

SUMMARY
Recent studies have revealed non-canonical activities of apoptotic caspases involving specific modulation of gene expression, such as limiting asymmetric divisions of stem-like cell types. Here we report that CED-3 caspase negatively regulates an epidermal p38 stress-responsive MAPK pathway to promote larval development in C. elegans. We show that PMK-1 (p38 MAPK) primes animals for encounters with hostile environments at the expense of retarding post-embryonic development. CED-3 counters this function by directly cleaving PMK-1 to promote development. Moreover, we found that CED-3 and PMK-1 oppose each other to balance developmental and stress-responsive gene expression programs. Specifically, expression of more than 300 genes is inversely regulated by CED-3 and PMK-1. Analyses of these genes showed enrichment for epidermal stress-responsive factors, including the fatty acid synthase FASN-1, anti-microbial peptides, and genes involved in lethargus states. Our findings demonstrate a non-canonical role for a caspase in promoting development by limiting epidermal stress response programs.
Graphical Abstract
In Brief
Weaver et al. describe a non-apoptotic role for CED-3 caspase during development in C. elegans. They show that a p38 MAPK pathway primes animals for stressful encounters by upregulating stress-responsive genes and slowing development. CED-3 antagonizes this stress response via proteolytic inactivation of p38, thus promoting development.
INTRODUCTION
Caspases are conserved proteases well known for their roles in initiating and executing apoptosis, mostly by proteolytic activation of target proteins (Conradt and Xue, 2005; Crawford and Wells, 2011). Recent studies across diverse phyla of metazoans have reported non-apoptotic functions for several apoptotic caspases, as recently reviewed (Bell and Megeney, 2017; Nakajima and Kuranaga, 2017). These functions include roles in differentiation, such as limiting stemness or promoting axonal regeneration (Dick et al., 2015; Fernando et al., 2002; Fujita et al., 2008; Pinan-Lucarre et al., 2012; Weaver et al., 2014). For instance, in C. elegans, we previously reported a non-apoptotic role for the CED-3 caspase in the LIN-28 pluripotency pathway. Inactivation of several key factors, including LIN-14, DISL-2(Dis3l2) and LIN-28, by CED-3 allows rapid cell fate specification of seam cells in C. elegans (Weaver et al., 2014, 2017). Analogous functions in limiting pluripotency have been shown in mammals, with caspase-3 inactivating Nanog in embryonic stem cells (Fujita et al., 2008) and Pax7 in muscle satellite cells (Dick et al., 2015). In addition, the caspase-3 ortholog, drICE, was shown to be required for sperm differentiation in Drosophila (Arama et al., 2003, 2007). Further, CED-3 caspase was recently shown to be necessary for the ability of neuroblasts to divide asymmetrically by size and counter their pig-1(MELK)-dependent mitotic potential prior to cell death (Mishra et al., 2018). Based on these and other ongoing studies, it has been suggested that caspases have conserved non-canonical roles in repressing expression of key stem factors to establish asymmetric cell divisions (Bell and Megeney, 2017).
One intriguing question that remains is whether or not caspases have wide-ranging non-apoptotic roles in regulating gene expression during development. The discovery of such global non-canonical roles contributing to cell vigor would redefine the physiological roles for these caspases. Ongoing efforts in Drosophila have made use of sophisticated reporter systems to reveal the widespread presence of caspase activities throughout multiple tissues in development (Baena-Lopez et al., 2018; Tang et al., 2015). Exactly what roles these caspases are performing remains to be determined.
We show here that CED-3 caspase acts in opposition to the activity of a p38 MAPK in the absence of stress, to promote larval development in C. elegans. Our findings further suggest that this p38 signaling pathway, acting at least partly through the epidermal-specific GATA factor ELT-3, normally primes animals against adverse environmental conditions, such as hyperosmolarity, as well as exposure to pathogens. The balance in gene expression programs between the caspase and p38 directs organismal physiology toward either stress resistance or rapid development. Our results thus reveal an unexpected regulatory paradigm that may be widely used to regulate gene expression and support robust development.
RESULTS
CED-3 Caspase Limits PMK-1(p38 MAPK) Pathway to Promote Larval Development
Using an RNAi enhancer screen to reveal genes that become essential for development when ced-3 caspase is not functional, we identified the dual specificity phosphatase (DUSP) vhp-1, well known for regulating MAPK stress-responsive signaling. When vhp-1 was knocked down by RNAi in ced-3(−) mutants during post-embryonic development, we observed a dramatic delay in larval development and associated larval lethality (Figure 1A). When synchronized first larval stage animals were subjected to vhp-1(RNAi), ced-3(−) animals did not progress beyond the third larval stage in the same time that allowed wild-type animals fed mock(RNAi) or vhp-1 (RNAi), as well as ced-3(−) animals fed mock(RNAi), to mature into young adults with offspring (Figure 1B; see STAR Methods). Although the vhp-1(−) mutant has a reported larval arrest phenotype (Mizuno et al., 2004), reducing vhp-1 by RNAi caused only a small but significant larval growth delay (Figures 1A and 1B), which is consistent with previous observations (Richardson et al., 2010). The synergistic larval growth delay phenotype was observed with three ced-3 mutants when treated with vhp-1(RNAi). These alleles included the n717 reference null splice-defective mutation, the n1286 early stop codon mutation, and the ok2734 large ced-3 locus deletion mutation (Figures 1B and S1A). These findings suggest that the delay phenotype is the consequence of loss of both ced-3 and vhp-1 functions in post-embryonic development. We also found that loss of either the upstream ced-3 activator, ced-4(Apaf1) (Figure S1B), or ced-3 catalytic activity (Figure S1C) recapitulated the larval delay phenotype when combined with vhp-1 (RNAi). Together these results support the hypothesis that CED-3 caspase has a physiological role related to VHP-1 and may impact a MAPK stress-responsive signaling pathway.
Figure 1. Caspase Represses a Non-Stress p38 MAPK Function that Delays Post-Embryonic Development.
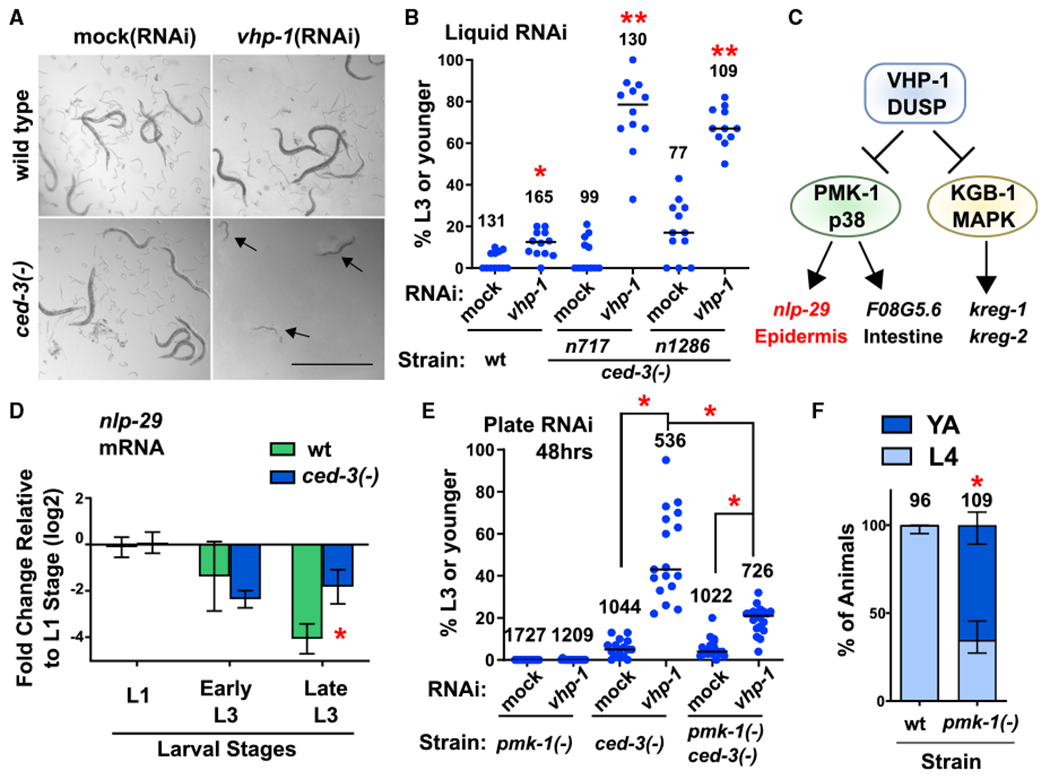
(A and B) Loss of both ced-3 and vhp-1 functions compromise viability. Images (A) and quantification (B) of worms in liquid RNAi cultures taken on a dissecting scope, showing a synergistic larval delay phenotype in ced-3(−) animals treated with vhp-1(RNAi). Magnification was the same throughout (scale bar indicates 1.0 mm). Arrows indicate P0 animals delayed in mid larval development. For all conditions, synchronous L1 stage P0 animals were added to the indicated food source (RNAi) and scored for developmental stage when negative control P0 animals had reached adulthood. Images in (A) show obvious presence of offspring (eggs and L1 stage larvae) in control wells. Quantified data are shown as dot plots indicating the percent of original P0 animals in the given well remaining at L3 stage or earlier, with total number of animals scored for each indicated condition. Data are pooled from three independent experiments and each dot corresponds to an individual well (four wells per experiment). Median values (horizontal bars). *Significant for wild-type treated with vhp-1(RNAi) compared to mock(RNAi), p = 0.0037, Mann-Whitney. **Significant for both ced-3(−) strains treated with vhp-1 (RNAi) compared to both mock(RNAi) of the given strain and wts train treated with vhp-1(RNAi), p < 0.0001, Mann-Whitney. For quantification of a ced-3(−) mutant generated by another lab, confirmation of ced-4(Apaf) requirement, and CED-3 caspase catalytic activity, see Figures S1A–S1C.
(C) The VHP-1 phosphatase is known to negatively regulate (black stops) PMK-1(p38 MAPK) and KGB-1 MAPK that activate (arrows) downstream genes (Kim et al., 2004; Richardson et al., 2010).
(D) qRT-PCR analysis demonstrates that CED-3 caspase limits the expression of epidermal PMK-1 downstream target nlp-29. *Significant for ced-3(−) strain compared to wt, p = 0.016, t test, standard deviation shown. Other PMK-1 and KGB-1 targets are not significantly upregulated by ced-3(−) mutation (Figure S1D).
(E) Loss of pmk-1 function helps restore larval development of ced-3(−) animals treated with vhp-1(RNAi) on agar plates. Data are shown as dot plots with total number of animals scored for each condition. Data are pooled from two independent experiments and each dot corresponds to an individual plate. Median values (thin horizontal bars). A significant restoration of development was observed with pmk-1(−) compared to animals with wild-type pmk-1 function. *Statistical comparisons indicated by brackets, p < 0.0001, Mann-Whitney.
(F) Effect of pmk-1(p38)(−) mutation alone on larval developmental rate. Wild-type and pmk-1(−) strains were assessed for the percent of animals at the fourth larval stage or adult stage 48 h after hatching. Proportion of animals for the given stage with standard deviation is shown, with total number of animals scored for each condition. *Significant, pmk-1(−) compared to wild-type, p < 0.0001, Fisher’s exact test. For confirmation of the PMK-1(p38) pathway and upstream factors in negatively repressing post-embryonic development in the absence of stress, see Figures S1E–S1G.
