

Abstract
Manganese (Mn) is an essential micronutrient. However, it is well established that Mn overexposure causes nervous system diseases. In contrast, there are few reports on the effects of Mn exposure on glomerular endothelium. In the present study, the potential effects of Mn exposure on glomerular endothelium were evaluated. Sprague Dawley rats were used as a model of Mn overexposure by intraperitoneal injection of MnCl2·H2O at 25 mg/kg body weight. Mn exposure decreased expression of vascular endothelial-cadherin, a key component of adherens junctions, and increased exudate from glomeruli in Sprague Dawley rats. Human renal glomerular endothelial cells were cultured with different concentration of Mn. Exposure to 0.2 mM Mn increased permeability of human renal glomerular endothelial cell monolayers and decreased vascular endothelial-cadherin expression without inducing cytotoxicity. In addition, Mn exposure increased phosphorylation of mothers against decapentaplegic homolog 2/3 and upregulated expression of zinc finger protein SNAI1, a negative transcriptional regulator of vascular endothelial-cadherin. Our data suggest Mn exposure may contribute to development of glomerular diseases by inducing permeability of glomerular endothelium.
Keywords: manganese, glomerular endothelial cells, VE-cadherin, adherens junction, glomerular filtration barrier
Graphical Abstract
Graphical Abstract.
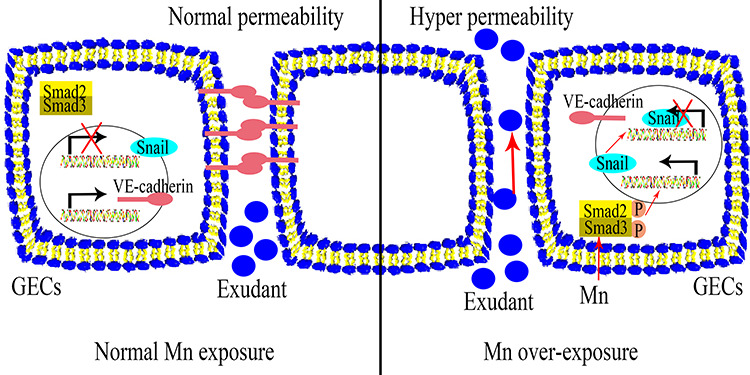
Introduction
Manganese (Mn) is an essential trace metal that participates in regulation of various physiological processes. Excessive environmental or occupational exposure to Mn is harmful [1]. Workers in the mining, welding, and smelting industries are continuously exposed to Mn and have higher risk of manganism [2]. Therefore, occupational exposure is likely to induce Mn overexposure, making Mn poisoning a common occupational disease. Mn poisoning leads to hepatic cirrhosis, polycythemia, hypermanganesemia, dystonia, and parkinsonism-like symptoms [3]. Mn poisoning primarily affects the central nervous system [4]; however, increased Mn deposition is also found in peripheral organs such as the kidney and liver [5]. Although the toxic effects of Mn on the nervous system have been thoroughly investigated, the effects of Mn exposure on other target tissues have received less attention.
Vascular endothelium is the primary channel for Mn transport. After gastrointestinal absorption, inhalation, intravenous injection, or skin exposure [1, 6], Mn is transported to different tissues through blood circulation. It is noteworthy that fetal health is also affected by maternal exposure of Mn. Through in utero exposure, average Mn concentration in umbilical cord blood is higher than in whole maternal blood [7]. There are few reports on the toxic effects of excessive Mn on vascular endothelium. Administration of physiological levels of Mn effectively inhibits injury to vascular endothelial cells induced by advanced glycation endproducts in patients with diabetes [8]. However, overexposure of Mn has been reported to lower diastolic blood pressure and impair cardiovascular function [9].
The kidney is also a target of metal toxicity, including Mn [10–12]. Roughly 5% of blood Mn is distributed in the kidneys [13]. Excess Mn in systemic circulation is filtrated by the glomerular filtration barrier (GFB) [14]. Glomerular endothelial cells (GECs) are specialized microvascular cells that contain fenestrations with endothelial glycocalyx on the cell membrane [15, 16]. GECs, together with glomerular basement membrane and podocytes, form the GFB that facilitates blood filtration to maintain body homeostasis [14]. Endothelial cells barrier function is important for maintaining the GFB. Loss of this function impairs the GFB and leads to proteinuria and hematuria [17]. It was reported that accidental ingestion of Mn causes acute renal failure [18]. However, there are fewer reports on the effects of Mn on the GFB.
Except for fenestrations, endothelial cells barrier function is also maintained in part by adherens junctions [19]. Vascular endothelial (VE)-cadherin is an adherens junction protein expressed specifically in endothelial cells. VE-cadherin interacts with β-catenin and connects indirectly with the actin cytoskeleton. Therefore, proper localization and function of VE-cadherin determine the permeability and function of the endothelial cell barrier [20]. Adherens junctions are dynamic and tightly regulated by extracellular stimuli and intracellular signaling pathways [21–23]. The transforming growth factor beta (TGF-β) mothers against decapentaplegic homolog 2/3 (Smad2/3) signaling pathway participates in regulation, localization, and expression of VE-cadherin in human renal glomerular endothelial cells (HRGECs) [24]. The effects of Mn exposure on endothelial barrier function and the Smad2/3 signaling pathway have not been reported.
We hypothesized that Mn affects the GFB by increasing GEC permeability. HRGECs were cultured and treated with different concentration of Mn. High concentration (5.0 mM) inhibited proliferation and induced apoptosis of HRGECs. Low concentration (0.2 and 1.0 mM) increased HRGEC permeability without affecting proliferation or causing apoptosis. In addition, Mn exposure increased Smad2/3 phosphorylation, upregulated expression of the zinc finger protein, SNAI1 (Snail), and reduced expression of VE-cadherin in HRGECs. To mimic Mn overexposure in vivo, Sprague Dawley (SD) rats were used. Although morphological changes of glomeruli were not observed in the Mn exposure group, we found increased protein exudate and downregulation of VE-cadherin in glomeruli. Our results suggest Mn overexposure causes GFB dysfunction by increasing permeability of GECs.
