

Abstract
Amyloidosis is a rare, generally multisystem disease that can also involve the heart. Infiltration of the myocardium with amyloid proteins is an important and underappreciated cause of heart failure with preserved ejection fraction in the elderly. We present the case of an 84-year-old man with chest tightness, dyspnoea, and ascites. He had a history of dyslipidaemia and ischaemic heart disease. Initial investigations showed severe diastolic dysfunction and elevated pulmonary artery systolic pressure on echocardiogram along with elevated serum natriuretic peptides. Further evaluation by a magnetic resonance imaging scan of the heart and endomyocardial biopsy confirmed the diagnosis of senile systemic amyloidosis. He made good progress after treatment with conventional heart failure drugs and is currently under consideration to start on specific medications to slow down the progression of amyloidosis. This case aims to increase clinicians’ awareness of senile amyloidosis as a cause of heart failure in the elderly.
Keywords: Diastolic dysfunction, Elderly, Heart failure, Infiltrative cardiomyopathy, Senile amyloidosis
1. Learning points
Cardiac amyloidosis is an underappreciated cause of heart failure in the elderly.
Evidence of ventricular thickening on echocardiography without a clear history of hypertension should point towards infiltrative cardiomyopathy.
Troponin and N-terminal pro-B-type natriuretic peptide elevation has a prognostic value in cardiac amyloidosis.
Cardiac magnetic resonance imaging can demonstrate pathognomonic late gadolinium enhancement suggestive of amyloid heart disease.
New treatments can slow down the progression of disease.
2. Introduction
Amyloidosis is a rare condition, characterised by infiltration of insoluble misfolded proteins in the extracellular matrix of the affected tissues. Cardiac involvement is the most important factor influencing the prognosis in the multisystem disease. The condition remains largely underdiagnosed, particularly in the elderly population where most of the literature on its prevalence and progression comes from the postmortem studies [1]. With the advent of new and specific treatment options and ongoing clinical trials, it is important to raise awareness amongst clinicians about this important disease entity [2].
3. Case report
An 84-year-old man was seen in the cardiology outpatient clinic with chest tightness, exertional dyspnoea, and abdominal distension. His past medical history included dyslipidaemia and myocardial infarction 5 years ago, for which he had percutaneous coronary intervention and stent insertion to the left circumflex artery. He was an ex-smoker and did not drink alcohol. His usual medications included aspirin, bisoprolol, ramipril, and atorvastatin.
On examination, his heart rate was 60 beats/min and his blood pressure was 126/84 mmHg. He had mild pedal oedema and elevated jugular venous pressure. Heart sounds were normal and chest was clear to auscultation. Abdominal examination revealed a palpable liver edge and moderate ascites. An electrocardiogram showed the sinus rhythm PR interval prolonged to 240 ms (normal range 120–200 ms) and no other abnormality. Routine blood tests revealed normal full blood count, renal, and thyroid function tests. Serum glucose was 5.7 mmol/L and haemoglobin A1c was 31 mmol/ mol. Liver function tests showed normal levels of bilirubin, albumin, and transaminases; alkaline phosphatase and gamma-glutamyl transferase were mildly elevated. N-terminal pro-B-type natriuretic peptide (NT-proBNP) was elevated at 6500 ng/L (normal <450 ng/L). An abdominal ultrasound scan confirmed hepatomegaly and a moderate amount of ascites. Diagnostic ascitic aspiration revealed it to be a transudate with albumin levels of 16 mg/L. Autoimmune and viral liver screen was negative. An echocardiogram showed normal sized left ventricle with moderate concentric hypertrophy and severe diastolic dysfunction (Fig. 1). The aortic valve had mild calcification with a normal valve area and forward flow velocity. The right ventricle was mildly dilated with normal systolic function. Estimated pulmonary artery systolic pressure was elevated at 77 mmHg. A trivial global pericardial effusion was also noted. CT scans of the thorax, abdomen, and pelvis showed no evidence of pulmonary embolism, significant lung disease, or malignancy. Serum and urine immunoglobulin electrophoresis found no abnormal bands and light chain assay was normal.