Because VHP-1 is known to negatively regulate both PMK-1 and KGB-1 and thereby affects the expression of several downstream genes (Figure 1C), we tested the steady-state mRNA levels of known downstream targets of MAPK signaling to reveal which, if any, were altered in ced-3(−) mutants. We found that ced-3(−) mutants exhibited a significant upregulation of the PMK-1-responsive, epidermally expressed anti-microbial peptide gene, nlp-29 (Figure 1D). In contrast, kreg-1 and kreg-2, regulated by KGB-1, and F08G5.6, an intestinally expressed target of PMK-1, were not significantly altered at the mRNA level in ced-3(−) mutants (Figure S1D). These results suggested that CED-3 caspase likely limits the epidermal branch of p38 stress-responsive signaling.
We then reasoned that if PMK-1 is a common downstream target for the CED-3 caspase and the VHP-1 phosphatase and if the synergistic developmental delay was due to dysregulation of PMK-1 during larval development, then loss of pmk-1 should suppress the synthetic larval delay phenotype of ced-3(−);vhp-1 (RNAi) animals. Indeed, in a pmk-1(−) ced-3(−) double mutant, the larval delay seen with vhp-1(RNAi) in ced-3(−) animals was partially suppressed and development was significantly restored (Figure 1E). These results are consistent with a model that both CED-3 and VHP-1 act to limit the effects of PMK-1(p38 MAPK) pathway signaling.
Our observations are consistent with previous studies demonstrating that hyperactivation of PMK-1 can have very deleterious consequences on development (Cheesman et al., 2016; Kim et al., 2016b; Pukkila-Worley et al., 2012). The activity of PMK-1 therefore needs to be tightly controlled during development. Based on our findings, we tested whether PMK-1 may have a role in limiting developmental rate in the absence of stress. Notably, we found that loss of pmk-1 function by either mutation (Figure 1F) or RNAi treatment in wild-type animals (Figures S1E and S1F) resulted in accelerated development. We observed that when the majority of wild-type animals fed normal food or mock(RNAi) were in the fourth larval stage, many pmk-1(−) mutant or pmk-1(RNAi)-treated animals had reached adulthood. In addition to PMK-1, we found that the upstream MAP2K and MAP3K also functioned to repress the rate of larval development by RNAi treatment of wild-type animals or mutant animals for those genes (Figures S1E–S1G). These data suggest that CED-3 caspase counters a p38 MAPK-dependent pathway function to finely regulate developmental speed in the absence of stress.
Caspase Blocks PMK-1(p38 MAPK)-Dependent Epidermal-Specific Stress-Responsive Program
Two PMK-1-dependent, tissue-specific GATA factors in C. elegans, namely ELT-2 in the intestine and ELT-3 in the epidermis, activate gene expression in response to various stressors (Block and Shapira, 2015) (Figure 2A). We reasoned that if CED-3 limits a PMK-1 activity in a tissue-specific manner, then we should expect to see a tissue-specific alteration in the levels of either one of the two PMK-1-dependent GATA factors and/or the established downstream target genes. We found that ced-3(−) mutants had upregulation of total protein levels of ELT-3::GFP but no obvious alteration of ELT-2::GFP protein levels (Figure S2A). Moreover, we found that the ELT-3::GFP signal was retained in epidermal nuclei of ced-3(−) mutants in the third larval stage, well beyond the time of its down-regulation when ced-3 function is intact (Figures 2B and 2C). Further, we found that loss of the elt-3 function restored larval growth of ced-3(−) mutants treated with vhp-1 (RNAi) to an extent comparable to loss of pmk-1 function (Figure S2B).
Figure 2. CED-3 Caspase Limits Expression of a p38-Dependent Anti-Microbial Peptide in the Epidermis.
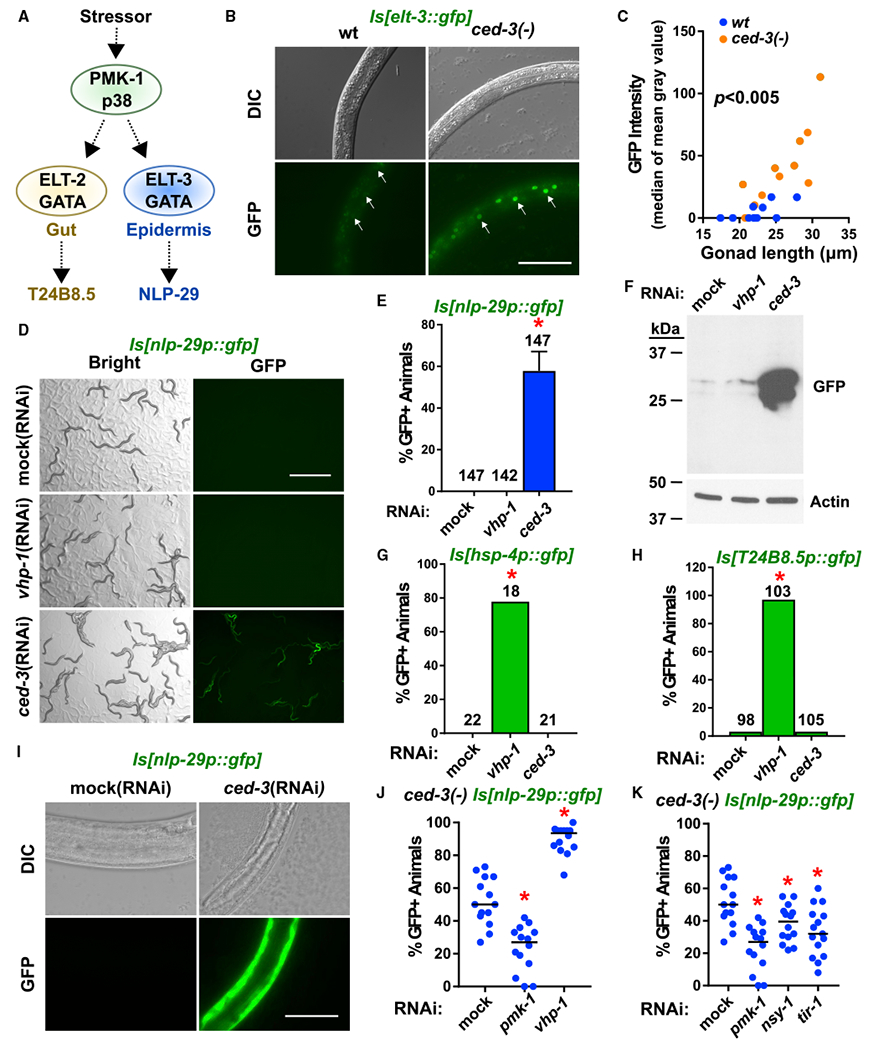
(A) Diagram of the stress-responsive p38 MAPK pathway and tissue-specific GATA transcription factors and transcriptional targets. Dashed arrows indicate downstream targets.
(B and C) Pseudo-colored images (B) and quantitation (C) of upregulation of the p38-dependent epidermal-specific GATA transcription factor ELT-3 in the ced-3(−) background (see also Figure S2A for western blot analyses of ELT-3 and ELT-2). Arrows indicate epidermal nuclei. Scale bar, 50 μm. The median of mean gray values is plotted as a function of gonad length to account for fine differences in developmental stage. Dots indicate individual animals. The ced-3(−) strain had significant upregulation compared to wt, p < 0.005, Mann-Whitney. Also see Figure S2B for suppression tests with pmk-1 and elt-3 mutants.
(D and E) Pseudo-colored images (D) and quantitation (E) of ced-3(RNAi) induction of nlp-29p::gfp expression. GFP protein expressed from a transgene with nlp-29 promoter and 5′ UTR is induced by ced-3(RNAi), but not by vhp-1(RNAi). Scale bar, 1 mm. Percentage of animals expressing GFP with standard deviation is shown with total number of animals scored for each condition. *Significant, mock (RNAi) versus ced-3(RNAi), p < 0.0001, Fisher’s exact test. Standard deviation shown.
(F) nlp-29p::gfp expression was confirmed by western blot. For additional lanes on right side, see Figure S2C for confirmation of comparable GFP induction by ced-3(−) null mutation with same exposure time and see Figure S2D for confirmation of GFP fluorescence by ced-3(n2433) caspase active site mutation.
(G and H) Intestinal reporters show upregulation by vhp-1(RNAi) but not by ced-3(RNAi). The number of GFP positive animals from the hsp-4p::GFP (G) and T24B8.5p::GFP (H) transgenes were scored in the blind. L4 larvae were put on the indicated RNAi plates and their L4 or young adult stage offspring were examined. *Significant for vhp-1(RNAi) versus either mock (RNAi) or ced-3(RNAi), p < 0.0001, Fisher’s exact test.
(I) Pseudo-colored GFP fluorescence and DIC microscopy show GFP expression caused by ced-3(RNAi) is visible only in the epidermis. L4 animals were put on the indicated RNA is and their L4 or young adult stage offspring were examined. Magnification was the same throughout (scale bar indicates 50 μm). Exposure time was the same throughout for GFP fluorescence (150 ms).
(J) Induction of nlp-29p::GFP in ced-3(−) mutants was blocked by pmk-1 (RNAi) and enhanced by vhp-1(RNAi). *Significant, mock (RNAi) versus pmk-1 (RNAi) and vhp-1(RNAi), p < 0.001, Mann-Whitney. Each dot represents a plate. Median values (thin horizontal bars).
(K) Confirmation that upregulation of nlp-29p::GFP expression by ced-3(−) mutation requires known factors upstream of PMK-1 in p38 epidermal pathway. All phenotypes were scored three times in the blind. *Significant, mock (RNAi) versus pmk-1(RNAi), nsy-1(RNAi), and tir-1(RNAi), p < 0.05, Mann-Whitney.
We also examined GFP protein expression from several established transgenes that reflect PMK-1 target genes, namely intestinal hsp-4 (Richardson et al., 2010; Urano et al., 2002), intestinal T24B8.5 (Shivers et al., 2009) and epidermal nlp-29 (Pujol et al., 2008a). We found that GFP protein expression from the nlp-29p::gfp reporter was strongly induced when treated with ced-3(RNAi) but not vhp-1(RNAi) (Figures 2D–2F). The observation we see for vhp-1 (RNAi) is consistent with previous findings (Zugasti et al., 2016). However, we found that the GFP protein expressed from the hsp-4p::gfp and T24B8.5p::gfp reporters were induced, predominately in the intestine, by vhp-1 (RNAi), as previously shown (Richardson et al., 2010), but not by ced-3(RNAi) (Figures 2G and 2H). The GFP expressed by the nlp-29p::gfp reporter due to ced-3(RNAi) was visible only in the epidermis (hypodermis) (Figure 2I). This induction was confirmed for the ced-3(−) null (Figure S2C) and ced-3 catalytic null (G360S) (Figure S2D) mutations.
Notably, nlp-29 induction during fungal infection is pmk-1-dependent (Pujol et al., 2008a, 2008b). We found that loss of pmk-1 suppressed the ced-3-dependent induction of nlp-29p::gfp with fewer GFP positive animals whereas vhp-1 (RNAi) enhanced the ced-3-dependent induction with more GFP positive animals (Figure 2J). We also confirmed the roles of factors upstream of PMK-1 for GFP expression from the nlp-29p::gfp transgene by showing a significant reduction in the percent of GFP positive animals due to ced-3(−) (Figure 2K). Our data suggest that the p38-dependent GATA factor, ELT-3, is responsible for upregulation of nlp-29p::gfp mRNA levels. Further, our findings confirmed the requirement for CED-3 to limit induction of the epidermal p38 stress-response pathway during development.