Materials and Methods
Animals and Mn exposure protocol
In our previous study, we described a rat model of Mn poisoning [25]. All procedures related to animals were performed in compliance with guidelines of the Animal Care and Use Committee of Shandong Academy of Medical Sciences. Briefly, healthy male SD rats aged 6–8 weeks and weighing roughly 200 g were housed in a specific pathogen-free animal facility (licensed No. SYXK20140012) at constant temperature and humidity, and with a 12-h light/dark cycle. A total of 12 male SD rats were divided into two groups. In the Mn exposure group, 25 mg/kg body weight MnCl2·H2O were injected intraperitoneally every 48 h for 3 months. In the control group, equal amounts of physiological saline were injected intraperitoneally. After 3 months, animals were sacrificed. Kidney tissues were harvested for the subsequent analyses. The Ethics Committee of Shandong Academy of Medical Sciences approved the experiments involving rats, which were performed in accordance with animal research reporting in vivo experiments (ARRIVE) guidelines, and with relevant experimental principles and standards.
Cell culture
HRGECs were obtained from ScienCell Research Laboratories and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Corning) with 10% fetal bovine serum (Lonza) and antibiotics (100 IU/ml penicillin and 100 mg/ml streptomycin) at 37°C in a humidified atmosphere (5% CO2/21% O2). MnCl2·H2O was obtained from Sigma-Aldrich. Varying concentrations of MnCl2·H2O (0.04, 0.2, 1.0, and 5.0 mM) were applied to HRGEC cultures for 24 h. Physiological saline was used as control.
Fluorescein isothiocyanate-dextran transwell assay
HRGECs were cultured in phenol red-free DMEM for fluorescein isothiocyanate (FITC)-dextran transwell assay. Transwell chambers were placed in 24-well plates and incubated in DMEM (phenol red free) for 1 h. In total, 600 μl medium was added to each well of 24-well plates, and 200 μl HRGEC suspensions (3.5 × 104 cells) were seeded on the insert of chambers and cultured until confluent. Different concentrations of MnCl2·H2O were added to each chamber and incubated for 24 h. Chambers were washed with hydroxyethyl piperazineethanesulfonic acid (HEPES) medium, 30 μl (10 mg/ml) FITC-dextran (Invitrogen) was added to the top chamber, and 600 μl phenol red-free DMEM was added to the outside of chamber. After incubation for 30 min, transwell chambers were removed and read in a fluorometer at excitation of 490 nm and emission of 520 nm.
Methyl thiazolyl tetrazolium (MTT) assay
MTT assay was performed to determine the effect of Mn on cell proliferation. Briefly, HRGECs were seeded in 96-well plates (5 × 103/well) and cultured overnight. The next day, various concentrations of MnCl2·H2O were added, and cells were incubated for 24 h at 37°C in a humidified atmosphere. Before detection, 10 μl MTT solutions (5 mg/ml) was added to each well and incubated for 4 h. Next, media was discarded, and formazan crystals were solubilized with dimethyl sulfoxide. Colorimetric intensity was analysed using a 96-well plate reader at wavelength of 570 nm.
Flow cytometry
Apoptosis of HRGECs treated with Mn was analysed by annexin V-FITC and propidium iodide (PI) staining in accordance with the manufacturer’s introduction (NeoBiosciences). Briefly, HRGECs were cultured to density of 90% and incubated with different concentrations of MnCl2·H2O for 24 h. HRGECs were then harvested by trypsinization, washed twice with ice-cold phosphate-buffered saline (PBS), and incubated with binding buffer containing annexin V-FITC (0.25%) and PI (1 μg/ml). A FACSAria II flow cytometer (BD Biosciences) was used to detect positively stained cells. Data were analysed with FACSDiva acquisition and analysis software version 11.5 (BD Biosciences).
Trypan blue staining
HRGECs were incubated with physiological saline or 0.2 mM MnCl2·H2O for 24 h. HRGECs were then digested with trypsin. Single-cell suspensions were prepared by diluting HRGECs with PBS. After cell counting, 1 × 106/ml HRGECs were incubated with 0.4% trypan blue dye, and the living and dead cells were counted. Cell viability (%) = number of living cells ÷ (number of living cells + number of dead cells) × 100%.
Hematoxylin and eosin staining and immunohistochemistry
Hematoxylin and eosin (H&E) staining and immunohistochemistry experiment were performed as previously described [26]. Briefly, the kidney tissue samples from the different groups of rats were fixed in 4% paraformaldehyde at room temperature for 24 h. Samples were dehydrated, cleared, and embedded in paraffin for sectioning. Paraffin sections were dewaxed and dehydrated using an alcohol gradient. For H&E staining, sections were immersed in 1% hematoxylin (Yuanmu Biotechnology Co., Ltd) for 5 min and washed in distilled water. A 1% hydrochloric acid–alcohol solution was used to clear residual dye, and then 0.5% eosin was used to stain the sections (Huihong Reagent Co., Ltd). Following gradient ethanol dehydration, sections were cleared in dimethylbenzene and sealed with resinene (Boster Biological Engineering). For immunohistochemistry, sections were heated, blocked with hydrogen peroxide at room temperature, and then incubated with primary antibody, rabbit anti-VE-cadherin (1:50, Invitrogen). After rinsing with PBS, sections were incubated with secondary antibody from the MaxVisionTM HRP-Polymer anti-Mouse/Rabbit IHC Kit (Maixin, China). Both H&E-stained sections and those for immunohistochemistry were observed using a BX51 microscopic imaging system (Olympus).
Quantitative real-time polymerase chain reaction
Total RNA was extracted with RNAiso Plus (TaKaRa), and cDNA was synthesized using a PrimeScript First Strand cDNA Synthesis Kit (TaKaRa) in accordance with the manufacturer’s instructions. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed by monitoring increase in fluorescence of SYBR green dye (Tiangen) using a CFX96TM Real-Time System (Bio-Rad). All PCR reactions were performed in triplicate. Relative expression of RNA was calculated using actin as an endogenous internal control. Primer sequences are shown in Table 1.
Table 1.
