferase (SULT) family. SULT is highly expressed in liver, and metabolically active or hormonally responsive tissues outside the liver. 49 The association between T2D and SULT2A1 has been reported in an MR study using the proteomics source from 5438 elderly Icelanders. 44 Several large GWAS also identified susceptibility loci at SULT2A1 associated with risk of cholelithiasis (gallstone disease). 29 , 50 , 51 Cholelithiasis is featured as the formation of one or more gallstones in the gallbladder, and bile acid dysregulation is one of the established mechanisms. 52 Insulin resistance can decrease the expression of bile acid synesthetic enzymes and increase the gallstone susceptibility. 53 Thus, alternation of SULT2A1 among T2D patients may change the hepatic sulfation of bile acid, bile acid metabolism and in turn increase the risk of developing cholelithiasis. 51
The putative role of ADH1B in linking T2D to cirrhosis was also noted. ADH1B is involved in ethanol metabolic pathway 54 and has potential role in insulin resistance. 55 , 56 Molecular studies suggested that elevated expression of ADH1B decreased the expression of an intracellular lipid transporter FABP4, which can stimulate β‐cells to secrete insulin to maintain glucose homeostasis. 55 ADH1B is predominately expressed in liver, and accumulating evidence suggested its association with cirrhosis, especially alcohol‐related liver cirrhosis. 57 , 58 However, no previous studies have revealed that this protein is associated with excessive cirrhosis caused by T2D. Nevertheless, the rs1229984 variant in the ADH1B has been strongly linked to T2D, potentially altering alcohol consumption and resulting in T2D development. 59 We could not rule out that the observed mediating effect of ADH1B in the association between T2D and cirrhosis might be driven by an upstream effect of alcohol consumption on T2D. Thus, future studies are needed to warrant our findings.
4.3. Strengths
This study is the first study to explore the protein pathway between T2D and GI disease and provided insights into the complex pathophysiological process of GI disease comorbidities in T2D. The MR study design has the advantages of reducing the confounding and reverse causation and facilitate the causal inference. Another notable strength of study is that we used multiple data sources and conducted a series sensitivity and replication analyses to validate our findings. In addition, combined evidence from colocalisation analysis, we were able to rule out the possible bias caused by linkage disequilibrium. The stringent replication criteria enhanced the reliability of our findings.
4.4. Limitations
The study also has some limitations. First, some proteins might be inadvertently neglected due to lack of cis‐pQTL signals. Second, the current study used data only from European ancestry. Although it can minimise the population stratification bias, it decreases the generalisability of our findings to other populations. Third, pleiotropic effect could not be fully ruled out. To minimise the bias, we removed the SNPs near the FTO gene that has been associated with obesity, a shared risk factor between T2D and GI diseases. 4 Fourth, although we used genome‐wide meta‐analysis of two large cohorts for the GI diseases, statistical power might still be inadequate for weak‐to‐moderate associations for some outcomes with a small number of cases. Further study using emerging larger GWAS is warranted.
5. CONCLUSIONS
In summary, the study identified many circulating proteins associated with T2D. SULT2A1 and ADH1B were suggested as vital protein biomarkers mediating the association between T2D and cholelithiasis and the association between T2D and cirrhosis respectively.
AUTHOR CONTRIBUTIONS
SY, ZD, XL and JG designed the study and initiated analysis plan. JG, XR, XW and SY contributed to the statistical analysis and wrote the draft of the manuscript. JG, XR, XW, XC, TF, DG, SB, JC, JFL, SCL, XL, ZD and SY contributed to discussion and revision of the manuscript. JG, XR, XW, XC, TF, DG, SB, JC, JFL, SCL, XL, ZD and SY critically reviewed the manuscript. JG and SY are the guarantors of this work and take responsibility for the integrity of the data and the accuracy of the data analysis.
