

Summary:
Regulatory networks that maintain functional, differentiated cell states are often dysregulated in tumor development. Here, we use single-cell epigenomics to profile chromatin state transitions in a mouse model of lung adenocarcinoma (LUAD). We identify an epigenomic continuum representing loss of cellular identity and progression towards a metastatic state. We define co-accessible regulatory programs and infer key activating and repressive chromatin regulators of these cell states. Among these co-accessibility programs, we identify a pre-metastatic transition, characterized by activation of RUNX transcription factors, which mediates extracellular matrix remodeling to promote metastasis and is predictive of survival across human LUAD patients. Together, these results demonstrate the power of single-cell epigenomics to identify regulatory programs to uncover mechanisms and key biomarkers of tumor progression.
Graphical Abstract
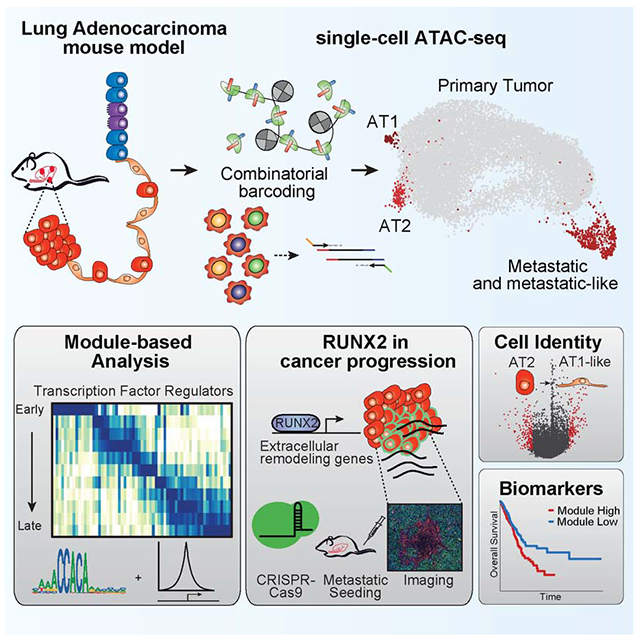
Introduction:
Cancer occurs through the acquisition of genetic mutations (Vogelstein et al., 2013) leading to the disruption of lineage-specifying transcription factors (TFs), among other effects (Bradner et al., 2017; Spitz and Furlong, 2012). Dysregulation of these regulatory programs is influenced by extensive selection in response to genetic, epigenetic, and environmental factors (Flavahan et al., 2017; Hanahan and Weinberg, 2000), which promote tumor development by altering lineage restriction and cell identity (Flavahan et al., 2017; Lee and Young, 2013; Spitz and Furlong, 2012; Sur and Taipale, 2016). These selective pressures begin during the initial transformation of normal cells and continue through all stages of tumor development, resulting in a diverse and heterogeneous regulatory landscape (Chen et al., 2014; Dagogo-Jack and Shaw, 2018).
Genetically-engineered mouse models (GEMMs) of cancer provide an opportunity to study tumor development in experimentally-defined conditions. Mice from the KrasLSL(lox-stop-lox)-G12D/+ Trp53fl/fl (KP) model develop LUAD and progress to metastasis in the absence of frequent additional driver somatic mutations (McFadden et al., 2016; Westcott et al., 2015; Birkbak and McGranahan, 2020). This model reproducibly mirrors human LUAD progression (Jackson et al., 2001, 2005), which is one of the leading causes of cancer-related deaths worldwide (Dietel et al., 2016; Herbst et al., 2018). During tumor development, KP cancer cells exhibit substantial transcriptional dysregulation including altered expression of the lung lineage factor Nkx2.1 (Chen et al., 2014; Winslow et al., 2011). However, the full repertoire of TFs driving the disruption of these regulatory programs has not been established. Further characterization of these regulatory transitions would provide mechanistic insights and opportunities for the identification of novel biomarkers and treatment strategies for LUAD patients.
Epigenomic analysis can help define regulatory state transitions dictating normal and altered cellular programs. For example, studies measuring DNA methylation (Klughammer et al., 2018), histone modifications (Dubuc et al., 2013; Noberini et al., 2018) and chromatin accessibility (Corces et al., 2018; Denny et al., 2016; Latil et al., 2017; Roe et al., 2017) have defined cell state changes during tumor progression and metastasis. However, these prior studies have predominantly measured bulk profiles that average over the genetic and regulatory heterogeneity of tumor cells present in the sample. Single-cell methods have provided new insights into the genetic and transcriptional states of primary tumors (Flavahan et al., 2017; Ren et al., 2018). Recent advances in single-cell epigenomics have opened new possibilities to further define regulatory states within single-cells (Buenrostro et al., 2015a; Cusanovich et al., 2015; Gaiti et al., 2019; Hou et al., 2016; Shema et al., 2019). In particular, methods to measure chromatin accessibility using single-cell Assay for Transposase Accessible Chromatin (scATAC-seq) provide an opportunity to map the activity of cis- and trans-regulators through tumor development (Buenrostro et al., 2018; Cusanovich et al., 2018).
Here, we use scATAC-seq to characterize tumor development from initiation to metastasis in the KP model. To do this, we first optimized single-cell combinatorial indexing ATAC-seq (sciATAC-seq) (Cusanovich et al., 2015) to profile normal lung cells and cancer cells derived from primary tumors and metastases. We utilize these single-cell profiles to identify modules of differential TF activity, assign relevant module-associated peaks to genes, and create a framework to explore TF-regulated chromatin accessibility patterns in KP tumor development. Overall, we characterize heterogeneous phenotypic landscapes that arise in cancer which can lead to plastic cell states and selection for epigenomic alterations, serving as drivers for tumor progression and metastasis.
Results
Epigenomic analysis of KP cancer cells at single-cell resolution
To generate single-cell epigenomic data from KP-derived cancer cells, we developed an improved protocol for sciATAC-seq, which utilizes dual barcoding during transposition and PCR (Figure 1A,B) (Cusanovich et al., 2015). This method provides a flexible platform for multiplexing samples using barcoded Tn5 transposition in each single-cell experiment. To increase the throughput and yield of previously described methods (Cusanovich et al., 2015; Pliner et al., 2018; Preissl et al., 2018), we i) removed fluorescence-activated cell sorting (FACS) of nuclei, ii) combined recent protocol developments (Chen et al., 2018; Lake et al., 2018; Preissl et al., 2018), iii) expanded the barcoding space, and iv) optimized fixation (0.1% formaldehyde), transposition, and reverse crosslinking conditions (Figure S1A-G, STAR Methods, Table S1). In species mixing experiments, this approach captured 33,663 fragments per cell, with a cell doublet rate of 7.3% (Figure 1C-E and Figure S1F-G).
Figure 1. An optimized single-cell ATAC-seq approach enabled analyses of single KP tumor cells.

(A) Schematic of alleles in the KPT model, LSL: lox-stop-lox; loxp (blue arrows); FRT site (orange arrows). Inset immunofluorescence (IF) image of a tdTom positive (tdTom+) tumor. (B) Schematic of sciATAC-seq strategy for single-cell profiling of tdTom+ cancer cells. (C) Unique fragments from species-mixing experiment of GM12878 (n = 1) and 3T3 cells (n = 1). (D) Estimated library sizes of published data (Cusanovich et al., 2015; Pliner et al., 2018; Preissl et al., 2018) and this study, derived from GM12878 cells. Box intervals represent 25% and 75% bounds. (E) FRIP by total fragments recovered from GM12878 and 3T3 cells. (F) IHC of a tumor-burdened lung at 30 weeks after tumor initiation in KPT model, representing H&E with Aiforia defined grades (top) and NKX2.1 IHC (scale bar; bottom, left 400 μm and right 100 μm). (G) Chromatin accessibility tracks generated from bulk ATAC-seq of a KPT tumor (red) (n = 1) and aggregated single-cell from a primary KPT tumor (n = 13,070; orange) at the S100 gene family locus. see also Fig. S1 and Table S1.
We then used this approach to study tumor development in the KP model. To isolate cancer cells by FACS, we introduced a Rosa26-LSL-tdTomato reporter allele into KP mice (KPT) (Figure 1A) (Madisen et al., 2010). We performed intratracheal instillation with adenovirus (Ad5-SPC-Cre) to initiate lung tumors in alveolar type II (AT2) cells where mice developed late-stage tumors and metastases 30-35 weeks post-infection. Analysis of late-stage histological sections using a deep learning-based algorithm (STAR Methods) identified extensive intratumoral heterogeneity, reflecting a range of tumor grades (grades 1-4) (Figure 1F) (DuPage et al., 2009). We also performed immunohistochemistry (IHC) on KPT tumors for the well-studied tumor progression markers NKX2.1 and HMGA2 (Winslow et al., 2011) as well as tdTomato to confirm tumors retain expression (Figure 1F; Figure S1H; Supplementary Data 1).
To investigate the diversity of cell states in the KPT model, we applied the improved sciATAC-seq protocol to normal lung and FACS-isolated tdTom+ cancer cells in late-stage tumors (Figure S2A) derived from either individually dissected tumors (Figure 1G), whole tumor-bearing lungs, or metastases (thymus, lymph node, liver) (Table S1). Altogether, we collected a total of 17,274 high-quality epigenomic profiles across 44 individual samples, comprising 13,670 cancer cells (13,070 tumor-derived and 600 metastatic cells) and 3,604 normal lung cells (STAR Methods). From these data, we obtained an average fraction of reads in peaks (FRIP) of 63.7% and 18,312 unique nuclear fragments per cell. We qualitatively confirmed the high-purity of cancer cells by visually inspecting the Trp53 locus (Figure S2B). To analyze cell state diversity, we first computed the chromatin accessibility across all possible sequence k-mers and TF motifs per cell (Schep et al., 2017), which we refer to as the “TF motif score” in the remainder of the text. We visualized these data using Uniform Manifold Approximation and Projection (UMAP) (STAR Methods) (McInnes et al., 2018) (Figure 2A and Figure S2C); single-cell data can be explored interactively using our web resource: https://buenrostrolab.shinyapps.io/lungATAC/). Single-cell grouping in UMAP space was not driven by FRIP or sequencing depth (Figure S2D,E).
Figure 2. Single-cell chromatin accessibility data defined heterogeneous normal and KP cell states.

(A) UMAP visualization of normal and KPT cancer cells profiled by sciATAC-seq. Individual samples are labeled by mouse number (M1-M12), primary tumor (T1-T5, pool), or metastatic tumor number (N1-N5); color codes represent normal (n = 2), immune-depleted normal lung (n = 1), tdTom+ cells isolated from lung tumors (n = 23), lymph node or thymic metastases (n = 15), and liver metastases (n = 3). Two examples highlighted in red of individual tumors are shown (right). (B) Schematic of approach to calculate gene scores using an exponential decay function. Individual fragments are weighted based on the inverse distance to the TSSs, then summed across the chosen window (9,212 bp) reflecting 1% of the total weight for the chosen exponential half-life (1 kb). (C-E) Example gene scores are shown on the UMAP for Cd45 (C), Cd19 (D) and Vim (E). (F) Chromatin accessibility tracks for normal cell clusters at lineage-defining marker genes; track with associated genomic location shown (bottom). (G) Normal cell-type cluster identities shown on the UMAP of single-cells, tumor and metastatic cells labeled in gray and red, respectively, with inset zoom of the metastatic-like cluster. (H) Fractions of cancer cells within individual tumors that cluster with metastatic cells (red) or with cells derived from the primary tumor (gray) (n = 35). (I) Images of NKX2.1, VIM and H&E staining of a representative grade 4 region (zoom 9.5X; scale bar, 100 μm). see also Fig. S2, Table S1 and Data S1.
Cell types and states across normal and cancer cells from the lung
In order to compare KPT cancer cells to normal cell states, we first annotated the normal cell population which clustered into 12 distinct subsets (Figure S2G and Table S1). To identify each cluster, we quantified chromatin accessibility surrounding annotated transcription start sites (TSSs) to infer differences around individual genes (hereafter referred to as “gene score”) using an exponential decay scoring function that weights aligned transposase fragments by their proximity to each gene’s TSS (Figure 2B) (Granja et al., 2019; Satpathy et al., 2019) (STAR Methods). To validate this approach, we found that transcriptional activity largely correlated with calculated gene scores in published bulk ATAC-seq and RNA-seq data from LUAD tumors (Figure S2F) (Corces et al., 2018). For visualization, we smoothed gene scores using a k-nearest-neighbor (k-NN) approach (k = 10; STAR Methods). Gene scores of known marker genes of distinct cell types followed expected patterns across clusters, including Cd45 (immune), Cd19 (B cells), and Vimentin (mesenchymal and macrophage cells) (Figure 2C-E). We then used gene scores for de novo assignment of cell identities for each cluster including Epcam-positive lung cells, namely, alveolar type II (AT2) (Sftpc), alveolar type I (AT1) (Hopx), club (Foxj1), and ciliated (Scgb1a1) cells (Figure 2F,G, Figure S2G,H and Table S1) (Cohen et al., 2018; Lambrechts et al., 2018; Treutlein et al., 2014) and confirmed the assignments by computationally matching these data to single-cell RNA-seq (scRNA-seq) data from normal lung tissue (Figure S2I) (Cohen et al., 2018).
The sciATAC-seq profiles of single cancer cells isolated from the KPT model largely spanned a continuous epigenomic progression from profiles reflecting normal AT2 cells (the presumed cell-of-origin in this model (Sutherland et al., 2014)) to profiles present in thymic, lymph node and liver metastases (Figure 2A,G). The epigenomic states present in cancer cells from KPT tumors were highly heterogeneous, while metastatic cancer cells exhibited reduced heterogeneity (Figure 2G and Figure S2J). Interestingly, rare primary tumor-derived populations across individual samples overlapped with cancer cells isolated from metastases and, strikingly, there were no apparent motif differences between the “metastatic-like” and distal metastatic cancer cells (Figure 2H and Figure S2K). IHC analyses confirmed that Grade 4 regions were commonly small, located at central regions of the tumor, and positive for VIM (a gene score feature that specifically marks the metastatic cluster) (Figure 2E,I). These results suggest that cells activate a metastatic regulatory program within the primary tumor, supporting data that metastatic seeding occurs late in KP progression and requires local remodeling prior to dissemination (Caswell et al., 2014). However, we cannot exclude a model in which these metastatic-like cells arise from reseeded metastatic cells. Altogether, our analysis of KPT tumors shows a high degree of heterogeneity, with cancer cells isolated from primary tumors occupying a continuum of epigenomic states from the cell-of-origin to cells with presumed metastatic potential.
Loss of lineage identity during tumor development
AT2 cells are believed to be a common cell-of-origin in LUAD, as evidenced predominantly by the finding that SPC-expressing cells can give rise to LUAD in GEMMs (Cheung and Nguyen, 2015; Lin et al., 2012; Mainardi et al., 2014; Sutherland et al., 2014; Xu et al., 2012). The alveolar differentiation hierarchy remains an active area of investigation, including studies describing rare bipotent progenitor cells as sharing expression of AT1 and AT2 markers (Figure 3A) (Treutlein et al., 2014). Notably, AT2 cells can transdifferentiate into AT1 cells in response to injury, cell death and altered WNT-signaling in the niche (Desai et al., 2014; Jain et al., 2015; Nabhan et al., 2018; Wang et al., 2018). In the UMAP embedding, we found a subset of KPT cells overlapped with normal AT2 cells and others with normal AT1 cells, motivating an analysis of cell identity during tumor development (Figure 2A,G).
Figure 3. KPT cancer cells reflected AT1 and AT2 epigenomic states.