CED-3 Caspase and PMK-1(p38 MAPK) Inversely Regulate the Expression of Multiple Epidermal Pathogen- and Stress-Response Factors
To examine potentially broad effects by CED-3 and p38 in regulating gene expression, we analyzed whole transcriptome steady-state mRNA and whole translatome ribosomal associated mRNAs from synchronous third larval stage hermaphrodites (Figures S3A and S3B). Following optimization, we isolated bona fide ribosomal protected fragments (RPFs) (Figures S3C and S3D). We collected two biological replicates for wild-type, ced-3(−) mutants, and pmk-1(−) mutants. The biological replicates for each strain clustered with their respective counterpart and differed from the other strains by multi-dimensional scaling analysis (Figure S3E). We found a strong correlation (nearly 100%) between the transcriptome and translatome data sets.
Because ced-3(−) and pmk-1(−) mutants displayed opposing phenotypes on post-embryonic growth and induction of the nlp-29 anti-microbial peptide, we focused our expression analyses on genes that were inversely affected by the caspase and p38 MAPK. We found that CED-3 and PMK-1 had opposing regulation of 313 genes (Figure 3A; Table S1). Gene ontology analyses showed that these genes were enriched for pathogen- and stress-responsive genes from the epidermis that function as part of innate immunity and structural integrity (Figure 3B). As examples, the nlp-29, nlp-30, and nlp-31 family of epidermal-specific anti-microbial peptides showed a strong trend toward negative regulation by CED-3 and positive regulation by PMK-1 (Figure S3F). This finding was consistent with our nlp-29p::gfp transgene reporter results (Figures 2D and 2I). The NLP-29-31 family has a well-established role in innate immunity (Pujol et al., 2008a, 2008b). Using an in-house class enrichment tool (Zugasti et al., 2016), we also found an over-representation of genes regulating sleep-like lethargus (Table S1), which may contribute to the regulation of the developmental rate we observed (Figure 1). We also analyzed genes both downregulated or both upregulated by ced-3(−) and pmk-1(−) and found these were enriched for intestinal immune and epithelial molting genes, respectively (Figure S4). We also did not find any alteration of ELT-3 mRNA levels demonstrating the effects of ced-3(−) mutation on ELT-3 levels were working at the protein level.
Figure 3. Caspase and p38 Inversely Regulate Expression of Epidermal Stress Resistance Genes Including Fatty Acid Synthase fasn-1.
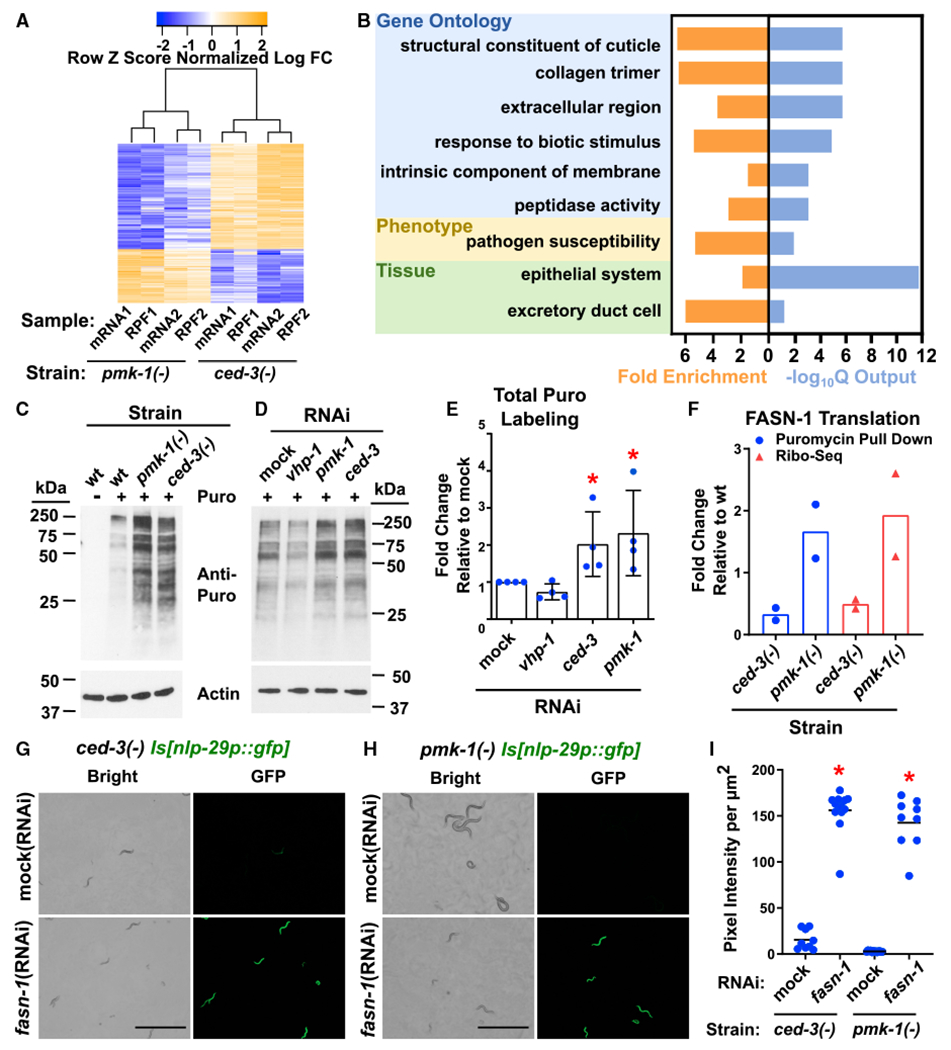
(A) Whole transcriptome and ribosome profiling analyses of L3 stage animals identified 313 genes that are inversely regulated by CED-3 and PMK-1 in two independent biological replicates. For ribosome profiling workflow, raw data, and selection of L3 stage, see Figure S3; Table S1; STAR Methods.
(B) Gene enrichment analysis of the 313 inversely regulated genes identified an enrichment for pathogen resistance genes expressed in the epidermis (hypodermis in nematodes). Fold enrichment and significance (output of the −log10Q function is shown). Also see Figure S4 and Table S1 for enrichment analysis of genes down- and up-regulated in both ced-3(−) and pmk-1(−) mutants.
(C and D) Western blot of puromycin-labeled peptides from wild type, ced-3(−) and pmk-1(−), or respective RNAi as indicated. Duplicates loaded with more protein on left (C) and right (D) sides, respectively removed for simplicity. Also see Figure S5B for additional western blots used in quantitation.
(E) Quantitation of replicates (Figures 3D and S5B) shows that pmk-1 (RNAi) and ced-3(RNAi) each have upregulated global translation. *Significant, mock (RNAi) versus ced-3(RNAi) and pmk-1(RNAi), p = 0.0286, Mann-Whitney. Standard deviation shown.
(F) FASN-1 translation is upregulated in pmk-1(−) mutants and downregulated in ced-3(−) mutants. Strains were normalized to wild-type expression. FASN-1 was identified by both ribosome profiling and puromycin labeling methods as a highly abundant target inversely regulated by pmk-1 and ced-3. For protein IDs, see Table S2. See Figure S5C and STAR Methods for replicates and details of puromycin-labeled peptides that were immunoprecipitated with an anti-puromycin antibody and analyzed by mass spectrometry.
(G–I) fasn-1 negatively regulates anti-microbial peptide gene expression likely downstream of ced-3 and pmk-1 functions. Pseudo-colored images and quantification of nlp-29p::gfp expression. Scale bars, 1.0 mm. (G) GFP protein induction by fasn-1(RNAi) is beyond ced-3(−) alone (mock(RNAi) and compare to Figure 4 ced-3(−) alone with similar pixel intensity). (H) The fasn-1(RNAi) overcomes pmk-1-dependence of nlp-29p::gfp expression (see Figure 2J) suggesting fasn-1 functions downstream of (or parallel to) pmk-1 function. (I) Quantitation of GFP intensity per area. *Significant, ced-3(−) mock (RNAi) versus fasn-1 (RNAi) and pmk-1(−) mock (RNAi) versus fasn-1(RNAi), p < 0.001, Mann-Whitney. Median values (thin horizontal bars).
Next, to identify altered translation of highly abundant genes, we used a previously established method to pulse label actively elongating peptides that were then identified with mass spectrometry (Arnold et al., 2014; Schmidt et al., 2009) as outlined in Figure S5A. We found that loss of either pmk-1 or ced-3 resulted in increased translation as seen by an increase in gross elongating peptides, whereas loss of vhp-1(DUSP) resulted in a minor decrease in translation (Figures 3C–3E and S5B). These results suggest that CED-3 and PMK-1 repress a subset of the translatome.
Analyses of the genes and/or proteins in both ribosome profiling (Figure S3) and puromycin labeling methods (Figure S5C) revealed that fasn-1/FASN-1, pod-2/POD-2, and hsp-60/HSP-60 were downregulated in ced-3(−) mutants. Further, fasn-1/FASN-1 was consistently up-regulated in pmk-1(−) mutants based on the findings of both methods (Figure 3F; Table S2). FASN-1 plays a role in lipid metabolism but has also been shown to have critical roles in regulating nlp-29 expression (Lee et al., 2010) and stress adaptation (Kim et al., 2016a). In both cases, it was found to also act in concert with POD-2. On the basis of these results, we hypothesized that CED-3 and PMK-1 may have critical but opposing roles in regulating the expression of pathogen response genes.
Induction of nlp-29 expression in the epidermis has previously been described for stress adaptation and innate immunity (Pujol et al., 2008b). Because fasn-1 has been shown to work in a p38-independent manner to induce nlp-29 (Lee et al., 2010) and we found that FASN-1 was a highly abundant gene inversely regulated by CED-3 and p38, we wanted to understand the relationship of fasn-1 to ced-3(−)-induced nlp-29p::gfp expression. We found that nlp-29p::gfp was induced by fasn-1 (RNAi) beyond the ced-3(−) mutation alone (Mock RNAi) (Figures 3G–3I). Also, the fasn-1(RNAi) completely overcame the PMK-1 dependence on nlp-29p::gfp expression (Figures 3H and 3I; see Figure 2J). The ability of fasn-1(RNAi) to activate nlp-29 independently of p38 in conjunction with the opposing effects of p38 and CED-3 on expression of fasn-1 suggests that fasn-1 may work downstream of p38 function to negatively regulate expression of nlp-29.
CED-3 Caspase Cleaves PMK-1(p38 MAPK) to Promote Development
We examined the possibility that the PMK-1(p38 MAPK) protein is a direct cleavage target of CED-3 caspase by using an established in vitro cleavage assay (Lee and Xue, 2014; Weaver et al., 2014). We found that CED-3 caspase cleaved PMK-1 at Asp327 but not at neighboring Asp residues (Figure 4A), suggesting that PMK-1 is a candidate cleavage target for CED-3 caspase. Mutation of other Asp residues also did not alter CED-3 cleavage, further supporting specificity of the recognition site (Figure S6).
Figure 4. CED-3 Caspase Targets PMK-1(p38 MAPK) to Promote Development and Limit Expression of Anti-Microbial Peptide.