Sequence information of the primers in this work
| Species | Gene | Sequence |
|---|---|---|
| Human | VE-cadherin | 5′-GCGACTACCAGGACGCTTTCA-3′ |
| 5′-CATGTATCGGAGGTCGATGGTG-3′ | ||
| Actin | 5′-TTGCCGACAGGATGCAGAA-3′ | |
| 5′-GCCGATCCACACGGAGTACT-3′ | ||
| Rat | VE-cadherin | 5′-ATGAGGTCGGTGCCCGTATT-3′ |
| 5′-CGTTGGTCTTGGGGTCTGTGA-3′ | ||
| Actin | 5′-GGGAAGGACGACAGGTAACG-3′ | |
| 5′-TTCTTTGACATCCCGCACGA-3′ |
Western blot
HRGECs were washed with ice-cold PBS and lysed in ice-cold RIPA containing protease inhibitors (Beyotime Biotechnology). Renal tissue was disrupted with Tissuelyser-24 (JingXin Inc.) and lysed in ice-cold RIPA containing protease inhibitors. Protein concentration was measured using a BCA Protein Assay Kit (Invitrogen). Western blot was performed as previously described [27]. Primary antibodies included rabbit anti-VE-cadherin (1:2000, Abcam), rabbit anti-phospho-Smad2 (pSer255) (1:1000, Abcam), rabbit anti-Smad2 (1:1000, Abcam), rabbit anti-phospho-Smad3 (pSer423/425), rabbit anti-Smad3, and mouse anti-Snail (1:1000, Cell Signaling Technology). α-Tubulin (1:3000, Abcam) was used as the loading control. Secondary antibodies were horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) and HRP-conjugated goat anti-mouse IgG (1:10 000, Proteintech).
Immunofluorescence
HRGECs were seeded in 35-mm glass bottom dishes (1 × 105/dish) and cultured overnight. The next day, 0.2 mM MnCl2·H2O was added and incubated at 37°C in a humidified atmosphere for 24 h. Media was aspirated and monolayers were washed twice with ice-cold PBS, then fixed with 4% paraformaldehyde, and washed thrice with PBS. Next, 0.2% Triton X-100 was added to increase cell permeability and 5% bovine serum albumin was used to block nonspecific antibody binding. Primary antibody, rabbit anti-VE-cadherin (1:200; Abcam), and an Alexa Fluor 488 goat anti-rabbit secondary antibody (1:200; Life Technologies) were used for immunofluorescence. 4′,6-diamidino-2-phenylindole (DAPI) was used to stain nuclei. Dishes were photographed using an Olympus LCX100 Imaging System (Olympus Corporation).
Statistical analysis
All experiments were repeated independently at least three times, and data are expressed as mean ± SEM. For animal experiments, there were six rats in each group. However, during tissue processing, one kidney sample from the Mn exposure group was lost. Therefore, five samples each from the control and Mn exposure groups were used for experiments. All statistical analyses were performed using GraphPad Pro Prism 6.0. Student’s t-test or one-way analysis of variance was used for statistical analysis. P < 0.05 was considered statistically significant.
Results
Effects of Mn exposure on rat glomeruli and VE-cadherin expression
To determine the effect of Mn exposure on permeability of GECs, healthy SD rats were used to perform in vivo Mn exposure analysis. As shown in Fig. 1, infiltration of inflammatory cells and hyperplasia were not observed in glomeruli from the Mn exposure group. There were no morphological differences in glomeruli between the control and Mn exposure groups. However, protein exudate from glomeruli was increased in the Mn exposure group (Fig. 1A). These observations suggest Mn exposure does not induce inflammation or morphological changes of glomeruli, although it increases accumulation of protein exudate. According to western blot and qRT-PCR, both VE-cadherin protein and mRNA levels were significantly decreased in rat renal tissue exposed to Mn (Fig. 1B–D; **P < 0.01). Moreover, immunohistochemistry of rat glomeruli showed that VE-cadherin expression was decreased in glomerular cells in the Mn exposure group (Fig. 1E–F). These results demonstrate that Mn exposure induces protein exudate accumulation in rat glomeruli by inhibiting expression of VE-cadherin in GECs and increasing their permeability.
Figure 1.
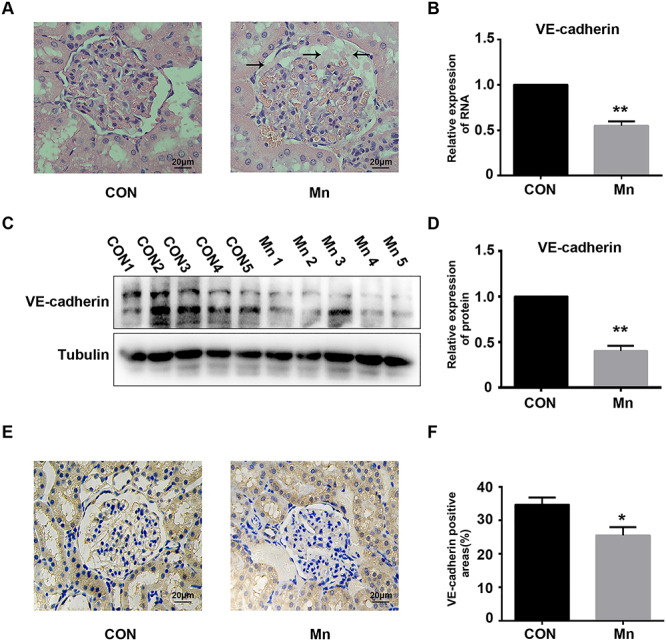
Effects of Mn exposure on VE-cadherin expression in a rat model. (A) Representative H&E stain of glomerular tissue from rats treated with or without Mn. Protein exudates in the Mn exposure group are indicated with black arrows. (B) Relative VE-cadherin mRNA expression in rat glomeruli measured by qRT-PCR (**P < 0.01 vs. control). (C) VE-cadherin expression in rat glomeruli was analysed by western blot. (D) Relative expression of VE-cadherin (**P < 0.01 vs. control). (E) Detection of VE-cadherin expression in rat glomeruli by immunohistochemistry. (F) Positive areas of VE-cadherin staining in glomeruli (*P < 0.05 vs. control).