(A) Schematic of epithelial cell types and alveolar differentiation hierarchy. (B) Hierarchical clustering of AT1 (n = 67) and AT2 (n = 186) cells based on top significant TF motif scores, labeled by AT1 and AT2 cluster identity (bottom). (C) Volcano plot of differential gene scores between AT1 versus AT2 cells. Genes with a differential gene score greater than 1.8 or less than −1.8 are highlighted in red with −log10 p value shown. (D) Correlation of each cancer cell to normal AT1 and AT2 cells using gene score signatures. Cells are colored by their Pearson r differential correlation coefficients. (E) Images of serial sections of early KP tumors (n = 2), late KP tumors (n = 2), and lymph node metastases (n = 2) stained for SFTPB (AT2 marker) and CAV1 (AT1 marker) (scale bar, 250 μm except Met tumor 2 125 μm; inset, 50 μm). (F) Fraction of single cancer cells per sample that resemble AT1-like, AT2-like or late-stage cells; red=AT1, orange=AT2 and gray=late (n = 23). (G) Multiplexed IHC in a late-stage tumor sample; whole lung and two individual tumors shown; red (SPC; AT2), yellow (NKX2-1), green (HOPX; AT1), and overlay with DAPI (scale bar; whole lung, 0.5x, 2000 μm; tumor 1; 7.5x, 200 μm; tumor 2; 4.5x, 200 μm). (H) Aiforia graded 8-week tumor-burdened lung (red=grade 1, green=grade 2, blue=grade 3, and orange=grade 4). (I) Multiplexed IHC staining of an exemplar lung lobe at 8 weeks post-initiation stained with SPC (red), NKX2-1 (yellow), HOPX (green) and overlaid channels with DAPI. tdTom+ cells from entire lung used for scATAC-seq profiling (scale bar; whole lung, 0.7x, 1000 μm; tumors; 10x, 100 μm). (J) scATAC-seq profiling and projection of early time point (ETP) cells (n = 4,610) onto the original UMAP clustering of all lung cells (gray points). ETP cells are colored by cluster density. see also Fig. S3 and Table S2.
In order to characterize KP epigenomic states with respect to alveolar identity, we first defined gene and TF motif score differences between normal AT1 and AT2 cells. Differential TF motif score analysis and hierarchical clustering identified higher CEBPA TF motif accessibility in AT2 cells, in contrast to higher TEAD and GATA TF motif scores in AT1 cells (Figure 3B and Figure S3A,B). Furthermore, differential analysis of gene scores between AT1 and AT2 cells revealed Cebpa (a lineage-defining TF) and Cav1 (a caveolae-associated protein) as the most significant AT2 and AT1-specific gene score markers, respectively (Figure 3C, Figure S3C and Table S2) (Campbell et al., 1999; Treutlein et al., 2014; Wang et al., 2018). We used AT1, AT2 and tumor gene score signatures (n = 1,393 genes) to compare KPT-derived cancer cells to alveolar cells. Interestingly, cancer cells at the left side of the continuum scored highly for both AT1- and AT2-like signatures, while cancer cells with late-stage features show reduced correlation, suggesting a global loss of lineage identity (Figure 3D-F and Figure S3D). KPT cancer cells expressed markers of both AT1 (HOPX, PDPN) and AT2 (SPC, SFTPB) cell identity, including at earlier time points, suggesting that transformation induces lineage infidelity (Figure 3G-I and Figure S3E-G) (Ge et al., 2017). Analysis of marker gene scores and TF motif scores, as well as scoring cancer cells with scRNA-seq signatures from normal lung development (Figure S3H-J), were consistent with this finding (Cohen et al., 2018; Mund et al., 2008). Together, these data show that KP cancer cells lose AT2 lineage identity through tumor progression.
Based on these findings, we propose that primary cancer cells adopt an altered identity arising from either (1) transformation of an immature cell, (2) transdifferentiation of AT2-like cells during tumor progression, or (3) dedifferentiation. To assess these possibilities, we performed a droplet-based scATACseq experiment on 4,610 cancer cells isolated at 8 weeks post-tumor initiation (Figure 3H-J) (Lareau et al., 2019) and projected these epigenomic profiles onto the coordinates of the original UMAP (Figure 3J). Cancer cells from the early time point exhibited epigenomic profiles that largely overlapped with normal AT2 cells, suggesting that early cancer cells maintain an AT2 identity and heterogeneity arises over time in the KPT model. To further validate this finding, we performed multiplexed IHC in 8-week tumors and found that KPT tumors were largely SPC-positive (Figure 3I and Figure S3G) (Mainardi et al., 2014; Sutherland et al., 2014; Xu et al., 2012). Altogether, we propose that an immature alveolar cell identity likely arises across tumor development, consistent with scRNA-seq analyses performed along a tumorigenesis time course in the KP model (Marjanovic et al., see accompanying paper).
Co-accessibility modules reveal epigenomic dysregulation in cancer cells
Epigenomic profiling of KPT-derived cancer cells identified a spectrum of cell states indicating substantial heterogeneity in tumors. To study the regulatory programs underlying these epigenomic states, we performed unsupervised hierarchical clustering of all cancer cells based on significant TF motif accessibility scores (n = 350 motifs) and found that cancer cells reflected differences across many TF motifs including NKX2.1, CEBPA, TEAD4, FOS and RUNX2 (Figure 4A and Figure S4C). Importantly, the NKX2.1 TF motif score clearly demarcated early versus late epigenomic states (Figure 4B). To determine the extent of chromatin change reflected by the NKX2.1 TF score at individual peaks, we grouped cells as “high” or “low” for the NKX2.1 TF motif score (defined as being above or below the median score, respectively). We next identified differential peaks across these two groups revealing extensive chromatin accessibility changes associated with tumor progression (n = 38,164 peaks; FDR < 10−6; Figure 4B-D).
Figure 4. Chromatin co-accessibility modules defined cell state transitions during tumor progression.

(A) Hierarchical clustering of cancer cells (n = 13,670) using significant TF motif scores (n = 350 motifs) associated with tumor progression score as calculated by a distance from a fit polynomial line (bottom). (B) UMAP of cancer cells colored by NKX2.1 TF motif score. (C) Histogram of NKX2.1 TF motif scores for all cancer cells. Cells are delineated as “high” or “low” based on the median motif score across cancer cells (blue dashed line). (D) Differential chromatin accessibility for each peak between NKX2.1 TF motif “high” or “low” cells. Peaks with a significant FDR (q < 10−6) calculated by a two-sample Student’s t-test are shown in dark blue. (E) Schematic depicting the co-accessibility module analysis workflow. (F) Clustering of differential TF motif associated peaks (n = 74,732 rows) using the log2 fold-change (FC) of mean accessibility between “high” versus “low” cell groups per TF motif (n = 67 columns). Clustering is performed based on the Louvain method. Peaks are hierarchically clustered per module for visualization. (G) UMAP plots highlighting single-cell module scores for cancer cells. see also Fig. S4 and Table S3.
The analysis of TF motif scores revealed numerous putative cancer cell regulators (Figure 4A); however, TFs typically function combinatorially to drive distinct regulatory programs (Gerstein et al., 2012). We therefore reasoned that classification of chromatin accessibility changes by multiple differential TF motifs may better serve to uncover regulatory programs underlying disease progression. As such, we developed a computational strategy to identify chromatin accessibility peaks that are co-accessible and concordant with changes in TF motif scores (STAR Methods). Briefly, we grouped cancer cells as TF “high” or “low” based on each TF motif score, and for each TF motif we tested all peaks for differential accessibility between the “high” versus “low” cells. Next, we repeated this procedure independently for each non-redundant and variable TF motif (n = 67; Figure 4D,E and Figure S4A,B). Finally, to define co-accessibility modules, we took the union of all differential peaks, resulting in 74,732 chromatin accessibility changes (FDR q < 10−6), and clustered these differential peaks using their fold-change in mean chromatin accessibility for each TF “high” versus “low” comparison. This approach resulted in 11 distinct clusters of peaks (henceforth referred to as ‘modules’) (Figure 4F) that demonstrated extensive reorganization within cancer cells across tumor evolution.
Next, we sought to determine the functional identity of each module in tumor development. To this end, using a similar approach to determining TF motif scores, we first computed the enrichment of accessibility for each module’s peaks across single cells (Schep et al., 2017), which we refer to as module scores (STAR Methods, Figure 4G). To determine the biological relevance of each module, we first performed de novo assignment of well-established KPT cancer progression markers (TF motif scores and gene scores) to modules (Table S3). In addition, we generated a set of module-associated genes by assigning gene scores to their most correlated modules. Together with further analyses later described in the manuscript, we assigned functional identities to the 11 modules (Figure 4F,G). Modules 6, 5, 11, and 7 were associated with alveolar identity and earlier stages of KP transformation, with modules 5 and 11 most associated with tumor cells isolated from the 8-week time point (Figure 4G and Figure S4D,E; see Table S3 for relevant gene scores and motifs used for module assignments). In addition, modules 1, 9, 2, and 4 exhibited features of late-stage tumor progression. Interestingly, modules 1 and 9 coincided most closely with loss of NKX2.1 TF motif accessibility and marked all late-stage-like cancer cells, while modules 2 and 4 were most closely associated with progression toward metastasis. To further delineate the identity of each module, we ranked genes by the correlation of their gene scores to module accessibility scores across all cancer cells and performed gene set enrichment analysis (GSEA) for each module-ranked gene list (Table S3). These analyses confirmed progressive enrichment of gene sets associated with TGF-beta signaling, secreted factors, and extracellular matrix (ECM) in later stage modules, with EMT hallmark genes among module 2- and 4-associated genes (GSEA FDR q = 0.001 and q ≤ 0.001, respectively) (Figure S4F,G) (Heldin et al., 2012; Katsuno et al., 2019). Modules 7 and 10 were associated with high HNF4A TF motif scores and gastric gene signatures, consistent with a known gastric-like state in the KP model (Figure S4F,H) (Snyder et al., 2013). Furthermore, module 8 was enriched for immune and senescence TF motifs (IRF1, SFPI1 and MITF) (Giuliano et al., 2010), while module 3 delineated a transition between early and late stages of cancer progression. Overall, we identified extensive gene regulatory alterations across the tumor progression spectrum that clearly delineates key protumorigenic programs.
Combined gene and motif scores reveal regulators of tumor progression
We next examined the relationship between gene activity and motif accessibility of TFs in an effort to determine their regulatory activity. Notably, using published bulk ATAC-seq and RNA-seq data (Corces et al., 2018), we found that gene scores for TFs had a stronger signal relative to gene expression (Figure S5A) compared to non-TF-encoding genes, likely because TF gene expression tends to be controlled by several layers of regulatory control (González et al., 2015). Aggregated single-cells with high activity for representative modules described above revealed significant chromatin accessibility changes surrounding TFs at different cell states, including the 8-week cancer cells (ETP), alveolar identity modules (modules 5, 6, and 11) and late-stage modules (modules 9, 2, and 4) (Figure 5A). To investigate the function of these TFs, we reasoned that correlation of TF motif scores to TF gene scores (referred to hereafter as TF motif-gene pairs) may identify activating (positive correlation) or repressive (negative correlation) regulators of chromatin accessibility genome-wide (STAR Methods). Indeed, correlating TF motif-gene pairs across cancer cells revealed 85 (n = 63 positive, n = 22 negative) putative TF regulators of chromatin accessibility (Figure 5B-D, Figure S5B and Table S4).
Figure 5. Regulatory analysis of cancer cells identified chromatin activators and repressors.

(A) Chromatin accessibility tracks for cells with high module scores and normal AT1/AT2 cells respectively at key transcription factors. Modules include early time point (ETP), early-stage (5, 11) and late-stage (9, 2, 4) modules. Module high was defined as two standard deviations above the mean module score across cells. (B-C) UMAP highlighting single-cell TF motif scores and motif logos (left), and gene scores (right) for FOSL1 (B) and RUNX2 (C) in cancer cells. (D) Correlation of TF motif scores with gene scores for each TF (n = 769) plotted against the TF motif score variability. Significantly variable TF motifs (motif score s.d. ≥ 1.2) correlated with their gene score (permutation p < 0.001) are shown in red; TFs with positive or negative correlation are highlighted as activators or repressors, respectively. Permutation p values were calcuated using a Z-test between the observed TF-motif gene correlation coefficient to the permuted correlation coefficients. TF motif scores signficiance was computed with deviation Z-scores across cells. (E) Normalized correlation (max/min normalized using Pearson r correlations) of TF gene scores to module scores delineated by activators (n = 58) and repressors (n = 14). (F) IHC of heterogeneous late-stage TFs stained for NKX2.1 (module 5), RUNX1 (module 9/2), RUNX2 (module 9/2), ONECUT2 (module 2), and ZEB1 (module 4) at 250 μm, inset 50 μm (n = 1). (G) Grade 4 regions stained for RUNX1, RUNX2 and HMGA2 (n = 1). (H) Lymph node tumors stain for RUNX2 and ZEB1 (250 μm, inset 50 μm; n = 1). see also Fig. S5 and Table S4.
The significantly correlated and anti-correlated TF motif-gene pairs included a number of known and novel regulators of KP cancer cell states. Among them were key lineage regulators of AT1 (Tead4), AT2 (Cebpa), lung (Nkx2.1, Gata6) and gastric (Hnf4a) development (Li et al., 2000; Treutlein et al., 2014; Zhang et al., 2007) as well as known tumor progression activators (Fosl1, Myc) and repressors (Zeb1) (Caramel et al., 2018; Gabay et al., 2014; Vallejo et al., 2017). We next assigned each significant TF to modules by correlating TF gene scores to module scores across cancer cells (Figure 5E). We identified novel Nkx-family activators (gain of non-lung-lineage Nkx-family members Nkx6.2 and Nkx2.9), likely reinforcing the NKX motif activity early in tumor progression (Figure 5E). Further, NKX2.1 motif accessibility was repressed prior to loss of the Nkx2.1 gene score, suggesting that the NKX2.1 TF motif score may be modulated by additional chromatin regulators or post-transcriptional regulation (Figure 4B and Figure S4H). Runx factors were associated with modules 9 and 2, suggesting RUNX-mediated changes occur late in cancer progression and metastasis (Figure 5E). The activators RUNX1 and RUNX2 have been found to be upregulated in several cancer types and are associated with metastatic progression, including in LUAD, breast, and prostate cancers (Bai et al., 2017; Li et al., 2013; Ramsey et al., 2018; Xie et al., 2016; Zheng et al., 2016). Onecut2 and Sox9 were associated predominantly with module 2, while Sox2 was most correlated with module 4 (Figure 5E). Onecut2 has been identified as a master regulator of androgen signaling and is a mediator of metastasis (Chuang et al., 2017; Guo et al., 2019; Ma et al., 2019; Rotinen et al., 2018) while Sox2 and Sox9 activity have been associated with the emergence of primitive epithelial programs during metastatic progression in studies of human LUAD (Laughney et al., 2020).
To validate the expression of these putative regulators, we performed IHC on advanced KP tumors and lymph node metastases. Similar to our gene score analysis, we found heterogeneous protein expression of RUNX1, RUNX2 and ONECUT2 in KP primary lung tumors, with near ubiquitous staining of these factors in late-stage cancer cells (defined as cells marked by ZEB1 or HMGA2 expression) (Figure 5F and Figure S5C). Regions in the KP primary tumors and lymph node metastases with low NKX2.1 and high HMGA2 expression exhibited the most robust staining of RUNX1 and RUNX2 expression, suggesting that these regulatory drivers initiate programs that mediate progression (Figure 5G,H). While RUNX expression was largely restricted to late-stage tumors, we also found RUNX1 expression in the airway (which lacked RUNX2 staining), suggesting differential RUNX expression patterns during normal lung development (Figure S5D). In addition, two of the predicted early-stage repressors, BATF and ZKSCAN5, were expressed in early-stage tumors, but not in late-stage tumors, suggesting that these repressors, among others, may restrict tumor progression (Figure S5E). Lastly, module analysis of microRNA (miRNA) gene scores, which function to modulate gene expression and are not detected from scRNA-seq analyses, identified known (Han et al., 2014; Kolesnikoff et al., 2014; Li et al., 2017) and novel miRNA regulators (Figure S5F,G). To date, the comprehensive identification of master regulator activators and repressors that drive tumor progression has been challenging; we suggest that these strategies outlined here may be used to identify tumor development regulators in other cancer subtypes.