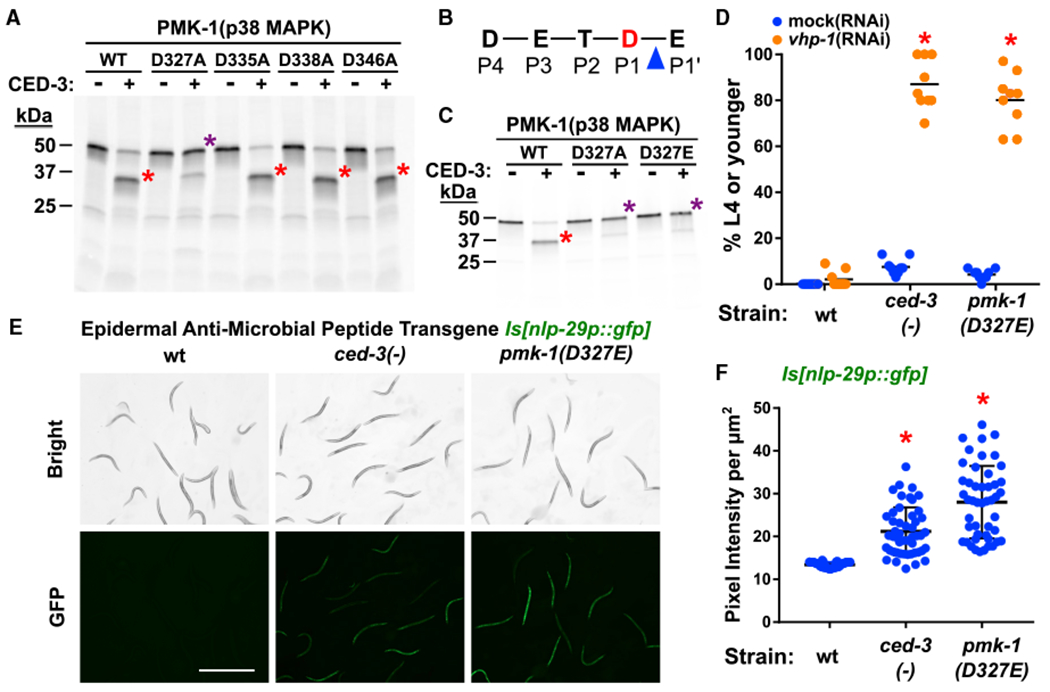
(A–C) In vitro cleavage analysis with purified CED-3 caspase identifies D327 of PMK-1 as a CED-3 cleavage site. (A) D327A blocks CED-3 cleavage. (B) DxxD cleavage site (P4–P1) residues in PMK-1. Blue triangle indicates scissile bond between the P1 residue, Asp327 (red D) and the P1′ residue, Glu328. (C) D327Aas well as D327E mutations abolish caspase cleavage. Red asterisk, main cleavage product. Purple asterisk, cleavage-resistant full-length protein. Also see Figure S6 for additional Asp to Ala mutants that do not block cleavage.
(D) ced-3(−) animals and pmk-1(D327E) animals show equivalent developmental delay when treated with vhp-1(RNAi). *Significant, mock (RNAi) versus vhp-1(RNAi), p < 0.001, Mann-Whitney. Median values (thin horizontal bars).
(E and F) The pmk-1(D327E) animals show upregulation of nlp-29p::gfp expression equivalent to ced-3(−) animals. Pseudo-colored images (E) and quantification (F) of nlp-29p::gfp expression. Scale bar, 1mm. *Significant, wt versus ced-3(−) and pmk-1(D327E), p < 0.001, Mann-Whitney. Each dot corresponds to an animal. Mean with standard deviation shown.
The Asp327 cleavage site conformed to an Asp-Xxx-Xxx-Asp P4-P1 site where Asp327 is the P1 residue N-terminal to the scissile bond (Figure 4B). Both an Asp327Glu and an Asp327Ala mutations blocked cleavage (Figure 4C). Because the negative charge at this position in MAPKs has recently been shown to be important for function in vivo (Taylor et al., 2019), we used the Asp327Glu charge-conservative mutation for our functional studies. We generated a pmk-1(D327E) cleavage-resistant mutation in vivo in the endogenous pmk-1 locus using CRISPR-Cas9 mutagenesis. We found that the pmk-1(D327E) mutant had a developmental delay comparable to ced-3(−) mutants when treated with vhp-1(RNAi) (Figure 4D). We also found that the pmk-1(D327E) mutant upregulated GFP expression when combined with the nlp-29p::gfp transgene to an extent comparable to ced-3(−) mutation (Figures 4E and 4F). The fact that pmk-1(D327E) phenocopies loss of ced-3 function in this context strongly suggests that PMK-1 activity is normally limited through cleavage by CED-3. We were unable to determine the effects of ced-3(−) on PMK-1 protein levels due to technical limitations.
Cleavage-Resistant PMK-1 Does Not Affect Cells Fated to Die by CED-3
To address the possibility that the perdurance of undead cells in ced-3(−) mutants during post-embryonic development is responsible for the phenotypes we observed, we used distinct methods to assay the effects of pmk-1(D327E) on ced-3-dependent programed cell death in early and late larval development. Using an established method (Zhou et al., 2001), we counted corpses of dying head cells using the ced-1(e1735) background to visualize cell deaths (Figures 5A and 5B). We found that pmk-1(D327E) had wild-type levels of corpses in first larval stage animals (Figure 5C). Using another established method (Reddien et al., 2007), we counted the number of undead P9-12 ventral nerve cord cells (VNCs) in young adults (Figures 5D and 5E). We observed that pmk-1(D327E) did not result in accumulation of undead VNCs, whereas a mild ced-3 reduction-of-function allele showed accumulation to a detectable extent (Figure 5F). These findings support that the action of CED-3 caspase on p38 MAPK at Asp327 is non-apoptotic in nature and not indirectly affecting development and gene expression because of the perdurance of undead cells.
Figure 5. Caspase-Cleavage-Resistant p38 MAPK Does Not Impact CED-3-Dependent Programmed Cell Death in Early or Late Larval Development.
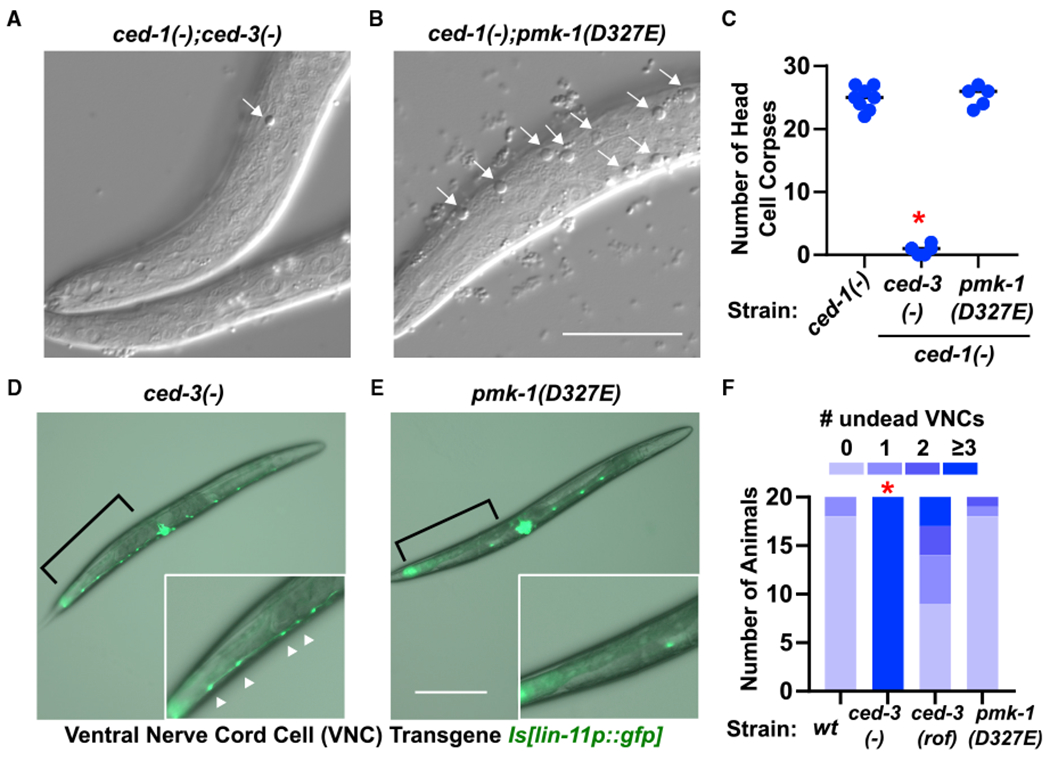
(A–C) Early first larval stage animals using ced-1(e1735) background to visualize head cell corpses of ced-3-dependent cells fated to die. The ced-3(−) (A) and pmk-1(D327E) (B) effects on accumulation of head corpses were evaluated. Representative images of a given focal plane fo rthe indicated genotypes with head corpses (arrows) from cells fated to die being engulfed by neighboring cell. Scale bar, 20 μm. (C) Total cell corpses found in heads of first larval stage animals. *Significant, ced-3(−) versus wt, p < 0.001, Mann-Whitney.
(D–F) First day adults using lin-11p::GFP to visualize undead ventral nerve cord cells (VNCs). The ced-3(−) (D) and pmk-1(D327E) (E) effects on undead VNCs were evaluated. Representative images of the indicated genotypes with area of P9-12 dead (no GFP) or undead VNCs (bright GFP) (brackets). Pseudo-colored GFP and transmitted light were over laid in each image to indicate anatomical position. Insets are digital zoom of the area indicated by brackets. Scale bar, 200 μm. (F) Summary of animals with indicated number of undead P9-12 cells by counting lin-11p::GFP positive VNCs. *Significant, ced-3(−) versus wt, p < 0.001, Fisher’s exact test.
PMK-1(p38 MAPK) Stress Priming Function Is Limited by CED-3 Caspase
The fungal pathogen Drechmeria coniospora infects nematodes by penetrating the cuticle and epidermis, eliciting expression of the anti-fungal peptide genes nlp-29 and nlp-31 that have established roles in the resistance to infection (Couillault et al., 2004; Pujol et al., 2008b). Because CED-3 and p38 inversely control expression of pathogen response genes, including the nlp-2931 cluster in the epidermis, we thus tested the extent of nlp-29p::gfp induction in animals with and without Drechmeria infection. In this assay, the STAT transcription factor family members sta-1 and sta-2 were used as negative and positive controls, respectively, for induction of nlp-29p::gfp with Drechmeria infection based on the findings of a previous study where sta-1 is not required for responsiveness but sta-2 is required (Dierking et al., 2011). We observed a heightened response in infected animals when ced-3 was reduced by RNAi, consistent with a role for CED-3 in limiting pathogen response (Figure 6A). We also observed an elevated response in pmk-1(D327E) mutants at the fourth larval stage (Figure 6B). As a possible explanation for the attenuated nlp-29p::gfp response observed for the ced-3(−) mutant, we observed numerous ced-3(−) animals delayed at the second larval stage with very high nlp-29p::gfp expression. The difference between the RNAi versus mutant is likely a reflection of transient versus complete loss of ced-3 function. Moreover, our original screen revealed that ced-3 has numerous other non-apoptotic functions during larval development.
Figure 6. Caspase Limits Diverse p38 MAPK-Dependent Stress Responses.
