

Abstract
Hepatic encephalopathy is a pathophysiological complication of acute liver failure, which may be triggered by hepatotoxic drugs such as acetaminophen (APAP). Although APAP is safe in therapeutic concentration, APAP overdose may induce neurotoxicity, which is mainly associated with oxidative stress. Caffeine is a compound widely found in numerous natural beverages. However, the neuroprotective effect of caffeine remains unclear during APAP intoxication. The present study aimed to investigate the possible modulatory effects of caffeine on brain after APAP intoxication. Mice received intraperitoneal injections of APAP (250 mg/kg) and/or caffeine (20 mg/kg) and, 4 h after APAP administration, samples of brain and blood were collected for the biochemical analysis. APAP enhanced the transaminase activity levels in plasma, increased oxidative stress biomarkers (lipid peroxidation and reactive oxygen species), promoted an imbalance in endogenous antioxidant system in brain homogenate and increased the mortality. In contrast, APAP did not induce dysfunction of the mitochondrial bioenergetics. Co-treatment with caffeine modulated the biomarkers of oxidative stress as well as antioxidant system in brain. Besides, survival assays demonstrated that caffeine protective effects could be dose- and time-dependent. In addition, caffeine promoted an increase of mitochondrial bioenergetics response in brain by the enhancement of the oxidative phosphorylation, which could promote a better energy supply necessary for brain recovery. In conclusion, caffeine prevented APAP-induced biochemical alterations in brain and reduced lethality in APAP-intoxicated mice, these effects may relate to the preservation of the cellular antioxidant status, and these therapeutic properties could be useful in the treatment of hepatic encephalopathy induced by APAP intoxication.
Keywords: caffeine, neurotoxicity, acetaminophen, oxidative damage, bioenergetics
Introduction
The brain is one of the tissues most susceptible to the oxidative damage and redox imbalance. Due to this susceptibility, brain damage may occur when other organs, such as liver, are affected [1, 2]. For instance, encephalopathy in acute liver failure has been demonstrated to be induced by high doses of acetaminophen (APAP) [2]. APAP is widely consumed because of its analgesic and antipyretic properties [3] and at high doses can induce a critical damage to the structure and cellular components of liver [4]. The neurotoxicity associated with APAP-induced acute liver failure may be related to the formation of the toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI), reactive species generation and accumulation of ammonia [2, 5, 6]. A common factor associated with this disorder is the redox imbalance, which could lead, at least in part, to the disruption of brain homeostasis, triggering a high mortality rate due to the deleterious effects to the central nervous system. Among these effects are development of brain edema and increased intracranial pressure, and often glutamate-mediated excitotoxicity, abrupt onset of delirium, seizures and coma [7, 8]. In addition, APAP is able to induce mitochondrial dysfunction, triggering reactive oxygen species (ROS) generation and mitochondrial transition permeability in brain [2].
As APAP hepatotoxicity is followed by significant enhance of oxidative stress, the consequent redox imbalance could be efficiently prevented by treatment with antioxidant [9, 10]. Caffeine (1,3,7-trimethylxanthine) is commonly found in beverages, food and in combination with APAP in popular analgesic products and cold medicine formulations. Caffeine has been studied due to anti-inflammatory, biochemical and physiological effects, which can protect the tissue during the liver injury [11, 12]. Moreover, it is able to prevent the oxidative damage induced by radiation, alcoholic and non-alcoholic hepatotoxicity [11, 13, 14]. In this context, caffeine have demonstrated modulatory effects on mitochondrial physiology, such as adaptation of mitochondrial glutathione (GSH) redox system, calcium homeostasis and bioenergetics parameters [15–17]. Many works have focused on the effects of caffeine on hepatic dysfunction associated to APAP intoxication [18, 19], whereas the caffeine effects on neurotoxicity associated to APAP overdose remains poorly understood.
Caffeine presents ergogenic properties, which is observed by the increase of mitochondrial metabolic rate [20]. As the brain have a high bioenergetics demand due to energetic consumption by the ATP-dependent Na+, K+-pump [2], the impairment of mitochondrial functioning could be fatal for the maintenance of normal brain functions. Mitochondrial bioenergetics maintenance is associated to the oxidative phosphorylation (OXPHOS) and the electron transport system [21, 22]. The combination of caffeine and APAP was demonstrated to enhance the APAP efficacy as well as the liver microsome metabolism, although this was accompanied by enhance of APAP hepatotoxicity [23]. On the other hand, some studies demonstrated that caffeine consumption could reduce the hepatotoxicity biomarkers in serum [24]. Thus, in order to understand the mechanism of caffeine during the neurotoxicity, this work aimed to evaluate the changes in the oxidative stress biomarkers and redox system, as well as the mitochondrial bioenergetics functioning in brain through the high-resolution respirometry (HRR) during the neurotoxicity associated with APAP intoxication.
Materials and Methods
Materials
Caffeine, thiobarbituric acid (TBA), 2′-7′-dichlorofluorescein diacetate (DCFH-DA), trichloroacetic acid (TCA) and nucleotides were purchased from Sigma Chemical Co. (St. Louis, MO, USA). All other chemicals were of analytical grade and obtained from standard commercial suppliers.
Animals
Seven-week-old male adult Swiss albino mice (30–40 g) from our own breeding colony were used. The animals were kept on a separate animal room, at 12-h light/dark cycle, and at temperature of 22 ± 2°C. Mice were acclimated for 7 days before initiation of any procedures. This study was approved by the Ethics and Animal Welfare Committee of Federal University of Santa Maria, Brazil (Permit Number: 3208150915). Animals had free access to water and rodent chow before initiation of any treatment. However, mice were fasted before treatment with APAP as indicated below.
Experimental protocol
Briefly, the mice were randomly divided into following four groups: control (vehicle), caffeine (Caf), APAP treated (APAP) and Caf + APAP. All the solutions were administered by the intraperitoneal (i.p.) route and prepared in saline 0.9%. Injections were administered at 9:00 a.m. in order to remove any confounding factors of circadian rhythm. Mice were fasted for 16 h before injection of APAP beginning with the removal of food at 5:00 p.m. on the previous day. First, mice in the control and caffeine groups received saline 0.9% (20 ml/kg) and caffeine 20 mg/kg, respectively, and the APAP and Caf + APAP groups received APAP 250 mg/kg (15 ml/kg). APAP and caffeine doses have been used in previous studies [25, 26] and animals received first caffeine and 30 min after APAP. At 4 h following APAP administration, animals were killed by cervical dislocation, brain was collected and blood also was collected by cardiac puncture using heparin-rinsed 1-ml syringes (20 gauge needles) and centrifuged. The plasma was used for determination of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activities using a commercial kit (Labtest®, Diagnostica S.A., Minas Gerais, Brazil).