Disruption of RUNX family transcription factors activate gene programs that drive tumor progression and metastasis
One striking finding of the module analyses was that the regulatory transition associated with loss of NKX-mediated regulation could be explained by a progressive gain of several late-stage co-accessibility modules. Importantly, we found that Runx1/2 gene scores and RUNX TF motif scores were strongly correlated with module 9 (Figure 4G and Figure 5C). Given the demonstrated role of RUNX TFs in tumor progression in other settings (Ge et al., 2016; Pratap et al., 2005, 2008), we next sought to functionally characterize the regulatory role of RUNX factors on chromatin accessibility surrounding genes associated with cancer progression. To establish a system to test this, we derived KP cancer cell lines from primary tumors and found increased expression of RUNX2 in metastatic (low NKX2.1 expression) compared to non-metastatic cell lines (high NKX2.1 expression) (Winslow et al., 2011) while RUNX1 was ubiquitously expressed (Figure S6A-C). To interrogate RUNX-mediated regulation in KP cancer cells, we engineered KP cell lines to express Cas9 and utilized CRISPR-based perturbation (knockout and activation) to modulate the expression of RUNX1, RUNX2, and RUNX3 (Figure 6A; STAR Methods). Perhaps because RUNX1 is already highly expressed in KP cell lines, we were unable to further increase RUNX1 expression via CRISPR activation. CRISPR knockout (KO) and CRISPR activation (CRISPRa) were achieved by transducing cell lines with guides targeting Runx1 and Runx2 and with truncated guides recruiting the HSF-MS2-p65 complex to the promoter of Runx2 and Runx3 (Horlbeck et al., 2016). Knockout was performed in RUNX high metastatic cell lines (n = 2) and overexpression was performed in a RUNX low non-metastatic cell line (n = 1) and a metastatic line (n = 1). We confirmed activation and knockout of RUNX proteins by western blot (Figure 6B,C and Figure S6D) and performed bulk ATAC-seq to determine the impact of RUNX family activity in KP cell lines.
Figure 6. CRISPR perturbation revealed RUNX TFs regulate extracellular matrix remodeling.

(A) Schematic of the strategy used to OE or KO TFs in tumor-derived KP cell lines. (B) Hierarchical (KO or OE vs control) RUNX TF motif scores (defined by RUNX perturbation score; top, bar plot) and associated differential gene scores (KO or OE vs control; bottom, heatmap) for each RUNX1, RUNX2, or RUNX3 KO (1183T3 and 860T3; metastatic) or OE (853T2; non-metastatic) bulk ATAC-seq experiment. RUNX perturbation score was determined using the slope from a linear regression. Samples include 1183T3: controls (n=10), RUNX1 KO (n = 10), RUNX2 KO (n = 6), RUNX3 KO (n = 1), RUNX2 OE (n = 2), RUNX3 OE (n = 3); 860T3: controls (n=10), RUNX1 KO (n = 15), RUNX2 KO (n = 7), RUNX3 KO (n = 3); 853T2 controls (n = 5), RUNX2 OE (n = 10), 853T2 RUNX3 OE (n = 3). (C) RUNX1 and RUNX2 expression in CRISPR KO cells from two independent guides as assessed by western blot; HSP90 shown as a loading control. (D) Log2 fold-change (RUNX KO vs control) of extracellular matrix proteins from a metastatic cell line (1183T3) with control (sgCON) (n = 1) or sgRunx2 (n = 1). Arrays with duplicate antibody spots and p values were determined by a Z-test (p<0.01*). (E) Chromatin accessibility tracks at differential RUNX genes (identified in panel B) for representative metastatic sgCON (control), metastatic sgRunx2 (KO), non-metastatic sgCON (control), and non-metastatic Runx2 (OE), and module-high cells (for comparison to KPT model). (F) Gene score for Lgals1 derived from cancer cells. (G) Multiplexed IHC for late-stage region in KPT tumor. Overlaid image (left), individual channel insets: green (NKX2-1), yellow (RUNX2), and red (LGALS1) (scale bar; whole tumor 2.4x, 500 μm; zoomed region, 7.5x, 200 μm). (H) Intravenous metastasis experiments with schematic for tail vein injection (left top). Exemplar IHC stains for example control and sgRUNX2 KO tumors (left bottom) (n = 5 per arm, repeated in triplicate). Survival curve (right) with log-rank p value (n = 5 per arm) with survival log-rank (Mantel-Cox) test. ** represents p<0.01. see also Fig. S6 and Table S5.
To analyze chromatin-induced changes associated with RUNX family perturbation, we performed TF motif and gene score regression analyses on RUNX-altered cell lines compared to sgRNA controls. Strikingly, overexpression (OE) guides increased the RUNX TF motif score and, in contrast, knockout guides decreased the score (Figure 6B). Importantly, RUNX protein OE and KO resulted in anti-correlated chromatin accessibility changes at RUNX TF motifs around relevant genes, suggesting that RUNX family members have functional overlap (Figure 6B and Figure S6E). We next defined differential gene scores associated with RUNX TF perturbation by correlating each gene score (normalized to controls) to the RUNX TF motif score across KO and OE conditions. GSEA of RUNX perturbation score associations against module-associated genes revealed that these gene signatures were significantly correlated with late-stage modules 9 and 4 and known oncogenic gene programs, including TGF-beta signaling (FDR q ≤ 0.001; Figure S6F-H and Table S5-6).
In further functional studies, we focused our attention on RUNX2 due to differential protein expression in tumor-derived cell lines. KP cancer cells have been found to secrete and differentially regulate ECM in late stages of the disease, which is thought to reshape the local environment and provide signals to adjacent cells (Brady et al., 2016; Gocheva et al., 2017; Reticker-Flynn and Bhatia, 2015). Therefore, we hypothesized that RUNX2 activity might affect extracellular secretion. To assess this directly, we analyzed the conditioned cell culture media from RUNX2-altered cell lines using extracellular protein antibody arrays (Figure 6D, Figure S6I,J and Table S5). We confirmed that RUNX2 KO cells have reduced secretion of well-studied ECM proteins, first identified using chromatin accessibility gene scores, including Lgals3 (Figure 6D). Genes associated with ECM components, cytoskeletal remodeling, and altered RUNX activity were upregulated in the single-cell data, including PODNL1 and LGALS1 (galectin-1), among others (Figure 6E,F and Figure S6K). Using multiplexed IHC, we found that RUNX2 positive tumor cells colocalized with LGALS1 expression, demonstrating the utility of chromatin accessibility studies to identify downstream targets of transcription factors (Figure 6G and Figure S6L-N). Finally, we performed tail vein injections of control and RUNX2 KO cancer cell lines to assess their metastatic potential. Notably, deletion of RUNX2 in metastatic KP cells resulted in significantly fewer lung metastases as well as increased survival of injected mice (Figure 6H and Figure S6O). This functional validation of RUNX2 biology demonstrates the utility of TF motif-gene analyses for discovering master regulators and provides an analytical framework for characterizing downstream changes induced by TF perturbations.
Regulatory networks derived from mouse KP cancer cells predict survival in human LUAD patients
We next investigated whether RUNX-mediated dysregulation might be relevant to human LUAD tumor progression. To this end, we performed IHC on human LUAD tissue microarrays (TMAs). Increased RUNX1 and RUNX2 staining was observed in higher grade lesions and confirmed in TMAs from the Human Protein Atlas, consistent with the role of RUNX in tumor progression (Figure 7A, Figure S7A, Figure S7B). We next tested whether module-associated gene scores defined in our study could represent new signatures with prognostic value in human LUAD patients. We determined representative gene signatures for each module by assigning the top 200 genes whose gene scores were most correlated with the module accessibility scores (Figure 7B, Figure S7C and Table S6). This analysis identified several expected gene associations, such as Sftpc with the AT2-like module 11 and EMT genes Vimentin and Twist1 with AT1-like module 6 (Figure 7B). To test if genes associated with each of the 11 co-accessibility modules were predictive of clinical outcome in human LUAD, we queried them against The Cancer Genome Atlas (TCGA) collection of bulk primary LUAD RNA-seq profiles (n = 506) (Figure 7B) (Campbell et al., 2016; Cancer Genome Atlas Research Network, 2014). We stratified patients by high versus low average expression of each module gene signature and tested for association with overall patient survival. Genes associated with late-stage module 3 and 9 were the most predictive of poor survival (logrank test p = 0.0031 and 0.0035, respectively) independent of patient genotype (Figure 7C,D and Figure S7D-F). We also found that modules highlighted in the early stages of tumor progression (module 11, 7, 5) were associated with better prognosis (p < 0.05), with module 11 having the greatest prognostic relevance (p = 2x10−6) (Figure 7C, Figure S7D). The module 9 gene signature outperformed NKX2.1 expression in predicting overall patient survival (Figure S7G), suggesting that regulatory analyses of single-cell epigenomics data can serve as surrogate markers for underlying processes defining tumor development and, thus, can more accurately predict survival in human patients.
Figure 7: Module-associated genes were predictive of survival across human LUAD cases.

(A) LUAD tumor microarray (TMA) map stained with RUNX1. Individual images of tumor sections with grade indicated on tumor image. (B) Schematic of human module survival analyses. Module-specific genes from mouse cancer cells were used to score RNA-seq data from primary human LUADs in TCGA (n = 506) to determine association with patient survival. (C) Significance of module-associated genes with overall survival (OS) based on a logrank test (dashed lines: logrank p = 0.01). Positive values denote decreased survival, negative values denote increased patient survival for patients with higher median module expression. For p value significance, **** represents p<0.0001, *** represents p<0.001, ** represents p<0.01, and Represents p<0.05. (D) Kaplan-Meier plots for human LUAD patients with respect to expression of module 11 (left) or module 9- (right) associated genes. Curves are shown comparing OS of high (red) versus low (blue) patient groups, determined based on the median module expression. p values determined by a logrank test. **** represents p<0.0001 and ** represents p<0.01. see also Fig. S7 and Table S6.
Discussion:
This single-cell epigenomics study adds to an increasing body of evidence that a common feature of tumor development is intratumoral heterogeneity, including at the chromatin level (Hinohara and Polyak, 2019; Lawson et al., 2018). Here we use a single-cell approach to determine the epigenomic evolution in a well-established GEMM of lung adenocarcinoma with limited confounding somatic variation. Together with Marjanovic et al. (see accompanying paper), we provide a deep characterization of the chromatin accessibility and transcriptional changes that drive cancer progression in this model. Single-cell epigenomic profiling provides a complementary approach to the study of gene regulation, as transcription factors are susceptible to technical drop-out in scRNA-seq approaches. In this study, we utilized the full epigenome as markers for cell state, rather than limiting our analysis to individual gene markers, powering robust cell state assignments. We identify heterogeneous cell states that reveal a diverse and continuous landscape of regulatory transitions. Within this diverse landscape, we find evidence of lineage infidelity (Ge et al., 2017) and cellular plasticity, as demonstrated by cells reflecting AT2, mixed, and AT1-like states through tumor progression, consistent with cell identities shown in the accompanying scRNA-seq study (Marjanovic et al.) and recently reported cell states in human LUAD tumors (Laughney et al., 2020). Furthermore, we find evidence of the high plasticity state described in Marjanovic et al., with chromatin state changes surrounding cell surface genes Slc4a11, Tigit, and Itga2 (Figure 7B).
Interestingly, we identified cells in primary tumors with regulatory states resembling those of cells isolated from metastatic sites. These metastatic-like cells exist within the primary tumors at a low frequency in a subset of tumors, indicating that transformation to this state is rare and stochastic. Cells isolated from metastases were less heterogeneous than primary tumors, consistent with the notion that cancer cells ultimately funnel toward a stable epigenomic state and add a regulatory context to prior reports demonstrating metastatic cells as more genetically homogeneous than primary tumors (Turajlic et al., 2018). Importantly, we also find that the heterogeneity observed across a collection of tumors was largely reproducible across individual tumors. These data support a model of a rather constrained set of cell state progressions that lead toward a metastatic state. Our data are also compatible with the emergence of non-productive paths in tumor development that do not ultimately result in metastasis.
In order to characterize this diverse regulatory landscape, we developed a computational framework for determining co-accessible modules using TF motif-driven chromatin changes. We also used gene scores to infer i) the upstream TF regulators of these modules and ii) the downstream target genes they regulate. This analytical approach allowed us to collapse the diverse spectrum of tumor states into 11 coherent co-accessibility programs–defined by the combinatorial activity of transcription factors–that we posit to represent meaningful regulatory transitions across the heterogeneous landscape. Altogether, our co-accessibility analysis largely uncovered developmental and lineage-identity regulators, adding to the concept that chromatin accessibility-mediated regulation is predominantly linked to developmental processes (González et al., 2015; Hnisz et al., 2013). By contrast, gene expression analysis integrates RNA processing, RNA stability, cell size and proliferation programs along with lineage identity (Shema et al., 2019). Altogether our data reveal massive reprogramming of the regulatory landscape within LUAD tumors, without direct genetic alteration of transcription factor function.
In this study, we define a co-accessibility module (module 9) representing a key and previously undefined transition between Nkx2.1 loss and EMT induction. We utilize CRISPR strategies to show that RUNX2 drives ECM-related gene expression and is a critical regulator of this module. Notably, remodeling of tumor-derived ECM is considered to be an important aspect of EMT in relation to tumor progression, promoting sequestration of cancer cells from immune responses and the microenvironment (Naba et al., 2012). Therefore, we propose that activation of RUNX2 functions in LUAD to initiate the expression of ECM proteins to develop a niche that sensitizes cells for EMT. We anticipate that further elucidation of this mechanism and other regulatory programs associated with key steps in tumor progression will reveal epigenetic and other cellular processes that could be targeted for intercepting the progression of human LUAD.
Single-cell technologies provide new opportunities to better understand primary tumor development. By improving experimental and computational workflows, our study has provided an atlas of the regulatory landscape of LUAD in a well-studied model system. However, additional work is needed to determine which selective pressures drive individual cells to undergo these regulatory state transitions; for example, the role of the tumor microenvironment (Altorki et al., 2019; Azizi et al., 2018) and of chromatin-modifying proteins (Rowbotham et al., 2018; Serresi et al., 2016; Zhang et al., 2017). To this end, we expect lineage-tracing approaches to be paired with single-cell approaches to determine how cells navigate these regulatory transitions toward productive or non-productive paths of cancer (Woodworth et al., 2017). Importantly, we find these regulation-derived co-accessibility modules can be used to score RNA expression from LUAD patients to provide highly predictive markers of survival. We anticipate additional efforts towards the characterization of direct epigenomic biomarkers using either ATAC-seq or DNA methylation will be valuable for the discovery of regulatory patterns across genes or regulatory elements providing a more robust strategy to define cell state regulators useful for the diagnosis and treatment of cancer.