Fig. 1.
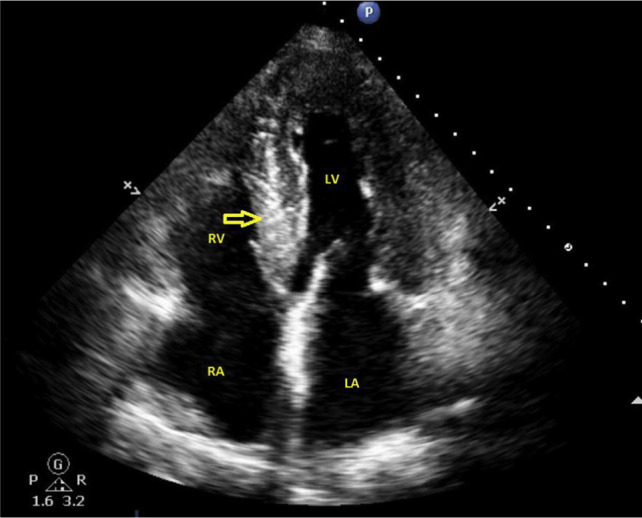
Transthoracic echocardiogram (four chamber view) showing generalised thickening of ventricular walls and echo-bright myocardium of the interventricular septum (arrow). LA = left atrium; RA = right atrium; LV = left ventricle; RV = right ventricle.
The patient was commenced on fluid and salt restriction, loop diuretics, and mineralocorticoid receptor antagonist which led to an improvement in his ascites, peripheral oedema, and shortness of breath. A coronary angiogram revealed no significant abnormality. Cardiac magnetic resonance imaging (MRI) showed moderate generalised thickening of the left and right ventricular walls, moderate bi-atrial enlargement, preserved systolic function, severe diastolic dysfunction, and bilateral pleural effusions (Fig. 2). Late post contrast images revealed multiple areas of diffuse circumferential gadolinium enhancement in both the left and right ventricular walls, highly indicative of amyloidosis (Fig. 3). Genetic testing did not show transthyretin (TTR) gene mutation. Endomyocardial biopsy revealed deposition of amyloid protein in the cardiac muscles. These findings confirmed the diagnosis of senile wild type TTR amyloidosis. His symptoms improved with conventional heart failure medications. He is currently being assessed for the use of diflunisal, which is a salicylic acid derivative known to slow down the progression of senile wild-type TTR amyloidosis.
Fig. 2.
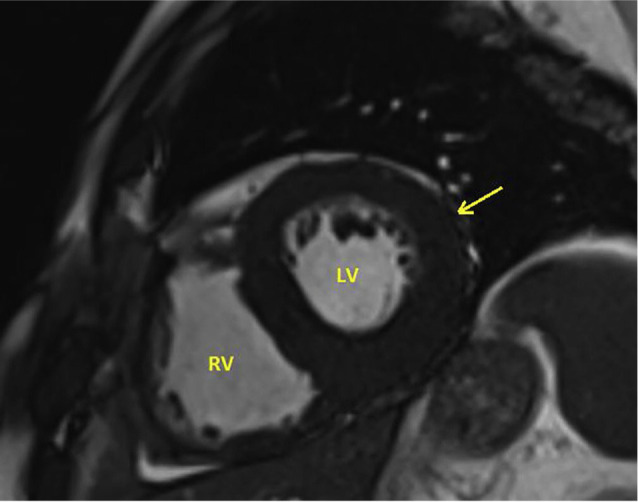
Cardiac magnetic resonance imaging scan (steady state free procession image) reveals diffuse thickening of the left ventricular wall (arrow). LV = left ventricle; RV = right ventricle.
Fig. 3.