FUNDING INFORMATION
D. G. is supported by the BHF Centre of Research Excellence (RE/18/4/34215) at Imperial College London. S. B. is supported by a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (award number 204623/Z/16/Z) and by the UK Research and Innovation Medical Research Council (MC_UU_00002/7). S. C. L. is supported by research grants from the Swedish Research Council (Vetenskapsrådet; grant no. 2019‐00977), the Swedish Heart Lung Foundation (Hjärt‐Lungfonden, grant number 20210351) and the Swedish Cancer Society (Cancerfonden). X. L. is funded by the Natural Science Fund for Distinguished Young Scholars of Zhejiang Province (LR22H260001) and the National Nature Science Foundation of China (82204019). Z. Y. D. is funded by the National Nature Science Foundation of China (82004353).
CONFLICT OF INTEREST STATEMENT
Dr. Ludvigsson has coordinated an unrelated study on behalf of the Swedish IBD quality register (SWIBREG). That study received funding from Janssen corporation. Dr. Ludvigsson has also received financial support from MSD developing a paper reviewing national healthcare registers in China. Dr. Ludvigsson has an ongoing research collaboration on celiac disease with Takeda.
PEER REVIEW
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1111/dom.16087.
ETHICS STATEMENT
All data used in the study were from the publicly available summary‐level datasets. All studies obtained the ethical permission from the relevant ethical committees.
Supporting information
Additional File 1. Supplementary Tables.
Table S1. Dataset description.
Table S2. Association of type 2 diabetes with 10 gastrointestinal diseases.
Table S3. Association of type 2 diabetes with deCODE plasma protein.
Table S4. Association of type 2 diabetes with UKB‐PPP plasma protein.
Table S5. Association of type 2 diabetes with Fenland plasma protein.
Table S6. Association of putative type 2 diabetes‐associated proteins in deCODE with gastrointestinal diseases.
Table S7. Association of putative type 2 diabetes‐associated proteins in UKB‐PPP with gastrointestinal diseases.
Table S8. Association of putative type 2 diabetes‐associated proteins in Fenland with gastrointestinal diseases.
Table S9. Colocalisation analysis of identified protein‐gastrointestinal pairs in deCODE, UKB‐PPP, and Fenland.
Table S10. Mediation analysis of identified protein between type 2 diabetes and gastrointestinal diseases in deCODE, UKB‐PPP, and Fenland.
Table S11. Appraisal of druggability.
Additional File 2: STROBE‐MR checklist.
ACKNOWLEDGEMENTS
We would like to thank all participants and investigators in the deCODE, Fenland study, FinnGen study and UK Biobank study for their great contributions and sharing of data.
Geng J, Ruan X, Wu X, et al. Network Mendelian randomisation analysis deciphers protein pathways linking type 2 diabetes and gastrointestinal disease. Diabetes Obes Metab. 2025;27(2):866‐875. doi: 10.1111/dom.16087
Jiawei Geng, Xixian Ruan, and Xing Wu are co‐first authors.
Contributor Information
Xue Li, Email: xueli157@zju.edu.cn.
Zhongyan Du, Email: duzhongyan@zcmu.edu.cn.
Shuai Yuan, Email: shuai.yuan@ki.se.
DATA AVAILABILITY STATEMENT
All data used in the current study were obtained from publicly released GWAS summary statistics. The SNP of T2D can be extracted from the study Vujkovic M et al. (2020, PMID32541925). The pQTL GWAS summary for deCODE, Fenland and UKB‐PPP study used in the analyses can be downloaded from https://www.decode.com/summarydata/, www.omicscience.org/apps/pgwas and http://ukb-ppp.gwas.eu respectively. Summary‐level data on these outcomes were obtained from the UK Biobank (https://www.leelabsg.org/resources) and FinnGen R9 release (https://www.finngen.fi/fi), and the genome‐wide meta‐analysis data are deposited in https://osf.io/kxehz/?view_only=e24c3bb0a59b4d89aaa226ea86566262. Database for drug and drug target are available at DrugBank (https://go.drugbank.com/), Dependency Map (https://depmap.org/portal/), Connectivity Map (https://repo-hub.broadinstitute.org/repurposing-app) and ChEMBL (https://www.ebi.ac.uk/chembl/).