Mn treatment induces hyperpermeability of HRGECs
Endothelial dysfunction increases vascular permeability and decreased glomerular filtration rate [28]. The permeability of confluent HRGEC monolayers exposed to different concentrations of Mn was examined by FITC-dextran transwell assay. Treatment with 0.2, 1.0, and 5.0 mM Mn for 24 h significantly and dose dependently increased fluorescence intensity compared with the control group (Fig. 2; *P < 0.05, **P < 0.01). However, 0.04-mM Mn treatment for 24 h did not change the permeability of HRGEC monolayers (Fig. 2). Mn exposure therefore dose dependently increases permeability of HRGECs.
Figure 2.
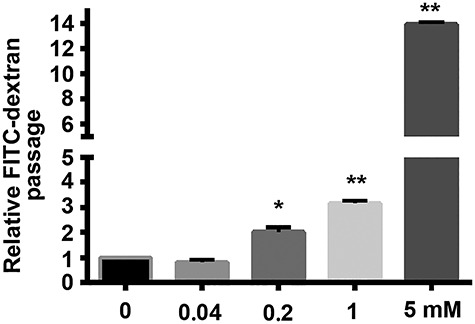
Effects of Mn on permeability of HRGEC monolayers. HRGEC monolayers were treated with 0, 0.04, 0.2, 1.0, and 5.0 mM Mn for 24 h, and then analysed by FITC-dextran transwell assay (n = 6, *P < 0.05, **P < 0.01 vs. control).
Effects of Mn treatment on HRGEC proliferation and apoptosis
To confirm that the increase in permeability of HRGECs was not caused by cell proliferation inhibition or induction of apoptosis, MTT assay was performed to assess proliferation. According to MTT assay, 0.04, 0.2, and 1-mM Mn treatment for 24 h did not affect cell growth, whereas 5.0-mM Mn treatment for 24 h significantly inhibited proliferation of HRGECs (Fig. 3C; **P < 0.01). HRGECs were stained with annexin V-FITC and PI. HRGEC apoptosis was detected by flow cytometry. According to the results, 0.04, 0.2, and 1.0-mM Mn treatment for 24 h did not increase the percentage of HRGECs in early and late apoptotic stages (Fig. 3A and B). However, 5.0-mM Mn treatment for 24 h increased the percentage of HRGECs in the Q1 region (Q1; 5.0 mM Mn, 14.15 ± 0.75% vs. control, 1.15 ± 0.25%; P < 0.01) and the percentage of HRGECs in early or late apoptotic stages (Q2 + Q4; 5.0 mM Mn, 5.20 ± 0.50% vs. control, 1.55 ± 0.25%; P < 0.05) compared with control (Fig. 3A). Therefore, 0.04, 0.2, and 1.0-mM Mn exposure for 24 h did not affect proliferation or apoptosis of HRGECs, whereas 0.2-mM Mn treatment increased permeability of HRGECs without affecting cell proliferation or apoptosis. Trypan blue staining was performed to evaluate cell membrane integrality. As showed in Supplementary Fig. 1, no significant difference was found between the control and 0.2-mM Mn treated group. This demonstrates that no other forms of cell death such as necroptosis and ferroptosis caused rupture of the cell membrane after 0.2-mM Mn treatment.
Figure 3.
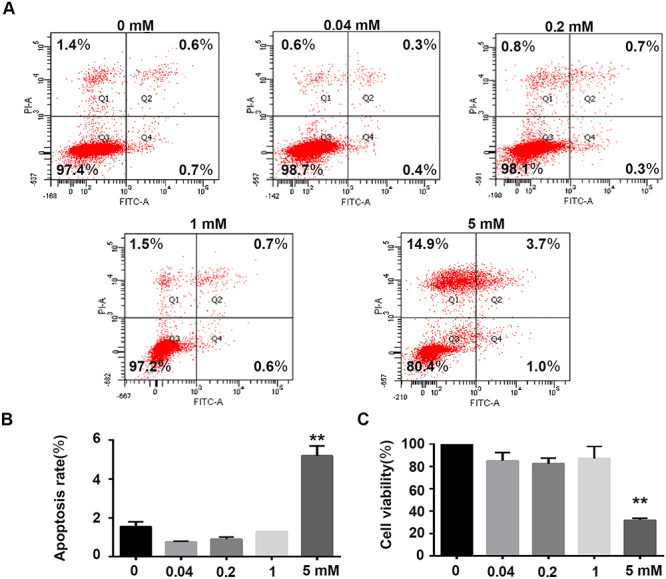
Effects of Mn on growth and apoptosis of HRGECs. (A) Representative image of flow cytometric analysis of HRGECs. Cells were stained with annexin V/PI following exposure with or without Mn for 24 h. (B) Rate of apoptosis of HRGECs (n = 3, **P < 0.01 vs. control). (C) HRGECs treated with 0, 0.04, 0.2, 1.0, and 5.0 mM Mn for 24 h, and then analysed by MTT assay (n = 6, **P < 0.01 vs. control).
Mn treatment inhibits expression of VE-cadherin in HRGECs
Adherens junctions are crucial for maintenance of endothelial cell permeability. We explored the effects of Mn on expression of VE-cadherin, a protein that maintains cell junctions. Using western blot, we found that 0.2, 1.0, and 5.0-mM Mn treatment decreased expression of VE-cadherin (Fig. 4A; **P < 0.01). qRT-PCR confirmed that Mn treatment decreased expression of VE-cadherin (Fig. 4B; *P < 0.05, **P < 0.01). The expression and localization of VE-cadherin were detected by immunofluorescence in HRGECs after 0.2-mM Mn treatment for 24 h. VE-cadherin was localized to cell–cell contacts in the control group, whereas Mn exposure reduced expression and localization of VE-cadherin at cell–cell contacts (Fig. 4C). Therefore, Mn treatment affects cell junctions of HRGECs by inhibiting expression of VE-cadherin.
Figure 4.