Brain homogenate preparation
At the end of the treatment period the animals were euthanized by cervical dislocation, perfused in petri dish with 5 ml of cold Tris buffer (10 mM Tris-HCl, pH 7.4) to remove the contaminating blood, in according to previously described protocol with some modifications [27]. So, a sagittal section was made in brain, cerebellum and brain stem were removed and one brain hemisphere was collected and homogenized in Tris buffer with a pre-cooled glass potter. Brain homogenates were centrifuged at 2000 × g for 10 min to separate large tissue fragments and yield the supernatant fractions with cellular components, which were used for the biochemical assays. The protein content of homogenate brain preparation was quantified by Bradford’s assay [28].
APAP survival challenge
For survival studies, mice were injected with caffeine and APAP in according to protocol described earlier, then 30 min later the mice were returned to their cages and fed with food and water ad libitum. To determine the effect of caffeine on mortality of APAP-administered mice, the survival rate after APAP administration was evaluated for 72 h.
Measurement of lipid peroxidation
Brain homogenate lipid peroxidation (LPO) was quantified measuring the malondiadehyde (MDA) levels. In summary, brain homogenate were incubated in 300 μl of a medium consisting of 175 mM KCl and 10 mM Tris-HCl, pH 7.4, and then were added to color reaction. TBA reactive substances levels were measured at 532 nm using a standard curve of MDA [29].
Measurement of ROS production
ROS generation was determined spectrofluorimetrically in vitro using H2DCF-DA levels as an index of the peroxide production by cellular components (1 μM) [30]. Briefly, the brain homogenate was added to standard medium and the fluorescence was determined at 488 nm for excitation and 525 nm for emission, with slit widths of 3 nm. Results are expressed as increase of fluorescence regarding fluorescence measured in control group, which was used as standard.
Determination of reduced (GSH) and oxidized glutathione (GSSG)
For the measurement of GSH and GSSG levels, the brain homogenate was treated with 0.5 ml of 13% TCA and centrifuged at 13 000 rpm for 10 min at 4°C. Aliquots (100 μl) of the supernatant were mixed with 2 ml of 100 mM NaH2PO4 buffer, pH 8.0, containing 5 mM EDTA. One hundred microliters of O-phthalaldehyde (1 mg/ml) was added and fluorescence was measured 15 min later using the 350/420 nm excitation/emission wavelength pair, with slit widths of 3 nm [31]. For measurement of GSSG levels, a 250 μl of the brain supernatant was incubated at room temperature with 100 μl of N-ethylmaleimide (0.04 M) for 30 min at room temperature, and after that 140 μl of the mixture were added to 1760 μl of NaOH (0.1 N) buffer, following of added 100 μl OPT and incubated for 15 min, using the procedure outlined above for GSH assay. Results are expressed in nmol of GSH and corrected by protein content of each sample.
Total superoxide dismutase activity
Brain homogenate total superoxide dismutase (SOD) activity was measured by the capacity of inhibiting auto-oxidation of adrenaline to adrenochrome at 480 nm [32]. The brain supernatant (5 mg protein) was added to a medium containing 2 mM EDTA, 50 mM NaHCO3/Na2CO3 buffer (pH 10.3) and adrenaline (4 mM). The SOD activity was analysed in spectrophotometer at 480 nm. The results were expressed in U SOD/mg protein.
Catalase activity
The catalase (CAT) enzyme activity was determined in brain homogenate in according to the method previously proposed [33]. Brain homogenate (5 mg protein) was added to a medium containing potassium phosphate buffer (50 mM KH2PO4, 50 mM K2HPO4; pH 7.4) and H2O2 (1 mM). The kinetic analysis of CAT was started after H2O2 addition. The CAT activity was determined using the molar extinction coefficient 36 M−1 cm−1 and the reaction was measured at 240 nm.
Glutathione-S-transferase activity
The glutathione-S-transferase (GST) activity was determined spectrophotometrically [34]. GST activity was quantified in brain homogenates (5 mg protein) in a reaction mixture containing 1 mM 1-chloro-2,4-dinitrobenzene (CDNB) and 1 mM glutathione as substrates in 0.1 M sodium phosphate buffer, pH 6.5, at 37°C. Enzyme activity was calculated by the change in the absorbance value from the slope of the initial linear portion of the absorbance time curve at 340 nm for 5 min. Enzyme activity was determined using the molar extinction coefficient 9.6 mM−1 cm−1 and expressed as nmol CDNB/min/mg protein.
Glutathione peroxidase activity
Glutathione peroxidase (GPx) activity was determined spectrophotometrically at 340 nm by NADPH consumption for 2 min at 30°C [35]. The liver homogenate supernatant (5 mg protein) was added to medium containing 0.1 M phosphate buffer (0.1 M KH2PO4, 0.1 M K2HPO4 and 5 mM EDTA, pH 7.0), 1 mM GSH, 0.15 mM NADPH, 0.1 U/ml glutathione reductase (GR) and 1 mM sodium azide. So, the reaction was initiated by adding the H2O2 to a final concentration of 0.4 mM. The GPx activity was determined using the molar extinction coefficient 6220 M−1 cm−1 and expressed as nmol/min/mg protein.
GR activity
For the measurement of GR activity, the liver homogenate supernatant (5 mg protein) was added to medium containing 0.15 M phosphate buffer (0.15 M K2HPO4 and 1.5 mM EDTA, pH 7.0) and 0.15 mM NADPH. The measurements were made at 340 nm and initiated with addition of 20 mM GSSG at 30°C for 2 min [36]. GR activity was determined using the molar extinction coefficient 6220 M−1 cm−1 and expressed as nmol/min/mg protein.