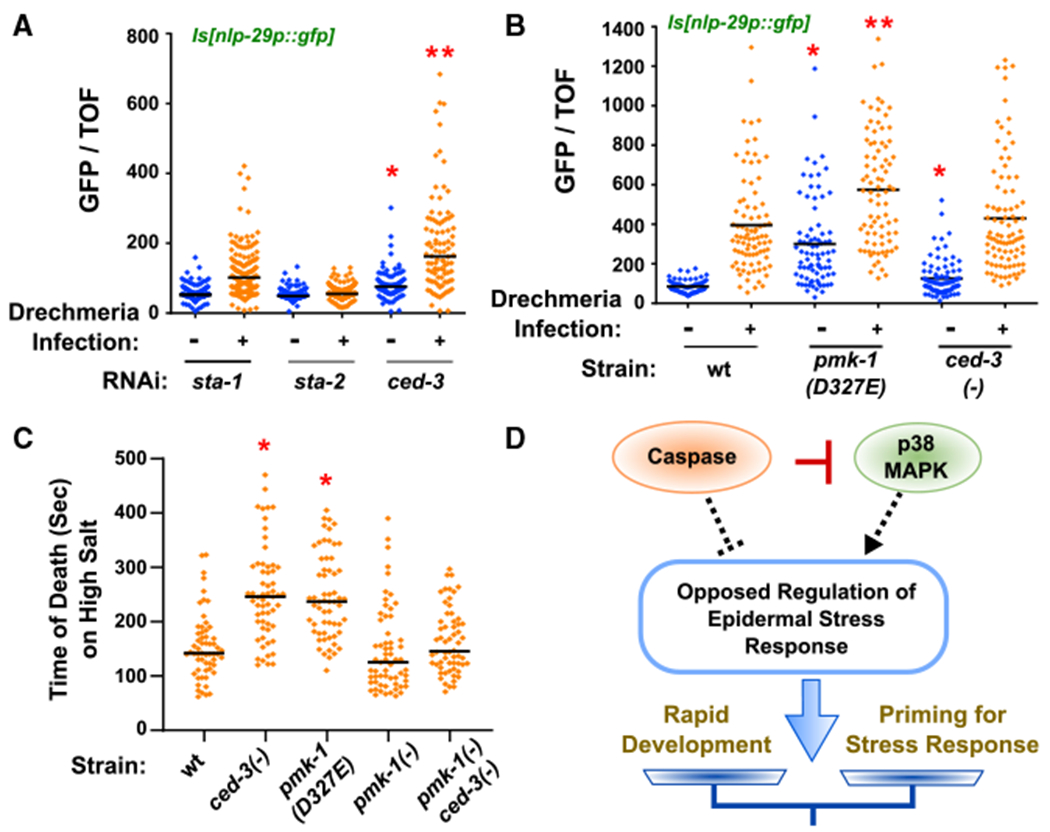
(A and B) Loss of ced-3 by RNAi and pmk-1(D327E) primes infected animals for expression of anti-microbial nlp-29p::gfp. Expression of nlp-29p::gfp in animals with and without Drechmeria infection and indicated RNAi treatments (A) or mutant strains (B). Vertical axis shows the intensity of green animals normalized by size (time of flight, TOF). (A) *Significant, ced-3(RNAi) versussta-1(RNAi) before infection, p < 0.0001, Mann-Whitney. *Significant, ced-3(RNAi) versus sta-1(RNAi) after infection, p < 0.0001, Mann-Whitney. Median values (thin horizontal bars). (B) *Significant, pmk-1(D327E) versus wt before infection, p < 0.0001, and ced-3(−) versus wt before infection, p = 0.0068, Mann-Whitney. **Significant, pmk-1(D327E) after infection, p < 0.0001, Mann-Whitney.
(C) Loss of ced-3 or loss of CED-3 cleavage site in pmk-1 primes animals for acute hyperosmolarity stress resistance. *Significant, wt versus ced-3(−) and pmk-1(D327E), p < 0.001, Mann-Whitney, median values (bars).
(D) Diagram of opposed regulation of ~300 genes by caspase and p38 to balance stress responses and rapid development. CED-3 directly blocks the action of p38 by proteolytic cleavage (red bar). By inversely regulating the epidermal stress response (dashed lines), ced-3 and pmk-1 fine tune the extent of stress priming. The net outcome (arrow) based on inputs alters the balance of stress response and developmental progression (scale).
We then considered if the CED-3 and PMK-1 stress priming functions are limited to pathogens only. We tested animals for survival with an acute high salt stress on agar plates in the absence of a food source. We decided to assay acute hyperosmolarity stress resistance because it does not involve an extended period to assay and therefore limits an adaptation opportunity. It therefore allowed us to test whether animals were primed for an acute stress prior to experiencing the stress. We observed a two-fold increase in survival time of mid-fourth larval stage animals when ced-3 was absent or with the cleavage-resistant pmk-1 mutation (Figure 6C). We observed suppression of the ced-3(−) mutant survival effect when combined with pmk-1(−) mutation. We also observed that pmk-1(−) mutants were most skewed with the median value below the mean, indicating higher sensitivity to the high salt (Figure 6C). Altogether, these findings demonstrate that p38 has a stress-priming function during post-embryonic development and the caspase acts to limit this responsiveness to promote development.
DISCUSSION
Caspase Limits Epidermal p38 Stress Surveillance Function
Previous studies have shown caspases, including CED-3, play a role in stress adaptation (Judy et al., 2013; Yee et al., 2014). The fact that the underlying mechanisms or pathways in those cases are still unclear underscores a potentially complex role for caspases in stress signaling. This complexity is further masked by compensatory regulatory mechanisms. Our findings demonstrate an intriguing role for a caspase in opposing a p38 activity that primes animals in advance of adverse environmental encounters. We showed that loss of pmk-1(p38 MAPK) significantly suppresses the larval delay for ced-3(−);vhp-1(RNAi) animals (Figure 1E). These data suggest a role for CED-3 in limiting the effects of the PMK-1 pathway in the epidermis during larval stages. Previous studies have already demonstrated that the expression of p38 target genes needs to be tightly regulated and coordinated across tissues during development (Kim et al., 2016b).
Based on our findings, p38 seems to delay development potentially to prime animals for adverse exposure to pathogens or other stressors. By limiting the p38-dependent stress priming program, CED-3 caspase promotes development. Lethality associated with expressing CED-3 from transgenes prevented us from testing its tissue-specific functions more precisely. Nonetheless, our findings that the CED-3 cleavage-resistant pmk-1 mutant phenocopied ced-3(−) mutants for larval growth and stress resistance phenotypes are consistent with a cell-autonomous function for the cleavage of PMK-1 by CED-3. Moreover, based on the observations that the epidermis was previously shown to be the relevant tissue for a PMK-1-dependent osmolarity stress resistance by osr-1 (Solomon et al., 2004) as well as PMK-1-dependent Drechmeria pathogen resistance (Polanowska et al., 2018), we suggest that ced-3 caspase likely acts to limit p38 signaling in the epidermis. However, as p38 is not critical to other aspects of osmotic stress adaptation (Pujol et al., 2008b; Rohlfing et al., 2010), ced-3 functions within the context of a complex and incompletely understood regulatory system. The relationship of vhp-1 and ced-3 is likely equally complex because loss of vhp-1 enhanced expression of intestinal stress-responsive reporters, whereas loss of ced-3 only enhanced an epidermal stress-responsive reporter. As another layer of complexity, ced-3 blocks expression of epidermal stress response genes but activates expression of intestinal immune response genes (Figures 3 and S4). It remains an area for continued investigation, how caspase impacts immune and stress signaling at the organismal level. The balance of gene expression programs supporting innate immunity and development may demonstrate an evolutionary trade-off between rapid maturation and durability in harsh environments (Figure 6D), reminiscent of the effect of population density on developmental speed (Ludewig et al., 2017).
Caspase Supports Broad Gene Expression Dynamics
Our previous work revealed specific targets in the LIN-28 pluripotency pathway for the CED-3 caspase in a non-apoptotic function that limited symmetric cell divisions (Weaver et al., 2014). Our current study of the ced-3 caspase demonstrates a larger role for non-canonical caspase functions in development. These distinct roles suggest the possibility of very diverse non-canonical caspase functions in development and homeostasis.
Genetic redundancy likely masks many significant biological roles of genes like ced-3 that have been functionally characterized through direct genetic approaches. Revealing roles for CED-3 in supporting homeostasis, in addition to its role in achieving cell death, not only expands the range of biological roles for apoptotic caspases, but also reveals a gene regulatory role that could be widely used to regulate gene expression. This unexpected function seems to be important in changing gene expression programs either in stemness decisions or stress surveillance. In both cases, the caspase facilitates the termination of one program and initiation of another.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to the Lead Contact Benjamin Weaver (Benjamin.Weaver@UTSouthwestern.edu). Unique and stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains and Rationale for Selection
The wild-type (wt) reference strain is N2. The ced-3(−) mutants bear either the ced-3(n717) allele, the ced-3(n1286) allele, or ced-3(ok2734) allele. These three ced-3(−) mutant strains were chosen based on the following rationale. The ced-3(n717) allele was identified in a screen for animals with complete loss of post-embryonic cell deaths and was also found to have semi-dominant suppression of heterozygous egl-1(n487) egg-laying defect (Ellis and Horvitz, 1986). Additionally, the ced-3(n717) allele is the reference allele for ced-3 loss-of-function mutants and results in defective mRNA splicing (loss of the splice acceptor site upstream of exon 7) (Shaham et al., 1999). The ced-3(n1286) allele was identified in a non-complementation screen for suppression of ectopic neuronal cell death and bears a premature stop codon (W428Opal) (Shaham et al., 1999; Yuan et al., 1993). Non-complementation screens have the power to identify lethal alleles. Additionally, the ced-3(ok2734) allele bears a large genomic deletion of the ced-3 gene including the active site (C. elegans Deletion Mutant Consortium, 2012) and was used to confirm the vhp-1(RNAi) genetic interaction (Figure S1A) as was the ced-3(n2433) catalytic site null mutant (Shaham et al., 1999) (Figure S1C). The pmk-1(−) strain had the pmk-1(km25) allele (Mizuno et al., 2004). The hsp-4::gfp(zcIs4) reporter strain (Richardson et al., 2010; Urano et al., 2002), the T24B8.5::gfp(agIs219) reporter strain (Shivers et al., 2009) and the nlp-29p::gfp(frIs7) reporter strain (Pujol et al., 2008a) were used as indicated. ELT-2::GFP (OP56) and ELT-3::GFP (OP75) (Sarov et al., 2006) were used where indicated.
METHOD DETAILS
RNAi Treatments
Unless otherwise noted, liquid RNAi and plate RNAi treatments were prepared as previously described (Fraser et al., 2000; Lehner et al., 2006; Timmons et al., 2001). In brief, cultures were grown for 15hr with shaking at 37°C with 150 μg/mL ampicillin. Cultures for plates were then spotted on NGM agar with 1 mM IPTG and 150 μg/mL ampicillin and allowed to grow lawns for several days in the upright position to maintain selection. Alternatively, cultures for liquid RNAi were grown out in 96 well plates, spun down at 1,000 xg, resuspended in complete media (Lehner et al., 2006) and synchronous L1 animals were added to wells (about 20 per well). Unless otherwise noted, strains were added to RNAi plates after synchronization in M9 for 18 hours at 20°C.
Microscopy
Images were taken with either a Leica MZ 16F dissecting scope and a Hamamatsu ORCA-ER C4742-95 digital camera (Figures 1, 2 and 3), a Zeiss Axiozoom V16 dissecting scope with a Zeiss Axiocam 506 mono camera (Figures 4 and 5), a fluorescent microscope with DIC (Nomarski) optics (Zeiss Axioplan 2) with a Zeiss Axiocam MRm (Figure 2), or a Zeiss AxioImager M2 with a Hamamatsu ORCA C13440 camera (Figure 5). All images were taken at the same magnification for the same amount of time for comparisons as indicated in the relevant figure legends.