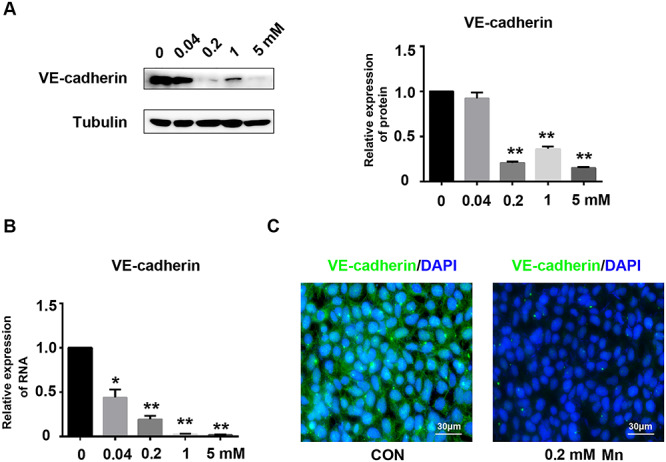
Mn treatment inhibits expression of VE-cadherin in HRGECs. (A) HRGECs were treated with 0, 0.04, 0.2, 1.0, or 5.0 mM Mn for 24 h. VE-cadherin expression was analysed by western blot (n = 3, **P < 0.01 vs. control). (B) Relative VE-cadherin mRNA expression measured by qRT-PCR in HRGEC monolayers exposure to different concentrations of Mn for 24 h (n = 3, *P < 0.05, **P < 0.01). (C) Immunofluorescence staining of VE-cadherin on HRGEC monolayers treated with or without 0.2 mM Mn for 24 h.
The effects of Mn treatment on Smad2/3-Snail signal pathway of HRGECs
The Smad2/3-Snail signaling pathway negatively regulates expression of VE-cadherin in HRGECs [24]. In this study, we examined whether 0.2-mM Mn exposure inhibits expression of VE-cadherin by activating the Smad2/3-Snail signaling pathway. Western blot shows that 0.2-mM Mn treatment induced phosphorylation of both Smad2 and Smad3 in HRGECs compared with control (Fig. 5A–C; **P < 0.01). Accordingly, expression of the downstream signaling molecule, Snail, was increased by Mn treatment (Fig. 5D–F; **P < 0.01). Therefore, Mn inhibits expression of VE-cadherin by activating the Smad2/3-Snail signaling pathway.
Figure 5.
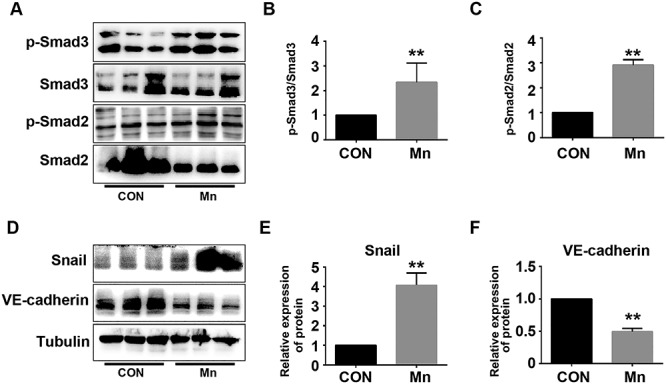
Mn treatment increases phosphorylation of Smad2/3 and inhibits VE-cadherin expression. (A) HRGECs were treated with 0.2 mM Mn for 24 h. Phosphorylation of Smad2/3 and total Smad2/3 were analysed by western blot. (B) Phospho-Smad3/total Smad3 (**P < 0.01 vs. control). (C) Phospho-Smad2/total Smad2 (n = 3, **P < 0.01 vs. control). (D) Expression of Snail and VE-cadherin were analysed by western blot. (E) Relative expression of Snail (**P < 0.01 vs. control). (F) Relative expression of VE-cadherin (n = 3, **P < 0.01 vs. control).
Discussion
Mn is an essential micronutrient that acts as cofactor for a number of enzymes that participate in cellular processes [29]. Excessive intake of Mn is toxic to cells. The kidney is one of five tissues with the highest accumulation of Mn in the body. Overall, 5% of Mn is distributed from plasma to the kidneys, which is much higher than in the brain (0.1%) [30]. The nervous system has the highest sensitivity to excessive Mn. It has been reported that excessive Mn induces brain hemorrhage in SD rat [31], which provides evidence for endothelium impairment from Mn overexposure. In this study, we explored the effects of Mn on GECs and glomeruli. We found that Mn exposure induced protein exudate from rat glomeruli, reduced VE-cadherin expression, and increased GEC permeability. This implies that increased GECs permeability from excessive Mn may affect the GFB, which is related to glomerular injury and induces renal diseases.
It is widely accepted that Mn exposure is associated with both neurodevelopment and neurodegenerative disorders [32]. The kidney is not considered to be a target for Mn. However, it has been reported that increased blood Mn is associated with chronic renal failure [33]. We found that 25 mg/kg Mn increased protein exudate from glomeruli, which was consistent with the previous study. Nascimento et al. [34] reported that blood Mn level in exposed children was significantly associated with oxidative damage and kidney function. Overexposure of Mn increased microalbuminuria levels and N-acetyl-β-D-glucosaminidase activity, which reflected glomerular injury and tubular impairment [34]. In our study, histological analysis revealed no morphological changes in glomeruli in response to Mn treatment. This was consistent with a previous report, which showed that 15 mg/kg Mn treatment over 4 consecutive weeks significantly reduced antioxidant capacity without causing morphological changes to kidney in male rats [5]. This implies that Mn exposure is associated with renal disease by affecting the GFB.
GECs maintain and regulate glomerular permeability through fenestrated pores and cell–cell junctions [35]. In our study, 0.2 mM Mn increased the permeability of HRGECs. It was reported that GECs form an ultrafiltration membrane as a component of the GFB, which permits water and small solutes to pass into Bowman’s space, while rejecting large proteins such as albumin [36]. Dysregulation of glomerular permeability leads to severe pathophysiological conditions such as pre-eclampsia [37]. GECs impairment is associated with dysfunction of glomerular permeability, which causes renal failure and proteinuria [38, 39]. Our in vivo experiments showed that protein exudate from glomeruli were increased by Mn exposure. Taken together, these observations suggest that Mn exposure impairs GEC barrier function and is associated with diseases related to the GFB.
