

Abstract
The host macrophage response to implants has shown to be affected by tissue location and physio-pathological conditions of the patient. Success in immunomodulatory strategies is thus predicated on the proper understanding of the macrophage populations participating on each one of these contexts. The present study uses an in vivo implantation model to analyze how immunomodulation via an IL-4 eluting implant affects distinct macrophage populations at the tissue-implant interface and how this may affect downstream regenerative processes. Populations identified as F4/80+, CD68+ and CD11b+ macrophages at the peri-implant space showed distinct susceptibility to polarize towards an M2-like phenotype under the effects of delivered IL-4. Also, the presence of the coating resulted in a significant reduction in F4/80+ macrophages, while other populations remained unchanged. These results suggests that the F4/80+ macrophage population may be predominant in the early stages of the host response at the surface of these implants, in contrast to CD11b+ macropahge populations which were either fewer in number or located more distant from the implant surface. Gene expression assays showed increased proteolytic activity and diminished matrix deposition as possible mechanisms explaining the decreased fibrotic capsule deposition and improved peri-implant tissue quality shown in previous studies using IL-4 eluting coatings. The pattern of M2-like gene expression promoted by IL-4 was correlated with glycosaminoglycan production within the site of implantation at early stages of the host response, suggesting a significant role in this response. These findings demonstrate that immunomodulatory strategies can be utilized to design and implement targeted delivery for improving biomaterial performance.
Graphical Abstract
Macrophage populations and gene expression of the host response were studied under the effects of IL-4 released from eluting implants.

Introduction
Performance of implanted biomaterials has recently shown to be highly influenced by early interactions with macrophages at the host-biomaterial interface1, 2. Although an oversimplification, macrophages have been described as existing in a spectrum of phenotypes ranging between two extremes - pro-inflammatory (M1) and anti-inflammatory, pro-regulatory (M2). In reality, they are capable of adopting diverse functional phenotypes that regulate a variety of cellular responses that include tissue homeostasis, pathogen removal, cancer and tissue regeneration3–7. While tissue-resident macrophages are acquired during embryogenesis and remain in the native tissue through processes of self-renewal, recruited macrophages originate largely from the bone marrow and transit to tissues via gradients of inflammatory cytokines8–10. The extent and phenotypic diversity of these macrophage populations have also been shown to vary according to tissue type and age11–13.
Many successful biodegradable materials have recently been associated with a more M2-like macrophage response at acute timepoints14–18. The host response to permanent implants, however, is most often predominated by an early and chronic pro-inflammatory (M1) response, which leads to the formation of scar tissue around the implanted material (encapsulation) or tissue degradation as a result19–22. These findings have increased research strategies aiming to control the host response by shifting macrophages from an initially M1 towards an M2-like phenotype during early stages of the host response23–29. In particular, delivery of M2-polarizing cytokines (e.g. IL-4, IL-10) have been shown to reduce wear particle-induced osteolysis23, 30 and mitigate the foreign body reaction25. Another study using sequential delivery of IFN-γ (an M1 cytokine) and IL-426 showed enhanced vascularization and bone healing, further demonstrating that both M1 and M2 macrophage responses are important in remodelling processes.
The ability to design more successful immunomodulatory strategies depends on an in-depth knowledge of host-implant interactions and macrophage phenotypes. Adding complexity to such designs, recent studies have demonstrated that the host response is affected by physiological and pathological changes in the patient. For example, aging has been shown to result in a delayed and dysregulated macrophage response that leads to an unresolved host response in the long term31–37. This phenomenon was observed to also affect the macrophage response at the host-tissue interface. The cells responding to the implanted mesh in vivo were shown to be dysfunctional and anergic to IL-4 mediated M2 polarization while in vitro testing of circulation derived macrophages in aged animals showed that their ability to polarize was largely intact. This led to a design utilizing a contextual delivery strategy including MCP-1 (macrophage chemoattractant protein-1) and IL-4 to first recruit circulating macrophages and then polarize them to anti-inflammatory phenotypes, respectively. This study showed that both MCP-1 and IL-4 were necessary to positively affect the host macrophage response in aged animals, suggesting that local tissue macrophages were anergic to stimulation while newly recruited cells were intact. These results were in contrast to delivery of IL-4 alone, shown to be the most effective regimen in younger animals38.
Biomaterial performance and the host response have also been recently studied in pathological tissues. Compared to healthy animals, increases in inflammatory factors and fibrosis have been observed in diabetes39–41, cancer42–44 and lupus-prone45, 46 animal models. In addition, implantation site has also been shown to distinctly influence the host response. One study using polyether polyurethane sponges implanted in the vaginal cavity showed increased inflammatory response compared to abdominal implants47. All these studies seem to suggest that the origin and profile of macrophages vary from location and physio-pathological conditions of the tissue. This is in agreement with studies showing gradual replacement of tissue-resident macrophages with bone marrow-derived macrophages during aging, and that the circulation derived populations function differently than tissue-resident48 Success in strategies based on modulating the macrophage host response is hence largely predicated on the proper understanding of the populations and profile of macrophages at the tissue-biomaterial interface on each of these contexts.