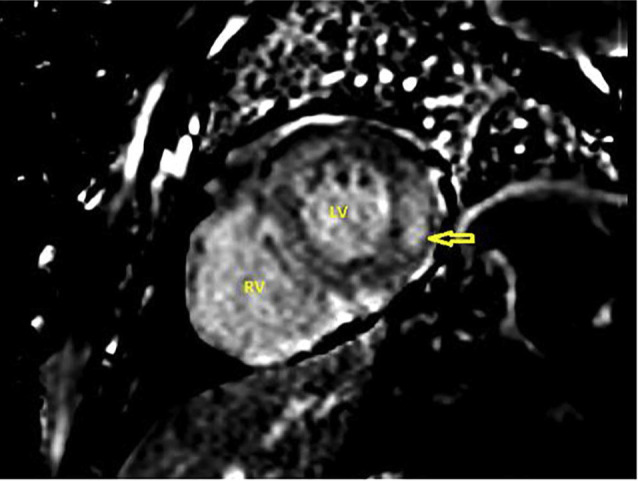
Cardiac magnetic resonance imaging scan (late gadolinium enhancement image) shows subendocardial areas of late gadolinium enhancement in the left ventricle (LV) free wall suggestive of myocardial infiltration with amyloid. Further, diffuse late gadolinium enhancement areas can be seen in the right ventricle (RV) wall, too.
4. Discussion
Based upon the type of infiltrating protein, amyloidosis can be divided into three groups, namely, light chain (AL) amyloidosis, hereditary amyloidosis (usually due to a mutation in TTR), and senile systemic amyloidosis (deposition of wild-type TTR monomers) [3]. Isolated atrial amyloidosis and secondary systemic amyloidosis are also seen. A recent autopsy study showed approximately 5% of elderly patients with heart failure with preserved ejection (HFpEF) have moderate to severe amyloid infiltration in their left ventricle [4]. The incidence of wild-type TTR amyloidosis in hospitalised elderly patients with HFpEF has been reported to be around 13% on scintigraphy [5].
Cardiac amyloidosis leads to progressive thickening and stiffening of the ventricular walls resulting in poor filling and patients present with symptoms of congestive heart failure including breathlessness and fatigue [6]. The signs of right ventricular failure, for example oedema, raised jugular venous pressure, hepatomegaly, and ascites predominate the clinical picture. In addition, features of systemic amyloidosis, for example macroglossia, periorbital purpura, petechial rash on the eyelids, carpal tunnel syndrome, autonomic neuropathy, cuta laxa, nail dystrophy, and weight loss, may also be present. Neuropathy is common in the hereditary variant, which can present with neurological manifestations only, despite significant cardiac involvement on imaging and histology [7]. AL amyloidosis tends to affect kidneys and can cause heavy proteinuria.
Standard laboratory investigations including full blood count, renal and liver function tests, and inflammatory markers form an essential component of the initial assessment. Immunoglobulin electrophoresis and serum free light chain assay can help detect circulating paraproteins. Elevation of specific cardiac biomarkers troponin and NT-proBNP usually points towards the presence of heart failure and is associated with poor outcomes [8]. Genetic testing may reveal mutations in the TTR gene in hereditary cases.
An electrocardiogram may be completely normal but can show low electrical voltage, AV conduction blocks, and arrhythmias. Atrial fibrillation and flutter are common findings in those with infiltration of the atria. A pseudo-infarct pattern with Q waves and a loss of R waves in the precordial lead is also seen in some patients [9].
Echocardiography is a readily available noninvasive imaging modality that can offer useful clues towards the diagnosis which include increased thickness of the myocardium, a restrictive filling pattern, and diastolic dysfunction with reduced cavity size. Bi-atrial enlargement, valve thickening, and pericardial effusion are also present in a significant proportion of patients.
Cardiac MRI offers the advantage of accurate structural measurements including ventricular walls and volumes, atrial size, and septal thickness, combined with tissue characterisation with late gadolinium enhancement and serves as an investigation of choice. Global subendocardial late gadolinium enhancement is considered to be the pathognomonic pattern in cardiac amyloidosis, however, localised, diffuse, and transmural patterns have also been reported [10].
