

Abstract
The brain renin-angiotensin system (RAS) plays an important role in the regulation of autonomic and neuroendocrine functions, and maintains cardiovascular homeostasis. Ang-II is the major effector molecule of RAS and exerts most of its physiological functions, including blood pressure (BP) regulation, via activation of AT1 receptors. Dysregulation of brain RAS in the central nervous system results in increased Ang-II synthesis that causes leading to sympathetic outflow and hypertension.
Brain angiotensin converting enzyme-2 (ACE2) was discovered two decades ago as a RAS component, exhibiting a counter-regulatory role and opposing the adverse cardiovascular effects produced by Ang-II. Studies using synthetic compounds that can sustain the elevation of ACE2 activity or genetically overexpressed ACE2 in specific brain regions found various beneficial effects on cardiovascular function. More recently, ACE2 has been shown to play critical roles in neuro-inflammation, gut dysbiosis and the regulation of stress and anxiety-like behaviors. In the present review we aim to highlight the anatomical locations and functional implication of brain ACE2 related to its BP regulation via modulation of the sympathetic nervous system and discuss the recent developments and future directions in the ACE2-mediated central cardiovascular regulation.
Keywords: Renin-Angiotensin System, ACE2, autonomic regulation
Introduction
The brain renin-angiotensin system (RAS) plays an important role in the regulation of autonomic and neuroendocrine functions, and maintains cardiovascular homeostasis [1]. Several studies have reported the presence of angiotensin (Ang) receptors in the central nervous system (CNS) and the synthesis of Ang-I, Ang-II, Ang-(1–7), Ang-(1–9) peptides and the enzymes that are involved in their formation, such as prorenin (PRR), angiotensin converting enzyme type 1 (ACE), type 2 (ACE2) and chymases [2]. Interestingly, many of the brain regions expressing these components are known to be involved in central cardiovascular regulation, including the subfornical organ (SFO), median preoptic area (MnPO), paraventricular nucleus of the hypothalamus (PVN), rostral ventrolateral medulla (RVLM), nucleus of the solitary tract (NTS) and area postrema (AP) [3]. Alteration or dysregulation of the brain RAS results in cardiovascular and metabolic diseases, such as hypertension, obesity, diabetes and heart failure.
The classical understanding of the brain RAS (Figure 1), follows the same dogma as seen in the circulating hormonal system of RAS, in terms of Ang-II production by enzymatic actions, that catalyze the conversion of angiotensinogen to Ang-I, and subsequent transformation into a biologically active peptide, Ang-II. All RAS components necessary to synthesize Ang-II are localized within the brain [1]. Ang-II is the major effector molecule that exerts its physiological actions via activation of Ang-II type 1 (AT1R) and type 2 (AT2R) receptors, throughout the CNS. The most influential responses observed with Ang-II upon binding to the AT1R include, increased sympathetic activity that facilitates vasoconstriction, increased cardiac contractility, arginine vasopressin release, inflammation, tissue fibrosis, the sensation of thirst and physiological responses to stress (Figure 1). In contrast, the activation of AT2R by Ang-II reverses all these physiological functions (Figure 1). The precise CNS mechanisms that lead to AT2R activation are not clear. However, previous reports suggest that increased circulating Ang-II overstimulates AT2R [4]. In addition, recent studies have highlighted the presence of AT2R on GABAergic neurons [5, 6] and activation of these neurons is thought to produce inhibitory cardiovascular effects. Thus, hyperactivity of RAS, in particular Ang-II/AT1R axis in the brain, is considered to be a potential contributor in the development of neurogenic hypertension. Of note, specific AT1R-blockers or ACE inhibitors are commonly prescribed drugs in the treatment of hypertension and cardiovascular-related diseases.
Figure 1. The Renin-Angiotensin System (RAS) cascade.
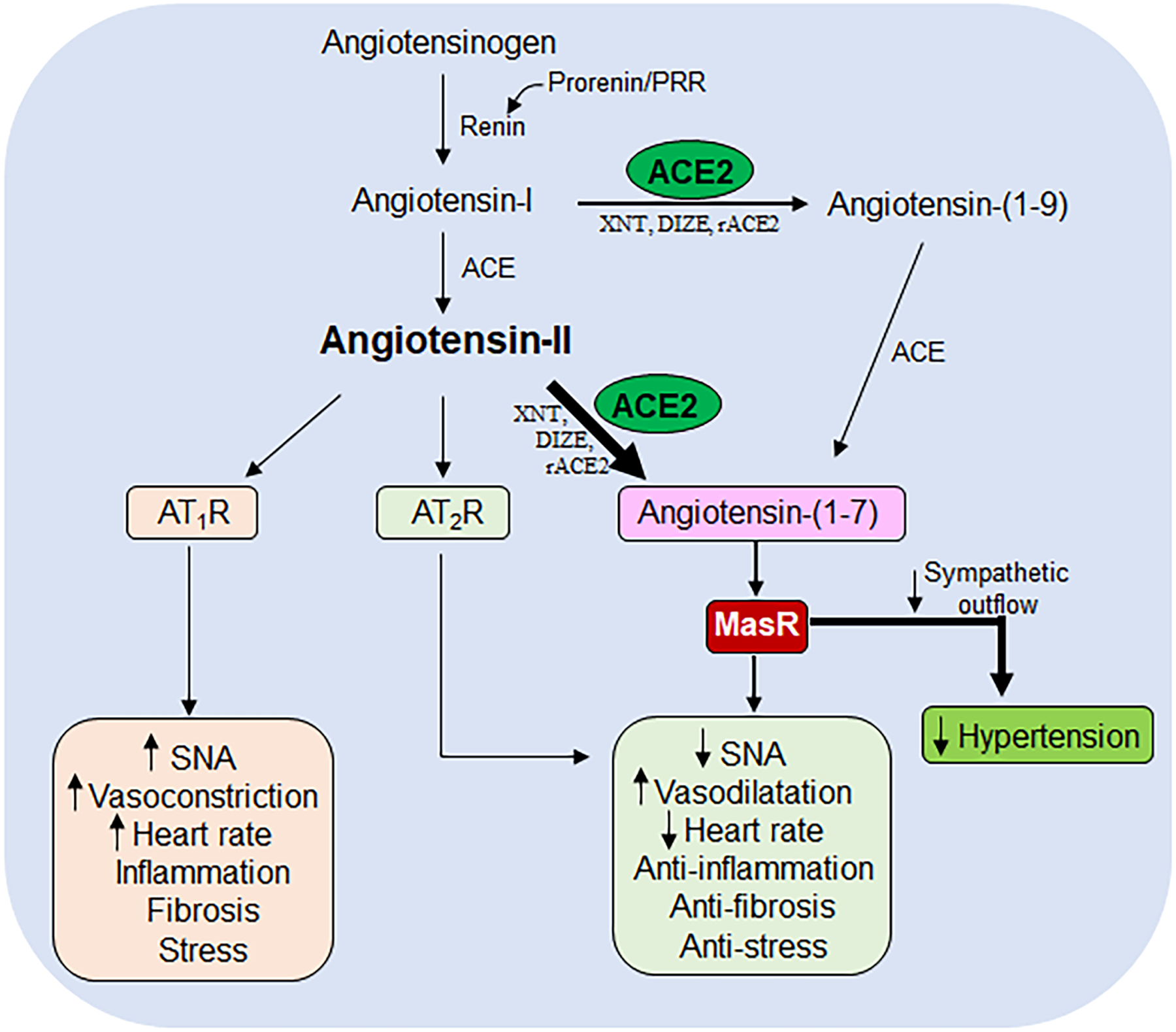
The renin-angiotensin system (RAS) cascade showing the sequence of events taking place during the enzymatic conversion of angiotensinogen to angiotensin-II and angiotensin-(1–7), their target receptors i.e., AT1R, AT2R and MasR, along with their physiological actions. ACE: angiotensin converting enzyme; ACE2: angiotensin converting enzyme-2; XNT: xanthenone; DIZE: diminazene aceturate; rACE2: recombinant angiotensin converting enzyme-2; AT1R: angiotensin II type 1 receptor; AT2R: angiotensin II type 2 receptor; MasR: mas receptor; SNA: sympathetic nerve activity.