HRR
For the respirometry determination, the mouse brain homogenate (400 mg) was weighed and transferred to 2 ml of cold homogenization buffer containing 5 mM Tris-HCl, 250 mM sucrose and 2 mM EGTA (pH = 7.4), and brain homogenate (0.1 mg/ml) was used to the HRR. Oxygraph-2 k (O2k, Oroboros Instruments, Innsbruck, Austria) was employed for all used for measurements of respiration. The experiments were performed in MiR05 [110 mM sucrose, 60 mM K-lactobionate, 0.5 mM EGTA, 3 mM MgCl2, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES and 0.1% bovine serum albumin (BSA) essentially fatty acid free, pH 7.1] [22, 37, 38]. All experiments were performed at 37°C using DatLab 4.0 software (Oroboros Inc., Austria), with continuous stirring at 750 rpm, and all experiments started by registering the endogenous substrate supported respiration, following protocols established in the literature [39]. Titration protocols of multiple substrates and inhibitors were used to assess mitochondrial function in terms of different respiration states. Complex I (CI)-mediated “leak” respiration was determined using 2 mM malate and 10 mM glutamate. CI-mediated “OXPHOS” was determined using saturating ADP (2.5 mM). The convergence electron flow, CI and complex II (CII) OXPHOS, was then determined by the addition of 10 mM succinate. CII-mediated “OXPHOS” was determined using 0.5 μM rotenone. Addition of 2.5 μM antimycin A (Ama) inhibited complex III, resulting in non-mitochondrial respiration with small contributions from electron leak in the uncoupled state. Cytochrome c oxidase activity was evaluated using 0.25 mM TMPD and 2 mM ascorbate.
Protein determination
Protein content was determined using BSA as standard [28].
Statistical analysis
Statistical analysis was performed using GraphPad (version 5.0 for Macintosh OSX, GraphPad Software, San Diego, CA, USA). Significance was assessed by two-way analysis of variance, followed by Newman–Keuls Test for post hoc comparison. Values of P ≤ 0.05 were considered statistically significant.
Results
Liver injury biomarkers
In order to analyse the liver damage, the transaminase activity in plasma was determined. APAP treatment induced a significant increase of ALT and AST activities compared with the control, and caffeine did not reduce the transaminase activities levels when compared with the APAP groups (Table 1).
Table 1.
Analyses of transaminase levels in plasma after ALF induced by APAP
| AST | ALT | |
|---|---|---|
| Control | 66.2 ± 1.9 n.s. | 45.6 ± 1.0 n.s. |
| Caffeine | 67.2 ± 1.9 n.s. | 48.5 ± 0.5 n.s. |
| APAP | 116.3 ± 11.9* | 98.0 ± 17.0* |
| Caf ± APAP | 115.9 ± 16.6* | 114.4 ± 21.1* |
Values are presented as means ± SEM. AST, aspartate aminotransferase (IU/l); ALT, alanine aminotransferase (IU/l); ALF, acute liver failure; n.s., not significant.
* P ≤ 0.05 from control group by Newman–Keuls Multiple Comparison Test.
# P ≤ 0.05 from APAP group by Newman–Keuls Multiple Comparison Test.
Markers of oxidative damage in brain
As the oxidative stress plays an important role in the development of several diseases, ROS generation and LPO was investigated in vitro. LPO levels were increased in APAP group as well as the Caf + APAP group when compared with control group (Fig. 1A). APAP group presented significant increase of ROS in relation to the control; on the other hand, the ROS generation levels remained unchanged in the Caf + APAP group (Fig. 1B).
Figure 1.
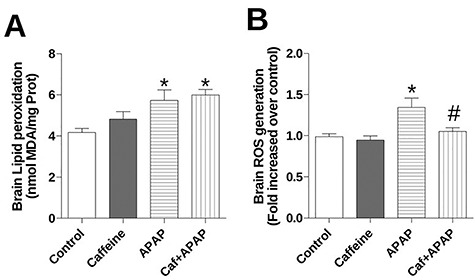
Effects of treatment with caffeine and APAP on oxidative damage markers in brain homogenate: (A) lipid peroxidation and (B) ROS generation. Mice were given caffeine (20 mg/kg; i.p.) and APAP (250 mg/kg, i.p.) and were killed at 4 h after the APAP treatment. Data are expressed as means ± SEM (n = 5). Significance was assessed by one-way analysis of variance, followed by Newman–Keuls Test for post hoc comparison. Significant differences are indicated by *P ≤ 0.05 when compared with control group. Significant difference is indicated by #P ≤ 0.05 when compared with APAP group.
CAT and SOD system in brain
Both CAT and SOD activities were significantly reduced during the APAP treatment when compared with control group, whereas the Caf + APAP group presented similar levels to the control group (Fig. 2A and B).
Figure 2.
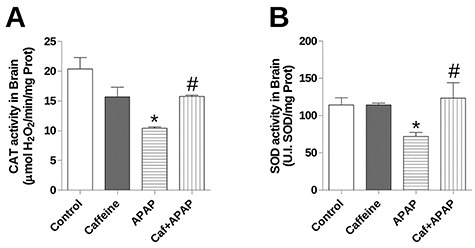
Effects of treatment with caffeine and APAP on antioxidant enzyme system in brain homogenate: (A) catalase (CAT) activity and (B) total superoxide dismutase (SOD) activity. Mice were given caffeine (20 mg/kg; i.p.) and APAP (250 mg/kg, i.p.) and were killed at 4 h after the APAP treatment. Data are expressed as means + SEM (n = 5). Significance was assessed by one-way analysis of variance, followed by Newman–Keuls Test for post hoc comparison. Significant differences are indicated by *P < 0.05 when compared with control group. Significant difference is indicated by #P < 0.05 when compared with APAP group.
Glutathione redox system in brain
Homeostasis of glutathione redox system was evaluated. APAP treatment did not change the GSH levels, but it was able to promote the increase of the GSSG levels in vitro, resulting in the reduction of brain GSH/GSSG ratio. GSH levels were reduced in the Caf + APAP group; in contrast, the GSSG levels were similar to the control as well as the GSH/GSSG ratio (Table 2). In addition, levels of GSSG (% of total GSH) were significantly higher than control in APAP group, whereas the Caf + APAP remain unchanged (Table 2).
Table 2.