Even though a positive impact between macrophage polarization and outcomes in tissue engineering and regenerative medicine has been found, the origin of macrophages and its impact upon immunomodulation strategies has not been well described. In the present study, we hypothesized that the delivery of immunomodulatory cytokines exerts distinct phenotypic changes in different macrophage populations. To answer these questions, we have used an animal implantation model to study modulation of phenotype on different macrophage subpopulations using IL-4 as an immunomodulatory cytokine released from a polypropylene mesh, an implant that without IL-4 is widely known to generate a high inflammatory M1 host response49, 50. When this implant is modified with an ultrathin eluting coating, IL-4 has been shown to release in a sustained manner for two weeks, resulting in a shift towards the M2 macrophage phenotype. This period of time is known as the acute phase of the host response, shown to be determinant for the downstream outcomes that include diminished fibrotic capsule deposition and implant integration, 90 days post-implantation25.
In the present work, immunofluorescent labelling was used to determine the degree of polarization of macrophages expressing common macrophage markers (F4/80, CD68, and CD11b). Additional gene expression analyses from granulation tissue harvested from the peri-implant region were performed to elucidate the changes in the host macrophage response promoted by IL-4 at the tissue-biomaterial interface, including representative genes for M1 and M2 macrophage responses, inflammation, tissue remodelling and angiogenesis. This study provides an analysis of how biomaterial immunomodulation affects distinct populations of macrophages within the remodelling zone and how this may affect a broad range of regenerative processes.
Materials and Methods
Materials.
A polypropylene mesh, Gynemesh® PS (Ethicon, Somerville, NJ) was used. Maleic anhydride, dermatan sulfate (chondroitin sulfate B), chitosan (low molecular weight, deacetylation degree 85%) and histologic staining materials were purchased from Sigma Aldrich (St. Louis, MO). Murine IL-4 was purchased from Peprotech (Rocky Hill, NJ). Rabbit anti-mouse arginase (Arg-1), inducible nitric oxide synthase (iNOS), CD11b, CD68, goat anti-mouse Arg-1 and donkey anti-rabbit IgG, anti-rat IgG, anti-mouse IgG, anti-goat IgG Alexa-fluor secondary antibodies were purchased from Abcam (Cambridge, MA). Rat anti-mouse F4/80 was purchased from ABD Serotec (Raleigh, NC). DAPI was purchased from Fisher Scientific (Pittsburgh, PA). RNALater, Qiagen RNEasy Mini Prep Kit, High Capacity RNA-to-cDNA kit, Taqman gene expression primers and Master Mix were purchased from Thermo Fisher (Pittsburgh, PA).
Fabrication of IL-4 eluting mesh implants.
A plasma procedure was performed to provide a negatively charged surface for further coating, as previously reported51. Briefly, polypropylene mesh implants were cleaned with a 1:1 acetone:isopropanol mixture for 10 minutes and then dried. Implants were placed into the plasma chamber at a distance of 8.5 cm from maleic anhydride powder. Plasma was generated using argon gas (30 s, 600W, 35 mL/min and 250 mTorr) using an Ion 40 Gas Plasma System (PVA Tepla America, Inc), then boiled for 20 minutes in milli-Q water. Layer-by-layer coating was performed as previously reported25. Dermatan sulfate (2 mg/mL) was incubated overnight at 4°C with IL-4 (1.5 μg/mL) prior to coating. First, 1 cm2 pieces of mesh implant were dipped in chitosan (2 mg/mL in 0.5% acetic acid) for 10 minutes at RT, then washed three times (10, 20 and 30 seconds) in milli-Q water. Next, implants were dipped in a dermatan sulfate solution (with or without IL-4) for 10 minutes. Implants were washed again three times in milli-Q water. This cycle was repeated to make a 10-bilayer core coating, followed by 40-bilayers containing IL-4. Eluting coated implants were ethylene oxide-sterilized and stored at −20°C until use.
Mouse implantation model.
An implantation model using C57BL/6J female mice, 8 – 10 weeks old was used, following all procedures in accordance with the Guidelines for Care and Use of Laboratory Animals of the University of Pittsburgh and approved by both the Institutional Animal Care and Use Committee (IACUC) of the University of Pittsburgh and the Animal Ethics Committee of the National Institutes of Health, National Research Council of the United States of America. A power analysis was performed to determine that 6 animals per group was required to maintain a statistical power of at least 80%, according to our previous studies25, 31. Briefly, a midline incision was made and a subcutaneous pocket was created in the abdomen of each mouse in order to implant a 1 cm2 piece of pristine, coated (no IL-4) and IL-4 eluting (40 bilayers) polypropylene meshes. PGCL sutures were used to close the incision, then 0.5 mg/kg of Baytril and 0.2 mg/kg of Buprenex were administered for 3 days as antibiotic and analgesic, respectively. Buprenorphine (Buprenex), an opioid analgesic, has been studied and shown not to exert any effects nor alterations in the immunological response, both acutely and chronically administered52, 53. After 3, 7, 14 or 90 days, mice were euthanized and skin/mesh/muscle tissue complexes were harvested. Half of this tissue was incubated overnight at RT in RNALater (Thermo Fisher, Pittsburgh, PA) for RNA extraction, while the other half was fixed for 72 hours in neutral buffered formalin. Healthy and SHAM groups were used as controls. Finally, fixed tissues were paraffin embedded and cross-sections of 7 μm were used for histological studies.