ACE2 was discovered at the turn of the millenium as a critical component of RAS [7, 8], which caught immense attention for having a counter-regulatory role on RAS thus opposing the adverse cardiovascular effects produced by Ang-II. ACE2 is a mono-carboxypeptidase and shares 42 % homology within the catalytic domain with ACE [8]. Unlike ACE, ACE2 cleaves Ang-II and produces Ang-(1–7), a vasodilatory peptide that binds specifically to the G-protein-coupled receptor Mas (MasR), which opposes the deleterious cardiovascular effects of Ang-II (Figure 1). Ang-(1–7)-mediated responses include, decreased sympathetic excitation leading to vasodilatation and lowering blood pressure (BP), reduced cardiac contractility, anti-inflammatory and anti-fibrosis effects, reduced physiological response to stress and anxiety (Figure 1). Because of these various beneficial roles associated with ACE2 on cardiovascular function, the ACE2/Ang-(1–7)/MasR axis is repeatedly termed as the protective arm of RAS in cardiovascular regulation.
In the present review, we detail ACE2 anatomical location in specific brain cardiovascular regions and its functional implications. In particular, we aim to highlight ACE2 activity in central cardiovascular regulation and its influence over the sympathetic nervous system that innervate the peripheral vasculature and heart. In addition, we summarize the recent developments related to ACE2 being involved in the brain-immune organs axis, the brain-gut axis, stress and the consequences for cardiovascular regulation.
Consequences of increased brain ACE2 activity
In the CNS, ACE2 acts as a neuromodulator whose activity antagonizes the effects of Ang-II in a paracrine signaling, either by degrading Ang-II into Ang-(1–7) or by decreasing activity of AT1R-expressing neurons [9]. The Ang-(1–7) peptide utilizes MasR as its target receptor in the brain that triggers a vasodilatory response and decrease BP (Figure 1). Recent studies highlighted that the neurons expressing MasR are mostly GABAergic [10, 11] and activation of these neurons produces sympatho-inhibition, which can decrease BP [12]. Others have indicated MasR to be also expressed in glutamatergic neurons and few studies have also reported AT2R involvement in Ang-(1–7)-mediated BP lowering effects [13–15].
Several studies have reported beneficial effects linked to ACE2 activation and synthetic compounds or small peptides that can sustain elevated ACE2 activity which could become potential future therapeutic drugs in cardiovascular diseases. Here we highlight some of these compounds (XNT, DIZE, rACE2) that have been investigated in vivo, and found to produce cardioprotective effects associated with increased ACE2 activity.
Xanthenone (XNT) is a small synthetic compound identified as ACE2 activator. Relatively, very few studies have investigated its beneficial roles in cardiovascular-related diseases, including hypertension [16–20]. Chronic XNT administration resulted in increased ACE2 mRNA expression [16], suggesting that it has additional effects beyond enzymatic activation. When administered in vivo, XNT dose-dependently reduced BP and heart rate [21]. Interestingly, these responses were found to be enhanced in spontaneously hypertensive rats (SHR), with improved cardiac function and reduced fibrosis in myocardial and perivascular tissues [16, 21]; similar to the effects produced by Ang-(1–7). These studies strongly suggested that XNT beneficial effects were due to increased ACE2 activity. In contrast, other groups, while corraborating the positive results, showed no significant difference in ACE2 activity following Ang-II infusions, when comparing XNT vs. vehicle-treated animals; and found no difference in plasma levels of Ang-II and Ang-(1–7) peptides [22]. Moreover, the XNT-induced BP lowering effect was also observed in ACE2 knockout animals, which suggested XNT-induced cardiovascular effects may be ACE2-independent. Further in vivo studies are required to elucidate the role of XNT in cardiovascular regulation.
Diminazene aceturate (DIZE) is an FDA-approved antiparasitic drug, reported in disease models to mediate various organ-protective functions that are attributed to the activation of ACE2 [23–25]. The experimental models include, pulmonary hypertension [25], hypertension [26], ischemic stroke [27], diabetes [28] and glaucoma [29]. Mecca et al., initially demonstrated the neuroprotective role of DIZE in ischemic stroke, in endothelin-1-induced middle cerebral artery occlusion in a rat model [24]. They showed that administration of Ang-(1–7) or DIZE significantly attenuated the stroke-induced infarct size, along with some neurological deficits. This study highlighted the involvement of DIZE in neuroprotection. However, others have also shown the involvement of nitric oxide (NO), NFκB and eNOS-dependent angiogenic pathways that also contribute to these neuroprotective effects [30, 31]. Interestingly, pretreatment with a MasR blocker (A-779) prevented DIZE-induced neuroprotective effects supporting a critical role for ACE2 activity [24].
DIZE was also implicated in cardioprotection, in addition to XNT, in pulmonary hypertension [25]. Raizada’s group has shown that chronic administration of DIZE increased mRNA expression of ACE2 and triggers vasodilatory effects, especially in pulmonary arteries, with improved cardiac function. In contrast, ACE2 inhibitors were found to abolish these effects and suggested DIZE to function via ACE2 activation. On the contrary, other studies found no change in ACE2 gene expression neither in the plasma content of Ang-II and Ang-(1–7) peptide when compared between disease controls vs. DIZE-treated animals [32]. One possibility might be that DIZE functions through AT1R antagonism that may likely contribute to cardioprotection, without affecting the levels of Ang-II or Ang-(1–7). These contradictory results further substantiated when BP lowering effects were observed even in ACE2 knockout mice. Further investigations are needed to clarify these contradictory reports.
Recombinant ACE2 (rACE2) is another approach devised to enhance ACE2 activity by effectively degrading Ang-II and producing Ang-(1–7) [22]. Ang-II is a strong vasoconstrictor that raises BP and contributes to the development of neurogenic hypertension. Studies in hypertension animal models showed impaired ACE2 activity [33–35]. Wysocki et al., showed that human rACE2 treatment increases plasma ACE2 and Ang (1–7) formation with a significant fall in Ang-II content [36]. This resulted in attenuated BP responses to Ang-II infusion and prevented hypertension [22]. Similar observations were reported by Ye et al., using a murine rACE2 [37]. In addition, these authors demonstrated that in their murine rACE2 model, a classic ACE2 inhibitor MLN-4760 significantly blunted all the beneficial cardiovascular changes. Pretreatment with the Ang-(1–7) receptor inhibitor (A779) along with Ang-II and human rACE2 showed no significant effect on BP with an increase in Ang-(1–7) peptide, suggested as a downstream effect of rACE2 after MasR blockade [37]. These studies suggests that rACE2 gene therapy approaches can effectively increase ACE2 activity and target the pathogenesis of hypertension and cardiovascular diseases.
Effects of increased ACE2 activity in the circumventricular organs (CVO)
Although the discovery of ACE2 in peripheral organs, such as heart [7], lungs [38], liver [39], kidney [40], pancreas [41] and gastrointestinal system [8] is well established, the initial identification of ACE2 mRNA expression in the brain was controversial [7, 8, 42]. Due to its low level of expression compared to other organs, very few studies initially highlighted the presence of ACE2 in the brain [43–45]. Our laboratory, was among the first to demonstrate the expression of ACE2 in different brain nuclei that are well-established in central cardiovascular regulation [46], a finding soon confirmed by others [47]. While ACE2-expressing neurons are found in many areas of the brain, the following highlights a functional role of ACE2 in three potential sensory circumventricular organs (CVO), i.e., vascular organ of lamina terminalis (OVLT), subfornical organ (SFO) and area postrema (AP), in central cardiovascular regulation.
There is substantial evidence that shows Ang-II regulate BP and fluid intake behavior through sensory CVO system in the brain [48–50]. The CVO are known to act as an interface that lack a blood brain barrier (BBB) through which small circulating peptides interact freely to their specific receptors and communicate with deep brain structures to mediate cardiovascular and behavioral functions. Since circulating Ang-II does not cross the BBB in normal conditions, much of its physiological functions rely on neurons that express Ang-II receptors in the CVO [51, 52]. A significant amount of AT1R, as well as ACE2, was found in these brain regions [46, 53, 54]. Earlier studies revealed that intracerebroventricular or systemic administration of Ang-II increases BP, releases vasopressin and increases water intake behavior through AT1R-dependent mechanisms [55–59]. Systemic Ang-II infusion was found to increase cFos expression, a neuronal activity marker, in the SFO and OVLT [50], as well as the nearby median preoptic area (MnPO) [60]. Focal microinjections of Ang-II directly into SFO or OVLT replicated the increases in BP and fluid intake behavior that were elicited by peripheral or central administration of Ang-II [48, 61]. In contrast, lesioning of SFO or OVLT impaired these peripherally-administered Ang-II-elicited cardiovascular and behavioral responses [62, 63].