The mechanisms of Mn-induced cell toxicity are well established in the nervous system, involving impairment of mitochondria and induction of reactive oxygen species, which causes apoptosis [29, 40, 41]. Previous studies showed that 0.3 mM Mn inhibited cell viability and induced apoptosis of differentiated rat pheochromocytoma (PC12) cells in a p53-dependent manner [42]. Another study showed that 0.5 mM Mn reduced viability and increased apoptosis in a human neuroblastoma cell line (SK-N-MC) [31]. In our study, only 5.0 mM Mn inhibited cell viability and induced apoptosis of HRGECs, as 0.2-mM Mn treatment had no effect on viability or apoptosis. It was reported that treatment with low concentration of Mn for 24 h did not affect viability of endothelial cells [43]. The discrepancy in results between HRGECs and cells of the nervous system in response to similar concentrations of Mn may be explained by different sensitivities to Mn stimulation. This is also consistent with the relatively low accumulation of Mn in the brain (only 0.1%), even though it primarily causes nervous system diseases [30]. Treatment with 0.2 mM Mn increased permeability of HRGECs without affecting cell viability or apoptosis.
VE-cadherin is a major component of endothelial adherens junctions, which maintain and regulate permeability of endothelial cells [44]. Intercellular VE-cadherin interacts with β-catenin, which binds to the cytoskeleton to mediate the distribution of adherens junctions [45]. Both reduction and redistribution of VE-cadherin induce endothelial cell hyperpermeability [21, 46]. Previously, we found that proinflammatory cytokines increased the permeability of HRGECs by reducing expression of VE-cadherin [21], and anti-inflammatory drugs increased expression of VE-cadherin in HRGECs [24]. Furthermore, we reported that short-term, low-dose cadmium (Cd) induced hyperpermeability of HRGECs through redistribution of VE-cadherin and β-catenin [47, 48]. In the present study, we found that 0.2 mM or higher concentration of Mn inhibited expression of VE-cadherin at both the mRNA and protein levels. In vivo analysis demonstrated that VE-cadherin was downregulated in kidneys of rats exposed to Mn. Both Cd and Mn induce permeability of GECs. Cd increases permeability of HRGECs partially via activating the p38 mitogen-activated protein kinase pathway, which modifies the distribution of VE-cadherin [47]. In addition, Cd affects endothelial cells by regulating expression of von Willebrand factor and complement factor H [49, 50]. However, as an essential trace element, Mn directly reduced VE-cadherin expression through increase expression of transcriptional factor, Snail. Our results suggest Mn induced permeability of HRGECs through a mechanism consistent with that of proinflammatory cytokines, not heavy metals.
Smad2 and Smad3 (Smad2/3) are intracellular effectors in the TGF-β signaling pathway. TGF-β activates Smad2/3, which in turn positively or negatively regulate a number of different processes [51]. It was reported that TGF-β downregulates expression of VE-cadherin, which led to impairment of barrier function in microvascular endothelial monolayers [52]. We found that 0.2 mM Mn increased phosphorylation of Smad2/3 in HRGECs, and upregulated expression of Snail, an epithelial-to-mesenchymal transition-associated protein, which is considered a downstream effector of the TGF-β-Smad2/3 signaling pathway [53]. Previous studies showed that Smad2/3 bind to the promoter region of Snail in epithelial cells and increased expression of Snail [54]. Recently, we found that the Snail promoter is regulated by Smad2/3 in HRGECs [24]. As a negative regulator of VE-cadherin, Snail binds to the proximal E-boxes (at −240) of the human VE-cadherin promoter and inhibits expression of VE-cadherin [53]. These observations suggest Mn may inhibit expression of VE-cadherin by activating the Smad2/3-Snail signaling pathway.
Conclusions
Mn exposure induces protein exudate from glomeruli in rats and increases permeability of HRGECs. Increase permeability was combined with reduced VE-cadherin expression, which is partially mediated by activation of the Smad2/3-Snail signaling pathway. Our data indicate that Mn affects permeability of the GEC barrier and may contribute to the development of renal diseases.
Supplementary Material
Acknowledgments
This work was supported by grants from the Medical Science and Technology Development Plan of Shandong Province (Grant No. 2018WS260), the Natural Science Foundation of Shandong Province (Grant No. ZR2019BH007), and the National Natural Science Foundation of China (Grant No. 81472933, 81972989, 81970615).
Contributor Information
Peng Gao, Laboratory of Microvascular Medicine, Medical Research Center, Shandong Provincial Qianfoshan Hospital, The First Affiliated Hospital of Shandong First Medical University, 16766 Jingshi Road, Jinan 250014, Shandong, China; Department of Nephrology, Shandong Provincial Qianfoshan Hospital, The First Affiliated Hospital of Shandong First Medical University, 16766 Jingshi Road, Jinan 250014, Shandong, China.
Yutian Tian, Shandong Academy of Occupational Health and Occupational Medicine, Shandong First Medical University & Shandong Academy of Medical Sciences, 18877 Jingshi Road, Jinan 250062, Shandong, China.
Qi Xie, Laboratory of Microvascular Medicine, Medical Research Center, Shandong Provincial Qianfoshan Hospital, The First Affiliated Hospital of Shandong First Medical University, 16766 Jingshi Road, Jinan 250014, Shandong, China.
Liang Zhang, College of Life Sciences, Shandong Provincial Key Laboratory of Animal Resistance Biology, Institute of Biomedical Sciences, Shandong Normal University, 88 East Wenhua Road, Jinan 250014, Shandong, China.
Yongjian Yan, Shandong Academy of Occupational Health and Occupational Medicine, Shandong First Medical University & Shandong Academy of Medical Sciences, 18877 Jingshi Road, Jinan 250062, Shandong, China.
Dongmei Xu, Department of Nephrology, Shandong Provincial Qianfoshan Hospital, The First Affiliated Hospital of Shandong First Medical University, 16766 Jingshi Road, Jinan 250014, Shandong, China.