Cardiac heart catheterisation is reserved for selected cases. Histological confirmation of amyloid deposits is required to tailor a specific treatment strategy and remains the gold standard to make the diagnosis [11]. In patients with systemic disease, subcutaneous fat and rectal mucosa may lend themselves for easy-to-access biopsy sites, while in those with focal cardiac disease, an endomyocardial biopsy is required [12]. Amyloid fibrils are detected with Congo red staining and further confirmed by their typical apple green birefringence under polarised light. However, there is some evidence that in patients with no evidence of monoclonal gammopathy, the diagnosis of cardiac amyloidosis can be reliably made by using bone scintigraphy without the need of biopsy [13].
General treatment for cardiac amyloidosis is aimed at managing the heart failure. Initial treatment with loop diuretics and mineralocorticoid receptor blockers leads to symptomatic improvement [14]. Patients with concurrent autonomic neuropathy are at high risk of postural hypotension and need close monitoring. Angiotensin-converting-enzyme inhibitors, angiotensin receptor blockers, and beta blockers are usually poorly tolerated in cardiac amyloidosis.
Atrial fibrillation in patients with cardiac amyloidosis is often poorly tolerated due to a rapid ventricular rate, irregular ventricular filling, and the loss of atrial contractility [15]. Rhythm control with amiodarone or catheter ablation, along with anticoagulation due to a high risk of embolisation, is a preferred choice. The risk of sudden death in patients with cardiac amyloidosis has been reported as high as 60% [16]. The risk is high in those with a history of light chain disease and syncope. An implantable cardiac defibrillator may be considered in those at highest risk [17]. Cardiac transplantation may be eventually needed for end-stage disease.
Specific treatments targeting the molecular mechanisms underpinning amyloid synthesis and deposition are available for some of the subtypes. These include bortezomib, dexamethasone, cyclophosphamide, lenalidomide, humanised monoclonal antibodies, and stem cell transplantation for AL amyloidosis and tafamidis, doxycyline, diflunisal, and patisiran for TTR-related amyloidosis [18]. Diflunisal is a nonsteroidal anti-inflammatory drug (NSAID) that has been shown to stabilise TTR tetramer both in vitro and in vivo [19]. In a carefully selected subgroup of older adults with biopsy proven ATTR cardiac amyloidosis, diflunisal is well tolerated and leads to a reduction in heart failure-related hospitalisations and improved survival [20,21]. Patients with hypersensitivity to NSAIDs, active or recent gastrointestinal bleeding, significant renal dysfunction, and expected survival less than 12 months were excluded from these trials [22]. Concurrent use of dual antiplatelets, NSAIDS, and anticoagulants are other contraindications to the use of diflunisal. The prognosis for untreated amyloid heart disease is poor; advancing age, elevated cardiac biomarkers (troponin T/NT-proBNP), pleural effusion, right ventricle systolic pressure, and right ventricular strain are important predictors of mortality [23].
5. Conclusion
Amyloidosis is a rare infiltrative disorder that can involve the heart resulting in severe cardiac failure with preserved ejection fraction; however, the condition remains underdiagnosed, particularly in older patients. A cardiac MRI scan with late gadolinium enhancement is a useful tool to establish the diagnosis. In an era of expanding treatment options for cardiac amyloidosis, delayed diagnosis is one of the most important factors leading to poor clinical outcomes [24].