Neuroanatomical studies have shown that SFO and OVLT interact with circulating Ang-II peptides and communicate first with the MnPO which lies inside of the BBB. Neurons in the MnPO send axonal projections to the PVN, RVLM and intermediolateral column (IML) that regulate autonomic, neuroendocrine and behavioral functions. Both excitatory (glutamatergic) and inhibitory (GABAergic) neurons are present in MnPO with AT1R and ACE2 expression, respectively [64]. Our group recently demonstrated that ACE2 is specifically expressed in GABAergic neurons [9, 10] which suggests that activation of ACE2-expressed GABAergic neurons in MnPO could potentially drive the inhibitory tone to downstream autonomic pathways, such as PVN, RVLM & IML (Figure 2), and potentially contribute to the reduction of arterial BP, similar to the results obtained following ACE2 overexpression in the SFO [65]. It was also shown in vitro and in vivo that hyperactivation of AT1R-containing glutamatergic neurons down-regulate ACE2 expression and activity through a sophisticated shedding mechanism [66–68] which tremendously contributes to the increased sympathetic outflow and hormonal release (vasopressin) during the development of neurogenic hypertension. In contrast, genetic deletion of AT1R specifically in the SFO or on glutamatergic PVN neurons showed significant attenuation in the cardiovascular changes [69, 70].
Figure 2. ACE2-mediated inhibitory regulation of PVN neuronal perikarya.
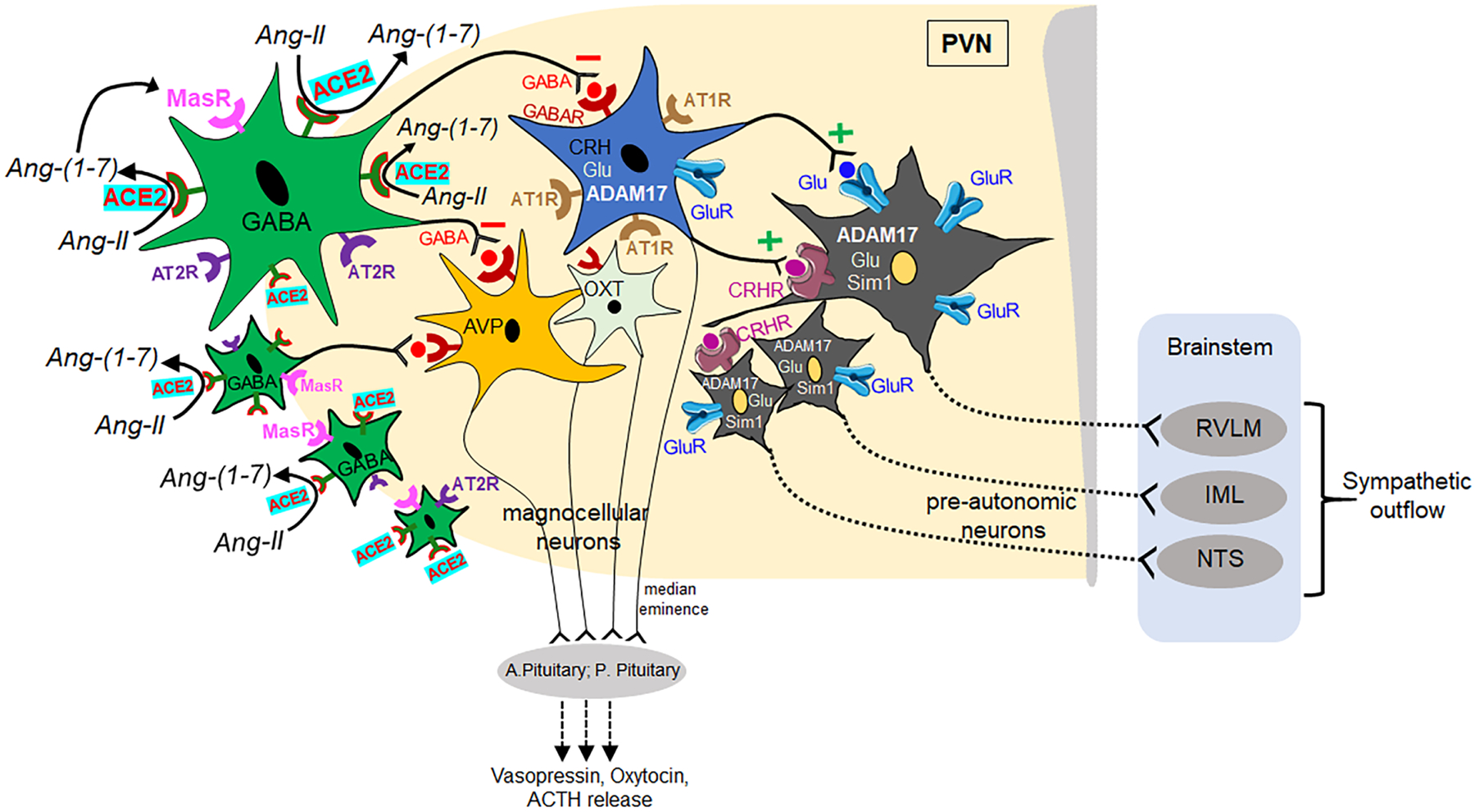
Diagram showing the ACE2-mediated inhibitory signaling in the PVN microcircuits. Stimulation of ACE2 activity in peri-PVN regions converts Ang-II into Ang-(1–7) and enhances GABAergic signaling to the PVN region that inhibits neuronal activity of autonomic and neuroendocrine functions. Upon stimulation, ACE2 blunts the activity of AT1R-containing neurons, which also contain CRH, ADAM17, Glutamate, and decreases the excitatory input to pre-autonomic neurons that project to the brainstem cardiovascular nuclei (RVLM, NTS) and IML, leading to decreased sympathetic outflow. Similarly, ACE2 also directly influences oxytocin and vasopressin neurons in the PVN and modulates their neuroendocrine release in the circulation. ACE2: angiotensin converting enzyme-2; AT1R: angiotensin II type 1 receptor; AT2R: angiotensin II type 2 receptor; CRH: corticotropic releasing hormone; Glu: glutamate; ADAM17: metallopeptidase domain 17; AVP: arginine vasopressin; OXT: oxytocin; CRHR: corticotropic releasing hormone receptor; ACTH: adrenocorticotropic hormone; sim1: single-minded 1; MasR: mas receptor; GABAR: GABA receptor; A. Pituitary: anterior pituitary; P. Pituitary: posterior pituitary; RVLM: rostral ventrolateral medulla; IML: intermediolateral column; NTS: nucleus tractus solitarius. Color coding: GABAergic neurons containing ACE2 (green), MasR (pink), AT2R (violet); Grey colored neurons represent pre-autonomic neurons containing ADAM17, glutamate and Sim1 along with GluR (blue), CRHR (violet).
Similarly, the area postrema (AP) is another CVO in the brainstem which also lacks BBB and contains AT1R that can be activated by circulating Ang-II levels [71]. AP is a complex structure that has long range axonal projections in the CNS, including NTS and RVLM. Our group was one of the first who initially demonstrated ACE2 expression in AP [46]. In spite of earlier studies showing that peripheral administration of Ang-II increases Fos-positive cells in AP [50] and local injection increases BP and reduced heart rate [72–76], suggesting AP is a critical area of the brain for Ang-II-induced hypertension, to date there has been no report of a specific role for ACE2 in this region. Nevertheless, similar to SFO, overexpression of ACE2 in AP could also potentially decrease AT1R-driven excitatory drive to the RVLM that may contribute in lowering sympathetic outflow and arterial BP.
ACE2 activity in the PVN regulates neuroendocrine and autonomic functions
The PVN, is well-known to contain pre-autonomic neurons and regulate cardiovascular and neuroendocrine functions (Figure 2). Activation of PVN neurons increases sympathetic outflow and triggers vasoactive peptides release, such as vasopressin and oxytocin, that causes vasoconstriction and raises arterial BP [6, 77, 78]. Several studies have shown that PVN neurons contain dense expression of AT1R, ACE2, AT2R and MasR [46, 79–82]. Interestingly, each receptor-specific neuronal activation produces distinct cardiovascular responses. For instance, activation of AT1R-containing PVN neurons produces a sympatho-excitatory response that raises arterial BP and heart rate [77, 83]. In contrast, the activation of ACE2 or AT2R-expressing neurons produces sympatho-inhibitory responses that decrease BP and heart rate. It has been postulated that the synergistic balance between these two groups of angiotensinergic PVN neurons dictates BP homeostasis.
Early studies showed that PVN neurons receive monosynaptic inputs from the osmotic-sensitive angiotensinergic neurons from the forebrain CVO, as well as from the MnPO [84]. Administration of Ang-II directly into SFO increased Fos activity in PVN [85], and raised arterial BP. This indicated that PVN integrate sympathetic outflow in response to osmotic balance mainly through the Ang-II receptors [84, 86] and lesioning of PVN neurons prevented this Ang-II elicited BP increase [77, 84, 87, 88].