Analyses of glutathione redox system in brain after ALF induced by APAP
| GSH | GSSG | GSH/GSSG ratio | GSSG (% of total GSH) | |
|---|---|---|---|---|
| Control | 36.8 ± 2.9 n.s. | 6.1 ± 0.8 n.s. | 5.0 ± 0.1 n.s. | 13.9 ± 0.4 n.s. |
| Caffeine | 29.7 ± 1.8 n.s. | 6.1 ± 0.5 n.s. | 5.0 ± 0.5 n.s. | 14.6 ± 1.0 n.s. |
| APAP | 34.5 ± 1.9 n.s. | 8.8 ± 0.6* | 3.5 ± 0.1* | 17.8 ± 0.5* |
| Caf ± APAP | 32.2 ± 0.6 n.s. | 6.4 ± 0.3# | 5.4 ± 0.3# | 14.2 ± 0.5# |
Values are presented as means ± SEM. GSH levels (nmol GSH/mg protein); GSSG levels (nmol GSSG/mg protein); ALF, acute liver failure; n.s., not significant.
* P ≤ 0.05 from control group by Newman–Keuls Multiple Comparison Test.
# P ≤ 0.05 from APAP group by Newman–Keuls Multiple Comparison Test.
Enzymes glutathione redox system in brain
In order to evaluate the enzymatic antioxidant system associated with GSH, GPx, GR and GST activities were determined in vitro. GPx and GST activities were higher than control in APAP group (Fig. 3A and C), whereas the GR activity presented a significant reduction (Fig. 3B). On the other hand, GPx, GR as well as GST remain similar to control levels in Caf + APAP group (Fig. 3A–C).
Figure 3.
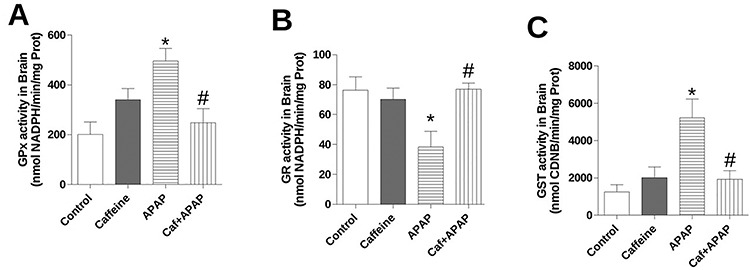
Effects of treatment with caffeine and APAP on enzymes of glutathione redox system in brain homogenate: (A) glutathione peroxidase (GPx) activity, (B) glutathione reductase (GR) activity and (C) glutathione S-transferase (GST) activity. Mice were given caffeine (20 mg/kg; i.p.) and APAP (250 mg/kg, i.p.) and were killed at 4 h after the APAP treatment. Data are expressed as means ± SEM (n = 5). Significance was assessed by one-way analysis of variance, followed by Newman–Keuls Test for post hoc comparison. Significant differences are indicated by *P ≤ 0.05 when compared with control group. Significant difference is indicated by #P ≤ 0.05 when compared with APAP group.
APAP survival challenge
Mice were monitored for 72 h to determinate the effects of treatment. In this way, mice that received APAP presented a significant mortality when compared with control, which was 100% in ~48 h, and the treatment with caffeine (20 mg/kg) demonstrated a significant extension of survival, which was ~33% in 72 h (Fig. 4).
Figure 4.
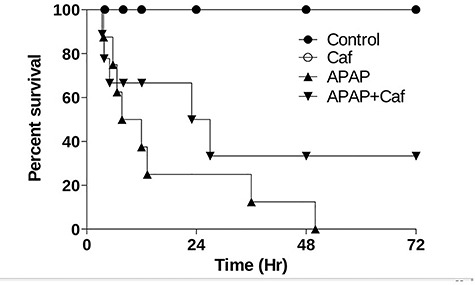
Mortality of the treatment with caffeine on APAP intoxication (250 mg/kg; i.p.). Survival curve in mice treated with low caffeine dose (20 mg/kg). Survival was followed for 72 h, n = 10 per group.
Mitochondrial bioenergetics parameters in brain
To evaluate the mitochondrial function, O2 flux consumption was determined by respiratory control ratio (RCR). In this way, APAP intoxication did not presented any significant effect on RCR levels (Table 3).
Table 3.
Analyses of bioenergetics parameters in brain after ALF induced by APAP
| Respiratory control ratio (RCR) for complex I | |
|---|---|
| Control | 3.7 ± 0.02 n.s. |
| Caffeine | 4.2 ± 0.1 n.s. |
| APAP | 4.0 ± 0.2 n.s. |
| Caf ± APAP | 3.6 ± 0.3 n.s. |
Values are presented as means ± SEM. ALF, acute liver failure; n.s., not significant.
In order to understand the possible changes in the mitochondrial bioenergetics parameters, the OXPHOS capacity was evaluated. APAP and Caf + APAP groups presented a significant increase of CI + CIIOxphos when compared with the control group (Fig. 5). Caffeine group promoted the enhance of OXPHOS capacity associated to the convergent electron flow simultaneously through complexes I and II (CI + CIIOxphos) when compared with the control, whereas the APAP and Caf + APAP groups remained unchanged (Fig. 5). On the other hand, both caffeine and APAP enhanced the CIIOxphos when compared with the control group, and the Caf + APAP group was significantly different from control (Fig. 5). The residual oxygen consumption (ROX) was determined with addition of Ama, the levels of ROX were increased in APAP and Caf + APAP groups (Fig. 5). The APAP intoxication did not present significant effects on COx activity; in contrast, Caf + APAP group had significant increase of COx activity when compared with the control group (Fig. 5).
Figure 5.

HRR representative in brain homogenate of the treatment with caffeine on APAP intoxication (250 mg/kg; i.p.). Mitochondrial function are presented with the abbreviation(s) of the complex(es) involved followed by the state of respiration measured in presence of glutamate + malate (CILeak), + ADP (CIOxphos), + succinate (CI + CIIOxphos), + rotenone (CIIOxphos), + antimycin A (Ama). Ama was used for correction of residual O2 consumption. Significance was assessed by one-way analysis of variance, followed by Newman–Keuls Test for post hoc comparison. Results are expressed as means ± SEM for four different preparations. *Significant difference is indicated by #P ≤ 0.05 when compared with APAP group.
Discussion
This work was performed with the purpose to understand the mechanism of caffeine during the neurotoxicity induced by APAP. To our knowledge, this is the first study demonstrating that caffeine is able to reduce the neurotoxicity induced by APAP, being the beneficial effects associated, at least in part, with the modulation of the redox homeostasis. In addition, the mitochondrial function remained unchanged during the neurotoxicity induced by APAP, but the caffeine treatment promoted a significant increase of CI + CIIOxphos and CIIOxphos, suggesting a bioenergetics adaptation in brain, which could be associated with the ergogenic properties of caffeine. It is interesting that according to our data mitochondrial respiration is not affected in brain homogenates from APAP-treated mice in 4 h, which would exclude the early mitochondrial dysfunction associated with APAP toxicity. However, as recognized by other authors, in vitro and in vivo experiments, the brain mitochondrial dysfunction had been noticed to be closely related with APAP doses and exposure time dependent, with a progressive caspase 3 activation and cytochrome c releases 6 h after APAP intoxication [2, 4, 40, 41].