Authors’ Contributions
P.G. and L.Z. contributed for investigation; P.G., Y.T., Q.X., and L.Z. for methodology; Y.T. for resources; Y.Y. and D.X. for supervision; P.G. for writing original draft; and Y.Y. and D.X. for writing, reviewing, and editing the article.
Conflict of Interest
The authors have declared that they have no conflicts of interest.
Abbreviations
Cd, Cadmium; DMEM, Dulbecco’s modified Eagle’s medium; FITC, Fluorescein isothiocyanate; GECs, Glomerular endothelial cells; GFB, Glomerular filtration barrier; H&E, Hematoxylin and eosin; HRGECs, Human renal glomerular endothelial cells; HRP, Horseradish peroxidase; IgG, Immunoglobulin G; Mn, Manganese; PBS, Phosphate-buffered saline; PC12, Pheochromocytoma; PI, Propidium iodide; qRT-PCR, Quantitative real-time polymerase chain reaction; SD, Sprague Dawley; Smad2, Mothers against decapentaplegic homolog 2; Smad3, Mothers against decapentaplegic homolog 3; Snail, Zinc finger protein SNAI1; TGF-β, Transforming growth factor beta; VE-cadherin, Vascular endothelial-cadherin.
References
- 1. Chen P, Bornhorst J, Aschner M. Manganese metabolism in humans. Front Biosci (Landmark Ed) 2018;23:1655–79. [DOI] [PubMed] [Google Scholar]
- 2. O'Neal SL, Zheng W. Manganese toxicity upon overexposure: a decade in review. Curr Environ Health Rep 2015;2:315–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Erikson KM, Aschner M. Manganese: its role in disease and health. Met Ions Life Sci 2019;19. doi: 10.1515/9783110527872-016. [DOI] [PubMed] [Google Scholar]
- 4. Liu K, Jing MJ, Liu C et al. Effect of trehalose on manganese-induced mitochondrial dysfunction and neuronal cell damage in mice. Basic Clin Pharmacol Toxicol 2019;125:536–47. [DOI] [PubMed] [Google Scholar]
- 5. Richter Schmitz CR, Eichwald T, Branco Flores MV et al. Sex differences in subacute manganese intoxication: oxidative parameters and metal deposition in peripheral organs of adult Wistar rats. Regul Toxicol Pharmacol 2019;104:98–107. [DOI] [PubMed] [Google Scholar]
- 6. Sahni V, Leger Y, Panaro L et al. Case report: a metabolic disorder presenting as pediatric manganism. Environ Health Perspect 2007;115:1776–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guan H, Wang M, Li X et al. Manganese concentrations in maternal and umbilical cord blood: related to birth size and environmental factors. Eur J Public Health 2014;24:150–7. [DOI] [PubMed] [Google Scholar]
- 8. Zhuang X, Pang X, Zhang W et al. Effects of zinc and manganese on advanced glycation end products (AGEs) formation and AGEs-mediated endothelial cell dysfunction. Life Sci 2012;90:131–9. [DOI] [PubMed] [Google Scholar]
- 9. Jiang Y, Zheng W. Cardiovascular toxicities upon manganese exposure. Cardiovasc Toxicol 2005;5:345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujishiro H, Hamao S, Isawa M et al. Segment-specific and direction-dependent transport of cadmium and manganese in immortalized S1, S2, and S3 cells derived from mouse kidney proximal tubules. J Toxicol Sci 2019;44:611–9. [DOI] [PubMed] [Google Scholar]
- 11. Chen X, Li J, Cheng Z et al. Low dose cadmium inhibits proliferation of human renal mesangial cells via activation of the JNK pathway. Int J Environ Res Public Health 2016;13:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen X, Xu Y, Cheng Z et al. Low-dose cadmium activates the JNK signaling pathway in human renal podocytes. Int J Mol Med 2018;41:2359–65. [DOI] [PubMed] [Google Scholar]
- 13. Leggett RW. A biokinetic model for manganese. Sci Total Environ 2011;409:4179–86. [DOI] [PubMed] [Google Scholar]
- 14. Miner JH. Glomerular basement membrane composition and the filtration barrier. Pediatr Nephrol 2011;26:1413–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Savage CO, Bogle R. Resident glomerular cells in glomerular injury: endothelial cells. Semin Nephrol 1991;11:312–9. [PubMed] [Google Scholar]
- 16. Dong F, Li L, Chen X et al. Glomerular endothelial cell IQGAP2 and filtration barrier function. Kidney Int 2016;89:1160–1. [DOI] [PubMed] [Google Scholar]
- 17. Ebefors K, Wiener RJ, Yu L et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int 2019;96:957–70. doi: 10.1016/j.kint.2019.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang WH, Lin JL. Acute renal failure following ingestion of manganese-containing fertilizer. J Toxicol Clin Toxicol 2004;42:305–7. [DOI] [PubMed] [Google Scholar]
- 19. Dejana E, Giampietro C. Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol 2012;19:218–23. [DOI] [PubMed] [Google Scholar]
- 20. Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 2013;26:441–54. [DOI] [PubMed] [Google Scholar]
- 21. Du L, Dong F, Guo L et al. Interleukin-1beta increases permeability and upregulates the expression of vascular endothelial-cadherin in human renal glomerular endothelial cells. Mol Med Rep 2015;11:3708–14. [DOI] [PubMed] [Google Scholar]
- 22. Komarova YA, Kruse K, Mehta D et al. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ Res 2017;120:179–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang C, Liu Q, Dong F et al. Catalpol downregulates vascular endothelial cadherin expression and induces vascular hyperpermeability. Mol Med Rep 2016;13:373–8. [DOI] [PubMed] [Google Scholar]
- 24. Li L, Chen X, Dong F et al. Dihydroartemisinin up-regulates VE-cadherin expression in human renal glomerular endothelial cells. J Cell Mol Med 2018;22:2028–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tian Y, Chen C, Guo S et al. Exploration of the establishment of manganese poisoning rat model and analysis of discriminant methods. Toxicology 2018;410:193–8. [DOI] [PubMed] [Google Scholar]