Interestingly, overexpression of ACE2 in PVN neurons decreases BP and reduces hypertension [80]. This is mostly due to decreased Ang-II levels and increased Ang-(1–7) peptide production by ACE2 in PVN. Mukerjee et al. highlighted that these small population of neurons are ACE2-expressing GABAergic neurons [10], anatomically localized at the boundaries of PVN, also called peri-PVN region, and send axonal projections to the ADAM17- and AT1R- containing glutamatergic neurons within the PVN (Figure 2). Blockade of GABA receptors in PVN elevated sympathetic outflow and increased arterial BP [10, 89]. This strongly suggests that peri-PVN ACE2-expressing neurons tonically inhibits the excitatory drive from PVN and contributes to reducing sympathetic outflow in hypertension. However, it is unclear whether pre-autonomic neurons contain Ang-II receptors, as it was recently suggested by De Kloet et al., that pre-autonomic neurons do not contain AT1R but that AT1R-containing neurons synapse into pre-autonomic neurons within the PVN and regulate sympathetic outflow [82]. In contrast, our group recently demonstrated that Sim1-expressing PVN neurons, which were suggested to have AT1R [90], harbor pre-autonomic neurons, as pretreatment with a ganglionic-blocker impaired the optogenetic-elicited BP increases. Further investigations are needed to clarify these descrepancies in the PVN microcircuits.
Central ACE2, stress and hypothalamus-pituitary-adrenal (HPA) axis
Several brain regions are activated in response to stress, as indicated by increased c-fos expression [91–95]. Many of these brain regions are directly linked to cardiovascular regulation, including medial prefrontral cortex, PVN, dorsomedial hypothalamus, amygdala, pariaqueductal gray, RVLM and NTS (Figure 3). Interestingly, the majority of these brain regions express AT1R [96], and binding of Ang-II to these receptors contributes to the stress-induced activation of sympathetic outflow and HPA axis. Previous studies suggested that stress increases both peripheral and brain Ang-II levels [97, 98], and binding of Ang-II on AT1R during stress can influence cardiovascular variables, i.e., increase arterial BP and heart rate, and vasoactive hormonal release, i.e. CRH, ACTH and vasopressin. Xang et al., demonstrated in conscious rats that under acute and chronic stress challenges there was a significant increase in Ang-II levels in plasma, cardiovascular tissues and autonomic brain regions. Accordingly, these authors suggested Ang-II as a stress hormone [97]. Castren et al., had also demonstrated earlier in rats that stress increases the density of Ang-II binding sites in cardiovascular brain regions and suggested brain Ang-II sites contribute to the regulation of stress responses [99]. In addition to the stress response, Saavedra et al. have also demonstrated that Ang-II receptors play a vital role in anxiety related behavior [91]. Blockade of these receptors, both peripheral and central, by pharmacological administration of AT1R-antagonists, prevented both hormonal and autonomic influences to stress and anxiety [91].
Figure 3. Proposed model for ACE2 role in immune regulation of hypertension.
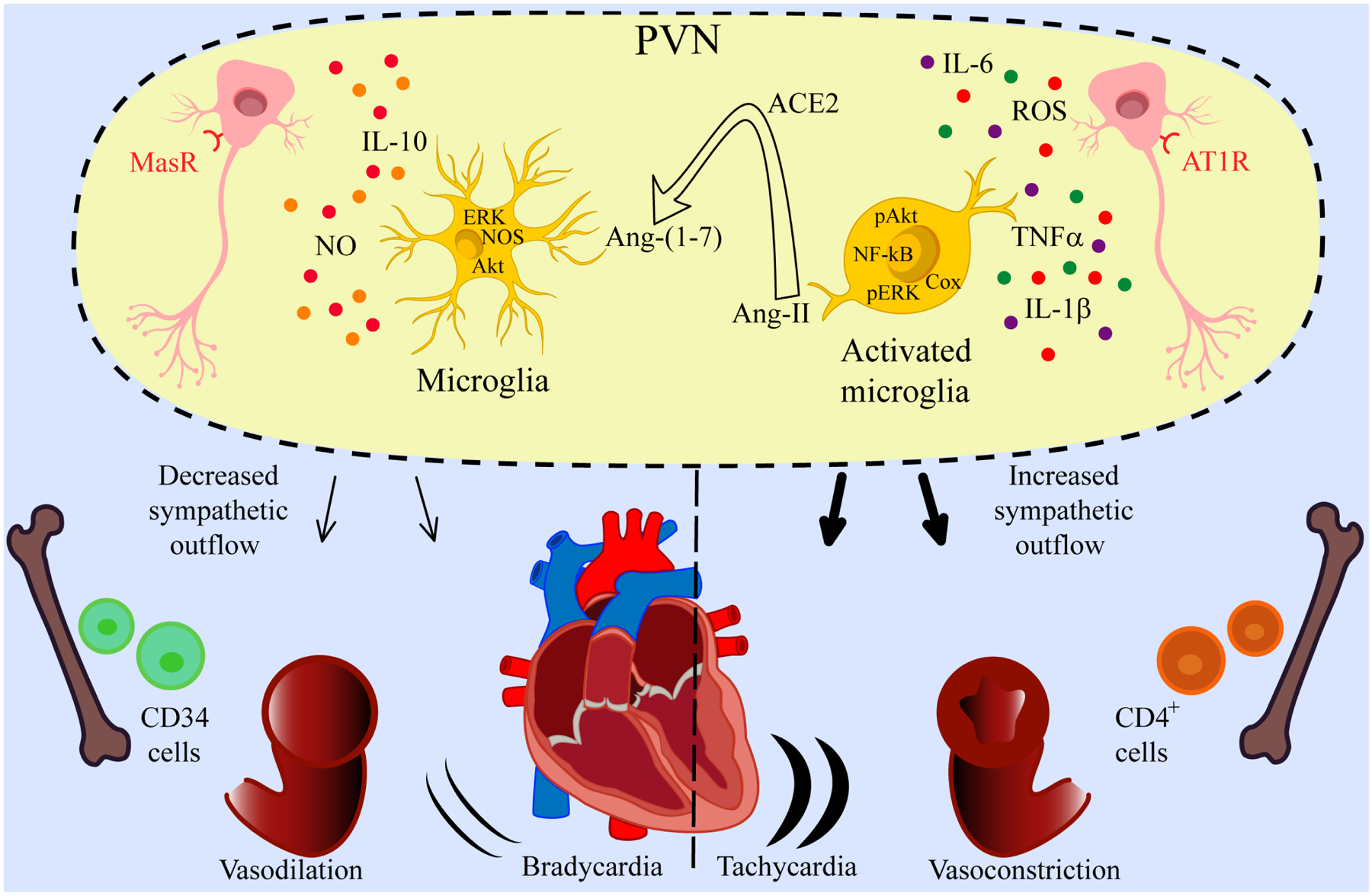
Ang-II-led microglial activation produces pro-inflammatory cytokines that affect neuronal activity in the PVN region and cause heightened sympathetic outflow resulting in vasoconstriction, tachycardia and hypertension. ACE2 converts Ang-II into Ang-(1–7) and decreases inflammation which results in decreased sympathetic outflow, vasodilatation, and decreased heart rate leading to a reduction in neurogenic hypertension. MasR: Mas receptor; PVN: paraventricular nucleus; AT1R: angiotensin II type 1 receptor; IL-1β: interleukin 1 beta; TNFα: tumor necrosis factor alpha; ROS: reactive oxygen species; IL-6: interleukin 6; ERK: extracellular signal-regulated kinases; Akt: protein kinases B; NOS: nitric oxide synthase; COX: cyclooxygenase; NO: nitric oxide; IL-10: interleukin 10.
ACE2 is part of a counter-regulatory arm of the RAS and has recently been highlighted as an experimental therapeutic target for stress-related disorders. ACE2 was found to express in many of the brain regions previously identified as stress-activated regions, which potentially contribute to central cardiovascular regulation [46]. Lima et al., demonstrated that increased ACE2 activity significantly attenuated air-jet-induced stress in conscious animals [20]. The authors suggested a sympatho-inhibitory mechanism for ACE2-mediated attenuation of stress responses, as they observed inhibition of renal sympathetic nerve activity, in vivo. In addition, in vitro studies have shown that pretreatment with Ang-(1–7) impairs norepinephrine release from the sympathetic terminals, and suggested ACE2 to mediate its action via sympatho-inhibition [100]. In contrast, Wang et al. recently overexpressed ACE2 specifically in CRH PVN neurons using the Cre-LoxP system in male mice and highlighted the role of ACE2 in the regulation of HPA axis and anxiety related behavior [101]. They concluded that increased expression of ACE2 in PVN neurons alone significantly dampens stress-induced HPA activations and reduced anxiety-like behavior in male mice. However, when ACE2 ubiquitously overexpressed both in central and peripheral tissues in female mice, a similar observation was found in anxiety-like behavior but without any significant effect on the HPA axis [102]. These new data suggests ACE2 to be a potential therapeutic target for stress related disorders, such as PTSD and hypertension.