REFERENCES
- 1. GBD . Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the global burden of disease study 2021. Lancet. 2023;402(10397):203‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang G, Wei J, Liu P, et al. Role of the gut microbiota in type 2 diabetes and related diseases. Metabolism. 2021;117:154712. [DOI] [PubMed] [Google Scholar]
- 3. Du YT, Rayner CK, Jones KL, Talley NJ, Horowitz M. Gastrointestinal symptoms in diabetes: prevalence, assessment, pathogenesis, and management. Diabetes Care. 2018;41(3):627‐637. [DOI] [PubMed] [Google Scholar]
- 4. Chen J, Yuan S, Fu T, et al. Gastrointestinal consequences of type 2 diabetes mellitus and impaired glycemic homeostasis: a mendelian randomization study. Diabetes Care. 2023;46(4):828‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fu T, Sun Y, Lu S, et al. Risk assessment for gastrointestinal diseases via clinical dimension and genome‐wide polygenic risk scores of type 2 diabetes: a population‐based cohort study. Diabetes Care. 2024;47(3):418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yuan S, Larsson SC. Genetically predicted adiposity, diabetes, and lifestyle factors in relation to diverticular disease. Clin Gastroenterol Hepatol. 2022;20(5):1077‐1084. [DOI] [PubMed] [Google Scholar]
- 7. Sun XM, Tan JC, Zhu Y, Lin L. Association between diabetes mellitus and gastroesophageal reflux disease: a meta‐analysis. World J Gastroenterol. 2015;21(10):3085‐3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao JH, Stacey D, Eriksson N, et al. Genetics of circulating inflammatory proteins identifies drivers of immune‐mediated disease risk and therapeutic targets. Nat Immunol. 2023;24(9):1540‐1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5(10):821‐834. [DOI] [PubMed] [Google Scholar]
- 10. Beijer K, Nowak C, Sundström J, Ärnlöv J, Fall T, Lind L. In search of causal pathways in diabetes: a study using proteomics and genotyping data from a cross‐sectional study. Diabetologia. 2019;62:1998‐2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Noordam R, van Heemst D, Suhre K, Krumsiek J, Mook‐Kanamori DO. Proteome‐wide assessment of diabetes mellitus in Qatari identifies IGFBP‐2 as a risk factor already with early glycaemic disturbances. Arch Biochem Biophys. 2020;689:108476. [DOI] [PubMed] [Google Scholar]
- 12. Gummesson A, Björnson E, Fagerberg L, et al. Longitudinal plasma protein profiling of newly diagnosed type 2 diabetes. EBioMedicine. 2021;63:103147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Niu L, Geyer PE, Wewer Albrechtsen NJ, et al. Plasma proteome profiling discovers novel proteins associated with non‐alcoholic fatty liver disease. Mol Syst Biol. 2019;15(3):e8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zenlander R, Fredolini C, Schwenk JM, et al. A wide scan of plasma proteins demonstrates thioredoxin reductase 1 as a potential new diagnostic biomarker for hepatocellular carcinoma. Scand J Gastroenterol. 2023;58(9):998‐1008. [DOI] [PubMed] [Google Scholar]
- 15. Torres J, Petralia F, Sato T, et al. Serum biomarkers identify patients who will develop inflammatory bowel diseases up to 5 years before diagnosis. Gastroenterology. 2020;159(1):96‐104. [DOI] [PubMed] [Google Scholar]
- 16. Bourgonje AR, Hu S, Spekhorst LM, et al. The effect of phenotype and genotype on the plasma proteome in patients with inflammatory bowel disease. J Crohns Colitis. 2022;16(3):414‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanderson E, Glymour MM, Holmes MV, et al. Mendelian randomization. Nat Rev Methods Primers. 2022;2(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomisation (STROBE‐MR): explanation and elaboration. BMJ. 2021;375:n2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elhadad MA, Jonasson C, Huth C, et al. Deciphering the plasma proteome of type 2 diabetes. Diabetes. 2020;69(12):2766‐2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yuan S, Xu F, Li X, et al. Plasma proteins and onset of type 2 diabetes and diabetic complications: proteome‐wide mendelian randomization and colocalization analyses. Cell Rep Med. 2023;4(9):101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen J, Xu F, Ruan X, et al. Therapeutic targets for inflammatory bowel disease: proteome‐wide mendelian randomization and colocalization analyses. EBioMedicine. 2023;89:104494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bourgault J, Abner E, Manikpurage HD, et al. Proteome‐wide mendelian randomization identifies causal links between blood proteins and acute pancreatitis. Gastroenterology. 2023;164(6):953‐965.e953. [DOI] [PubMed] [Google Scholar]