Despite the importance of APAP hepatic metabolism for the hepatotoxicity [2], previous studies demonstrated that brain cytochrome P450 plays a significant role during neurotoxicity induced by APAP [42], resulting in oxidative damage. The increase of ROS and LPO is associated with the inhibition of CAT and SOD activities, which could enhance the oxidative damage in brain during the neurotoxicity induced by APAP. In contrast, the caffeine treatment was able to reduce the ROS generation with maintenance of the CAT and SOD activities, essentials for the ROS detoxification. In addition, the antioxidant properties of caffeine have been highlighted and the efficacy to scavenger ROS was demonstrated to be similar or higher than classical antioxidants [43, 44]. Pharmacokinetic properties of caffeine, such as fast absorption and widely distribution among the organism, could be an advantage for the treatment against the neurotoxicity induced by APAP [45]. The metabolism of caffeine promote the generation of paraxanthine and theophylline, which could contribute to the benefit action [46]. In addition, previous work demonstrated that chronic treatment with caffeine (20 mg/kg/day) reduces the LPO and promotes the upregulation of antioxidant enzymes [47].
Another important point is the impairment of GSH redox homeostasis due to the direct and/or indirect effects of reactive metabolite generation, which induce the oxidative damage in cellular components including protein and lipids in cellular membrane [48]. Here, treatment with APAP increased GST and GPx activities. The inhibition of GR activity in group treated with APAP could be associated with the higher levels of GSSG, which indicates the inability of the brain to restore the GSH pool. Caffeine consumption has demonstrated significant biochemical adaptive response in brain, such as increase of antioxidant enzyme activity (SOD and GR) and GSH levels [49]. Undoubtedly, these beneficial effects increase the cytoplasmic antioxidant capacity, reducing the impact of ROS generation and redox imbalance associated with the neurotoxicity induced by APAP.
In addition, our results demonstrated that the treatment with one dose of caffeine were able to extent the survival of mice treated with APAP. Other study demonstrates that caffeine modulates hyperammonemia-induced behavioral changes in patients with liver cirrhosis, decreasing subjective sleepiness and helping improve the vigilance state [50]. However, literature evidence demonstrated that ammonia levels in plasma were elevated only after 15 h to the acute liver failure induced by APAP (250 mg/kg) [51]. Beside that caffeine inhibits production of pro-inflammatory cytokines and promotes production of anti-inflammatory cytokines [52, 53], this could improve survival in APAP intoxication. Previous works have demonstrated the pro-oxidant activity of some caffeine concentrations, which can react with metals and, consequently, would contribute for oxidative damage, exacerbating the APAP toxicity. Caffeine is a non-selective adenosine receptors antagonist; recently, a protective role of moderate chronic caffeine consumption against neurodegenerative diseases has been discussed [54, 55]. The antagonization of caffeine on adenosine receptors modulates inflammation pathways in a concentration-dependent way [26]. Caffeine effects also can include reduction of oxygenated hemoglobin and increase of deoxygenated hemoglobin in cerebral blood flow [56].
Our results demonstrated that the caffeine concentration should be considered important during the neurotoxicity induced by APAP. In addition, higher concentration of caffeine could affect the CYP450 activity, accelerating the metabolism of APAP in liver and causing the enhance of NAPQI generation responsible for hepatoxicity and its consequences for organism [18].
Although APAP intoxication caused an increase in oxidative damage, our results did not demonstrate mitochondrial bioenergetics dysfunction, and besides cerebral oxidative stress, other factors may contribute to APAP-induced mortality. In this context, it has been demonstrated that the neurotoxicity induced by APAP is a process associated to the increase of toxic metabolite generation [6]. This factor could be important to cause the mitochondrial dysfunction, but the glutathione system helps to reduce the brain toxicity induced by APAP intoxication, increasing the transaminase levels and inflammation markers at liver [5]. While there is no direct evidence for normal ammonia levels in the brain under the experimental conditions of the present study, according to other references low doses of APAP, such as used in this study, are not able to increase the ammonia levels in blood serum [57], although there are evidences in literature demonstrating liver toxicity [58].
HRR analysis of brain mitochondrial function revealed that caffeine caused the stimulation of OXPHOS respiration when the convergent pathway (CI + CIIOXPHOS) and CIIOXPHOS were activated. In addition, the CIV activity was increased by caffeine treatment, CIV is a terminal oxidase of mitochondrial respiratory chain, which can regulate the aerobic ATP generation. Based upon these data, we postulate that concentration of caffeine (20 mg/kg) produces a bioenergetics adaptation and the respiration depends on the substrate applied, since the smallest stimulation was noticed when OXPHOS respiration was stimulated with only CI substrates (glutamate + malate), these changes may be associated with increased substrate oxidation in mitochondrial respiratory chain. Caffeine increases the oxidative metabolism without inducing a futile cycle associated with the mitochondrial uncoupling. It has been suggested that caffeine mobilizes the intracellular calcium concentration in brain [59, 60]. This intracellular signal modulates the regulatory enzymes of tricarboxylic acid cycle, increasing the NADH and FADH2 production by dehydrogenases, and, consequently, leading to increase of the electron flow and H+-pumping in mitochondrial respiratory chain culminating with the mitochondrial membrane potential (∆Ψm). Given the lack of evidence demonstrating caffeine-induced increase of mitochondrial OXPHOS, this is the first work demonstrating that caffeine plays an important role for brain metabolism increasing the bioenergetics capacity. In this way, caffeine could be a useful tool for the re-establishment of cellular energy, since caffeine is able to increase the mitochondrial OXPHOS.
In conclusion, caffeine presents a modulatory effect on brain during the APAP intoxication. In addition, the APAP intoxication does not seems to induce bioenergetics impairment in brain; however, caffeine treatment promotes an increase in brain mitochondrial OXPHOS, which is an indicator of metabolic adaptation important for maintenance of vital functions in this tissue.
Declarations
Conflict of Interest
None declared.