Central ACE2 and neuroinflammation
Several physiological and molecular mechanisms underlie the onset and progression of neurogenic hypertension. These include neuroinflammation (the inflammatory response in the CNS) in BP-relevant brain areas as a consequence of resident microglial activation and infiltration of precursors from bone marrow [103], overstated sympathetic outflow, oxidative stress [104–106] and vascular inflammation [107, 108]. Neuroinflammation in hypertension plays a key role as it accounts for the brain increase in inflammatory mediators and cytokines produced by neurons and glia. Several studies showed the presence of inflammation in cardioregulatory brain centers (PVN, RVLM, NTS) in different animals models through glial activation in these regions [109].
Ang-II plays an important role in the development of neuroinflammation in the PVN activating microglia and its production of pro-inflammatory cytokines as wells as a central inflammatory status (Figure 3). Recent work showed that infusion of Ang-II induced early resident microglia activation in the PVN and later an increase in bone marrow sympathetic activity; this event increased the production of IL-17 and CD4+T cells, and the infiltration of inflammatory cells in the PVN exacerbating the neuroinflammation, leading to an increase in BP [110]. Accordingly, several works were carried out to investigate the role of ACE2 as a neuroprotective molecule in the brain. Sharma et al. showed that systemic overexpression of ACE2 protects against neuroinflammation and decrease microglia activation in the PVN [111], possibly through the protective action of Ang-(1–7) on microglia by reducing the release of pro-inflammatory cytokines [112]. Also, Sriramula et al. showed that an overexpresion of ACE2 in the PVN decreased AT1R, ACE and pro-inflammatory cytokines (IL-1β, IL-6, TNFα, MCP-1) mRNA expression, while increasing AT2R and MasR [80]. Later work also showed that ACE2 overexpression increases eNOS and nNOS expression levels while decreasing COX1, COX2, pAkt and pERK1/2 MAPK in the PVN of a DOCA-salt hypertension model [113].
Neurons and glial cells have been shown (in autonomic brain regions) to have metabolic imbalances that cause neuroinflammation and oxidative stress leading to hypertension. Indeed, Ang-II through AT1R is involved in the development of neurogenic hypertension as it can modulate glucose uptake by astrocytes affecting its bioenergetics [114] leading to a malfunction in the role of astrocytes in neuronalglial communication [115, 116]. Ang-II also stimulates the production of reactive oxygen species (ROS) during hypertension and when Ang-II binds to AT1R, it triggers the phosphorylation of ERK1/2 MAPK [117, 118] which stimulate the production of pro-inflammatory cytokines. On the other hand, ACE2 is expressed throughout the brain, including in brain centers which regulate BP [46]. ACE2/Ang-(1–7)/MasR axis, is widely expressed in the brain parenchyma cells and exerts protection by counteracting the ACE/Ang-II/AT1R axis, so the activation of ACE2/Ang-(1–7)/MasR axis has been seen to favor the improvement of neurological pathologies since it acts as a powerful antioxidant and anti-inflammatory effector. Indeed, Abdel-Fattah et al. measured the activity of brain ACE and ACE2 as well as pro-inflammatory markers (caspase-3, MDA, iNOS, TNFα, IL-6 and IL-10) and concluded that there is a threshold level of brain ACE/ACE2 activity beyond which they observed production of oxidative and inflammatory mediators [119]. In addition, our group previously showed that ACE2 overexpression targeted to the SFO [65]or to all neurons [120], led to a decrease in AT1R expression, a reduction of the downstream NF-kB signaling pathway and a decrease in oxidative stress. These results suggest that ACE2 has beneficial properties as reduce oxidative stress and pro-inflammatory cytokines.
In conclusion, the activated neuron-glia increases sympathetic nerve outflow, increases local and general inflammatory status, and alters the vasculature leading to hypertension and neurogenic hypertension. In addition, increased sympathetic nerve outflow to the bone marrow leads to the production of inflammatory cells and pro-inflammatory cytokines which infiltrate the brain and contribute to neuroinflammation. In other words, both resting microglia and BM-derived microglia are activated to release pro-inflammatory cytokines, chemokines, and ROS which will result in established hypertension. Brain ACE2 is thought to down-regulate COX, pERK1/2 MAPK, pAkt, AT1R, ACE, microglia activation as well as pro-inflammatory mediators, while up-regulating NOS, AT2R, MasR and Il-10. In addition, since glial cells are the primary source of pro-inflammatory cytokines in the brain, and ACE2 was shown to reduce pro-inflammatory cytokines in the brain, this may be indicating that ACE2 contributes to microglia cells remaining in a “resting” status.
ACE2 and brain-gut axis
Harmer et al. first reported that the gut has high levels of ACE2 expression [43]. Although ACE2 is a well-established catalytic protein, it plays an important non-catalytic role in the gut. Gut ACE2 is associated in a protein-protein relation with system-B0 Na+-neutral amino acid co-transporter (B°AT1) [121, 122]. So, gut ACE2 is pivotal for the expression, stabilization, intracellular trafficking and functionality maintenance of B°AT1 in the luminal surface of intestinal enterocytes [121, 123], being a key for the regulation in absorption and transport of amino acids (like tryptophan) across enterocytes apical membrane [124]. In addition, gut ACE2 acts as an anti-inflammatory agent by increasing Ang(1–7) [125]. On the other hand, it has been observed that a deficit of ACE2 deregulates the integrity of the intestinal barrier, which leads to a release of the metabolites produced by the gut microbiota in the circulation [126]; impairs the tryptophan homeostasis causing colitis and inflammation [122]; decreases antimicrobial peptide expression which leads to gut dysbiosis; and decreases the intracellular activity of mTOR [122]. Moreover, some bacteria produce ACE and renin inhibitors as well as antioxidant metabolites which can trigger hypertension [127], and may thus also produce ACE2 inhibitors. The gut is connected to, and therefore can influence, the brain via several pathways including: the enteric, sympathetic, parasympathetic nervous systems, and the hypothalamic-pituitary-adrenal axis. Several molecules are implicated in this axis like neurotransmitters, hormones, immunomodulators, and a wide variety of gut microbiome metabolites [128, 129].
Conclusion and Perspective
Although ACE2 was discovered 20 years ago, recent work continues to increase our knowledge of the brain ACE2 in the regulation of sympathetic outflow and vasopressin release related to BP control. It has been shown in number of functional studies that brain ACE2 activity can modulate experimentally-induced heightened sympathetic activity and lower arterial BP, which is now thought to involve GABAergic signalling, in hypertension. Several brain regions are known to contain ACE2 which supports a role for ACE2 activity in a number of autonomic physiological functions. Because of its various cardioprotective beneficial roles, ACE2 is critical within the protective arm of the RAS. Previous work has shown that pharmacological or genetic approaches can enhance the activity of ACE2 for prolonged periods of time associated with beneficial cardiovascular effects which strongly suggested ACE2 to be an potential future therapeutic target for cardiovascular diseases. However, in vivo studies are still needed to resolve confounding results with regards to synthetic compounds.
In addition to cardiovascular regulation, recent data show that ACE2 also potentially contributes in the regulation of HPA axis during stress and anxiety. It is likely that the next decade will unmask additional roles for brain ACE2 in cardiovascular regulation but also other CNS-related conditions.
Sources of Funding
This work was supported in part by research grants from the National Institutes of Health (HL150592 to E.L.) and the Department of Veterans Affairs Merit review Awards (BX004294 to E.L.).
Footnotes
Conflict of Interest/Disclosure
These authors have no conflict of interest to disclose.