- 23. Sun J, Zhao J, Jiang F, et al. Identification of novel protein biomarkers and drug targets for colorectal cancer by integrating human plasma proteome with genome. Genome Med. 2023;15(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vujkovic M, Keaton JM, Lynch JA, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi‐ancestry meta‐analysis. Nat Genet. 2020;52(7):680‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kamat MA, Blackshaw JA, Young R, et al. PhenoScanner V2: an expanded tool for searching human genotype–phenotype associations. Bioinformatics. 2019;35(22):4851‐4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferkingstad E, Sulem P, Atlason BA, et al. Large‐scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53(12):1712‐1721. [DOI] [PubMed] [Google Scholar]
- 28. Sun BB, Chiou J, Traylor M, et al. Plasma proteomic associations with genetics and health in the UK biobank. Nature. 2023;622(7982):329‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pietzner M, Wheeler E, Carrasco‐Zanini J, et al. Mapping the proteo‐genomic convergence of human diseases. Science. 2021;374(6569):eabj1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yao C, Chen G, Song C, et al. Genome‐wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat Commun. 2018;9(1):3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sudlow C, Gallacher J, Allen N, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well‐phenotyped isolated population. Nature. 2023;613(7944):508‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985‐1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bowden J, Hemani G, Davey SG. Invited commentary: detecting individual and global horizontal pleiotropy in mendelian randomization—a job for the humble heterogeneity statistic? Am J Epidemiol. 2018;187(12):2681‐2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carter AR, Sanderson E, Hammerton G, et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur J Epidemiol. 2021;36(5):465‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074‐D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mendez D, Gaulton A, Bento AP, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47(D1):D930‐D940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Le TN, Nestler JE, Strauss JF 3rd, Wickham EP 3rd. Sex hormone‐binding globulin and type 2 diabetes mellitus. Trends Endocrinol Metab. 2012;23(1):32‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moin ASM, Nandakumar M, Diboun I, et al. Hypoglycemia‐induced changes in complement pathways in type 2 diabetes. Atheroscler Plus. 2021;46:35‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gudmundsdottir V, Zaghlool SB, Emilsson V, et al. Circulating protein signatures and causal candidates for type 2 diabetes. Diabetes. 2020;69(8):1843‐1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhong W, Edfors F, Gummesson A, Bergström G, Fagerberg L, Uhlén M. Next generation plasma proteome profiling to monitor health and disease. Nat Commun. 2021;12(1):2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cronjé HT, Mi MY, Austin TR, et al. Plasma proteomic risk markers of incident type 2 diabetes reflect physiologically distinct components of glucose‐insulin homeostasis. Diabetes. 2023;72(5):666‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Slieker RC, Donnelly LA, Akalestou E, et al. Identification of biomarkers for glycaemic deterioration in type 2 diabetes. Nat Commun. 2023;14(1):2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adewuyi EO, Porter T, O'Brien EK, Olaniru O, Verdile G, Laws SM. Genome‐wide cross‐disease analyses highlight causality and shared biological pathways of type 2 diabetes with gastrointestinal disorders. Commun Biol. 2024;7(1):643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xie Y, Xie W. The role of sulfotransferases in liver diseases. Drug Metab Dispos. 