Reverse Transcription and Quantitative PCR
Total RNA was extracted using TRIzol™ Reagent (Invitrogen, 15596-026) from animals with different genotypes and developmental stages. For cDNA synthesis, 1μg of total RNA was used as input for all samples. The SuperScript™ III Reverse Transcriptase (Invitrogen, 18080-044) and Oligo dT (Invitrogen, 18418-020) system was used for cDNA synthesis. The Mx3000P system (Agilent Technologies) with ABsolute Blue qPCR SYBR Green Low ROX Mix (Thermo Scientific, AB4323/A) was used for qPCR analysis. Each condition was tested with 4 technical replicates and 3 biological replicates in 20μL reactions. rpl-4 was used as the reference gene for normalization in all experiments. Oligos P1-P12 were used for qRT-PCR analyses (Table S3).
Assays for Development
For liquid culture, unless specified otherwise in the figure legends, animals were scored for developmental stages after 48 hr at 20°C following addition of synchronous first larval stage animals on the indicated food source as previously established (Weaver et al., 2014). To obtain gravid adults, mixed populations were maintained on 60 mm NGM plates seeded with OP50 E. coli lawns at 20°C. For liquid cultures, gravid adults were treated with alkaline hypochlorite solution (1% sodium hypochlorite and 500 mM sodium hydroxide in water) to obtain eggs that were washed four times in M9 salt solution and incubated at 20°C overnight for hatching. Synchronous L1 animals were then placed on indicated food sources. For solid media, gravid young adults were allowed to lay eggs for 3 hours and removed. Animals were incubated at 20°C and staged at the indicated time intervals. Prior to testing, animals were maintained under stress-free conditions at 20°C for multiple generations, including no starvation, and no obvious contaminations.
Preparation of L3 Larval Stage Samples for RNA Sequencing
Ribosome profiling was done with two biological replicates that were independently generated, collected and processed. Synchronized day 1 young adult wild type (N2), ced-3(n717) and pmk-1(km25) worms were bleached and then the eggs were incubated in M9 for 24 hours to obtain synchronized L1s. L1s were plated on OP50 for 18hrs at 20°C until >90% of wild type worms reached L3 stage (Figure S3B).
Isolation of Ribosomal Protected Fragments and Total mRNA
The ribosomal profiling protocol was modified based on a published protocol (Aeschimann et al., 2015) and the TruSeq® Ribo Profile Protocol (Illumina). For our method, L3 staged worms were washed off plates, counted and collected in M9. About 165,000 L3 worms per strain were used for one Ribosome Profiling experiment. Worms were washed 2 more times in 10mL of M9 with a final wash was done in 10mL Buffer A (20mM Tris-Hcl pH7.9,140mM KCl, 1.5mM MgCl2, 0.5% (v/v) NP-40). After washing, centrifugation and aspirating the buffer, the worm pellet was flash frozen in liquid nitrogen. Frozen pellet was then thawed on ice for 5min before adding 300mL of Assay Buffer (20mM Tris-Hcl pH7.9, 140mM KCl, 1.5mM MgCl2, 0.5% (v/v) NP-40, 2% (w/v) polyoxyethylene-10-tridecylether, 1%(w/v) sodiumdeoxycholate monohydrate,1mM DTT,0.1mM cycloheximide). Worms thawed in the Assay Buffer were then added in drops into a mortar that was pre-cooled with liquid N2. Using pre-cooled pestle, worm pellets were crushed and grinded into fine powder. After thawing in the mortar, lysate was transferred into 1.5mL tube and centrifuged for 800xg for 3min followed by 12,000xg for 8min. The supernatant was transferred to a fresh tube and A260 was measured at 1:100 dilution. For each genotype, two A260 units of RNA in 100uL volume were used with 50 units of RNAseI (Ambion, AM2294) in digestion at room temperature with shaking at 250 rpm for 45 minutes. 5μL of SUPERase-In RNase Inhibitor (Ambion, AM2694) was used to stop the reaction. The RNA Clean & Concentrator-25 Kit (Zymo Research, R1017) was used to purify both the ribosomal protected fragments (RPFs), after RNAse digestion, and total RNA. 10μg of RNA were then treated with DNase I (NEB, M0303S) at 37°C for 10min, followed by purification with RNA Clean & Concentrator-25 Kit (Zymo Research, R1013). Total RNA was then rRNA depleted with Ribo-Zero Gold rRNA Removal Kit (Illumina, MRZG12324) and purified with RNA Clean & Concentrator-5 Kit (Zymo Research, R1015). RPFs were then purified with 15% TBE Urea gel (Invitrogen, Novex™ EC6885BOX) (Figure S3). 2830 nt fragments (RPFs) were cut, eluted and purified according to Illumina TruSeq® Ribo Profile Protocol.
RNA Library Preparation and Sequencing
The 3’ adaptor ligation, reverse transcription, gel purification, cDNA circularization and PCR amplification steps were performed according to the Illumina TruSeq® Ribo Profile Protocol.
For final PCR amplification of library, circular cDNA for total mRNA input was 1 μL and circular cDNA input for RPF was 5μL.
The average fragment size of the libraries was determined using the High Sensitivity D1000 screentape (Agilent, 5067-5584) on a Tapestation 2200 system (Agilent, G2964A). Library sizes ranged between 158-179bp, consistent with ribosomal profiling libraries. Individual library concentrations were determined by qPCR using the KAPA library quantification kit (KAPA, KK4824). Sequencing libraries were pooled together in equal molar quantities. The final library pool had an average size of 166bp and the final pool concentration was determined to be 2.22nM by using the Qubit 3 fluorometer (Invitrogen). Libraries were sequenced on the Illumina Next-Seq 500 (SN: NB501447)at 1.7pM with 1% phiX control (Illumina, FC-110-3001). Paired-end 37 bp reads were obtained using a Next-seq 500/550 High Output 75-cycle sequencing kit v2 (Illumina).
RNA Sequencing Data Analyses
RNA-seq and ribosomal profiling data were processed with the following workflow. Adaptors were removed using SeqPurge (Sturm et al., 2016). rRNA reads were removed using bowtie2 (Langmead and Salzberg, 2012). Reads were mapped to the C. elegans genome using STAR (Dobin et al., 2013) allowing only uniquely mapped reads. Mapped reads were counted using HTSeq (Anders et al., 2015) and gene expression profile was analyzed using edgeR (Robinson et al., 2010). Gene set enrichment was done with the Wormbase Enrichment Tool (Angeles-Albores et al., 2016).
Puromycin Labeling and Mass Spectrometry
Puromycin pulse labeling was done in multiple biological replicates that were independently generated, collected and processed. Mass spectrometry ID was performed on two independent biological replicates with 6 conditions in each replicate. Synchronized day 1 young adult wild type (N2), ced-3(n717) and pmk-1(km25) worms were bleached and eggs were incubated in M9 for 24 hours to obtain synchronized L1s. L1s were plated on OP50 for 18hrs at 20°C until >90% wild type reach L3 stage (Figure S5C). L3 stage worms were transferred onto plates with OP50 supplemented with 0.5mg/mL puromycin. Worms were pulse labeled for 4 hrs at 20°C. After labeling, worms were harvested by washing off the plates using M9 and rinsing 3 times with M9 to remove bacteria and the pellets were flash frozen in liquid N2.
The frozen pellet was then sonicated into lysis buffer (50mM Tris PH 8.0,150mM NaCl, 1%NP-40, 0.2%SDS, 0.5% deoxycholate and 1x Halt Protease inhibitor). Sonication was performed using Branson SFX150 Cell disruptor (Branson Ultrasonics) with 5 times 6 sec pulse at 50% Amp. Lyates were centrifuged at 10,000xg for 1min and then protein concentration of the supernatant was determined using BCA assay (Thermo Scientific 23227). Immunoprecipitation was performed at 4°C for 4 hours. Lysate concentration was at 1.5μg/μL with 750μg total protein for each condition. Each sample was adjusted to the same volume and protein concentration prior to immunoprecipitation. Puromycin antibody was used at 5ug per sample. Pierce MS-Compatible Magnetic IP Kit (Protein A/G) (Thermo Scientific, 90409) was used for pulling down puromycin antibody labeled proteins and subsequent elution step. For each sample, 30μL of beads was incubated with lysate containing antibody bound protein at room temperature for 1 hr followed by washing 3 times with wash buffer (50mM Tris PH 8.0, 150mM NaCl) and eluting with 50μL elution buffer provided by the kit.
Samples from immunoprecipitation were resuspended in 0.1-M ammonium bicarbonate (ABC), 0.1% sodium deoxycholate, reduced and alkylated using 5 mM TCEP20 mM chloroacetamide at 70°C for 15 min in the dark. Samples were, then trypsinized with 0.25 μg of sequencing grade modified trypsin (Promega) at 42°C for 4 hours. Sodium deoxylcholate was removed by phase-transfer toethylacetate. Tryptic peptides were desalted using in-house StageTips with 3M Empore SDB-RPS membrane, and dried using vacuum centrifugation. The peptides were reconstituted in 15 μL of Buffer A (0.1% formic acid in water), of which 5 μL was subjected to LC-MS/MS analysis.
Tryptic peptides were resolved using a Waters nanoACQUITY UPLC system in a single pump trap mode. The peptides were loaded onto a nanoACQUITY 2G-V/MTrap 5μm Symmetry C18 column (180 μm × 20 mm) with 99.5% Buffer A and 0.5% Buffer B (0.1% formic acid in acetonitrile) at 15 μL/min for 3 min. The trapped peptides were eluted and resolved on a BEH C18 column (130 Å, 1.7 μm × 75 μm × 250 mm) using gradients of 3 to 5% buffer B (0-3 min) and 8 to 28% buffer B (3-185 min) at 0.3 μL/min. MS/MS was performed on a LTQ Orbitrap Velos mass spectrometer, scanning precursor ions between 300 and 1800 m/z (1 × 106 ions, 60,000 resolution) and selecting the 20 most intense ions for MS/MS with 180s dynamic exclusion, 10 ppm exclusion width, repeat count = 1, and 30 s repeat duration. Ions with unassigned charge state and MH+1 were excluded from the MS/MS. Maximal ion injection times were 500 ms for FT (one microscan) and 250 ms for LTQ, and the AGC was 1 × 104. The normalized collision energy was 35% with activation Q 0.25 for 10 ms.
MaxQuant/Andromeda (version 1.5.2.8) was used to process raw files from LTQ-orbitrap, and search the peak lists against protein databases consisting of the UniProt C.elegans proteome (total 27,124 entries, downloaded at 7/20/2018). The search allowed trypsin specificity with maximum two missed-cleavages, and carbamidomethyl modifications of cysteines were set as fixed whereas protein N-terminal acetylation and oxidation of methionine were set as variable. MaxQuant used 4.5 ppm main search tolerance for precursor ions, 0.5 Da MS/MS match tolerance, searching top 8 peaks per 100 Da. False discovery rates for both protein and peptide were 0.01 with minimum seven amino acid peptide length. A label-free quantification was enabled with minimum 2 LFQ ratio counts and a fast LFQ option.
In Vitro Caspase Cleavage Assay
CED-3 cleavage reactions were performed using previously established conditions (Lee and Xue, 2014; Weaver et al., 2014, 2017) with the following modifications. In brief, 35S-labeled substrates were synthesized fresh before cleavage reactions using coupled transcription-translation system (Promega). Point mutations were generated using Q5 mutagenesis kit (NEB) and sequence verified. Substrates were freshly synthesized using Easy Tag 35S (PerkinElmer) for 1 hour. 10 μL cleavage reactions were set up with 1 μL of resulting substrate, 0.2 μL of purified CED-3, 25 mM Tris-HCl pH8.0, 0.25 mM EDTA, 0.25 mM sucrose, and 2.5% glycerol for 2 to 4 hours at 30°C and stopped by adding 3 volumes of 2x Laemmli sample buffer and heating to 85°C. Samples were resolved by SDS-PAGE on 4-16% gradient gels and dried before imaging.