Bibliography
- 1.Nakagawa P, et al. , The Renin-Angiotensin System in the Central Nervous System and Its Role in Blood Pressure Regulation. Current hypertension reports, 2020. 22(1): p. 7–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xia H and Lazartigues E, Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem, 2008. 107(6): p. 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapleau MW and Abboud FM, Neuro-cardiovascular regulation: from molecules to man. Introduction. Ann N Y Acad Sci, 2001. 940: p. xiii–xxii. [PubMed] [Google Scholar]
- 4.Saavedra JM, Brain Angiotensin II: New Developments, Unanswered Questions and Therapeutic Opportunities. Cell Mol Neurobiol, 2005. 25(3): p. 485–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Légat L, Smolders IJ, and Dupont AG, Investigation of the Role of AT2 Receptors in the Nucleus Tractus Solitarii of Normotensive Rats in Blood Pressure Control. Front Neurosci, 2019. 13: p. 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohammed M, et al. , Angiotensin AT2 receptors in the solitary tract nucleus lower blood pressure via inhibition of GABA signaling. The FASEB Journal, 2020. 34(S1): p. 1–1. [Google Scholar]
- 7.Donoghue M, et al. , A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res, 2000. 87(5): p. E1–E9. [DOI] [PubMed] [Google Scholar]
- 8.Tipnis SR, et al. , A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem, 2000. 275 (43): p. 33238–33243. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, et al. , Activation of ADAM17 (A Disintegrin and Metalloprotease 17) on Glutamatergic Neurons Selectively Promotes Sympathoexcitation. Hypertension, 2019. 73(6): p. 1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukerjee S, et al. , ACE2 and ADAM17 Interaction Regulates the Activity of Presympathetic Neurons. Hypertension, 2019. 74(5): p. 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, et al. , Increasing brain angiotensin converting enzyme 2 activity decreases anxiety-like behavior in male mice by activating central Mas receptors. Neuropharmacology, 2016. 105: p. 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh Y, Singh K, and Sharma PL, Effect of combination of renin inhibitor and Mas-receptor agonist in DOCA-salt-induced hypertension in rats. Mol Cell Biochem, 2013. 373(1–2): p. 189–94. [DOI] [PubMed] [Google Scholar]
- 13.Rowe BP, et al. , Angiotensin-(1–7) binding at angiotensin II receptors in the rat brain. Regul Pept, 1995. 56(2–3): p. 139–46. [DOI] [PubMed] [Google Scholar]
- 14.de Kloet AD, Steckelings UM, and Sumners C, Protective Angiotensin Type 2 Receptors in the Brain and Hypertension. Curr Hypertens Rep, 2017. 19(6): p. 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sumners C, et al. , Anti-fibrotic mechanisms of angiotensin AT(2) -receptor stimulation. Acta Physiol (Oxf), 2019. 227(1): p. e13280. [DOI] [PubMed] [Google Scholar]
- 16.Ferreira AJ, et al. , Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med, 2009. 179(11): p. 1048–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferreira AJ, et al. , Angiotensin-converting enzyme 2 activation protects against hypertension-induced cardiac fibrosis involving extracellular signal- regulated kinases. Exp Physiol, 2011. 96(3): p. 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh N, et al. , ACE2/Ang-(1–7)/Mas axis stimulates vascular repair-relevant functions of CD34+ cells. Am J Physiol Heart Circ Physiol, 2015. 309(10): p. H1697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murça TM, et al. , Oral administration of an angiotensin-converting enzyme 2 activator ameliorates diabetes-induced cardiac dysfunction. Regul Pept, 2012. 177(1–3): p. 107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martins Lima A, et al. , Activation of angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas axis attenuates the cardiac reactivity to acute emotional stress. Am J Physiol Heart Circ Physiol, 2013. 305(7): p. H1057–67. [DOI] [PubMed] [Google Scholar]
- 21.Hernández Prada JA, et al. , Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension, 2008. 51(5): p. 1312–7. [DOI] [PubMed] [Google Scholar]
- 22.Haber PK, et al. , Angiotensin-converting enzyme 2-independent action of presumed angiotensin-converting enzyme 2 activators: studies in vivo, ex vivo, and in vitro. Hypertension, 2014. 63(4): p. 774–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulemina LV and Ostrov DA, Prediction of off-target effects on angiotensin-converting enzyme 2. J Biomol Screen, 2011. 16(8): p. 878–85. [DOI] [PubMed] [Google Scholar]
- 24.Mecca AP, et al. , Cerebroprotection by angiotensin-(1–7) in endothelin-1-induced ischaemic stroke. Exp Physiol, 2011. 96(10): p. 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shenoy V, et al. , Diminazene Attenuates Pulmonary Hypertension and Improves Angiogenic Progenitor Cell Functions in Experimental Models. Am J Respir Crit Care Med, 2013. 187(6): p. 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marilda LADM, et al. , Anti-hypertensive Effects of Diminazene Aceturate: An Angiotensin- Converting Enzyme 2 Activator in Rats. Protein Pept Lett, 2016. 23(1): p. 9–16. [DOI] [PubMed] [Google Scholar]
- 27.Der Sarkissian S, et al. , ACE2: A novel therapeutic target for cardiovascular diseases. Prog Biophys Mol Bio, 2005. 91(1–2): p. 163–198. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, et al. , Upregulation of Angiotensin (1–7)-Mediated Signaling Preserves Endothelial Function Through Reducing Oxidative Stress in Diabetes. Antioxid Redox Signal, 2015. 23(11): p. 880–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foureaux G, et al. , Antiglaucomatous effects of the activation of intrinsic Angiotensin-converting enzyme 2. Invest Ophthalmol Vis Sci, 2013. 54(6): p. 4296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang T, et al. , Suppressing inflammation by inhibiting the NF-κB pathway contributes to the neuroprotective effect of angiotensin-(1–7) in rats with permanent cerebral ischaemia. Br J Pharmacol, 2012. 167(7): p. 1520–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang F, et al. , Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat Rev Cardiol, 2014. 11(7): p. 413–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajapaksha IG, et al. , The small molecule drug diminazene aceturate inhibits liver injury and biliary fibrosis in mice. Sci Rep, 2018. 8(1): p. 10175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crackower MA, et al. , Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature, 2002. 417(6891): p. 822–828. [DOI] [PubMed] [Google Scholar]
- 34.Yamazato Y, et al. , Prevention of Pulmonary Hypertension by Angiotensin- Converting Enzyme 2 Gene Transfer. Hypertension, 2009. 54(2): p. 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia H, et al. , Angiotensin II Type 1 Receptor-Mediated Reduction of Angiotensin-Converting Enzyme 2 Activity in the Brain Impairs Baroreflex Function in Hypertensive Mice. Hypertension, 2009. 53: p. 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wysocki J, et al. , Targeting the Degradation of Angiotensin II With Recombinant Angiotensin-Converting Enzyme 2: Prevention of Angiotensin II-Dependent Hypertension. Hypertension, 2010. 55(1): p. 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye M, et al. , Murine Recombinant Angiotensin-Converting Enzyme 2: Effect on Angiotensin II-Dependent Hypertension and Distinctive Angiotensin-Converting Enzyme 2 Inhibitor Characteristics on Rodent and Human Angiotensin-Converting Enzyme 2. Hypertension, 2012. 60(3): p. 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gembardt F, et al. , Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides, 2005. 26 (7): p. 1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paizis G, et al. , Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. 2005. 54 (12): p. 1790–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye M, et al. , Increased ACE2 and decreased ACE protein in renal tubules from diabetic mice: a renoprotective combination? Hypertension, 2004. 43(5): p. 1120–1125. [DOI] [PubMed] [Google Scholar]
- 41.Fang HJ and Yang JK, Tissue-specific pattern of angiotensin-converting enzyme 2 expression in rat pancreas. J Int Med Res, 2010. 38(2): p. 558–569. [DOI] [PubMed] [Google Scholar]
- 42.Harmer D, et al. , Quantitative mRNA expression profiling of ACE2, a novel homologue of angiotensin converting enzyme. FEBS lett, 2002. 203: p. 631–637. [DOI] [PubMed] [Google Scholar]
- 43.Harmer D, et al. , Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett, 2002. 532 (1–2): p. 107–110. [DOI] [PubMed] [Google Scholar]
- 44.Gallagher PE, et al. , ACE2 expression in brain: angiotensin II down- regulates ACE2 in astrocytes. Abstract presented at the 57th Annual Fall Conference and Scientific sessions of the Council for High Blood Pressure Research, 2003. [Google Scholar]