2020;48(9):742‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ferkingstad E, Oddsson A, Gretarsdottir S, et al. Genome‐wide association meta‐analysis yields 20 loci associated with gallstone disease. Nat Commun. 2018;9(1):5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Joshi AD, Andersson C, Buch S, et al. Four susceptibility loci for gallstone disease identified in a meta‐analysis of genome‐wide association studies. Gastroenterology. 2016;151(2):351‐363.e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reshetnyak VI. Concept of the pathogenesis and treatment of cholelithiasis. World J Hepatol. 2012;4(2):18‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Biddinger SB, Haas JT, Yu BB, et al. Hepatic insulin resistance directly promotes formation of cholesterol gallstones. Nat Med. 2008;14(7):778‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Polimanti R, Gelernter J. ADH1B: from alcoholism, natural selection, and cancer to the human phenome. Am J Med Genet B Neuropsychiatr Genet. 2018;177(2):113‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morales LD, Cromack DT, Tripathy D, et al. Further evidence supporting a potential role for ADH1B in obesity. Sci Rep. 2021;11(1):1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Winnier DA, Fourcaudot M, Norton L, et al. Transcriptomic identification of ADH1B as a novel candidate gene for obesity and insulin resistance in human adipose tissue in Mexican Americans from the veterans administration genetic epidemiology study (VAGES). PLoS One. 2015;10(4):e0119941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hoang YTT, Nguyen YT, Vu LT, et al. Association of ADH1B rs1229984, ADH1C rs698, and ALDH2 rs671 with alcohol abuse and alcoholic cirrhosis in people living in Northeast Vietnam. Asian Pac J Cancer Prev. 2023;24(6):2073‐2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ayuso P, García‐Martín E, Cornejo‐García JA, Agúndez JA, Ladero JM. Genetic variants of alcohol metabolizing enzymes and alcohol‐related liver cirrhosis risk. J Personal Med. 2021;11(5):409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yuan S, Larsson SC. An atlas on risk factors for type 2 diabetes: a wide‐angled Mendelian randomisation study. Diabetologia. 2020;63(11):2359‐2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional File 1. Supplementary Tables.
Table S1. Dataset description.
Table S2. Association of type 2 diabetes with 10 gastrointestinal diseases.
Table S3. Association of type 2 diabetes with deCODE plasma protein.
Table S4. Association of type 2 diabetes with UKB‐PPP plasma protein.
Table S5. Association of type 2 diabetes with Fenland plasma protein.
Table S6. Association of putative type 2 diabetes‐associated proteins in deCODE with gastrointestinal diseases.
Table S7. Association of putative type 2 diabetes‐associated proteins in UKB‐PPP with gastrointestinal diseases.
Table S8. Association of putative type 2 diabetes‐associated proteins in Fenland with gastrointestinal diseases.
Table S9. Colocalisation analysis of identified protein‐gastrointestinal pairs in deCODE, UKB‐PPP, and Fenland.
Table S10. Mediation analysis of identified protein between type 2 diabetes and gastrointestinal diseases in deCODE, UKB‐PPP, and Fenland.
Table S11. Appraisal of druggability.
Additional File 2: STROBE‐MR checklist.
Data Availability Statement
All data used in the current study were obtained from publicly released GWAS summary statistics. The SNP of T2D can be extracted from the study Vujkovic M et al. (2020, PMID32541925). The pQTL GWAS summary for deCODE, Fenland and UKB‐PPP study used in the analyses can be downloaded from https://www.decode.com/summarydata/, www.omicscience.org/apps/pgwas and http://ukb-ppp.gwas.eu respectively. Summary‐level data on these outcomes were obtained from the UK Biobank (https://www.leelabsg.org/resources) and FinnGen R9 release (https://www.finngen.fi/fi), and the genome‐wide meta‐analysis data are deposited in https://osf.io/kxehz/?view_only=e24c3bb0a59b4d89aaa226ea86566262. Database for drug and drug target are available at DrugBank (https://go.drugbank.com/), Dependency Map (https://depmap.org/portal/), Connectivity Map (https://repo-hub.broadinstitute.org/repurposing-app) and ChEMBL (https://www.ebi.ac.uk/chembl/).