Acknowledgments
This study was supported by the CNPq/FAPERGS/DECIT/SCTIE-MS/PRONEM #11/2029-1. The authors acknowledge CNPq, Brazil (404603/2015-7, 482313/2013-7), FAPERGS (2380-2551/14-8) and PROPESQ-Unipampa for the financial support. J.L.F. is recipient of a CNPq research fellowship (310861/2014-4). F.A.S. received a fellowship by CNPq.
Contributor Information
Débora F Gonçalves, Departamento de Bioquímica e Biologia Molecular, Centro de Ciências Naturais e Exatas, Universidade Federal de Santa Maria, Santa Maria, Rio Grande do Sul, Brazil.
Cintia C Tassi, Departamento de Bioquímica e Biologia Molecular, Centro de Ciências Naturais e Exatas, Universidade Federal de Santa Maria, Santa Maria, Rio Grande do Sul, Brazil.
Guilherme P Amaral, Departamento de Bioquímica e Biologia Molecular, Centro de Ciências Naturais e Exatas, Universidade Federal de Santa Maria, Santa Maria, Rio Grande do Sul, Brazil.
Silvio T Stefanello, Departamento de Bioquímica e Biologia Molecular, Centro de Ciências Naturais e Exatas, Universidade Federal de Santa Maria, Santa Maria, Rio Grande do Sul, Brazil.
Cristiane L Dalla Corte, Departamento de Bioquímica e Biologia Molecular, Centro de Ciências Naturais e Exatas, Universidade Federal de Santa Maria, Santa Maria, Rio Grande do Sul, Brazil.
Félix A Soares, Departamento de Bioquímica e Biologia Molecular, Centro de Ciências Naturais e Exatas, Universidade Federal de Santa Maria, Santa Maria, Rio Grande do Sul, Brazil.
Thais Posser, Centro Interdisciplinar de Pesquisas em Biotecnologia – CIPBIOTEC, Universidade Federal do Pampa, Campus São Gabriel, Rio Grande do Sul, Brazil.
Jeferson L Franco,, Centro Interdisciplinar de Pesquisas em Biotecnologia – CIPBIOTEC, Universidade Federal do Pampa, Campus São Gabriel, Rio Grande do Sul, Brazil.
Nélson R Carvalho, Departamento de Bioquímica e Biologia Molecular, Centro de Ciências Naturais e Exatas, Universidade Federal de Santa Maria, Santa Maria, Rio Grande do Sul, Brazil; Centro Interdisciplinar de Pesquisas em Biotecnologia – CIPBIOTEC, Universidade Federal do Pampa, Campus São Gabriel, Rio Grande do Sul, Brazil; Instituto Federal Farroupilha, Campus Santo Ângelo, Rio Grande do Sul, Brazil.
References
- 1. Comim CM, Rezin GT, Scaini G et al. Mitochondrial respiratory chain and creatine kinase activities in rat brain after sepsis induced by cecal ligation and perforation. Mitochondrion 2008;8:313–8. doi: 10.1016/j.mito.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 2. Silva MH, Rosa EJF, Carvalho NR et al. Acute brain damage induced by acetaminophen in mice: effect of diphenyl diselenide on oxidative stress and mitochondrial dysfunction. Neurotox Res 2012;21:334–44. doi: 10.1007/s12640-011-9288-1. [DOI] [PubMed] [Google Scholar]
- 3. Larson A, Polson J, Fontana R. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005;42:1364–72. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 4. Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 2012;44:88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nassini R, Materazzi S, Andrè E et al. Acetaminophen, via its reactive metabolite N -acetyl- p -benzo-quinoneimine and transient receptor potential ankyrin-1 stimulation, causes neurogenic inflammation in the airways and other tissues in rodents. FASEB J 2010;24:4904–16. doi: 10.1096/fj.10-162438. [DOI] [PubMed] [Google Scholar]
- 6. Brusilow SW, Cooper AJL. Encephalopathy in acute liver failure resulting from acetaminophen intoxication: new observations with potential therapy. Crit Care Med 2011;39:2550–3. doi: 10.1097/CCM.0b013e31822572fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Norenberg MD, Jayakumar AR, Rama Rao KV. Oxidative stress in the pathogenesis of hepatic encephalopathy. Metab Brain Dis 2004;19:313–29. doi: 10.1023/b:mebr.0000043978.91675.79. [DOI] [PubMed] [Google Scholar]
- 8. Bosoi CR, Rose CF. Oxidative stress: a systemic factor implicated in the pathogenesis of hepatic encephalopathy. Metab Brain Dis 2013;28:175–8. doi: 10.1007/s11011-012-9351-5. [DOI] [PubMed] [Google Scholar]
- 9. Brown JM, Ball JG, Hogsett A et al. Temporal study of acetaminophen (APAP) and S-adenosyl-L-methionine (SAMe) effects on subcellular hepatic SAMe levels and methionine adenosyltransferase (MAT) expression and activity. Toxicol Appl Pharmacol 2010;247:1–9. doi: 10.1016/j.taap.2010.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carvalho NR, Rosa EF, Silva MH et al. New therapeutic approach: diphenyl diselenide reduces mitochondrial dysfunction in acetaminophen-induced acute liver failure. PLoS One 2013;8:e81961. doi: 10.1371/journal.pone.0081961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lv X, Chen Z, Li J et al. Caffeine protects against alcoholic liver injury by attenuating inflammatory response and oxidative stress. Inflamm Res 2010;59:635–45. doi: 10.1007/s00011-010-0176-6. [DOI] [PubMed] [Google Scholar]
- 12. Chen S, Teoh NC, Chitturi S et al. Coffee and non-alcoholic fatty liver disease: brewing evidence for hepatoprotection? J Gastroenterol Hepatol 2014;29:435–41. doi: 10.1111/jgh.12422. [DOI] [PubMed] [Google Scholar]
- 13. Asadullina NR, Gudkov SV, Bruskov VI. Caffeine modifies effects of X-ray action on mice after exposure to radiation and exhibits radioprotective properties. Dokl Biochem Biophys 2012;442:22–5. doi: 10.1134/S1607672912010073. [DOI] [PubMed] [Google Scholar]
- 14. Dubroqua S, Yee BK, Singer P. Sensorimotor gating is disrupted by acute but not chronic systemic exposure to caffeine in mice. Psychopharmacology (Berl) 2014;231:4087–98. doi: 10.1007/s00213-014-3548-8 [DOI] [PubMed] [Google Scholar]