CRISPR Mutagenesis
We generated the pmk-1(D327E) mutation in the endogenous pmk-1 locus using established reagents for CRISPR/Cas9 mutagenesis in C. elegans (Dickinson et al., 2013; Paix et al., 2014). Briefly, an sgRNA with the gatttgaatgatgatgtaa spacer was generated from pDD162 as prescribed (Dickinson et al., 2013). The resulting sgRNA vector (pDD162_C768, Key Resources Table) was combined with a rescue oligo bearing the D327E mutation and synonymous mutations (capitalized) for the spacer sequence (ctatggagcatgaa-tatctggctgcttatcacgatgaaactgaGgagccGattgcagaagaaatggatttgaatgatgaCgtCCgTgcagatacaattgatgaatggaagaaaattatttggg). Resulting mutants were sequence-confirmed and outcrossed two times before analyses.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GFP (clone JL-8) antibody | Clontech | Cat# 632380; RRID: AB_10013427 |
| Rabbit polyclonal anti-Actin antibody | Sigma-Aldrich | Cat# A2066; RRID: AB_476693 |
| Anti-Puromycin Antibody, clone 12D10 | Millipore | Cat# MABE343 RRID: AB_2566826 |
| Anti mouse IgG, HRP linked Antibody | Cell Signaling Technology | Cat#7076S RRID: AB_330924 |
| Anti rabbit IgG, HRP linked Antibody | Cell Signaling Technology | Cat#7074S RRID: AB_2099233 |
| Bacterial and Virus Strains | ||
| C. elegans ORF RNAi library Collection | Dharmacon | Car# RCE1181 |
| OP50 E coli Strain | Caenorhabditis Genetics Center | OP50 |
| Drechmeria coniospora | J. Ewbank Lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| C. elegans CED-3 | Weaver et al., 2014 | N/A |
| TRIzol® Reagent | Thermo Fisher Scientific | Cat#15596026 |
| EasyTag™ L-[35S]-Methionine | PerkinElmer | Cat# NEG709A500UC |
| Critical Commercial Assays | ||
| ABsolute Blue qPCR SYBR Green Low ROX Mix | Thermo Fisher Scientific | Cat# AB-4322/B |
| TruSeq Ribo Profile (Mammalian) Library Prep Kit | Illumina | Cat# RPHMR12126 |
| Ribo-Zero Gold rRNA Removal Kit (H/M/R) | Illumina | Cat# MRZG12324 |
| Pierce MS-Compatible Magnetic IP Kit (Protein A/G) | Thermo Scientific | Cat# 90409 |
| SuperScript™ III Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18080044 |
| TnT® Quick Coupled Transcription/Translation System | Promega | Cat# L1170 |
| Deposited Data | ||
| RNA-seq and ribosome profiling data | GEO Database | GSE145983 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Strain MT1522:ced-3(n717) | Caenorhabditis Genetics Center | WB Strain: MT1522 |
| C. elegans: Strain KU25:pmk-1(km25) | Caenorhabditis Genetics Center | WB Strain: KU25 |
| C. elegans: Strain MT3002:ced-3(n1286) | Caenorhabditis Genetics Center | WB Strain: MT3002 |
| C. elegans: Strain RB2071:ced-3(ok2734) | Caenorhabditis Genetics Center | WB Strain: RB2071 |
| C. elegans: Strain MT2547:ced-4(n1162) | Caenorhabditis Genetics Center | WB Strain: MT2547 |
| C. elegans: Strain MT6347:ced-3(n2433) | Caenorhabditis Genetics Center | WB Strain: MT6347 |
| C. elegans: Strain KU4:sek-1(km4) | Caenorhabditis Genetics Center | WB Strain: KU4 |
| C. elegans: Strain AU1:sek-1(ag1) | Caenorhabditis Genetics Center | WB Strain: AU1 |
| C. elegans: Strain AU3:nsy-1(ag3) | Caenorhabditis Genetics Center | WB Strain: AU3 |
| C. elegans: Strain MH5935:pmk-1(km25) ced-3(n717) | This Study | MH5935 |
| C. elegans: Strain MH5936:ced-3(n717);frIs7[nlp-29p::gfp] | This Study | MH5936 |
| C. elegans: Strain IG274 frIs7 [nlp-29p::GFP + col-12p::DsRed] | Caenorhabditis Genetics Center | WB Strain: IG274 |
| C. elegans: Strain BPW47 ced-3(n717);vpIs1 [lin-15(+) + elt-3::GFP] | This Study | BPW47 |
| C. elegans: Strain JG5 vpIs1 [lin-15(+) + elt-3::GFP] | Caenorhabditis Genetics Center | WB Strain: JG5 |
| C. elegans: Strain BPW24 pmk-1(miy7 [pmk-1(D327E)]) frIs7 [nlp-29p::GFP + col-12p::DsRed] IV | This Study | BPW24 |
| C. elegans: Strain:BPW45 ced-1(e1735);pmk-1(miy7[pmk-1 (D327E)]) | This Study | BPW45 |
| C. elegans: Strain: BPW47 ced-1(e1735) | This Study | BPW47 |
| C. elegans: Strain: MT5811 ced-1(e1735) I; ced-3(n717) IV | Caenorhabditis Genetics Center | WB Strain: MT5811 |
| C. elegans: Strain:BPW3 ced-3(n2427) IV; nIs106 [lin-11::GFP + lin-15(+)] X | This Study | BPW3 |
| C. elegans: Strain:BPW49 nIs106 [lin-11::GFP + lin-15(+)] X | This Study | BPW49 |
| C. elegans: Strain: BPW50 ced-3(n717)IV; nIs106 [lin-11::GFP + lin-15(+)] X | This Study | BPW50 |
| C. elegans: Strain: BPW51 pmk-1(miy7 [pmk-1 (D327E)])IV; nIs106 [lin-11::GFP + lin-15(+)]X | This Study | BPW51 |
| C. elegans: Strain: SJ4005 zcIs4 [hsp-4::GFP] V. | Caenorhabditis Genetics Center | WB Strain: SJ4005 |
| C. elegans: Strain: AU78 agIs219 [T24B8.5p::GFP::unc-54-3′ UTR + ttx-3p::GFP::unc-54-3′ UTR] III | Caenorhabditis Genetics Center | WB Strain: AU78 |
| C. elegans: Strain: BPW18 ced-3(n2433);frIs7 [nlp-29p::GFP + col-12p::DsRed] IV | This Study | BPW18 |
| Oligonucleotides | ||
| Primers 1-12 | This Study | See Table S3 |
| Recombinant DNA | ||
| pDD162_C743 | This Study | dpy-10 sgRNA |
| pDD162_C768 | This Study | pmk-1 sgRNA |
| pWE592 | This Study | pTNT_wt pmk-1 |
| pWE593 | This Study | pTNT_pmk-1 D99A |
| pWE594 | This Study | pTNT_pmk-1 D112A |
| pWE595 | This Study | pTNT_pmk-1 D241A |
| pWE596 | This Study | pTNT_pmk-1 D327A |
| pWE597 | This Study | pTNT_pmk-1 D335A |
| pWE598 | This Study | pTNT_pmk-1 D338A |
| pWE599 | This Study | pTNT_pmk-1 D346A |
| pWE601 | This Study | pTNT_pmk-1 D327E |
| Software and Algorithms | ||
| Fiji (ImageJ) | http://fiji.sc | Ver 1.50i; RRID: SCR_002285 |
| Graphpad Prism | http://www.graphpad.com/ | Ver 6.07; RRID: SCR_002798 |
| WormBase | RRID: SCR_003098 | |
| OMIM | http://www.omim.org | RRID: SCR_006437 |
| UniProt | http://www.uniprot.org/ | RRID: SCR_002380 |
| SeqPurge | https://github.com/imgag/ngs-bits | N/A |
| Bowtie 2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | RRID: SCR_005476 |
| STAR | https://github.com/alexdobin/STAR | RRID: SCR_015899 |
| HTSeq | https://htseq.readthedocs.io/en/release_0.10.0/ | RRID: SCR_005514 |
| edgeR | http://bioconductor.org/packages/release/bioc/html/edgeR.html | RRID: SCR_012802 |
| WormBase Enrichment Analysis |
https://wormbase.org/tools/enrichment/tea/tea.cgi |
N/A |
Cell Death Assays
A previously established assay (Zhou et al., 2001) for ced-3-dependent programmed cell death was used to measure extent of cell death in early first larval stage by counting head corpses visualized in the ced-1(e1735) background with Nomarski optics. In brief, early L1 stage larvae just hatched from tri-fold stage embryos were scored for number of head corpses through focal planes. Midplane images were taken for representative animals. A previously established assay (Reddien et al., 2007) for ced-3-dependent programmed cell death was used to measure the extent of cell death in late larval stage. Briefly, first day adults were picked using a non-fluorescent microscope and then scored using a fluorescence dissecting scope for number of undead P9-12 ventral nerve cord cells visualized by the lin-11p::GFP reporter.
Drechmeria coniospora Infection and Assay for nlp-29p::gfp Induction
Eggs prepared by the standard bleach method were allowed to hatch in 50 mM NaCl in the absence of food at 25 °C. Synchronized L1 larvae were transferred to RNAi plates, and cultured at 25 °C until the young adult stage (44 hours). Worms were then infected with Drechmeria coniospora and cultured at 25 °C before being analyzed with the COPAS Biosort after 7 or 24 hours.
Hyperosmolarity Assay
Mid-fourth larval stage animals with obvious Christmas stage vulva were placed on high salt NGM agar plates (600 mM NaCl final concentration) without food and scored for survival time. Animals were scored as dead based on the absence of both crawling and pharyngeal pumping.
Drechmeria coniospora Infection and Survival Assay
Infection and survival assays were conducted based on a previously described protocol (Polanowska et al., 2018). Briefly, synchronized L1 larvae were cultured on OP50 plates at 25°C until L4 stage and then infected with D. coniospora for 24 h. Groups of 25 worms were then transferred to wells in 12-well plates (4 wells per condition), and images of each well collected automatically at regular intervals (roughly every 20 minutes). The images were examined, and worms scored as dead when they no longer showed sign of any movement between successive images.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification methods, statistical tests, sample sizes, and p values were reported throughout the study and detailed in figure legends.
DATA AND CODE AVAILABILITY
RNA-seq and ribosome profiling sequence data were deposited in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE145983. All software analyses were done with open source code as indicated in Key Resources Table.
Supplementary Material
Highlights.