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Ueda M, Horibata Y, Shono M, Misumi Y, Oshima T, Su Y, et al. Clinicopathological features of senile systemic amyloidosis: an ante- and post-mortem study. Mod Pathol. 2011;24:1533–44. doi: 10.1038/modpathol.2011.117. [DOI] [PubMed] [Google Scholar]
- 2.Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure. Arch Intern Med. 2005;165:1425–9. doi: 10.1001/archinte.165.12.1425. [DOI] [PubMed] [Google Scholar]
- 3.Tuzovic M, Yang EH, Baas AS, Depasquale EC, Deng MC, Cruz D, et al. Cardiac amyloidosis: diagnosis and treatment strategies. Curr Oncol Rep. 2017;19:46. doi: 10.1007/s11912-017-0607-4. [DOI] [PubMed] [Google Scholar]
- 4.Mohammed SF, Mirzovev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2:113–22. doi: 10.1016/j.jchf.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.González-López E, Gallego-Delgado M, Guzzo-Merello G, de Harodel Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–94. doi: 10.1093/eurheartj/ehv338. [DOI] [PubMed] [Google Scholar]
- 6.Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12:91–102. doi: 10.1038/nrcardio.2014.165. [DOI] [PubMed] [Google Scholar]
- 7.Gertz MA. Immunoglobulin light chain amyloidosis: 2018 Update on diagnosis, prognosis, and treatment. Am J Hematol. 2018;93:1169–80. doi: 10.1002/ajh.25149. [DOI] [PubMed] [Google Scholar]
- 8.Palladini G, Lavatelli F, Russo P, Perlini S, Perfetti V, Bosoni T, et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107:3854–8. doi: 10.1182/blood-2005-11-4385. [DOI] [PubMed] [Google Scholar]
- 9.Fikrle M, Paleček T, Kuchynka P, Němeček E, Bauerová L, Straub J, et al. Cardiac amyloidosis: a comprehensive review. Cor Vasa. 2013;55:e60–75. [Google Scholar]
- 10.Falk RH, Quarta CC, Dorbala S. How to image cardiac amyloidosis. Circ Cardiovasc Imaging. 2014;7:552–62. doi: 10.1161/CIRCIMAGING.113.001396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benson MD, Dasgupta MR. Amyloid cardiomyopathy. J Am Coll Cardiol. 2016;68:25–8. doi: 10.1016/j.jacc.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Bradshaw SH, Veinot JP. Cardiac amyloidosis. Curr Opin Cardiol. 2012;27:143–7. doi: 10.1097/HCO.0b013e32834fdc7e. [DOI] [PubMed] [Google Scholar]
- 13.Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–12. doi: 10.1161/CIRCULATIONAHA.116.021612. [DOI] [PubMed] [Google Scholar]
- 14.Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29:1924–33. doi: 10.1200/JCO.2010.32.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.John RM. Arrhythmias in cardiac amyloidosis. J Innov Card Rhythm Manag. 2018;9:3051–7. doi: 10.19102/icrm.2018.090301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dubrey SW, Cha K, Skinner M, LaValley M, Falk RH. Familial and primary AL cardiac amyloidosis: echocardigraphically similar disease with different outcomes. Heart. 1997;78:74–82. doi: 10.1136/hrt.78.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kisten AV, Dengler TJ, Hegebart U, Schonland SO, Goldschmidt H, Sack FU, et al. Prophylactic implantation of cardioverter defibrillator in patients with severe cardiac amyloidosis and high risk of sudden cardiac death. Heart Rhythm. 2008;5:235–40. doi: 10.1016/j.hrthm.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 18.Milani P, Palladini G, Merlini G. New concepts in the treatment and diagnosis of amyloidosis. Expert Rev Hematol. 2018;11:117–27. doi: 10.1080/17474086.2018.1424534. [DOI] [PubMed] [Google Scholar]
- 19.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–6. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 20.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–49. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 21.Rosenblum H, Castano A, Alvarez J, Goldsmith J, Helmke S, Maurer MS. TTR (transthyretin) stabilisers are associated with improved survival in patients with TTR cardiac amyloidosis. Circ Heart Fail. 2018;11:e004769. doi: 10.1161/CIRCHEARTFAILURE.117.004769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikram A, Donnelly JP, Sperry BW, Samaras C, Valent J, Hanna M. Diflunisal tolerability in transthyretin cardiac amyloidosis: a single center’s experience. Amyloid. 2018;25:197–202. doi: 10.1080/13506129.2018.1519507. [DOI] [PubMed] [Google Scholar]
- 23.Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97:75–84. doi: 10.1136/hrt.2009.190405. [DOI] [PubMed] [Google Scholar]
- 24.Bishop E, Brown EE, Fajardo J, Barouch LA, Judge DP, Halushka MK. Seven factors predict a delayed diagnosis of cardiac amyloidosis. Amyloid. 2018;25:174–9. doi: 10.1080/13506129.2018.1498782. [DOI] [PubMed] [Google Scholar]