STAR Methods.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tyler Jacks (tjacks@mit.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The datasets generated during this study are available at Gene Expression Omnibus (GEO) under GSE134812, GSE145192, and GSE151403. The raw single-cell ATAC-sequencing files and processed data files generated in this study are available in GEO under the super series GSE145194. Single-cell combinatorial indexing data is available under GSE134812 and ETP data is available under GSE145192. Bulk ATAC-sequencing raw and processed files are available under accession GSE151403. The R Shiny-based web application for data visualization is accessible here: https://buenrostrolab.shinyapps.io/lungATAC/
UCSC genome browser tracks associated with this study are made available with the following weblinks: Normal cells cluster: http://genome.ucsc.edu/s/lmlafave/normal_lung_scATAC KPT modules: http://genome.ucsc.edu/s/lmlafave/KPT_modules
Code used for the analysis of scATAC-seq data in this study is available on Github (https://github.com/buenrostrolab/lungATAC_analysis_code).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mouse experiments described in this study were approved by the Massachusetts Institute of Technology Institutional Animal Care and Use Committee (IACUC) (institutional animal welfare assurance no. A-3125-01). LSL-KrasG12D/+; Trp53fl/fl mice have been described previously (Jackson et al., 2001, 2005). Mice were crossed with the tdTomato Ai9 reporter allele from Jackson laboratory (stock 007905) to generate LSL-KrasG12D/+; Trp53fl/fl; Rosa26tom/+ mice. All mice were maintained on a mixed C57BL/6-129/Sv background. Mice with appropriate genotypes were aged 8-12 weeks and randomly selected for tumor initiation studies. Mice were infected intratracheally with Adenoviral SPC-Cre (Ad5-SPC-Cre) virus (Iowa) as described with viral titers 1x108 or 2.5x107 TTU to allow for the development of metastases (Sutherland et al., 2011). Normal lungs were collected from wild-type mice from a mixed C57BL/6-129/Sv background in mice aged to 6-8 weeks. Mice of both sexes were used for experiments, but predominantly male mice were profiled for single-cell analyses.
Isolation of normal lung and primary lung adenocarcinoma cells from mice
LUAD cells were isolated from mice as described previously, with a few modifications (Tammela et al., 2017). Genotyping primers are listed in Table S7. Briefly, KrasG12D/+; Trp53−/−; Rosa26tom/+ mice were euthanized 30-35 weeks after tumor initiation. Whole tumor burdened lungs or individually plucked tumors were dissociated with fine scissors and then proteolytic digestion was performed using the Lung Dissociation kit (Miltenyi Biotech) following the manufacturer’s instructions. Dissociated cells were then incubated at 37°C for 20 minutes with rotation, then filtered using a 100-μm strainer. Red blood cells were lysed using ACK buffer (Thermo Scientific) and stained with APC-conjugated CD45 (BD, 559864), CD11b (eBioscience 17-0112-82), CD31 (Biolegend, 102510), Ter119 (BD, 557909), and DAPI (Sigma-Aldrich). FACS of immuno-stained primary cells was performed using a FACSAria sorter (BD) to isolate tdTomato+; DAPI-; APC- tumor cells for sciATAC-seq. Normal lungs from mice were processed similarly to tumor-burdened tissue. CD45 depletion was conducted using CD45 microbeads (Miltenyi Biotec) following ACK Lysis.
For early time-point analyses, mice were euthanized 8 weeks following tumor initiation in an isoflurane chamber. Lungs were inflated by injecting digestion buffer (Adv DMEM/F12, 5uM HEPES, DNase, 1mg/mL Collagenase, 0.36mM CaCl2) into the trachea. Lungs were dissociated with fine scissors and proteolytic digestion was performed using the lung digestion buffer. Dissociated cells were incubated at 37° for 1 hour with rotation. Cells were washed in 1x PBS and red blood cells were lysed using ACK lysis buffer (Thermo Scientific) for 3 minutes at room temperature. Cells were filtered using a 100um strainer and stained with DAPI (Sigma-Aldrich). FACS was performed using a FACSAria sorter (BD) to isolate tdTomato+ cells for single-cell droplet ATAC-seq.
Cell culture and cell line generation
Individual tumors were dissected, digested in an enzymatic buffer (1X HBSS, 5mM HEPES, DNasel, Collagenase IV), and incubated with rotation at 37°C for 30 minutes. The enzymatic buffer was quenched with DMEM and spun at 1000 rpm. Cell pellets were resuspended in DMEM and plated in 6-well plates to allow for attachment. Cell lines were genotyped for Kras, p53, and tomato after 5 passages in culture. The cell lines used in this study were established from mouse LUAD over the course of the study. All lines were grown in DMEM, 10% FBS, and 1% pen-strep. KP cell lines have not been authenticated because the cell lines are not found in established databases. The KP cell lines were tested for mycoplasma and found to be negative. GM12878 cells were grown in DMEM, 10% FBS, and 1% pen-strep and 3T3 cells were grown in RPMI 1640, 10% FBS, and 1% pen-strep. GM12878 cells were authenticated by STR Profiling Service from ATCC.
METHOD DETAILS
Lentiviral vectors and sgRNA cloning for CRISPR and CRISPRa
For CRISPR knockout experiments, guides were cloned into the lentiCRISPR-V2 lentiviral vector (Joung et al., 2017). The lentiCRISPR-V2 vector was digested with Fast Digest EspI and ligated with EspI-compatible annealed oligonucleotides for sgRNAs. KP cell lines were infected with constructs containing guides and selected with puromycin after 48 hours. After puromycin selection, guide performance was tested by western blotting. For CRISPR activation (CRISPRa) experiments, the Lenti-Sam-puro construct was used (developed in the Jacks lab), an adaption of the previously published Lenti-Sam activation construct (Pentinmikko et al., 2019). EspI-compatible cloning was completed and cells were infected with constructs containing guides based on CRISPRa prediction software. Truncated guides of 15 bp were cloned into a lentiviral based expression construct which also encodes for a transcriptional activation complex (MS2-P65-HSF1) and a puromycin selection cassette. Non-metastatic and metastatic cell lines were engineered to express Cas9 following stable selection of a Cas9-Blast construct.
Western blots
Cells were lysed in RIPA buffer supplemented with protease inhibitors (Halt™, Thermo Scientific) and phosphatase inhibitors (Thermo Scientific) and incubated at 4°C for 20 minutes and were then cleared by spinning maximum speed for 10 minutes. The protein concentration of lysates was determined using the Pierce BCA Protein Assay (Thermo Scientific). Total protein concentrations of 40 μg were run on NuPage 4-12% Bis-Tris gradient gels (Thermo Scientific) by SDS-PAGE and transferred to nitrocellulose membranes. All western blots were imaged with a BioRad ChemiDoc MP imager. The following antibodies were used for immunoblotting: anti-Hsp90 (1:10000); BD Biosciences 610418, anti-Runx1 (1:1000), Cell Signaling Technology, 8529S, anti-Runx2 (1:1000), Cell Signaling Technology, 12556S, and anti-Runx3 (1:1000) Abcam, ab23981.
Immunohistochemistry
Individual lung tumors were fixed overnight in zinc formalin and embedded in paraffin. Tissue sections were dewaxed using a Thermo Autostainer 360. All sections from the same tumor regions were serially sectioned. Slides were then stained using antibodies against Nkx2.1 (1:1000), Hmga2 (1:1000), Onecut2, Proteintech, 21916-1-AP, 1:500 (in 1X PBST); Runx1, Cell Signaling Technology, 8529S, 1:500; Runx2, Cell signaling,12556S, 1:1000; Cav1, Sigma, C3237, 1:1000; Sftpb, ThermoFisher, PA5-42000, 1:200; Zeb1, Abcam, ab87280, 1:500; BATF, Sigma, SAB4500122, 1:100; Zfp95, Novus Biologicals, NBP2-20947, 1:200; RFP, Rockland, 600-401-379, 1:400; Fra1, ThermoFisher, PA5-40361, 1:100; CD45, Abcam, ab10558, 1:1000, Cell signaling technology 12556S, 1:1500; Sftpc, Millipore sigma AB3786, 1:5000; LGALS1, Cell Signaling Technology, 1388S, 1:1000. Slides were also counterstained with haematoxylin.
Opal Four-Color anti-Rabbit Manual Immunohistochemistry
Lung tissue was fixed overnight in zinc formalin and embedded in paraffin. Tissue sections were dewaxed using a Thermo Austostainer 360 and then fixed in 10% neutral buffered formalin for 20 minutes. Slides were stained sequentially using antibodies against Nkx2.1, Abcam ab76013, 1:250 (in Perkin Elmer Antibody Diluent/Block); Runx2, Cell Signaling Technology 12556S, 1:250; Pdpn, Abcam ab109059, 1:250; Hopx, Proteintech 11419-1-AP, 1:100; SPC, Millipore Sigma AB3786, 1:400; Lgals1, Cell Signaling Technology 13888S, 1:100. After detection with an Opal fluorophore (1:100 in Perkin Elmer 1X Amplification Diluent), the primary and secondary antibodies were stripped using a pressure cooker, followed by another round of staining. Slides were counterstained with DAPI and coverslipped using ProLong Diamond Antifade Mountant (Thermofisher). Slides were scanned using Pannoramic 250 Flash III at 20X or 40X.
Human lung adenocarcinoma arrays and Human Protein Atlas
The human lung adenocarcinoma microarrays used were LC1005A and LC2083 (Biomax) and were stained as described above in immunohistochemistry. Representative tissue sections from patients on the Human Protein Atlas (http://proteinatlas.org) were also included for RUNX1 and RUNX2 (Uhlén et al., 2015). Images can be found online at the following links (RUNX1; 2438; https://images.proteinatlas.org/4176/12280_B_1_1.jpg); (RUNX1; 2403; https://images.proteinatlas.org/4176/12280_B_3_4.jpg), (RUNX2; 4883; https://images.proteinatlas.org/22040/140406_B_1_8.jpg), (RUNX2; 4873; https://images.proteinatlas.org/22040/140406_B_2_4.jpg), (RUNX3; 4866; https://images.proteinatlas.org/25416/151928_B_1_2.jpg), (RUNX3; 1327; https://images.proteinatlas.org/25416/151928_B_1_4.jpg) available at v19.proteinatlas.org.
Quantitative PCR
RNA was isolated from cells using the RNeasy Plus kit (Qiagen) as specified by the manufacturer’s instructions. cDNA was synthesized from 1 μg of RNA using the High-Capacity cDNA reverse transcription kit (Thermo Scientific) and RNase inhibitor (Thermo Scientific). qPCR experiments were performed in triplicate with SYBR fast master mix (Kapa Biosystems) on a Roche Lightcycler 480 qPCR machine. Expression was normalized to Actb. All experiments were performed with three replicates.
Bulk-ATAC
For bulk-ATAC, the previously published ATAC-seq protocol was adapted from (Buenrostro et al., 2013; Ludwig et al., 2019). Briefly, 25,000-50,000 cells were trypsinized and washed twice in PBS. Pelleted cells were then directly transposed using an all-in-one transposition buffer (Tris pH 7.5, MgCl2, DMF 5%, PBS 0.3X, NP-40 0.1%, Illumina Tn5 1X, ddH20 to 50 uL). The transposition reaction was completed with thermomixing at 37°C for 30 minutes at 300 rpm on a thermoshaker. Transposed DNA was purified with MinElute column cleanup (Qiagen), then minimally amplified for sequencing as previously described (Buenrostro et al., 2015b). Prepared libraries were purified with MinElute column clean-up (Qiagen) and digested with ExoI (NEB). Libraries were quantified with a Qubit dsDNA HS Assay kit (Invitrogen) and sequenced on the Next-seq platform (Illumina) using a 75-cycle kit. Bulk ATAC-seq data was processed as previously described (Buenrostro et al., 2015a). Briefly, reads were trimmed and aligned using Bowtie 2 (v2.3.3.1) and the same peakset was utilized for single-cell ATAC sequencing experiments.
Extracellular secreted protein array
Extracellular protein antibody arrays were conducted using an L-308 mouse protein array (RayBiotech) following manufacturer instructions. Briefly, cells were seeded at a density of 1 x 106 in DMEM with 10% FCS for 48 hours. The media was then replaced with DMEM containing 0.2% FCS and collected after 48 hours of incubation. Supernatants were centrifuged at 1000 x g for 10 minutes and dialyzed overnight in dialysis buffer (2.6 mM KCl, 137 mM NaCl, 1.5 mM KH2PO4, and 8.1 mM Na2HPO4, pH=8) using dialysis vials (RayBiotech). The dialyzed media was then quantified and protein was labeled with biotin based on protein concentration. Excess biotin was removed from the media via spin filtration. Filtered biotin-labeled protein was incubated on arrays overnight. The arrays were then blocked and incubated with HRP-Streptavidin. Antibody arrays were imaged with the BioRad ChemiDoc MP imager and quantified using the protein microarray plugin on ImageJ (v1.52k) (Carpentier 2010, Schneider et al., 2012) with two replicates. Log2fold change was calculated for each spot on the array (in duplicate) and standard deviation across duplicate spots.
Aiforia
Histological quantification of mouse lung tumor grade was performed by an automated deep neural network (unpublished) developed by Aiforia Technologies in collaboration with the Jacks lab, and in consultation with veterinarian pathologist Dr. Roderick Bronson. We trained a convolutional neural network (CNN) for semantic multi-class segmentation using the Aiforia(R) platform. The CNN was trained to classify and detect lung parenchyma, NSCLC tumors, and NSCLC tumor grades (grade 1-4). For supervised training, we used selected areas from 93 hematoxylin and eosin stained slides. The algorithm performed consistently and with high correlation with human graders across multiple validation datasets independent of the training dataset. For grade calling, the NSCLC_v25 algorithm was used.
Tail vein injections
B6129SF1/J (Jackson lab, stock 101043) male mice were injected with between 100K-150K cells intravenously via the tail vein. Experiment in triplicate with control guides and RUNX2 guides. Mice were euthanized at experiment endpoint (3-4 weeks following cell line injection) and tumor burden was determined by organ weight and immunohistochemistry. Experiment was replicated three times with one replicate presented. Tumor volume was quantified using percentage of total tumor tissue area divided by normal tissue area.
Methods for sciATAC-seq
sciATAC-seq sample processing
Fixation:
Normal or tumor-derived lung cells were transferred to centrifuge tubes that were pre-coated with 7.5% BSA. Cells were centrifuged at 300g for 5 min, washed once in PBS, and resuspended to 1 million cells/ml. Cells were then fixed with 0.1% formaldehyde and incubated at room temperature for 5 min. The fixation was stopped by adding glycine to the final concentration of 125 mM. The sample was incubated at room temperature for 5 min and then centrifuged at 500g for 5 min to move supernatant. The cell pellet was washed twice with 1 ml of PBS and centrifuged at 500g for 5 min between washes. The cells were resuspended to 1-2 million cells/ml in PBS.
Transposition:
All the oligonucleotides used in this protocol can be found in Table S1. The 100 μM Ad1 or Ad2 oligos that have unique barcodes were annealed with an equal amount of 100 μM blocked ME-compliment oligo by heating at 85°C for 2 min and slowly cooling down to 20°C at a ramp rate of −1 °C/min. The annealed oligos were mixed with an equal volume of cold glycerol and stored at -80°C until use. In-house produced Tn5 (Picelli et al., 2014), was mixed with an equal volume of dilution buffer (50 mM Tris, 100 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 0.1% NP-40, and 50% glycerol). The diluted Tn5 was then mixed with an equal volume of annealed oligos and incubated at room temperature for 30 min before transposition.