PMK-1 (p38 MAPK) has a stress surveillance function that limits development
CED-3 caspase represses PMK-1 by proteolytic cleavage, promoting development
CED-3 and PMK-1 inversely regulate epidermal stress response factors
Protein stability, gene expression, and metabolism are coordinated in development
ACKNOWLEDGMENTS
We thank D. Xue and the CGC (funded by NIH Office of Research Infrastructure Programs [P40OD010440])for materials; D. Xue, W. Wood, A. Sewell, N. Pujol, M. Cobb, Han lab members, and Weaver lab members for helpful discussions; WormBase and OMIM databases for accessible information; and J. Belougne for worm sorting at the French National Functional Genomics platform, supported by the GIS IBiSA and Labex INFORM. The authors wish to thank T. Lee with the Central Analytical and Mass Spectrometry Laboratory at the University of Colorado Boulder for sample preparation and LC-MS/MS analysis of the samples with equipment purchased using funding from a W.M. Keck Foundation grant. The authors also wish to thank A. Scott and K. Hammond with the University of Colorado BioFrontiers Institute Next-Gen Sequencing Core Facility, which performed the Illumina sequencing. This work is supported in part by a Welch Foundation grant I-2022-20190330 (B.P.W.), National Institutes of Health grant R35GM133755-01 (B.P.W.), National Institutes of Health grant 5R01GM047869 (M.H.), the Howard Hughes Medical Institute (M.H.), the CNRS, INSERM, AMU, and the ANR (ANR-16-CE15-0001-01, ANR-11-LABX-0054, and ANR-11-IDEX-0001-02 to J.J.E.). B.P.W. is the Virginia Murchison Linthicum Scholar in Medical Research at UT Southwestern Medical Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2020.03.015.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aeschimann F, Xiong J, Arnold A, Dieterich C, and Großhans H (2015). Transcriptome-wide measurement of ribosomal occupancy by ribosome profiling. Methods 85, 75–89. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeles-Albores D, N Lee RY, Chan J, and Sternberg PW (2016). Tissue enrichment analysis for C. elegans genomics. BMC Bioinformatics 17, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arama E, Agapite J, and Steller H (2003). Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila. Dev. Cell 4, 687–697. [DOI] [PubMed] [Google Scholar]
- Arama E, Bader M, Rieckhof GE, and Steller H (2007). A ubiquitin ligase complex regulates caspase activation during sperm differentiation in Drosophila. PLoS Biol. 5, e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold A, Rahman MM, Lee MC, Muehlhaeusser S, Katic I, Gaidatzis D, Hess D, Scheckel C, Wright JE, Stetak A, et al. (2014). Functional characterization of C. elegans Y-box-binding proteins reveals tissue-specific functions and a critical role in the formation of polysomes. Nucleic Acids Res. 42, 13353–13369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baena-Lopez LA, Arthurton L, Bischoff M, Vincent JP, Alexandre C, and McGregor R (2018). Novel initiatorcaspase reporters uncover previously unknown features of caspase-activating cells. Development 745, dev170811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RAV, and Megeney LA (2017). Evolution of caspase-mediated cell death and differentiation: twins separated at birth. Cell Death Differ. 24, 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block DH, and Shapira M (2015). GATA transcription factors as tissue-specific master regulators for induced responses. Worm 4, e1118607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Deletion Mutant Consortium (2012). Large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 (Bethesda) 2, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheesman HK, Feinbaum RL, Thekkiniath J, Dowen RH, Conery AL, and Pukkila-Worley R (2016). Aberrant activation of p38 MAP kinase-dependent innate immune responses is toxic to Caenorhabditis elegans. G3 (Bethesda) 6, 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt B, and Xue D (2005). Programmed cell death. In WormBook, The C. elegans Research Community, WormBook., ed., pp. 1–13, 10.1895/wormbook.1.32.1. http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- Couillault C, Pujol N, Reboul J, Sabatier L, Guichou JF, Kohara Y, and Ewbank JJ (2004). TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat. Immunol 5, 488–494. [DOI] [PubMed] [Google Scholar]
- Crawford ED, and Wells JA (2011). Caspase substrates and cellular remodeling. Annu. Rev. Biochem 80, 1055–1087. [DOI] [PubMed] [Google Scholar]
- Dick SA, Chang NC, Dumont NA, Bell RA, Putinski C, Kawabe Y, Litchfield DW, Rudnicki MA, and Megeney LA (2015). Caspase 3 cleavage of Pax7 inhibits self-renewal of satellite cells. Proc. Natl. Acad. Sci. USA 112, E5246–E5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, and Goldstein B (2013). Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 10, 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierking K, Polanowska J, Omi S, Engelmann I, Gut M, Lembo F, Ewbank JJ, and Pujol N (2011). Unusual regulation of a STAT protein by an SLC6 family transporter in C. elegans epidermal innate immunity. Cell Host Microbe 0, 425–435. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 20, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis HM, and Horvitz HR (1986). Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829. [DOI] [PubMed] [Google Scholar]
- Fernando P, Kelly JF, Balazsi K, Slack RS, and Megeney LA (2002). Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA 99, 11025–11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, and Ahringer J (2000). Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408, 325–330. [DOI] [PubMed] [Google Scholar]
- Fujita J, Crane AM, Souza MK, Dejosez M, Kyba M, Flavell RA, Thomson JA, and Zwaka TP (2008). Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judy ME, Nakamura A, Huang A, Grant H, McCurdy H, Weiberth KF, Gao F, Coppola G, Kenyon C, and Kao AW (2013). A shift to organismal stress resistance in programmed cell death mutants. PLoS Genet. 9, e1003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Liberati NT, Mizuno T, Inoue H, Hisamoto N, Matsumoto K, and Ausubel FM (2004). Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc. Natl. Acad. Sci. USA 101, 10990–10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HE, Grant AR, Simic MS, Kohnz RA, Nomura DK, Durieux J, Riera CE, Sanchez M, Kapernick E, Wolff S, and Dillin A (2016a). Lipid biosynthesis coordinates a mitochondrial-to-cytosolic stress response. Cell 166, 1539–1552. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Thakur N, Piggott CA, Omi S, Polanowska J, Jin Y, and Pujol N (2016b). Coordinated inhibition of C/EBP by tribbles in multiple tissues is essential for Caenorhabditis elegans development. BMC Biol. 14, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ES, and Xue D (2014). Caspase protocols in Caenorhabditis elegans. Methods Mol. Biol 1133, 101–108. [DOI] [PubMed] [Google Scholar]
- Lee KZ, Kniazeva M, Han M, Pujol N, and Ewbank JJ (2010). The fatty acid synthase fasn-1 acts upstream of WNK and Ste20/GCK-VI kinases to modulate antimicrobial peptide expression in C. elegans epidermis. Virulence 1, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B, Tischler J, and Fraser AG (2006). RNAi screens in Caenorhabditis elegans in a 96-well liquid format and their application to the systematic identification of genetic interactions. Nat. Protoc 1, 1617–1620. [DOI] [PubMed] [Google Scholar]
- Ludewig AH, Gimond C, Judkins JC, Thornton S, Pulido DC, Micikas RJ, Döring F, Antebi A, Braendle C, and Schroeder FC (2017). Larval crowding accelerates C. elegans development and reduces lifespan. PLoS Genet. 13, e1006717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N, Wei H, and Conradt B (2018). Caenorhabditis elegans ced-3 caspase is required for asymmetric divisions that generate cells programmed to die. Genetics 210, 983–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Hisamoto N, Terada T, Kondo T, Adachi M, Nishida E, Kim DH, Ausubel FM, and Matsumoto K (2004). The Caenorhabditis elegans MAPK phosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. EMBO J. 23, 2226–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima YI, and Kuranaga E (2017). Caspase-dependent non-apoptotic processes in development. Cell Death Differ. 24, 1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Wang Y, Smith HE, Lee CY, Calidas D, Lu T, Smith J, Schmidt H, Krause MW, and Seydoux G (2014). Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics 198, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinan-Lucarre B, Gabel CV, Reina CP, Hulme SE, Shevkoplyas SS, Slone RD, Xue J, Qiao Y, Weisberg S, Roodhouse K, et al. (2012). The core apoptotic executioner proteins CED-3 and CED-4 promote initiation of neuronal regeneration in Caenorhabditis elegans. PLoS Biol. 10, e1001331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanowska J, Chen JX, Soulé J, Omi S, Belougne J, Taffoni C, Pujol N, Selbach M, Zugasti O, and Ewbank JJ (2018). Evolutionary plasticity in the innate immune function of Akirin. PLoS Genet. 14, e1007494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol N, Cypowyj S, Ziegler K, Millet A, Astrain A, Goncharov A, Jin Y, Chisholm AD, and Ewbank JJ (2008a). Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr. Biol 18,481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol N, Zugasti O, Wong D, Couillault C, Kurz CL, Schulenburg H, and Ewbank JJ (2008b). Anti-fungal innate immunity in C. elegans isenhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 4, e1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukkila-Worley R, Feinbaum R, Kirienko NV, Larkins-Ford J, Conery AL, and Ausubel FM (2012). Stimulation of host immune defenses by a small molecule protects C. elegans from bacterial infection. PLoS Genet. 8, e1002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW, Andersen EC, Huang MC, and Horvitz HR (2007). DPL-1 DP, LIN-35 Rb and EFL-1 E2F act with the MCD-1 zinc-finger protein to promote programmed cell death in Caenorhabditis elegans. Genetics 175, 1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CE, Kooistra T, and Kim DH (2010). An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature 463, 1092–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlfing AK, Miteva Y, Hannenhalli S, and Lamitina T (2010). Genetic and physiological activation of osmosensitive gene expression mimics transcriptional signatures of pathogen infection in C. elegans. PLoS One 5, e9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarov M, Schneider S, Pozniakovski A, Roguev A, Ernst S, Zhang Y, Hyman AA, and Stewart AF (2006). A recombineering pipeline for functional genomics applied to Caenorhabditis elegans. Nat. Methods 3, 839–844. [DOI] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, and Pierre P (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277. [DOI] [PubMed] [Google Scholar]
- Shaham S, Reddien PW, Davies B, and Horvitz HR (1999). Mutational analysis of the Caenorhabditis elegans cell-death gene ced-3. Genetics 753, 1655–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivers RP, Kooistra T, Chu SW, Pagano DJ, and Kim DH (2009). Tissue-specific activities of an immune signaling module regulate physiological responses to pathogenic and nutritional bacteria in C. elegans. Cell Host Microbe 6, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon A, Bandhakavi S, Jabbar S, Shah R, Beitel GJ, and Morimoto RI (2004). Caenorhabditis elegans OSR-1 regulates behavioral and physiological responses to hyperosmotic environments. Genetics 767, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm M, Schroeder C, and Bauer P (2016). SeqPurge: highly-sensitive adapter trimming for paired-end NGS data. BMC Bioinformatics 77, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HL, Tang HM, Fung MC, and Hardwick JM (2015). In vivo CaspaseTracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci. Rep 5, 9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C.A.t., Cormier KW, Keenan SE, Earnest S, Stippec S, Wichaidit C, Juang YC, Wang J, Shvartsman SY, Goldsmith EJ, and Cobb MH (2019). Functional divergence caused by mutations in an energetic hotspot in ERK2. Proc. Natl. Acad. Sci. USA 776, 15514–15523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, DL C, Court DL, and Fire A (2001). Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263, 103–112. [DOI] [PubMed] [Google Scholar]
- Urano F, Calfon M, Yoneda T, Yun C, Kiraly M, Clark SG, and Ron D (2002). A survival pathway for Caenorhabditis elegans with a blocked unfolded protein response. J. Cell Biol 758, 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BP, Weaver YM, Mitani S, and Han M (2017). Coupled caspase and N-end rule ligase activities allow recognition and degradation of pluripotency factor LIN-28 during non-apoptotic development. Dev. Cell 47, 665–673. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BP, Zabinsky R, Weaver YM, Lee ES, Xue D, and Han M (2014). CED-3 caspase acts with miRNAs to regulate non-apoptotic gene expression dynamics for robust development in C. elegans. eLife 3, e04265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C, Yang W, and Hekimi S (2014). The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 757, 897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, and Horvitz HR (1993). The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 75, 641–652. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hartwieg E, and Horvitz HR (2001). CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 704, 43–56. [DOI] [PubMed] [Google Scholar]
- Zugasti O, Thakur N, Belougne J, Squiban B, Kurz CL, Soulé J, Omi S, Tichit L, Pujol N, and Ewbank JJ (2016). A quantitative genome-wide RNAi screen in C. elegans for antifungal innate immunity genes. BMC Biol. 74, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and ribosome profiling sequence data were deposited in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE145983. All software analyses were done with open source code as indicated in Key Resources Table.