- 45.Sakima A, et al. , Impaired heart rate baroreflex in older rats: role of endogenous angiotensin-(1–7) at the nucleus tractus solitarii. Hypertension, 2005. 46(2): p. 333–40. [DOI] [PubMed] [Google Scholar]
- 46.Doobay MF, et al. , Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol - Regul Integr Comp Physiol, 2007. 292(1): p. R373–R381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin Z, et al. , RNA interference shows interactions between mouse brainstem angiotensin AT1 receptors and angiotensin-converting enzyme 2. Exp Physiol, 2008. 93(5): p. 676–84. [DOI] [PubMed] [Google Scholar]
- 48.Simpson JB and Routtenberg A, Subfornical organ: site of drinking elicitation by angiotensin II. Science, 1973. 181(4105): p. 1172–5. [DOI] [PubMed] [Google Scholar]
- 49.Simpson JB, The circumventricular organs and the central actions of angiotensin. Neuroendocrinology, 1981. 32(4): p. 248–256. [DOI] [PubMed] [Google Scholar]
- 50.Badoer E and McKinlay D, Effect of intravenous angiotensin II on Fos distribution and drinking behavior in rabbits. Am J Physiol, 1997. 272(5 Pt 2): p. R1515–24. [DOI] [PubMed] [Google Scholar]
- 51.Volicer L and Loew CG, Penetration of angiotensin II into the brain. Neuropharmacology, 1971. 10(5): p. 631–6. [DOI] [PubMed] [Google Scholar]
- 52.Schelling P, et al. , Impermeability of the blood-cerebrospinal fluid barrier for angiotensin II in rats. Clin Sci Mol Med Suppl, 1976. 3: p. 399s–402s. [DOI] [PubMed] [Google Scholar]
- 53.Sinnayah P, et al. , Genetic Ablation of Angiotensinogen in the Subfornical Organ of the Brain Prevents the Central Angiotensinergic Pressor Response. Circ Res, 2006. 99(10): p. 1125–1131. [DOI] [PubMed] [Google Scholar]
- 54.Sakai K, et al. , Local production of angiotensin II in the subfornical organ causes elevated drinking. J Clin Invest, 2007. 117(4): p. 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shoji M, et al. , Role of intracerebral angiotensin receptors in the regulation of vasopressin release and the cardiovascular system. Neuroendocrinology, 1986. 43(2): p. 239–44. [DOI] [PubMed] [Google Scholar]
- 56.Rohmeiss P, et al. , NaCl injections in brain induce natriuresis and blood pressure responses sensitive to ANG II AT1 receptors. Am J Physiol, 1995. 269(2 Pt 2): p. F282–8. [DOI] [PubMed] [Google Scholar]
- 57.Rohmeiss P, et al. , Osmotically induced natriuresis and blood pressure response involves angiotensin AT1 receptors in the subfornical organ. J Hypertens, 1995. 13(12 Pt 1): p. 1399–404. [PubMed] [Google Scholar]
- 58.Nakano-Tateno T, et al. , Prolonged effects of intracerebroventricular angiotensin II on drinking, eating and locomotor behavior in mice. Regul Pept, 2012. 173(1–3): p. 86–92. [DOI] [PubMed] [Google Scholar]
- 59.Campbell DJ, Do intravenous and subcutaneous angiotensin II increase blood pressure by different mechanisms? Clin Exp Pharmacol Physiol, 2013. 40(8): p. 560–70. [DOI] [PubMed] [Google Scholar]
- 60.Bealer SL, Metcalf CS, and Heyborne R, Increased dietary sodium alters Fos expression in the lamina terminalis during intravenous angiotensin II infusion. Exp Neurol, 2007. 204(1): p. 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mangiapane ML and Simpson JB, Subfornical organ: forebrain site of pressor and dipsogenic action of angiotensin II. 1980. 239(5): p. R382–R389. [DOI] [PubMed] [Google Scholar]
- 62.Hendel MD and Collister JP, Contribution of the subfornical organ to angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol, 2005. 288(2): p. H680–5. [DOI] [PubMed] [Google Scholar]
- 63.Collister JP, et al. , Lesion of the OVLT markedly attenuates chronic DOCA-salt hypertension in rats. Am J Physiol Regul Integr Comp Physiol, 2018. 315(3): p. R568–r575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McKinley MJ, et al. , The median preoptic nucleus: front and centre for the regulation of body fluid, sodium, temperature, sleep and cardiovascular homeostasis. Acta Physiol (Oxf), 2015. 214(1): p. 8–32. [DOI] [PubMed] [Google Scholar]
- 65.Feng Y, et al. , Angiotensin-Converting Enzyme 2 Overexpression in the Subfornical Organ Prevents the Angiotensin II-Mediated Pressor and Drinking Responses and Is Associated With Angiotensin II Type 1 Receptor Downregulation. Circ Res, 2008. 102(6): p. 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Queiroz TM, et al. , α-Lipoic acid reduces neurogenic hypertension by blunting oxidative stress-mediated increase in ADAM17. Am J Physiol Heart Circ Physiol, 2015. 309(5): p. H926–H934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xia H, et al. , Brain ACE2 Shedding Contributes to the Development of Neurogenic Hypertension. Circ Res, 2013. 113: p. 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu J, et al. , AT1 Receptor on Glutamatergic Neurons Regulate Autonomic Function Through Modulation of Neuronal Excitability and Sympathetic Outflow. Hypertension, 2017. 70: p. A088. [Google Scholar]
- 69.Hilzendeger AM, et al. , Angiotensin Type 1a Receptors in the Subfornical Organ Are Required for Deoxycorticosterone Acetate-Salt Hypertension. Hypertension, 2013. 61(3): p. 716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu J, et al. , Clinical Relevance and Role of Neuronal AT1 Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ Res, 2017. 121(1): p. 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Joy MD and Lowe RD, Evidence that the area postrema mediates the central cardiovascular response to angiotensin II. Nature, 1970. 228(5278): p. 1303–4. [DOI] [PubMed] [Google Scholar]
- 72.Casto R and Phillips MI, Cardiovascular actions of microinjections of angiotensin II in the brain stem of rats. Am J Physiol, 1984. 246(5 Pt 2): p. R811–6. [DOI] [PubMed] [Google Scholar]
- 73.Joy MD and Lowe RD, Abolition of the central cardiovascular effects of angiotensin by ablation of the area postrema. Clin Sci, 1970. 39(1): p. 3p. [DOI] [PubMed] [Google Scholar]
- 74.Ferrario CM, Gildenberg PL, and McCubbin JW, Cardiovascular effects of angiotensin mediated by the central nervous system. Circ Res, 1972. 30(3): p. 257–62. [DOI] [PubMed] [Google Scholar]
- 75.Barnes KL and Ferrario CM, Location of the area postrema pressor pathway in the dog brain stem. Hypertension, 1984. 6(4): p. 482–8. [DOI] [PubMed] [Google Scholar]
- 76.Fink GD, Bruner CA, and Mangiapane ML, Area postrema is critical for angiotensin-induced hypertension in rats. Hypertension, 1987. 9: p. 355–361. [DOI] [PubMed] [Google Scholar]
- 77.Basting T, et al. , Glutamatergic neurons of the paraventricular nucleus are critical contributors to the development of neurogenic hypertension. The Journal of physiology, 2018. 596(24): p. 6235–6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yosten GL and Samson WK, Neural circuitry underlying the central hypertensive action of nesfatin-1: melanocortins, corticotropin-releasing hormone, and oxytocin. Am J Physiol Regul Integr Comp Physiol, 2014. 306(10): p. R722–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Becker LK, et al. , Immunofluorescence localization of the receptor Mas in cardiovascular-related areas of the rat brain. Am J Physiol Heart Circ Physiol, 2007. 293(3): p. H1416–1424. [DOI] [PubMed] [Google Scholar]
- 80.Sriramula S, et al. , ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II-induced hypertension. Cardiovasc Res, 2011. 92(3): p. 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Kloet AD, et al. , Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system. Brain Struct Funct, 2016. 221(2): p. 891–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Kloet AD, et al. , A unique ‘angiotensin sensitive’ neuronal population coordinates neuroendocrine, cardiovascular and behavioral responses to stress. J Neurosci, 2017. 37(13): p. 3478–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cato MJ and Toney GM, Angiotensin II excites paraventricular nucleus neurons that innervate the rostral ventrolateral medulla: an in vitro patch-clamp study in brain slices. Journal of neurophysiology, 2005. 93(1): p. 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen QH and Toney GM, AT(1)-receptor blockade in the hypothalamic PVN reduces central hyperosmolality-induced renal sympathoexcitation. Am J Physiol Regul Integr Comp Physiol, 2001. 281(6): p. R1844–53. [DOI] [PubMed] [Google Scholar]