Fixed cells (1 μl) and 7 μl of 1.25x transposition buffer (41.25 mM Tris-acetate, 82.5 mM K-acetate, 12.5 mM Mg-acetate, 20% DMF, 0.125% NP-40, 0.5% Protease Inhibitor Cocktail) were distributed onto a 96-well plate. The plate was incubated at room temperature for 10 min. The assembled Tn5 was diluted with an equal volume of 1.25x transposition buffer. 1 μl of diluted Tn5 containing Ad1 oligo and 1 μl of diluted Tn5 containing Ad2 oligo were distributed onto a 96-well plate. The transposition was carried out at 37°C for 30 min with shaking at 300 rpm. The reaction was stopped by adding 1 μl of 0.5 M EDTA and incubated at 37°C for 15 minutes with gentle shaking at 300 rpm. All the cells were then pooled and 38.4 μl of 1 M MgCl2 was added to the pooled sample. The sample was centrifuged at 500g for 2 min and then washed with 1 ml of EB buffer (Qiagen) with 0.1% Triton X-100. The sample was resuspended to 0.5 ml of EB buffer with 0.1% Triton X-100. The sample was passed through a 50 μm filter to remove clumps and diluted to 6.7 or 13.3 cell/μl with the same buffer.
Reverse crosslinking and PCR:
2 μl of the sample was re-distributed onto another 96-well plate with 1.5 μl sample on each well. 2.5 μl of 2x reverse crosslinking buffer (100 mM Tris pH 8.0, 400 mM NaCl, 2 mM EDTA pH 8.0, 2% SDS, and 40 μg/ml proteinase K), 0.5 μl of 10 μM P2 oligo, and 0.5 μl of 10 μM P1 oligo were added to each well. The plate was incubated at 55°C for 16 hours for reverse crosslinking. 5 μl of 10% Tween-20 was then added to quench SDS. 12.5 μl 2x NEBnext PCR mix and 2.5 μl H2O were added to each well.
The PCR reaction was carried out at the following conditions:
72°C for 5 min (extension), 98°C for 5 min, and then thermocycling at 98°C for 10 s, 70°C for 30 s and 72°C for 1 min. After thermocycling for 5 cycles, we took a 5 μl sample from a few randomly selected wells and added 10 μl of PCR cocktail with 0.6x SYBRgreen. The 15 μl reactions were amplified to saturation to determine the number of cycles required for the remaining samples on the plate. Libraries were amplified for 13-14 cycles in total. Libraries were pooled and purified using Qiagen MinElute PCR purification column. The libraries were quantified using KAPA library quantification kit (Buenrostro et al., 2013). Libraries were sequenced on the Next-seq platform (Illumina) using a 150-cycle kit (Read 1: 47 cycles, Index 1: 36 cycles, Index 2: 36 cycles, Read 2: 47 cycles).
Read alignment and pre-processing
Base calls were converted to fastq format using bcl2fastq. Raw sequencing reads were trimmed using custom python scripts to remove adapter sequences. The reads were aligned to hg19 or mm10 genome using Bowtie2 (Langmead et al. 2012) with maximum fragment length set to 2 kb, and all other default settings (bowtie2 - X2000 --rg-id). The data were demultiplexed tolerating one mismatched base within barcodes. Mitochondrial, discordant and low quality reads were removed using SAMtools v1.9 (Li et al. 2009) (samtools view -b -q 30 -f 0x2). Duplicate sequences were removed using the picard toolkit (2.14.1-SNAPSHOT) (http://broadinstitute.github.io/picard/).
Peak calling
sciATAC-seq profiles for all cells were first merged into a single alignment (.bam) file and used as input for peak calling with MACS v2.1.2 (MACS2) (Zhang et al., 2008). All default options were used, with the following flags explicitly set: --nomodel, --nolambda, --keep-dup all, --call-summits. This returned a list of single base pair peak summits with associated significance scores (corresponding to log FDR q-value from MACS2). Only peak summits with FDR < 0.01 were retained. Next, a previously described iterative filtering approach was implemented to obtain a list of significant, non-overlapping fixed-width peak windows (Lareau et al., 2019). Briefly, the called peak summits were first padded with 150 base pairs (bp) at either end to generate evenly sized 301 bp window peak regions. Peaks were then sorted in decreasing order of their significance scores. Keeping the most significant peak, overlapping peak windows that had lower significance scores were identified and then removed. This was repeated for the next most significant peak window. Through this iterative process, lower significance overlapping peak regions were filtered out, resulting in 285,956 disjoint 301 bp peak windows.
sciATAC-seq counts generation and QC
Using the generated peak region list, the number of reads overlapping a given peak window (n = 285,956 peaks) was determined for each unique cell barcode tag. This generated a peak by cell counts matrix corresponding to ATAC reads in peaks for each cell profiled. Only cells having FRIP ≥ 0.4 and a minimum of 2000 unique nuclear reads per cell were retained for downstream analyses, resulting in a total of 17,274 cells.
ETP single-cell droplet ATAC-seq and analysis
ETP cells were profiled using the Whole Cell Tagmentation protocol as described previously (Lareau et al., 2019) using the SureCell ATAC-Seq Library Prep Kit (17004620, Bio-Rad). Briefly, cells were washed with 1mL 1x PBS + 0.1% BSA and resuspended in cold Whole-Cell Tagmentation Mix (ATAC Tagmentation Buffer, ATAC Tagmentation Enzyme, 0.5% Digitonin, 5% Tween-20, nuclease-free water). The cell suspension was incubated at 37° for 30 minutes with shaking. Barcode Suspension Mix and Enzyme Suspension Mix were prepared and kept on ice for droplet encapsulation. Tagmented nuclei were resuspended in the Enzyme Suspension Mix. Droplet encapsulation was performed using the Bio-Rad ddSEQ Single-Cell isolator. The encapsulated samples were transferred to a chilled 96-well plate for barcoding and amplification. The incubation protocol was as follows: 37° for 30 minutes, 85° for 10 minutes, 72° for 5 minutes, 98° for 30 seconds, eight cycles of 98° for 10 seconds, 55° for 30 seconds, 72° for 60 seconds, then 72° for 5 minutes. Emulsions were broken with the Droplet Disruptor and fragments were purified using AMPureXP beads. Barcoded fragments were amplified using the ATAC PCR Supermix and ATAC Primer Mix with the following incubations: 98° for 30 seconds, 7 cycles of 98° for 10 seconds, 55° for 30 seconds, 72° for 60 seconds, then 72° for 5 minutes. PCR products were cleaned up a second time using AMPure XP beads. scATAC-seq paired-end reads were first debarcoded using the bap-barcode utility as part of the bead-based ATAC-seq processing (BAP) pipeline (v0.5.9i; https://github.com/caleblareau/bap), allowing for 1 base mismatch. The resulting sequencing read files were aligned to the mm10 mouse reference genome assembly using BWA v0.7.15, and the corresponding alignment files processed to handle droplet bead multiplet merging using bap, with the following parameter specifications: -r mmio -bf 500 -bt XB. The same peak set derived using the mouse lung sciATAC-seq data was used to generate a reads in peaks counts matrix for ETP cells. Only cells with FRIP ≥ 0.4, unique nuclear fragments > 2,000 and a sequence duplication rate of at least 40% were retained (n = 4,610 cells).
SciATAC-seq data analysis and visualization
TF motif and k-mer scoring in single cells using chromVAR
TF motif and sequence k-mer accessibility scores were computed for single cells using chromVAR (Schep et al., 2017). The filtered accessibility counts matrix of peaks (n = 285,956) by cells (n = 17,274) was used as input data, along with binary overlap annotation matrices of either peaks by TF motifs (for TF motif scores) pertaining to a curated list of mm10 cisBP motifs (n = 797) or all possible 6-mers (n = 2,080; for k-mer scores) as previously described (Schep et al., 2017). Background peaks were sampled (n = 250 iterations) to adjust for GC bias and overall accessibility across all cells for each peak, and were used to compute motif and k-mer accessibility deviation Z-scores using the computeDeviations function in chromVAR (v0.2.0).
Single-cell clustering and visualization
The matrix of k-mer accessibility deviation Z-scores was first column-scaled and centered (using the scale function in R v.3.5.3) (R Core Team, 2019), and run through a principal component analysis (PCA) dimensionality reduction. The Uniform Manifold Approximation and Projection (UMAP) algorithm (McInnes et al., 2018) was then applied to project single cells in two dimensions using the k-mer PC scores for the first 20 PCs (implemented using the uwot package (v0.1.4) in R with the following non-default clustering parameters: n_neighbors = 20, min_dist = 0.4, metric = “cosine”). To further cluster the normal lung cellular populations into distinct subgroups, we applied the same PCA and UMAP clustering strategy, using only the k-mer accessibility Z-scores for normal lung cells (n = 3,604 cells). The Louvain method for network community detection (Blondel et al., 2008) was then applied on a k-nearest neighbor (k-NN) graph built using the normal cell PC scores (k = 50), identifying 12 distinct normal cell clusters that were then annotated and visualized in the original UMAP space (see Table S1).
Early time point (ETP) single-cell projection
Projection of ETP cells was performed using k-mer accessibility Z-scores derived from scATAC-seq data generated for 8 weeks tumor ETP cells, and the k-mer PC coefficients from the PCA run of all the lung cells used to produce the original UMAP clustering (Figure 2A). First, the matrix of ETP cell k-mer Z-scores (n = 2,080 k-mers and 4,610 cells) was multiplied with the PC coefficients matrix (2,080 k-mers x 20 PCs) to get a cells by PC scores matrix. We then projected the ETP cell PC scores onto the UMAP space of all lung cells using the umap_transform function in the uwot package in R.
Chromatin module definitions and single-cell scoring
Modules or groups of peaks exhibiting similar changes in accessibility with respect to TF motif deviation across tumor and metastatic cells (n = 17,274) were defined as follows. First, TFs were first grouped into ‘bags’ based on their motif sequence correlation levels (Pearson r cut-off = 0.7), with bag leaders determined as the TF with the most variable accessibility Z-score (from chromVAR) within a given TF bag. We then applied a jackstraw PCA (Chung and Storey, 2015) approach to filter for only those TFs whose motif accessibility significantly contribute to the systematic variation captured by PCA (as implemented using the Jackstraw function in the Seurat package (Satija et al., 2015) for gene filtering). Jackstraw PC coefficients were determined by randomly sampling 20% of the TF bag leader motifs and running PCA on their chromVAR deviation Z-scores, with motif accessibility scores independently permuted across all tumor and metastatic cells (i.e. any association of the feature sets and cells is distorted), keeping only the first 20 PCs. Doing this for n = 1000 iterations, permutation p values for each TF motif and each PC are determined by comparing PC coefficients from running PCA on the true unaltered dataset with the jackstraw PCA coefficients. Only TF motifs with p value < 0.1 among the first 10 PCs were kept, resulting in 67 TFs. Next, for each of these TF motifs, cells were binned as either motif-“high” or “low” based on whether their motif scores were above or below the median motif accessibility Z-score for all tumor cells, respectively. Counts of single-cell reads in peaks, normalized by the mean counts per cell across all peaks, were then used to test for differential accessibility between the high vs low cells for each peak using a two-sample Student’s t-test. Peaks that were significantly differentially accessible at FDR q < 10−6 for each TF were then retained to yield a set of 74,732 unique peaks. Log-2 fold-change of the mean accessibility for each of these peaks between the high and low groups was then computed for each motif. The resulting matrix of fold-changes of peaks (rows) across motifs (columns) was converted to a k-NN graph (k = 30) which was used to cluster the peaks using the Louvain method. This yielded 11 unique peak clusters (which we refer to as ‘modules’), which were used as peak annotations for chromVAR (see Table S3), along with scATAC-seq reads in peaks counts to score all single cells based on their enriched chromatin accessibility within module-specific peaks. For tracks, cells were defined as ‘module-high’ if their module accessibility score was greater than 2 standard deviations above the mean module score across all cells.
Gene activity scoring in single cells
Single-cell chromatin accessibility signal around gene TSSs was used to compute gene scores. TSS annotation pertaining to the RefSeq mm10 genome build (http://genome-euro.ucsc.edu/cgi-bin/hgTables) was obtained and processed into single base-pair, strand-aware coordinates (n = 35,856 genes). Scores were then computed per gene TSS as previously described, with slight modifications (Lareau et al., 2019). Briefly, an exponential decay function with a half-life of 1 kb was used to weight aligned sciATAC-seq reads based on the distance of aligned fragment centers to the TSS for a given gene. The total distance considered was set to 4,606 bp on either side of the TSS, determined to be the distance at which the decay weight equals 1%. These weights are then summed per cell for all fragments overlapping the 9,212 bp window around the TSS, to give the gene score for each cell. The equation below summarizes how gene score gaX for gene a in cell X is computed:
N : Total number of aligned fragments overlapping the TSS window for cell X
di : Distance (in bp) of the ith fragment center to the TSS
wi: Weight of ith fragment
Single-cell gene scores were then normalized to the mean gene score per cell, and used for downstream analysis. For visualization of gene scores in single cells (UMAP plots), normalized gene scores for cells were smoothed based on their nearest-neighbors (k = 10) defined using the k-mer PC scores for all cells being clustered.
Gene-module associations and gene set enrichment analyses
For each module (k = 1 to 11), the correlation coefficient (Pearson r) was computed between their module accessibility Z-scores and the gene scores for all TSSs (n = 35,856 genes) for all tumor and metastatic cells (n = 13,670). For module-wise gene set enrichment analyses, these gene-module correlations were first ranked based on the correlation coefficient per module. Mouse gene symbols were then lifted over to human HGNC symbols using the biomaRt R package (v2.34) specifying the ensembl mart database. The resulting ranked lists of mapped human gene identifiers and their correlation values (see Table S3) were then used to perform a pre-ranked gene set enrichment analysis per module using GSEA (Subramanian et al., 2005) against hallmark (h), canonical pathway (c2cp), chemical and genetic perturbation (c2cgp), and oncogenic (c6) annotated gene sets included in the molecular signature database (MSigDB v7.0) (Liberzon et al., 2015). To derive module-specific gene signatures, each gene was assigned to the module with the largest Pearson correlation coefficient. The top 200 genes for each module were retained (n = 2,200 genes), and were mapped to their human orthologs using biomaRt as described above (see Table S6). These mapped module gene signatures were then used for survival analysis, and for interrogation against RUNX TF perturbation effects (see below).
TF activator and repressor analysis
To calculate correlation between TF motif scores and TF gene scores, we first matched gene names to obtain 769 TF motif-gene feature pairs. Mean-normalized gene scores and TF motif scores (see methods above for how these were computed) for these TF genes were then used to compute the Pearson correlation coefficient between matched TF motif scores and TF gene scores across all cancer cells and normal AT1 and AT2 cells, reflecting a total of 13,923 cells. To calculate the statistical significance of the correlation, a permutation test was performed whereby the cell labels were permuted (n = 100 permutations with replacement). Permutation p values were calculated using a Z-test comparing the observed TF motif-gene correlation coefficient to the permuted correlation coefficients. TF motif variability was computed by taking the standard deviation of the TF motif scores across cells (n = 13,923). TF motif-gene pairs with p value < 0.001 and TF motif variability of >= 1.2 are considered significant. Activators and repressors are defined as TF motif-gene pairs where correlations are either positive or negative, respectively.
TCGA Survival and mutation analysis
Survival and normalized RNA-seq gene expression data for primary LUADs profiled as part of The Cancer Genome Atlas (TCGA) were obtained using Firehose for the July 15th, 2016, release as previously described (Kartha et al., 2018). Module-specific gene signatures were determined as described above. Then, for each module, the average expression of genes was computed for TCGA LUADs having paired RNA-seq and survival outcome information (n = 506). Patients were grouped as either module “high” or “low” if their module expression was above or below the median, respectively, and the overall survival (OS) of patients was compared between the two groups using a logrank test. Kaplan-Meier curves comparing OS in high versus low module groups for highlighted modules were generated using the survival (v2.41-3) and ggfortify (v0.4.10) packages in R. To test for association between module scores and KRAS and TP53 mutation status, binary somatic mutation calls for TCGA LUADs were obtained as previously described (Kartha et al., 2019). Standardized module expression Z-scores were then compared between LUADs with (n = 23) and without (n = 198) any KRAS and TP53 mutations using a Wilcoxon rank sum test.