- 26. Cheng Z, Qi R, Li L et al. Dihydroartemisinin ameliorates sepsis-induced hyperpermeability of glomerular endothelium via up-regulation of occludin expression. Biomed Pharmacother 2018;99:313–8. [DOI] [PubMed] [Google Scholar]
- 27. Gao P, Zhang Y, Liu Y et al. Signal transducer and activator of transcription 5B (STAT5B) modulates adipocyte differentiation via MOF. Cell Signal 2015;27:2434–43. [DOI] [PubMed] [Google Scholar]
- 28. Obeidat M, Obeidat M, Ballermann BJ. Glomerular endothelium: a porous sieve and formidable barrier. Exp Cell Res 2012;318:964–72. [DOI] [PubMed] [Google Scholar]
- 29. Choi EK, Nguyen TT, Gupta N et al. Functional analysis of SLC39A8 mutations and their implications for manganese deficiency and mitochondrial disorders. Sci Rep 2018;8:3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Horning KJ, Caito SW, Tipps KG et al. Manganese is essential for neuronal health. Annu Rev Nutr 2015;35:71–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bahar E, Kim JY, Yoon H. Quercetin attenuates manganese-induced neuroinflammation by alleviating oxidative stress through regulation of apoptosis, iNOS/NF-kappaB and HO-1/Nrf2 pathways. Int J Mol Sci 2017;18:1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lucchini RG, Aschner M, Landrigan PJ et al. Neurotoxicity of manganese: indications for future research and public health intervention from the Manganese 2016 conference. Neurotoxicology 2018;64:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanchez-Gonzalez C, Lopez-Chaves C, Gomez-Aracena J et al. Association of plasma manganese levels with chronic renal failure. J Trace Elem Med Biol 2015;31:78–84. [DOI] [PubMed] [Google Scholar]
- 34. Nascimento S, Baierle M, Goethel G et al. Associations among environmental exposure to manganese, neuropsychological performance, oxidative damage and kidney biomarkers in children. Environ Res 2016;147:32–43. [DOI] [PubMed] [Google Scholar]
- 35. Haraldsson BS. The endothelium as part of the integrative glomerular barrier complex. Kidney Int 2014;85:8–11. [DOI] [PubMed] [Google Scholar]
- 36. Deen WM. What determines glomerular capillary permeability? J Clin Invest 2004;114:1412–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ponnuchamy B, Khalil RA. Cellular mediators of renal vascular dysfunction in hypertension. Am J Physiol Regul Integr Comp Physiol 2009;296:R1001–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev 2008;88:451–87. [DOI] [PubMed] [Google Scholar]
- 39. Wang PR, Kitamura H, Shimizu A et al. Glomerular damage in experimental proliferative glomerulonephritis under glomerular capillary hypertension. Kidney Blood Press Res 2015;40:188–99. [DOI] [PubMed] [Google Scholar]
- 40. Porte Alcon S, Gorojod RM, Kotler ML. Regulated necrosis orchestrates microglial cell death in manganese-induced toxicity. Neuroscience 2018;393:206–25. [DOI] [PubMed] [Google Scholar]
- 41. Milatovic D, Zaja-Milatovic S, Gupta RC et al. Oxidative damage and neurodegeneration in manganese-induced neurotoxicity. Toxicol Appl Pharmacol 2009;240:219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wan C, Ma X, Shi S et al. Pivotal roles of p53 transcription-dependent and -independent pathways in manganese-induced mitochondrial dysfunction and neuronal apoptosis. Toxicol Appl Pharmacol 2014;281:294–302. [DOI] [PubMed] [Google Scholar]
- 43. Kaji T, Fujiwara Y, Yamamoto C et al. Stimulation by zinc of cultured vascular endothelial cell proliferation: possible involvement of endogenous basic fibroblast growth factor. Life Sci 1994;55:1781–7. [DOI] [PubMed] [Google Scholar]
- 44. Broman MT, Kouklis P, Gao X et al. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ Res 2006;98:73–80. [DOI] [PubMed] [Google Scholar]
- 45. Hatchigian EA, Santos JI, Broitman SA et al. Vitamin A supplementation improves macrophage function and bacterial clearance during experimental salmonella infection. Proc Soc Exp Biol Med 1989;191:47–54. [DOI] [PubMed] [Google Scholar]
- 46. Dong F, Guo F, Li L et al. Cadmium induces vascular permeability via activation of the p38 MAPK pathway. Biochem Biophys Res Commun 2014;450:447–52. [DOI] [PubMed] [Google Scholar]
- 47. Li L, Dong F, Xu D et al. Short-term, low-dose cadmium exposure induces hyperpermeability in human renal glomerular endothelial cells. J Appl Toxicol 2016;36:257–65. [DOI] [PubMed] [Google Scholar]
- 48. Zhang H, Li L, Wang Y et al. NF-kappaB signaling maintains the survival of cadmium-exposed human renal glomerular endothelial cells. Int J Mol Med 2016;38:417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen X, Li L, Liu F et al. Cadmium induces glomerular endothelial cell-specific expression of complement factor H via the −1635 AP-1 binding site. J Immunol 2019;202:1210–8. [DOI] [PubMed] [Google Scholar]
- 50. Wang X, Dong F, Wang F et al. Low dose cadmium upregulates the expression of von Willebrand factor in endothelial cells. Toxicol Lett 2018;290:46–54. [DOI] [PubMed] [Google Scholar]
- 51. Gaarenstroom T, Hill CS. TGF-beta signaling to chromatin: how Smads regulate transcription during self-renewal and differentiation. Semin Cell Dev Biol 2014;32:107–18. [DOI] [PubMed] [Google Scholar]
- 52. Maroni D, Davis JS. TGFB1 disrupts the angiogenic potential of microvascular endothelial cells of the corpus luteum. J Cell Sci 2011;124:2501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kataoka H, Murayama T, Yokode M et al. A novel snail-related transcription factor Smuc regulates basic helix-loop-helix transcription factor activities via specific E-box motifs. Nucleic Acids Res 2000;28:626–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smith AP, Verrecchia A, Faga G et al. A positive role for Myc in TGFbeta-induced Snail transcription and epithelial-to-mesenchymal transition. Oncogene 2009;28:422–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.