- 15. Cechella JL, Leite MR, Dobrachinski F et al. Moderate swimming exercise and caffeine supplementation reduce the levels of inflammatory cytokines without causing oxidative stress in tissues of middle-aged rats. Amino Acids 2014;46:1187–95. doi: 10.1007/s00726-014-1679-1. [DOI] [PubMed] [Google Scholar]
- 16. Sardão VA, Oliveira PJ, Moreno AJM. Caffeine enhances the calcium-dependent cardiac mitochondrial permeability transition: relevance for caffeine toxicity. Toxicol Appl Pharmacol 2002;179:50–6. doi: 10.1006/taap.2001.9334. [DOI] [PubMed] [Google Scholar]
- 17. Dragicevic N, Delic V, Cao C et al. Caffeine increases mitochondrial function and blocks melatonin signaling to mitochondria in Alzheimer’s mice and cells. Neuropharmacology 2012;63:1368–79. doi: 10.1016/j.neuropharm.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 18. Raińska-Giezek T. Influence of caffeine on toxicity and pharmacokinetics of paracetamol. Ann Acad Med Stetin 1995;41:69–85. http://www.ncbi.nlm.nih.gov/pubmed/8615554(10 March 2020, date last accessed). [PubMed] [Google Scholar]
- 19. Palmer H, Graham G, Williams K et al. A risk-benefit assessment of paracetamol (acetaminophen) combined with caffeine. Pain Med 2010;11:951–65. doi: 10.1111/j.1526-4637.2010.00867.x. [DOI] [PubMed] [Google Scholar]
- 20. Vaughan RA, Vaughan R, Randi Garcia-Smith M et al. Effects of caffeine on metabolism and mitochondria biogenesis in rhabdomyosarcoma cells compared with 2,4-dinitrophenol. Nutr Metab Insights 2012;5:59. doi: 10.4137/NMI.S10233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carvalho NR, Tassi CC, Dobraschinski F et al. Reversal of bioenergetics dysfunction by diphenyl diselenide is critical to protection against the acetaminophen-induced acute liver failure. Life Sci 2017;180:42–50. doi: 10.1016/j.lfs.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 22. Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol 2009;41:1837–45. doi: 10.1016/J.BIOCEL.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 23. DiPetrillo K, Wood S, Kostrubsky V et al. Effect of caffeine on acetaminophen hepatotoxicity in cultured hepatocytes treated with ethanol and isopentanol. Toxicol Appl Pharmacol 2002;185:91–7. doi: 10.1006/taap.2002.9535. [DOI] [PubMed] [Google Scholar]
- 24. Lee CA, Thummel KE, Kalhorn TF, et al. Inhibition and activation of acetaminophen reactive metabolite formation by caffeine. Roles of cytochromes P-450IA1 and IIIA2. Drug Metab Dispos 1991;19:348–53. http://www.ncbi.nlm.nih.gov/pubmed/1676635 (10 March 2020, date last accessed). [PubMed] [Google Scholar]
- 25. Tonge RP, Kelly EJ, Bruschi SA et al. Role of CYP1A2 in the hepatotoxicity of acetaminophen: investigations using Cyp1a2 null mice. Toxicol Appl Pharmacol 1998;153:102–8. doi: 10.1006/taap.1998.8543. [DOI] [PubMed] [Google Scholar]
- 26. Ohta A, Lukashev D, Jackson EK, et al. 1,3,7-trimethylxanthine (caffeine) may exacerbate acute inflammatory liver injury by weakening the physiological immunosuppressive mechanism. J Immunol 2007;179:7431–8. http://www.ncbi.nlm.nih.gov/pubmed/18025187 (4 September 2015, date last accessed). [DOI] [PubMed] [Google Scholar]
- 27. Burtscher J, Zangrandi L, Schwarzer C et al. Differences in mitochondrial function in homogenated samples from healthy and epileptic specific brain tissues revealed by high-resolution respirometry. Mitochondrion 2015;25:104–12. doi: 10.1016/j.mito.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 28. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54. http://www.ncbi.nlm.nih.gov/pubmed/942051 (9 July 2014, date last accessed). [DOI] [PubMed] [Google Scholar]
- 29. Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979;95:351–8. http://www.ncbi.nlm.nih.gov/pubmed/36810 (19 December 2016, date last accessed). [DOI] [PubMed] [Google Scholar]
- 30. Dionisio N, Garcia-Mediavilla MV, Sanchez-Campos S et al. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J Hepatol 2009;50:872–82. doi: 10.1016/j.jhep.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 31. Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem 1976;74:214–26. http://www.ncbi.nlm.nih.gov/pubmed/962076 (4 September 2015, date last accessed). [DOI] [PubMed] [Google Scholar]
- 32. Misra HP, Fridovich I. The generation of superoxide radical during the autoxidation of hemoglobin. J Biol Chem 1972;247:6960–2. http://www.ncbi.nlm.nih.gov/pubmed/4673289 (10 September 2016, date last accessed). [PubMed] [Google Scholar]
- 33. Aebi H. Catalase in vitro. Methods Enzymol 1984;105:121–6. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 34. Morin C, Zini R, Simon N et al. Low glucocorticoid concentrations decrease oxidative phosphorylation of isolated rat brain mitochondria: an additional effect of dexamethasone. Fundam Clin Pharmacol 2000;14:493–500. doi: 10.1111/J.1472-8206.2000.TB00432.X. [DOI] [PubMed] [Google Scholar]
- 35. Flohé S, Dominguez Fernández E, Ackermann M et al. Endotoxin tolerance in rats: expression of TNF-alpha, IL-6, IL-10, VCAM-1 and HSP 70 in lung and liver during endotoxin shock. Cytokine 1999;11:796–804. doi: 10.1006/cyto.1998.0490. [DOI] [PubMed] [Google Scholar]
- 36. Carlberg I, Mannervik B, Glutathione reductase. Methods Enzymol 1985;113:484–90. http://www.ncbi.nlm.nih.gov/pubmed/3003504 (29 August 2017, date last accessed). [DOI] [PubMed] [Google Scholar]
- 37. Krumschnabel G, Fontana-Ayoub M, Sumbalova Z et al. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol Biol 2015;1264:245–61. doi: 10.1007/978-1-4939-2257-4_22. [DOI] [PubMed] [Google Scholar]
- 38. Schöpf B, Schäfer G, Weber A et al. Oxidative phosphorylation and mitochondrial function differ between human prostate tissue and cultured cells. FEBS J 2016;283:2181–96. doi: 10.1111/febs.13733. [DOI] [PubMed] [Google Scholar]