RUNX TF perturbation analyses
To determine changes in chromatin accessibility induced by either overexpression or knockout of Runx2, perturbations were first normalized to their respective controls. Controls represent bulk ATAC-seq for guides targeting tdTomato for each cell line. For TF motif scores, the difference between the perturbation and control was determined. However, gene scores were first quantile normalized, then the difference between perturbation and controls was computed. To ensure the efficacy of the perturbations, we confirmed that i) every validated guide either increased or reduced the RUNX TF motif score as expected (see Figure 6B), and ii) that the perturbation was specific to the RUNX TF motif score (see Figure S6E). Next, we reasoned that CRISPRa overexpression would induce different levels of RUNX protein activation, therefore to determine differential gene scores, the RUNX TF motif score was used as a measure of the efficacy of the perturbation. The effect size (slope) from a linear regression between the differential RUNX TF motif score and each gene score was used to determine differential gene scores associated with RUNX perturbation. To determine gene set enrichments, gene scores were ranked by the calculated effect size. Following ranking, gene set enrichment was performed as described above. All experiments completed were shown with technical replicates.
Gene accessibility score and RNA expression correlations in TCGA LUAD
To investigate the relation between gene accessibility score and RNA expression estimates in human primary LUADs, bulk tumor ATAC-seq profiles generated for a subset of the TCGA LUADs, for whom paired RNA-seq information also existed (n = 21) (Corces et al., 2018) were obtained. This ATAC-seq data comprised a total of 139,135 peaks, with the reads in peaks counts matrix quantile-normalized. Gene activity scores were then computed using the normalized counts as described earlier (see section “Gene activity scoring in single cells”), with the following modifications: i) gene annotations corresponding to the hg38 genome build were used; ii) peaks overlapping the fixed window per TSS (9212 bp), and the corresponding read counts per peak were used to compute weighted gene scores per sample. To contrast gene expression and activity profiles for genes encoding TFs relative to non-TF-encoding genes, the top 10,000 genes were selected based on either total RNA expression or gene score across the samples assessed. The intersect of the two ranked gene lists (n = 6,191 genes) was then used to determine the fold-change of either mean gene score or mean RNA expression levels for TF (n = 228) versus non-TF (n = 5,963) annotated genes (determined by whether the gene was part of the human_pwms_v2 motif list from the chromVARmotifs package in R). To measure the association between gene expression and gene activity for genes of different gene activity levels, all genes that have expression and gene activity in at least 1 sample (n = 14,380 genes) were considered; the Pearson correlation of gene activity score to RNA expression per gene was then calculated. Correlation values were then visualized for different gene score percentiles (10 percentile bins).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Methods
All of the statistical details for experiments can be found in the figure legends as well as the Method Details section. For all comparisons of independent observations between two groups, two-tailed t-tests were performed, with p values unless otherwise specified. Z-tests were used to describe variance across groups. For all figures, **** represents p<0.0001, *** represents p<0.001, ** represents p<0.01, and * represents p<0.05. Additional details are described below.
Reads in peaks counts for ATAC-seq data
To generate count matrixes for all single-cell and bulk ATAC-seq data, the number of reads overlapping a given peak window in the determined peak set (see Method Details) was calculated for each unique cell barcode (sciATAC-seq and ETP data) or sample (cell line bulk ATAC-seq). FRIP was computed as the fraction of the total number of sequenced reads per cell that fall in peaks and was used, along with total unique nuclear reads per cell, to filter scATAC-seq cell barcodes.
TF motif scores, gene scores, and module associations
Quantification of chromatin accessibility features associated with sequence k-mers (used for single cell UMAP projection), TF motifs (used for annotating cell clusters and peak modules), and modules was performed using chromVAR (Schep et al., 2017), and is described under the Method Details sections and figure legends. For all these features, accessibility deviation Z-scores across mouse lung cells (for scATAC-seq), or cell line (for bulk ATAC-seq) were used. Gene scores were computed for single cells or cell lines as described earlier (Method Details). Gene scores were normalized by dividing by the mean gene score per cell (scATAC-seq), or quantile-normalized (bulk ATAC-seq), prior to downstream analyses. The significance of TF-motif gene score correlations was determined using permutation tests. Permutation p values were calculated using a Z-test comparing the TF-motif gene correlation coefficient to the permutated correlation coefficients. TF motif-gene pairs with permutation p value < 0.001 and TF motif variability of ≥ 1.2 are considered significant. Activator and repressor TFs were represented with max/min-normalized correlation of TF gene scores to module scores. Differential genes scores (AT1 and AT2 cells comparison) were represented as gene scores with absolute fold-change value greater than 1.8. Peak modules were determined using tumor and metastatic cell sciATAC-seq data as described under the Methods Detail section, and clustered and visualized using the log fold-change in mean module peak accessibility between motif-high vs motif-low cell groups. Module-gene associations were determined by assigning each gene to the module with the highest Pearson r correlation (gene score to module Z-score correlation). Gene signatures per module were obtained by selecting the top 200 genes basked on their associated correlation coefficients in a given module.
scATAC-seq matching to scRNA-seq datasets
Analysis of published scRNA-seq data was performed using the described meta-clusters (n = 260) representing clusters of cells across different lung developmental time points (Cohen et al., 2018). To match epigenomic profiles to these meta-clusters, scATAC-seq data were first filtered for highly variable gene scores and gene expression. The coefficient of variation (CV) of each gene was computed for each data set and filtered for genes with a CV > 1 in both data sets, resulting in a total of 6,888 genes. To match scATAC cells to meta-clusters, the most correlated (Pearson r) for each scATAC-seq cell was determined by matching gene scores to gene expression across the two data sets.
GSEA analyses and survival analysis
Gene set enrichment analysis was carried out using the pre-ranked GSEA mode as part of publicly available GSEA software (v3.0) (http://www.broadinstitute.otherg/gsea/index.jsp), with default settings. For module enrichment analyses, ranked lists of human gene identifiers and their correlation values (Pearson correlation of gene scores to module Z-scores) were used as input to test for enrichments per module against annotated gene sets included in the MsigDB database. For CRISPR perturbation enrichment analyses, slope coefficients of gene scores associated with differential perturbations were used to rank genes, and were queried against either MsigDB gene sets or module gene signatures. For module-associated survival analysis in TCGA LUADs, gene signatures per module were first averaged, and then tested for association with overall patient survival using a log-rank test comparing high vs low patient groups (determined based on the median module expression level). For testing association with oncogene mutational status, a Wilcoxon rank-sum test was used to compare module expression Z-scores between TCGA LUADs with and without KRAS and TP53 mutations.
Immunohistochemistry quantification
Immunohistochemistry images were converted to .tif format using CaseViewer (v2.2.1). Each image was split into multiple non-overlapping tiles and corrected for background fluorescence using a 2D Gaussian filter via a custom MATLAB script. Tiles with DAPI staining were then processed using Ilastik Pixel + Object Classification (v1.3.3) to generate nuclear segmentation masks (Berg et al., 2019). The resulting masks were loaded back into MATLAB (v2019a) and used to quantify the fluorescence within defined nuclear regions for the other protein markers. For each nucleus, the pixel values for each marker were summed and log normalized before visualization of the overall tissue distribution.
Tail vein experiments.
Each tail vein experiment was conducted with n = 5 animals in each group (control and RUNX2 KO). Survival significance was calculated using the survival log-rank (Mantel-Cox) test. Tumor burden studies were conducted with n = 5 animals in each group (control and RUNX2 KO). Total tumor burden was calculated using the Aiforia machine learning algorithm and significance was determined using Student t-tests.
Supplementary Material
Table S2. AT1 and AT2 gene signatures, Related to Figure 3.
Table S3. Definition of co-accessibility modules, Related to Figure 4.
Table S4. Transcription factor module regulators, Related to Figure 5.
Table S5. CRISPR-based perturbation of chromatin and protein expression, Related to Figure 6.
Table S6. Gene score to module associations, Related to Figure 7.
Table S7. Oligonucleotide sequences, Related to STAR Methods.
Data S1. Immunohistochemistry analysis of KP tumors, Related to Figure 2. Immunohistochemistry analysis of KPT tumors.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NKX2.1 | Abcam | Cat# ab76013; RRID:AB_1310784 |
| RUNX2 | Cell Signaling Technology | Cat# 12556S; RRID:AB_2732805 |
| LGALS1 | Cell Signaling Technology | Cat# 13888S; RRID:AB_2798338 |
| HMGA2 | Cell Signaling Technology | Cat# 8179S; RRID:AB_11178942 |
| ZEB1 | Abcam | Cat# ab87280; RRID:AB_2040541 |
| RUNX1 | Cell Signaling Technology | Cat# 8529S; RRID:AB_10950225 |
| SFTPC | Millipore Sigma | Cat# AB3786; RRID:AB_91588 |
| SFTPB | ThermoFisher | Cat# PA5-42000; RRID:AB_2609628 |
| BATF | Sigma Aldrich | Cat# SAB4500122; RRID:AB_10745033 |
| CAV1 | Sigma Aldrich | Cat# C3237; RRID:AB_476842 |
| HOPX | Proteintech | Cat# 11419-1-AP; RRID:AB_10693525 |
| HSP90 | BD Biosciences | Cat# 610418; RRID:AB_397798 |
| RUNX3 | Abcam | Cat# ab135248; RRID:AB_2848183 |
| RFP | Rockland | Cat# 600-401-379; RRID:AB_2209751 |
| Zfp795 | Novus Biologicals | Cat# NBP2-20947; RRID:AB_2848184 |
| Fra1 | ThermoFisher | Cat# PA5-40361; RRID:AB_2609389 |
| Onecut2 | Proteintech | Cat# 21916-1-AP; RRID:AB_2848180 |
| PDPN | Abcam | Cat# ab109059; RRID:AB_2848181 |
| RUNX2 | Abcam | Cat# ab23981; RRID:AB_777785 |
| CD45 | Abcam | Cat# ab10558; RRID:AB_442810 |
| CD11B-APC | eBioscience | Cat# 7-0112-82; RRID:AB_469344 |
| TER119-APC | BD Biosciences | Cat# 557909; RRID:AB_398635 |
| CD45-APC | BD Biosciences | Cat# 559864; RRID:AB_398672 |
| CD31-APC | Biolegend | Cat# 102510; RRID:AB_312917 |
| Bacterial and Virus Strains | ||
| Ad5-Sftpc-Cre | University of Iowa viral vector core facility | Cat# VVC-Berns-1168 |
| Biological Samples | ||
| Lung adenocarcinoma, 75 cases, tumor and matched NAT*, unstained slide | biomax | Cat# HLugA150CS02 |
| Lung cancer progression tissue array, including TNM, clinical stage and pathology grade, 100 cases/100 cores, replacing LC1005 | biomax | Cat # LC1005a |
| Lung disease spectrum (pulmonary cancer progression) tissue array, 193 cases/208 cores | biomax | Cat# LC2083 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NP-40 Surfact-Amps Detergent Solution | Thermo Scientific Nalgene | Cat# 28324 |
| Thermo Scientific Pierce Sequencing Grade Dimethylformamide | Thermo Fisher Scientific | Cat# 20673 |
| 0.5M EDTA pH 8.0 | Thermo Fisher Scientific | Cat# 15575-020 |
| Triton X-100 | Sigma Aldrich | Cat# T8787-50ML |
| HEPES (1M) | Life Technologies | Cat# 15630-080 |
| Tween(R)20, SigmaUltra | Sigma-Aldrich | Cat# P7949-100ML |
| NuPAGE MOPS SDS Running Buffer | Invitrogen | Cat# NP0001 |
| TBS Buffer 20X Liquid, 4L | Amresco | Cat# J640-4L |
| RIPA Buffer | Thermo Fisher Scientific | Cat# 89900 |