- 85.Saxena A, et al. , Angiotensin II type 1a receptors in subfornical organ contribute towards chronic intermittent hypoxia-associated sustained increase in mean arterial pressure. Am J Physiol Heart Circ Physiol, 2015. 308(5): p. H435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ramchandra R, et al. , Central angiotensin type 1 receptor blockade decreases cardiac but not renal sympathetic nerve activity in heart failure. Hypertension, 2012. 59(3): p. 634–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.LaGrange LP, Toney GM, and Bishop VS, Effect of intravenous angiotensin II infusion on responses to hypothalamic PVN injection of bicuculline. Hypertension, 2003. 42(6): p. 1124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li DP and Pan HL, Glutamatergic inputs in the hypothalamic paraventricular nucleus maintain sympathetic vasomotor tone in hypertension. Hypertension, 2007. 49(4): p. 916–25. [DOI] [PubMed] [Google Scholar]
- 89.Martin DS, Segura T, and Haywood JR, Cardiovascular responses to bicuculline in the paraventricular nucleus of the rat. Hypertension, 1991. 18(1): p. 48–55. [DOI] [PubMed] [Google Scholar]
- 90.de Kloet AD, et al. , Angiotensin Type 1a Receptors in the Paraventricular Nucleus of the Hypothalamus Protect against Diet-Induced Obesity. J Neurosci, 2013. 33(11): p. 4825–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saavedra JM, et al. , Anti-stress and anti-anxiety effects of centrally acting angiotensin II AT1 receptor antagonists. Regul Pept, 2005. 128(3): p. 227–38. [DOI] [PubMed] [Google Scholar]
- 92.Kataoka N, et al. , Psychological stress activates a dorsomedial hypothalamus-medullary raphe circuit driving brown adipose tissue thermogenesis and hyperthermia. Cell Metab, 2014. 20(2): p. 346–58. [DOI] [PubMed] [Google Scholar]
- 93.Lin X, et al. , c-Fos mapping of brain regions activated by multi-modal and electric foot shock stress. Neurobiol Stress, 2018. 8: p. 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kovács L, et al. , Both Basal and Acute Restraint Stress-Induced c-Fos Expression Is Influenced by Age in the Extended Amygdala and Brainstem Stress Centers in Male Rats. Front Aging Neurosci, 2018. 10: p. 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Numa C, et al. , Social defeat stress-specific increase in c-Fos expression in the extended amygdala in mice: Involvement of dopamine D1 receptor in the medial prefrontal cortex. Sci Rep, 2019. 9(1): p. 16670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sumners C, et al. , Brain angiotensin type-1 and type-2 receptors: cellular locations under normal and hypertensive conditions. Hypertens Res, 2020. 43(4): p. 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang G, et al. , Changes in circulating and tissue angiotensin II during acute and chronic stress. Biol Signals, 1993. 2(3): p. 166–72. [DOI] [PubMed] [Google Scholar]
- 98.Yang G, Wan Y, and Zhu Y, Angiotensin II--an important stress hormone. Biol Signals, 1996. 5(1): p. 1–8. [DOI] [PubMed] [Google Scholar]
- 99.Castren E and Saavedra JM, Repeated stress increases the density of angiotensin II binding sites in rat paraventricular nucleus and subfornical organ. Endocrinology, 1988. 122: p. 370–372. [DOI] [PubMed] [Google Scholar]
- 100.Gironacci MM, et al. , Neuromodulatory role of angiotensin-(1–7) in the central nervous system. Clin Sci (Lond), 2013. 125(2): p. 57–65. [DOI] [PubMed] [Google Scholar]
- 101.Wang LA, et al. , Coupling corticotropin-releasing-hormone and angiotensin converting enzyme 2 dampens stress responsiveness in male mice. Neuropharmacology, 2018. 133: p. 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Kloet AD, et al. , Overexpression of angiotensin converting enzyme 2 reduces anxiety-like behavior in female mice. Physiol Behav, 2020. 224: p. 113002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li P, et al. , Angiotensin-(1–7) enhances the effects of angiotensin II on the cardiac sympathetic afferent reflex and sympathetic activity in rostral ventrolateral medulla in renovascular hypertensive rats. J Am Soc Hypertens, 2015. 9(11): p. 865–77. [DOI] [PubMed] [Google Scholar]
- 104.Zimmerman MC and Zucker IH, Mitochondrial dysfunction and mitochondrial-produced reactive oxygen species: new targets for neurogenic hypertension? Hypertension, 2009. 53(2): p. 112–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu KL, Chan SH, and Chan JY, Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation, 2012. 9(1): p. 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Peterson J, Sharma R, and Davisson R, Reactive oxygen species in the neuropathogenesis of hypertension. Current Hypertension Reports, 2006. 8(3): p. 232–241. [DOI] [PubMed] [Google Scholar]
- 107.Zubcevic J, et al. , Autonomic-immune-vascular interaction: an emerging concept for neurogenic hypertension. Hypertension, 2011. 57(6): p. 1026–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Waki H, et al. , Contributions of vascular inflammation in the brainstem for neurogenic hypertension. Respir Physiol Neurobiol, 2011. 178(3): p. 422–8. [DOI] [PubMed] [Google Scholar]
- 109.Donertas Ayaz B and Zubcevic J, Gut microbiota and neuroinflammation in pathogenesis of hypertension: A potential role for hydrogen sulfide. Pharmacol Res, 2020. 153: p. 104677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ahmari N, et al. , Elevated bone marrow sympathetic drive precedes systemic inflammation in angiotensin II hypertension. American Journal of Physiology-Heart and Circulatory Physiology, 2019. 317(2): p. H279–H289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sharma RK, et al. , Microglial Cells Impact Gut Microbiota and Gut Pathology in Angiotensin II-Induced Hypertension. Circ Res, 2019. 124(5): p. 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu M, Shi P, and Sumners C, Direct anti-inflammatory effects of angiotensin-(1–7) on microglia. J Neurochem, 2016. 136(1): p. 163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sriramula S, et al. , Brain-Targeted Angiotensin-Converting Enzyme 2 Overexpression Attenuates Neurogenic Hypertension by Inhibiting Cyclooxygenase-Mediated Inflammation. Hypertension, 2015. 65(3): p. 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tang W, et al. , Angiotensin II increases glucose uptake and glucose transporter-1 mRNA levels in astroglia. Am J Physiol, 1995. 268(3 Pt 1): p. E384–90. [DOI] [PubMed] [Google Scholar]
- 115.Stern JE, et al. , Astrocytes Contribute to Angiotensin II Stimulation of Hypothalamic Neuronal Activity and Sympathetic Outflow. Hypertension, 2016. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim KJ, et al. , Astrocyte contributions to flow/pressure-evoked parenchymal arteriole vasoconstriction. J Neurosci, 2015. 35(21): p. 8245–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mehta PK and Griendling KK, Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol, 2007. 292(1): p. C82–97. [DOI] [PubMed] [Google Scholar]
- 118.Xiao L, Haack KKV, and Zucker IH, Angiotensin II regulates ACE and ACE2 in neurons through p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling. Am J Physiol Cell Physiol, 2013. 304(11): p. C1073–C1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Abdel-Fattah MM, Messiha BAS, and Mansour AM, Modulation of brain ACE and ACE2 may be a promising protective strategy against cerebral ischemia/reperfusion injury: an experimental trial in rats. Naunyn Schmiedebergs Arch Pharmacol, 2018. 391(9): p. 1003–1020. [DOI] [PubMed] [Google Scholar]
- 120.Feng Y, et al. , Brain-Selective Overexpression of Human Angiotensin-Converting Enzyme Type 2 Attenuates Neurogenic Hypertension. Circ Res, 2010. 106(2): p. 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Camargo SM, et al. , Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology, 2009. 136(3): p. 872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hashimoto T, et al. , ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature, 2012. 487(7408): p. 477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kowalczuk S, et al. , A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J, 2008. 22(8): p. 2880–7. [DOI] [PubMed] [Google Scholar]
- 124.Broer S, Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev, 2008. 88(1): p. 249–86. [DOI] [PubMed] [Google Scholar]
- 125.Khajah MA, et al. , Anti-Inflammatory Action of Angiotensin 1–7 in Experimental Colitis. PLoS One, 2016. 11(3): p. e0150861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Duan Y, et al. , Bone Marrow-Derived Cells Restore Functional Integrity of the Gut Epithelial and Vascular Barriers in a Model of Diabetes and ACE2 Deficiency. Circ Res, 2019. 125(11): p. 969–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dave LA, et al. , Human gut endogenous proteins as a potential source of angiotensin-I-converting enzyme (ACE-I)-, renin inhibitory and antioxidant peptides. Peptides, 2016. 76: p. 30–44. [DOI] [PubMed] [Google Scholar]
- 128.Carabotti M, et al. , The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol, 2015. 28(2): p. 203–209. [PMC free article] [PubMed] [Google Scholar]
- 129.Holzer P and Farzi A, Neuropeptides and the microbiota-gut-brain axis. Adv Exp Med Biol, 2014. 817: p. 195–219. [DOI] [PMC free article] [PubMed] [Google Scholar]