- 39. Kuznetsov AV, Schneeberger S, Seiler R et al. Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am J Physiol Heart Circ Physiol 2004;286:H1633–41. doi: 10.1152/ajpheart.00701.2003. [DOI] [PubMed] [Google Scholar]
- 40. McGill MR, Williams CD, Xie Y et al. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol 2012;264:387–94. doi: 10.1016/j.taap.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Posadas I, Santos P, Blanco A et al. Acetaminophen induces apoptosis in rat cortical neurons. PLoS One 2010;5:e15360. doi: 10.1371/journal.pone.0015360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferguson CS, Tyndale RF. Cytochrome P450 enzymes in the brain: emerging evidence of biological significance. Trends Pharmacol Sci 2011;32:708–14. doi: 10.1016/j.tips.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Devasagayam TP, Kamat JP, Mohan H et al. Caffeine as an antioxidant: inhibition of lipid peroxidation induced by reactive oxygen species. Biochim Biophys Acta 1996;1282:63–70. http://www.ncbi.nlm.nih.gov/pubmed/8679661 (13 June 2016, date last accessed). [DOI] [PubMed] [Google Scholar]
- 44. Shi X, Dalal NS, Jain AC. Antioxidant behaviour of caffeine: efficient scavenging of hydroxyl radicals. Food Chem Toxicol 1991;29:1–6. http://www.ncbi.nlm.nih.gov/pubmed/1847890 (6 July 2017, date last accessed). [DOI] [PubMed] [Google Scholar]
- 45. Xu K, Xu Y-H, Chen J-F et al. Neuroprotection by caffeine: time course and role of its metabolites in the MPTP model of Parkinson’s disease. Neuroscience 2010;167:475–81. doi: 10.1016/j.neuroscience.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martínez-López S, Sarriá B, Baeza G et al. Pharmacokinetics of caffeine and its metabolites in plasma and urine after consuming a soluble green/roasted coffee blend by healthy subjects. Food Res Int 2014;64:125–33. doi: 10.1016/j.foodres.2014.05.043. [DOI] [PubMed] [Google Scholar]
- 47. Mukhopadhyay S, Mondal A, Poddar MK. Chronic administration of caffeine: effect on the activities of hepatic antioxidant enzymes of Ehrlich ascites tumor-bearing mice. Indian J Exp Biol 2003;41:283–9. http://www.ncbi.nlm.nih.gov/pubmed/15255635 (10 March 2020, date last accessed). [PubMed] [Google Scholar]
- 48. Chia AJL, Goldring CE, Kitteringham NR et al. Differential effect of covalent protein modification and glutathione depletion on the transcriptional response of Nrf2 and NF-kappaB. Biochem Pharmacol 2010;80:410–21. doi: 10.1016/j.bcp.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Abreu RV, Silva-Oliveira EM, Moraes MFD et al. Chronic coffee and caffeine ingestion effects on the cognitive function and antioxidant system of rat brains. Pharmacol Biochem Behav 2011;99:659–64. doi: 10.1016/j.pbb.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 50. Casula EP, Bisiacchi PS, Corrias M et al. Acute hyperammonaemia induces a sustained decrease in vigilance, which is modulated by caffeine. Metab Brain Dis 2015;30:143–9. doi: 10.1007/s11011-014-9590-8. [DOI] [PubMed] [Google Scholar]
- 51. Thiel C, Thiel K, Etspueler A et al. A reproducible porcine model of acute liver failure induced by intrajejunal acetaminophen administration. Eur Surg Res 2011;46:118–26. doi: 10.1159/000323411. [DOI] [PubMed] [Google Scholar]
- 52. Horrigan LA, Kelly JP, Connor TJ. Immunomodulatory effects of caffeine: friend or foe? Pharmacol Ther 2006;111:877–92. doi: 10.1016/j.pharmthera.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 53. Kempf K, Herder C, Erlund I et al. Effects of coffee consumption on subclinical inflammation and other risk factors for type 2 diabetes: a clinical trial. Am J Clin Nutr 2010;91:950–7. doi: 10.3945/AJCN.2009.28548. [DOI] [PubMed] [Google Scholar]
- 54. Nabbi-Schroeter D, Elmenhorst D, Oskamp A et al. Effects of long-term caffeine consumption on the adenosine A1 receptor in the rat brain: an in vivo PET study with [18F]CPFPX. Mol Imaging Biol 2018;20:284–91. doi: 10.1007/s11307-017-1116-4. [DOI] [PubMed] [Google Scholar]
- 55. Tellone E, Galtieri A, Russo A et al. Protective effects of the caffeine against neurodegenerative diseases. Curr Med Chem 2019;26:5137–51. doi: 10.2174/0929867324666171009104040. [DOI] [PubMed] [Google Scholar]
- 56. Dodd FL, Kennedy DO, Riby LM et al. A double-blind, placebo-controlled study evaluating the effects of caffeine and L-theanine both alone and in combination on cerebral blood flow, cognition and mood. Psychopharmacology (Berl) 2015;232:2563–76. doi: 10.1007/s00213-015-3895-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gupta S, Rogers LK, Taylor SK et al. Inhibition of carbamyl phosphate synthetase-I and glutamine synthetase by hepatotoxic doses of acetaminophen in mice. Toxicol Appl Pharmacol 1997;146:317–27. doi: 10.1006/taap.1997.8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Michael Brown J, Ball JG, Wright MS et al. Novel protective mechanisms for S-adenosyl-l-methionine against acetaminophen hepatotoxicity: improvement of key antioxidant enzymatic function. Toxicol Lett 2012;212:320–8. doi: 10.1016/j.toxlet.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shmigol A, Kirischuk S, Kostyuk P et al. Different properties of caffeine-sensitive Ca2+ stores in peripheral and central mammalian neurones. Pflugers Arch 1994;426:174–6. doi: 10.1007/bf00374686. [DOI] [PubMed] [Google Scholar]
- 60. McPhersonx PS, Kim Y-K, Valdivia H et al. The brain ryanodine receptor: a caffeine-sensitive calcium release channel. Neuron 1991;7:17–25. doi: 10.1016/0896-6273(91)90070-G. [DOI] [PubMed] [Google Scholar]