| Halt Phosphatase Inhibitor | Thermo Scientific | Cat# PI-78420 |
| Halt Protease Inhibitor Cocktail (100X) | Thermo Scientific | Cat# 78430 |
| NuPAGE LDS Sample Buffer (4X) | Life Technologies | Cat# NP0007 |
| NuPAGE Sample Reducing Agent (10X) | Invitrogen | Cat# NP0009 |
| NuPAGE Transfer Buffer (20X) | Life Technologies | Cat# NP0006-1 |
| Blotting-Grade Blocker | Bio-Rad | Cat# 170-6404 |
| Ponceau S | Sigma Aldrich | Cat# P7170-1L |
| NuPAGE Novex 4-12% Bis-Tris Protein Gels, 1.5mm, 10 well | Life Technologies | Cat# NP0335BOX |
| Amersham ECL Prime Western Blotting Detection Reagent | GE Healthcare | Cat# RPN2232 |
| Dual Endogenous Enzyme Blocking Kit | Agilent Technologies | Cat# S200389-2 |
| 2.5% Normal Horse Serum Blocking Solution | Vector Laboratories | Cat# S-2012 |
| ImmPRESS HRP Anti-Rabbit IgG (Peroxidase) Polymer | Vector Laboratories | Cat# mp-7401 |
| ACK lysing buffer | Thermo Fisher Scientific | Cat# a10492-01 |
| DMEM with L-Glutamine | VWR | Cat# 10-013-CV (45000-304) |
| 0.25% Trypsin-EDTA (1X) Phenol Red | Invitrogen | Cat# 25200-114 |
| RPMI 1640 | VWR | Cat# 15-040-CV |
| Tet System Approved FBS | Clontech | Cat# 631106 |
| Penicillin-Streptomycin | VWR | Cat# 45000-652 |
| S-MEM | Life Technologies | Cat# 11380-037 |
| RNase inhibitor | Thermo Fisher Scientific | Cat# N8080119 |
| DPBS, 1X without calcium and magnesium | VWR Scientific Inc | Cat# 21-031-CV |
| 10X PBS | VWR | Cat# AAJ67653-AP |
| Bovine Albumin Fraction V (7.5% solution) | Thermo Fisher Scientific | Cat# 15260037 |
| Glycine | VWR | Cat# 97061-128 |
| Collagenase from Clostridium histolyticum | Sigma-Aldrich | Cat# C9407-500MG |
| MgCl2 (1M) | Thermo Fisher Scientific | Cat# AM9530G |
| NaCl, 5M | ThermoFisher Scientific | Cat# AM9759 |
| Tris, Hydrochloride | Santa Cruz Biotechnology | Cat# sc-216106A |
| Sodium Dodecyl Sulfate | Bio-Rad | Cat# 161-0302 |
| Sybr Fast 2X MM LC480 | Kapa Biosystems | Cat# KK4611 |
| Thermo Scientific Pierce Methanol free Formaldehyde Ampules | Thermo Fisher Scientific | Cat# 28908 |
| Proteinase K, Recombinant, PCR grade solution | Sigma-Aldrich | Cat# 3115828001 |
| HBSS, no Calcium, no Magnesium, no Phenol Red | Thermo Fisher Scientific | Cat# 14175-079 |
| Invitrogen DNase I | Thermo Fisher Scientific | Cat# 18-047-019 |
| Collagenase, Type 4 | Worthington Biochemical | Cat# LS004189 |
| FastDigest Esp3I | Thermo Fisher Scientific | Cat# FD0454 |
| Puromycin | Invitrogen | Cat# a11138-02 |
| Zinc formalin fixative, pH 6.25 | Electron Microscopy Sciences | Cat# 21516.375 |
| Exonuclease I | New England Biolabs (NEB) | Cat# M0293S |
| Collagenase from Clostridium histolyticum | Sigma-Aldrich | Cat# C9407-500MG |
| ProLong Glass Antifade Mountant | Thermo Fisher Scientific | Cat# P36980 |
| Digitonin | Promega | Cat# G9441 |
| Critical Commercial Assays | ||
| Opal 4-Color Manual IHC Kit 50 slides | Akoya Biosciences | Cat# NEL810001KT |
| DAB Peroxidase Substrate Kit | Vector Labs | Cat# SK-4100 |
| NEBNext High-Fidelity 2X PCR Master Mix | New England Biolabs (NEB) | Cat# M0541L |
| Pierce BCA Protein Assay Kit | Thermo Fisher -- Pierce | Cat# 23227 |
| KAPA Library Quant for Illumina Sequencing Platforms | Kapa Biosystems | Cat# KK4824 |
| MinElute PCR Purification Kit | Qiagen | Cat# 28006 |
| Lung Dissociation Kit, mouse | Miltenyi Biotec | Cat# NC0315167 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| High-Capacity cDNA reverse transcription kit | Thermo Fisher Scientific | Cat# 4368814 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32854 |
| QIAGEN Plasmid Plus Midi Kit (25) | Qiagen | Cat# 12943 |
| QIAquick Gel Extraction Kit (250) | Qiagen | Cat# 28706 |
| SMARTer ThruPLEX DNA-Seq Kit - 24 Rxns | Takara Bio | Cat# R400674 |
| ECM Cell Adhesion Array Kit, colorimetric | Millipore Sigma | Cat# ECM540 |
| CD45 microbeads mouse | Miltenyi Biotec | Cat# 130-052-301 |
| Mouse L308 Array, Membrane | RayBiotech | Cat# AAM-BLM-1A-2 |
| Nextera DNA Library Preparation Kit | Illumina | Cat# FC-121-1030 |
| NextSeq 500/550 High Output Kit v2 | Illumina | Cat# FC-404-2002 |
| NextSeq | Illumina | Cat# FC-404-2005 |
| SureCell ATAC-Seq Library Preparation Kit | Bio-Rad | Cat# 17004620 |
| Agencourt AMPure XP | Beckman Coulter | Cat# A63880 |
| SureCell ddSEQ Index Kit | Bio-Rad | Cat# 12009360 |
| Agilent High Sensitivity DNA Kit | Agilent | Cat# 5067-4626 |
| TC20 Cell Counting Kit, with Trypan Blue | Bio-Rad | Cat# 1450003 |
| Deposited Data | ||
| ScATAC-seq data | This manuscript | GSE134812 |
| Early time point single cell data | This manuscript | GSE145192 |
| Bulk-ATACseq | This manuscript | GSE151403 |
| Visualization of UMAP scATAC-seq | This manuscript | https://buenrostrolab.shinyapps.io/lungATAC/ |
| UCSC genome browser tracks for normal cells | This manuscript | http://genome.ucsc.edu/s/lmlafave/normal_lung_scATAC |
| UCSC genome browser tracks for tumor modules | This manuscript | http://genome.ucsc.edu/s/lmlafave/KPT_modules |
| Experimental Models: Cell Lines | ||
| 860T3 KP cell line | This manuscript | N/A |
| 1183T3 KP cell line | This manuscript | N/A |
| 853T2 KP cell line | This manuscript | N/A |
| 860T1 KP cell line | This manuscript | N/A |
| 1183T4 KP cell line | This manuscript | N/A |
| 932T2 KP cell line | This manuscript | N/A |
| 932T3 KP cell line | This manuscript | N/A |
| 932LN KP cell line | This manuscript | N/A |
| 779T1 KP cell line | This manuscript | N/A |
| 779T2 KP cell line | This manuscript | N/A |
| 779LN KP cell line | This manuscript | N/A |
| Experimental Models: Organisms/Strains | ||
| KP mouse | Jackson et al., 2001, 2005 | stock 008179, stock 008462 |
| Tomato mouse (Ai9) | Jackson Labs | stock 007905 |
| B6129SF1/J | Jackson Labs | stock 101043 |
| Oligonucleotides | ||
| Genotyping primers | Table S7 | N/A |
| RUNX2 control g1f: CACCGGGCCACGAGTTCGAGATCGA | This manuscript | N/A |
| RUNX2 control g1r: AAACTCGATCTCGAACTCGTGGCCC | This manuscript | N/A |
| RUNX2 OE sg1a: CACCGGAGGAGGAAATCGA | This manuscript | N/A |
| RUNX2 OE sg1b: AAACTCGATTTCCTCCTCC | This manuscript | N/A |
| RUNX2 OE sg2a: CACCGGGCGGAGTCTGCTG | This manuscript | N/A |
| RUNX2 OE sg2b: AAACCAGCAGACTCCGCCC | This manuscript | N/A |
| RUNX1 KO sg1a: CACCGAGGAGTACCTTGAAAGCGAT | This manuscript | N/A |
| RUNX1 KO sg1b: AAACATCGCTTTCAAGGTACTCCTC | This manuscript | N/A |
| RUNX1 KO sg4a: CACCGTAGCGAGATTCAACGACCTC | This manuscript | N/A |
| RUNX1 KO sg4b: AAACGAGGTCGTTGAATCTCGCTAC | This manuscript | N/A |
| RUNX2 KO sg3a: CACCGTGCGGACCAGTTCGGCCGGG | This manuscript | N/A |
| RUNX2 KO sg3b: AAACCCCGGCCGAACTGGTCCGCAC | This manuscript | N/A |
| RUNX2 KO sg4a: CACCGGCCCTCGGAGAGGTACCAGA | This manuscript | N/A |
| RUNX2 KO sg4b: AAACTCTGGTACCTCTCCGAGGGCC | This manuscript | N/A |
| RUNX3 KO sg1a: CACCGGGACGTGCTGGCCGACCACG | This manuscript | N/A |
| RUNX3 KO sg1b:AAACCGTGGTCGGCCAGCACGTCCC | This manuscript | N/A |
| Recombinant DNA | ||
| lentiCRISPR-V2-puro | Joung et al., 2017 | Addgene #98290 |
| Lenti-Sam-puro | This manuscript | N/A |
| Lenti-Cas9-blast | Sanjana et al., 2014 | Addgene #52962 |
| Software and Algorithms | ||
| Aiforia (NSCLC_v25 algorithm) | This manuscript | https://www.aiforia.com/ |
| R (v3.5.3) | R Core Team, 2019 | https://www.R-project.org |
| chromVAR R package (v0.2.0) | Schep et al., 2017 | https://github.com/GreenleafLab/chromVAR |
| uwot R package (v0.1.4) | McInnes et al., 2018 | https://github.com/jlmelville/uwot |
| survival R package (2.41-3) | Therneau and Grambsch, 2000 | https://cran.r-project.org/web/packages/survival/index.html |
| GSEA (v3.0) | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ImageJ (v1.52k) | Schneider, et al., 2012 | https://imagej.net/ |
| ImageJ Protein Array Analyzer (v1.1.c) | Carpentier, 2010 | https://imagej.net/macros/toolsets/Protein%20Array%20Analyzer.txt |
| FlowJo (v10.6.2) | N/A | www.flowjo.com |
| CaseViewer (v2.2.1) | N/A | https://www.3dhistech.com |
| MATLAB (v2019a) | Higham and Higham, 2016 | https://www.mathworks.com |
| Ilastik (v1.3.3) | Berg et al., 2019 | https://www.ilastik.org |
| MSigDB (v7.0) | Liberzon et al., 2015 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| bowtie2 (v2.3.3.1) | Langmead et al., 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 (v2.1.2) | Zhang et al., 2008 | https://github.com/taoliu/MACS/ |
| samtools (v1.9) | Li et al., 2009 | http://samtools.sourceforge.net |
| Picard toolkit (2.14.1-SNAPSHOT) | N/A | http://broadinstitute.github.io/picard |
| biomaRt (v2.34) | Durinck et al., 2005 | https://bioconductor.org/packages/release/bioc/html/biomaRt.html |
| ggfortify R package (v0.4.10) | Tang et al., 2016 | https://github.com/sinhrks/ggfortify |
| BAP (v0.5.9i) | Lareau et al., 2019 | https://github.com/caleblareau/bap |
| BWA (v0.7.15) | Li, 2013 | https://github.com/lh3/bwa |
| QuPath (0.1.2) | Bankhead et al., 2017 | https://qupath.github.io |
| Code generated for this manuscript | This study | https://github.com/buenrostrolab/lungATAC_analysis_code |
Significance:
Here we describe a generalizable framework to leverage single-cell chromatin accessibility data to investigate nuanced cell state transitions across tumor evolution in a mouse model of lung adenocarcinoma. Using an improved combinatorial indexing approach to study chromatin reorganization, we found that epigenomic state changes across cancer progression occupy a continuum, rather than discrete transitions, which makes characterization of these cell states particularly challenging. We developed a module-based approach to assess coordinated regulatory programs which are mediated by aberrant transcription factor activity. We elucidate a pre-metastatic cell state that arises in primary tumors prior to metastasis. Importantly, these epigenomic profiles serve as clear prognostic signatures in human malignancies, and these strategies can be adapted to other cancer studies.
Highlights.
Cancer progression is marked by a continuum of heterogeneous epigenomic states
Lung cancer cells adopt features of other cell identities across tumor evolution
RUNX2 transcription factor activity is associated with a pre-metastatic cell state
Regulatory programs defined from mouse models are useful predictive biomarkers
Using a generalizable framework to leverage single-cell chromatin accessibility data to investigate cell state transitions across tumor evolution in a mouse model of lung adenocarcinoma, LaFave et al. elucidate a pre-metastatic cell state that arises in primary tumors prior to metastasis.
Acknowledgments:
We thank the members of the Jacks and Buenrostro labs for their critical reading of the manuscript and helpful discussions, including Tongtong Zhao, Rodrigo Romero, Leanne Li, Alex Jaeger, Amanda Cruz, and Carla Concepcion. We also thank Carman Li and Vasilena Gocheva for helpful conversations. We thank Amir Giladi for help with the interpretation of the lung development data. We thank Paul Chamberlain for the support on tail vein assays. We thank Tuomas Tammela for the Aiforia algorithm. We thank the Histology and Flow Cytometry cores at the Swanson Biotechnology Center, the Walk-up sequencing core at the Broad Institute and the Bauer sequencing core at Harvard. We are grateful to the Zhang lab for providing Tn5. L.M.L. is supported by the Damon Runyon Cancer Foundation postdoctoral fellowship. J.D.B. acknowledges support from the Allen Distinguished Investigator Program, through The Paul G. Allen Frontiers Group. T.J. is a Howard Hughes Medical Institute Investigator and a Daniel K. Ludwig Scholar. This work was supported in part by grant PO1-CA42063 from the National Institutes of Health, and partially by Cancer Center Support (core) grant P30-CA14051 from the National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific. He is also a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. None of these affiliations represent a conflict of interest with respect to the design or execution of this study or interpretation of data presented in this manuscript. T.J. laboratory currently also receives funding from the Johnson & Johnson Lung Cancer Initiative and The Lustgarten Foundation for Pancreatic Cancer Research, but this funding did not support the research described in this manuscript. J.D.B. holds patents related to ATAC-seq and single-cell ATAC-seq and serves on the Scientific Advisory Board of CAMP4 Therapeutics and seqWell. A.R. is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and an SAB member of ThermoFisher Scientific, Syros Pharmaceuticals, Asimov, and Neogene Therapeutics.
References:
- Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, McGraw T, and Mittal V (2019). The lung microenvironment: an important regulator of tumour growth and metastasis. Nat. Rev. Cancer 19, 9–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Meng L, Sun H, Li Z, Zhang X, and Hua S (2017). MicroRNA-196b Inhibits Cell Growth and Metastasis of Lung Cancer Cells by Targeting Runx2. Cell. Physiol. Biochem 43, 757–767. [DOI] [PubMed] [Google Scholar]
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci. Rep 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, et al. (2019). ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Birkbak NJ, and McGranahan N (2020). Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 37, 8–19. [DOI] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment 2008, P10008. [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional Addiction in Cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady JJ, Chuang C-H, Greenside PG, Rogers ZN, Murray CW, Caswell DR, Hartmann U, Connolly AJ, Sweet-Cordero EA, Kundaje A, et al. (2016). An Arntl2-Driven Secretome Enables Lung Adenocarcinoma Metastatic Self-Sufficiency. Cancer Cell 29, 697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015a). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015b). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol 109, 21.29.1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Ryan Corces M, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, and Greenleaf WJ (2018). Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et al. (2016). Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet 48, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell L, Hollins AJ, Al-Eid A, Newman GR, von Ruhland C, and Gumbleton M (1999). Caveolin-1 expression and caveolae biogenesis during cell transdifferentiation in lung alveolar epithelial primary cultures. Biochem. Biophys. Res. Commun 262, 744–751. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramel J, Ligier M, and Puisieux A (2018). Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 78, 30–35. [DOI] [PubMed] [Google Scholar]
- Caswell DR, Chuang C-H, Yang D, Chiou S-H, Cheemalavagu S, Kim-Kiselak C, Connolly A, and Winslow MM (2014). Obligate progression precedes lung adenocarcinoma dissemination. Cancer Discov. 4, 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier G (2010). Protein Array Analyzer for ImageJ. https://imagej.net/macros/toolsets/Protein%20Array%20Analyzer.txt
- Chen X, Miragaia RJ, Natarajan KN, and Teichmann SA (2018). A rapid and robust method for single cell chromatin accessibility profiling. Nat. Commun 9, 5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Fillmore CM, Hammerman PS, Kim CF, and Wong K-K (2014). Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev. Cancer 14, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung WKC, and Nguyen DX (2015). Lineage factors and differentiation states in lung cancer progression. Oncogene 34, 5771–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang C-H, Greenside PG, Rogers ZN, Brady JJ, Yang D, Ma RK, Caswell DR, Chiou S-H, Winters AF, Grüner BM, et al. (2017). Molecular definition of a metastatic lung cancer state reveals a targetable CD109-Janus kinase-Stat axis. Nat. Med 23, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung NC, and Storey JD (2015). Statistical significance of variables driving systematic variation in high-dimensional data. Bioinformatics 31, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Giladi A, Gorki A-D, Solodkin DG, Zada M, Hladik A, Miklosi A, Salame T-M, Halpern KB, David E, et al. (2018). Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 175, 1031–1044.e18. [DOI] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. (2018). The chromatin accessibility landscape of primary human cancers. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, and Shendure J (2015). Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, et al. (2018). A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagogo-Jack I, and Shaw AT (2018). Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol 15, 81–94. [DOI] [PubMed] [Google Scholar]
- Denny SK, Yang D, Chuang C-H, Brady JJ, Lim JS, Grüner BM, Chiou S-H, Schep AN, Baral J, Hamard C, et al. (2016). Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 166, 328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai TJ, Brownfield DG, and Krasnow MA (2014). Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507, 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietel M, Bubendorf L, Dingemans A-MC, Dooms C, Elmberger G, García RC, Kerr KM, Lim E, López-Ríos F, Thunnissen E, et al. (2016). Diagnostic procedures for non-small-cell lung cancer (NSCLC): recommendations of the European Expert Group. Thorax 71, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez-Lago M, Kool M, Jones DTW, Unterberger A, Morrissy AS, et al. (2013). Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 125, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc 4, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, and Huber W (2005). BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440. [DOI] [PubMed] [Google Scholar]
- Flavahan WA, Gaskell E, and Bernstein BE (2017). Epigenetic plasticity and the hallmarks of cancer. Science 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay M, Li Y, and Felsher DW (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiti F, Chaligne R, Gu H, Brand RM, Kothen-Hill S, Schulman RC, Grigorev K, Risso D, Kim K-T, Pastore A, et al. (2019). Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 569, 576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C, Zhao G, Li Y, Li H, Zhao X, Pannone G, Bufo P, Santoro A, Sanguedolce F, Tortorella S, et al. (2016). Role of Runx2 phosphorylation in prostate cancer and association with metastatic disease. Oncogene 35, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Gomez NC, Adam RC, Nikolova M, Yang H, Verma A, Lu CP-J, Polak L, Yuan S, Elemento O, et al. (2017). Stem Cell Lineage Infidelity Drives Wound Repair and Cancer. Cell 169, 636–650.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan K-K, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. (2012). Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano S, Cheli Y, Ohanna M, Bonet C, Beuret L, Bille K, Loubat A, Hofman V, Hofman P, Ponzio G, et al. (2010). Microphthalmia-Associated Transcription Factor Controls the DNA Damage Response and a Lineage-Specific Senescence Program in Melanomas. Cancer Research 70, 3813–3822. [DOI] [PubMed] [Google Scholar]
- Gocheva V, Naba A, Bhutkar A, Guardia T, Miller KM, Li CM-C, Dayton TL, Sanchez-Rivera FJ, Kim-Kiselak C, Jailkhani N, et al. (2017). Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proc. Natl. Acad. Sci. U. S. A 114, E5625–E5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González AJ, Setty M, and Leslie CS (2015). Early enhancer establishment and regulatory locus complexity shape transcriptional programs in hematopoietic differentiation. Nat. Genet 47, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja JM, Klemm S, McGinnis LM, Kathiria AS, Mezger A, Corces MR, Parks B, Gars E, Liedtke M, Zheng GXY, et al. (2019). Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol 37, 1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ci X, Ahmed M, Hua JT, Soares F, Lin D, Puca L, Vosoughi A, Xue H, Li E, et al. (2019). ONECUT2 is a driver of neuroendocrine prostate cancer. Nat. Commun 10, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HS, Son S-M, Yun J, Jo YN, and Lee O-J (2014). MicroRNA-29a suppresses the growth, migration, and invasion of lung adenocarcinoma cells by targeting carcinoembryonic antigen-related cell adhesion molecule 6. FEBS Lett. 588, 3744–3750. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Heldin C-H, Vanlandewijck M, and Moustakas A (2012). Regulation of EMT by TGFβ in cancer. FEBS Letters 586, 1959–1970. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Morgensztern D, and Boshoff C (2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. [DOI] [PubMed] [Google Scholar]
- Higham DJ, and Higham NJ (2016). MATLAB Guide, Third Edition. [Google Scholar]
- Hinohara K, and Polyak K (2019). Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol. 29, 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andrá V, Sigova AA, Hoke HA, and Young RA (2013). Super-Enhancers in the Control of Cell Identity and Disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, et al. (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu X, Wen L, Tang F, Huang Y, et al. (2016). Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 26, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, and Jacks T (2005). The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 65, 10280–10288. [DOI] [PubMed] [Google Scholar]
- Jain R, Barkauskas CE, Takeda N, Bowie EJ, Aghajanian H, Wang Q, Padmanabhan A, Manderfield LJ, Gupta M, Li D, et al. (2015). Plasticity of Hopx type I alveolar cells to regenerate type II cells in the lung. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, and Zhang F (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartha VK, Alamoud KA, Sadykov K, Nguyen B-C, Laroche F, Feng H, Lee J, Pai SI, Varelas X, Egloff AM, et al. (2018). Functional and genomic analyses reveal therapeutic potential of targeting β-catenin/CBP activity in head and neck cancer. Genome Med. 10, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartha VK, Sebastiani P, Kern JG, Zhang L, Varelas X, and Monti S (2019). CaDrA: A Computational Framework for Performing Candidate Driver Analyses Using Genomic Features. Front. Genet 10, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno Y, Meyer DS, Zhang Z, Shokat KM, Akhurst RJ, Miyazono K, and Derynck R (2019). Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klughammer J, Kiesel B, Roetzer T, Fortelny N, Nemc A, Nenning K-H, Furtner J, Sheffield NC, Datlinger P, Peter N, et al. (2018). The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med 24, 1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikoff N, Attema JL, Roslan S, Bert AG, Schwarz QP, Gregory PA, and Goodall GJ (2014). Specificity protein 1 (Sp1) maintains basal epithelial expression of the miR-200 family: implications for epithelial-mesenchymal transition. J. Biol. Chem 289, 11194–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, et al. (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol 36, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwé H, Pircher A, Van den Eynde K, et al. (2018). Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med 24, 1277–1289. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau CA, Duarte FM, Chew JG, Kartha VK, Burkett ZD, Kohlway AS, Pokholok D, Aryee MJ, Steemers FJ, Lebofsky R, et al. (2019). Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol [DOI] [PMC free article] [PubMed]
- Latil M, Nassar D, Beck B, Boumahdi S, Wang L, Brisebarre A, Dubois C, Nkusi E, Lenglez S, Checinska A, et al. (2017). Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell 20, 191–204.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallée V-P, Xie Y, Masilionis I, Carr AJ, Kottapalli S, et al. (2020). Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med 26, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, and Werb Z (2018). Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol 20, 1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, and Young RA (2013). Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2. [Google Scholar]
- Li H, Zhou R-J, Zhang G-Q, and Xu J-P (2013). Clinical significance of RUNX2 expression in patients with nonsmall cell lung cancer: a 5-year follow-up study. Tumour Biol. 34, 1807–1812. [DOI] [PubMed] [Google Scholar]
- Li J, Ning G, and Duncan SA (2000). Mammalian hepatocyte differentiation requires the transcription factor HNF-4alpha. Genes Dev. 14, 464–474. [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang H, Dong Y, Fan Y, Li Y, Zhao C, Wang C, Liu J, Li X, Dong M, et al. (2017). MiR-146b-5p functions as a suppressor miRNA and prognosis predictor in non-small cell lung cancer. J. Cancer 8, 1704–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Song H, Huang C, Yao E, Gacayan R, Xu S-M, and Chuang P-T (2012). Alveolar type II cells possess the capability of initiating lung tumor development. PLoS One 7, e53817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, Li LH, Pelka K, Ge W, Oren Y, Brack A, et al. (2019). Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, 1325–1339.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Wu K, Li H, Li H, Zhu Y, Hu G, Hu L, and Kong X (2019). ONECUT2 overexpression promotes RAS-driven lung adenocarcinoma progression. Sci. Rep 9, 20021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi S, Mijimolle N, Francoz S, Vicente-Duenas C, Sanchez-Garcia I, and Barbacid M (2014). Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proceedings of the National Academy of Sciences 111, 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, et al. (2016). Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc. Natl. Acad. Sci. U. S. A 113, E6409–E6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, Saul N, and Großberger L (2018). UMAP: Uniform Manifold Approximation and Projection. Journal of Open Source Software 3, 861. [Google Scholar]
- Mund SI, Stampanoni M, and Schittny JC (2008). Developmental alveolarization of the mouse lung. Dev. Dyn. 237, 2108–2116. [DOI] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, and Hynes RO (2012). The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics 11, M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, and Desai TJ (2018). Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359, 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noberini R, Osti D, Miccolo C, Richichi C, Lupia M, Corleone G, Hong S-P, Colombo P, Pollo B, Fornasari L, et al. (2018). Extensive and systematic rewiring of histone post-translational modifications in cancer model systems. Nucleic Acids Res. 46, 3817–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentinmikko N, Iqbal S, Mana M, Andersson S, Cognetta AB, Suciu RM, Roper J, Luopajärvi K, Markelin E, Gopalakrishnan S, et al. (2019). Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund AK, Reinius B, Sagasser S, Winberg G, and Sandberg R (2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A, et al. (2018). Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 71, 858–871.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap J, Javed A, Languino LR, van Wijnen AJ, Stein JL, Stein GS, and Lian JB (2005). The Runx2 osteogenic transcription factor regulates matrix metalloproteinase 9 in bone metastatic cancer cells and controls cell invasion. Mol. Cell. Biol 25, 8581–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap J, Wixted JJ, Gaur T, Zaidi SK, Dobson J, Gokul KD, Hussain S, van Wijnen AJ, Stein JL, Stein GS, et al. (2008). Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Res. 68, 7795–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissl S, Fang R, Huang H, Zhao Y, Raviram R, Gorkin DU, Zhang Y, Sos BC, Afzal V, Dickel DE, et al. (2018). Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat. Neurosci 21, 432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2019). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). https://www.R-project.org/. [Google Scholar]
- Ramsey J, Butnor K, Peng Z, Leclair T, van der Velden J, Stein G, Lian J, and Kinsey CM (2018). Loss of RUNX1 is associated with aggressive lung adenocarcinomas. J. Cell. Physiol 233, 3487–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Kang B, and Zhang Z (2018). Understanding tumor ecosystems by single-cell sequencing: promises and limitations. Genome Biol. 19, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reticker-Flynn NE, and Bhatia SN (2015). Aberrant glycosylation promotes lung cancer metastasis through adhesion to galectins in the metastatic niche. Cancer Discov. 5, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe J-S, Hwang C-I, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi K, et al. (2017). Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 170, 875–888.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotinen M, You S, Yang J, Coetzee SG, Reis-Sobreiro M, Huang W-C, Huang F, Pan X, Yáñez A, Hazelett DJ, et al. (2018). ONECUT2 is a targetable master regulator of lethal prostate cancer that suppresses the androgen axis. Nat. Med 24, 1887–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowbotham SP, Li F, Dost AFM, Louie SM, Marsh BP, Pessina P, Anbarasu CR, Brainson CF, Tuminello SJ, Lieberman A, et al. (2018). H3K9 methyltransferases and demethylases control lung tumor-propagating cells and lung cancer progression. Nat. Commun 9, 4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 37, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serresi M, Gargiulo G, Proost N, Siteur B, Cesaroni M, Koppens M, Xie H, Sutherland KD, Hulsman D, Citterio E, et al. (2016). Polycomb Repressive Complex 2 Is a Barrier to KRAS-Driven Inflammation and Epithelial-Mesenchymal Transition in Non-Small-Cell Lung Cancer. Cancer Cell 29, 17–31. [DOI] [PubMed] [Google Scholar]
- Shema E, Bernstein BE, and Buenrostro JD (2019). Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet 51, 19–25. [DOI] [PubMed] [Google Scholar]
- Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, Wang DG, Crowley D, Whittaker CA, Meyerson M, Kimura S, et al. (2013). Nkx2-1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol. Cell 50, 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, and Furlong EEM (2012). Transcription factors: from enhancer binding to developmental control. Nature Reviews Genetics 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur I, and Taipale J (2016). The role of enhancers in cancer. Nat. Rev. Cancer 16, 483–493. [DOI] [PubMed] [Google Scholar]
- Sutherland KD, Proost N, Brouns I, Adriaensen D, Song J-Y, and Berns A (2011). Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19, 754–764. [DOI] [PubMed] [Google Scholar]
- Sutherland KD, Song J-Y, Kwon MC, Proost N, Zevenhoven J, and Berns A (2014). Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc. Natl. Acad. Sci. U. S. A 111, 4952–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammela T, Sanchez-Rivera FJ, Cetinbas NM, Wu K, Joshi NS, Helenius K, Park Y, Azimi R, Kerper NR, Wesselhoeft RA, et al. (2017). A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 545, 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Horikoshi M, and Li W (2016). ggfortify: Unified Interface to Visualize Statistical Results of Popular R Packages. The R Journal 8, 474. [Google Scholar]
- Therneau TM, and Grambsch PM (2000). Modeling Survival Data: Extending the Cox Model. Statistics for Biology and Health.
- Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, and Quake SR (2014). Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, Nicol D, O’Brien T, Larkin J, Horswell S, et al. (2018). Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 173, 581–594.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Vallejo A, Perurena N, Guruceaga E, Mazur PK, Martinez-Canarias S, Zandueta C, Valencia K, Arricibita A, Gwinn D, Sayles LC, et al. (2017). An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer. Nat. Commun. 8, 14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, and Kinzler KW (2013). Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang Z, Huang H, Li J, Wang Z, Yu Y, Zhang C, Li J, Dai H, Wang F, et al. (2018). Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. U. S. A 115, 2407–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott PMK, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, Delrosario R, Jen K-Y, Gurley KE, Kemp CJ, et al. (2015). The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 517, 489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow MM, Dayton TL, Verhaak RGW, Kim-Kiselak C, Snyder EL, Feldser DM, Hubbard DD, DuPage MJ, Whittaker CA, Hoersch S, et al. (2011). Suppression of lung adenocarcinoma progression by Nkx2-1. Nature 473, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodworth MB, Girskis KM, and Walsh CA (2017). Building a lineage from single cells: genetic techniques for cell lineage tracking. Nat. Rev. Genet. 18, 230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Yu F, Li D, Zhu X, Zhang X, and Lv Z (2016). MicroRNA-218 regulates cisplatin (DPP) chemosensitivity in non-small cell lung cancer by targeting RUNX2. Tumor Biol. 37, 1197–1204. [DOI] [PubMed] [Google Scholar]
- Xu X, Rock JR, Lu Y, Futtner C, Schwab B, Guinney J, Hogan BLM, and Onaitis MW (2012). Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proceedings of the National Academy of Sciences 109, 4910–4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Fillmore Brainson C, Koyama S, Redig AJ, Chen T, Li S, Gupta M, Garcia-de-Alba C, Paschini M, Herter-Sprie GS, et al. (2017). Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2. Nat. Commun 8, 14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Rath N, Hannenhalli S, Wang Z, Cappola T, Kimura S, Atochina-Vasserman E, Lu MM, Beers MF, and Morrisey EE (2007). GATA and Nkx factors synergistically regulate tissue-specific gene expression and development in vivo. Development 134, 189–198. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q-W, Zhou Y-L, You Q-J, Shou F, Pang Q-F, and Chen J-L (2016). WWOX inhibits the invasion of lung cancer cells by downregulating RUNX2. Cancer Gene Ther. 23, 433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. AT1 and AT2 gene signatures, Related to Figure 3.
Table S3. Definition of co-accessibility modules, Related to Figure 4.
Table S4. Transcription factor module regulators, Related to Figure 5.
Table S5. CRISPR-based perturbation of chromatin and protein expression, Related to Figure 6.
Table S6. Gene score to module associations, Related to Figure 7.
Table S7. Oligonucleotide sequences, Related to STAR Methods.
Data S1. Immunohistochemistry analysis of KP tumors, Related to Figure 2. Immunohistochemistry analysis of KPT tumors.
Data Availability Statement
The datasets generated during this study are available at Gene Expression Omnibus (GEO) under GSE134812, GSE145192, and GSE151403. The raw single-cell ATAC-sequencing files and processed data files generated in this study are available in GEO under the super series GSE145194. Single-cell combinatorial indexing data is available under GSE134812 and ETP data is available under GSE145192. Bulk ATAC-sequencing raw and processed files are available under accession GSE151403. The R Shiny-based web application for data visualization is accessible here: https://buenrostrolab.shinyapps.io/lungATAC/
UCSC genome browser tracks associated with this study are made available with the following weblinks: Normal cells cluster: http://genome.ucsc.edu/s/lmlafave/normal_lung_scATAC KPT modules: http://genome.ucsc.edu/s/lmlafave/KPT_modules
Code used for the analysis of scATAC-seq data in this study is available on Github (https://github.com/buenrostrolab/lungATAC_analysis_code).