Immunolabeling of histological sections.
Paraffin-embedded tissue cross-sections were deparaffinized and hydrated in a series of xylene/alcohol/water. Incubation with proteinase K (1X) for 10 minutes at 37°C was performed to retrieve antigens. After 3 washes in water, samples were incubated at 37°C in 50 mM of CuSO4 in 10 mM NH4Ac buffer pH = 5, to reduce tissue background fluorescence. Slides were washed twice in TBST (25 mM Tris buffer + 0.1% tween 20). Then, a 5% donkey serum + 2% BSA + 0.1% tween 20 + 0.1% triton X-100 solution was used as blocking agent (2 hours, RT). To immunolabel M2 macrophages, arginase-1 (1:100) with F4/80 (1:50), CD68 (1:150) or CD11b (1:100) primary antibodies were used (overnight at 4°C), followed by anti-rabbit Alexa Fluor 594 (1:200) and anti-rat Alexa Fluor 488 (1:100) secondary antibodies (40 min at RT) in blocking buffer. To immunolabel M1 macrophages, iNOS (1:100) with F4/80 (1:50), CD68 (1:150) or CD11b (1:100) primary antibodies were used (overnight at 4°C), followed by anti-rabbit Alexa Fluor 594 (1:100) and anti-rat Alexa Fluor 488 (1:100) secondary antibodies (40 min at RT) in blocking buffer. A list of macrophage markers indicating population and function is included in Table 1. Vectashield with DAPI mounting media (Vector laboratories, Burlingame, CA) was used to stain nuclei and mount. Images of centered single fibers (3 different single fibers per sample, N = 6 each group) were taken on a Nikon Eclipse E600 microscope equipped with epi-fluorescence at 40X and cell counts within ~ 50 μm from the implant surface were analyzed using ImageJ (version 1.51a, NIH).
Table 1.
Macrophage markers used in the present study, population and function.
| Marker | Macrophage population | Function |
|---|---|---|
| Inducible nitric oxide synthase (iNOS) | Pro-inflammatory (M1) | Nitric oxide production54–56. |
| Arginase-1 (Arg-1) | Regulatory (M2) | Catalyzes the hydrolysis of arginine to ornithine and urea54, 55, 57. |
| F4/80 | Predominantly tissue resident | Macrophage-specific adhesion-G-protein-coupled receptor58, 59. |
| CD68 | Monocyte-derived, circulatory and tissue resident | Glycoprotein, lysosome-associated membrane protein family60–62. |
| CD11b | Monocyte-derived and circulatory | Regulates leukocyte adhesion and migration to mediate the inflammatory response54,63. |
Histological stainings.
Paraffin-embedded tissue cross-sections were used for immunolabelling and Alcian Blue staining. Alcian Blue stained tissue sections were imaged on a Nikon Eclipse E600 microscope (Tokyo, Japan) at 20X. ImageJ (version 1.51a, NIH) equipped with a color deconvolution plug-in (version 1.5) was used to quantify the area of glycosaminoglycan deposition on single mesh fibers at 7, 14 and 90 days (3 different single fibers per sample, N = 6 each group). Quantification of GAGs at 3 days was not included due to absent or aberrant staining due to the presence of platelet aggregation, low cell count and tissue swelling during hemostasis.
Gene expression analysis.
Transient inflammatory tissue from mesh explants was isolated by removing the attached skin and muscle, leaving only the tissue surrounding the implant. RNA extraction was done using Qiagen RNeasy MiniPrep RNA Isolation Columns following standard kit protocol. RNA was quantified using a NanoDrop Spectrophotometer. cDNA templates were created using Invitrogen High Capacity RNA-to-cDNA kits. Taqman Gene Expression Assays (rt-qPCR) were performed for the markers specified on Table 2, using B2M as endogenous control. Gene expression was normalized against the healthy group (no surgery), using the connective tissue between the skin and muscle (fascia).
Table 2.
Gene expression markers used in the present study and role in macrophages and the host response.
| Marker | Gene name | Role in macrophages and the host response |
|---|---|---|
| Arg-1 | Arginase-1 | M2-macrophage marker. Catalyzes the hydrolysis of arginine to ornithine and urea54, 55, 57. |
| IL-10 | Interleukin-10 | Anti-inflammatory cytokine that promotes resolution of inflammation. Highly expressed in M2 macrophages64–66. |
| YM-1 | Chitinase-like 3 | M2-macrophage marker. Contributes to the digestion of glycosaminoglycans67, 68. |
| Fizz-1 | Resistin like alpha | M2-macrophage marker. Highly expressed in M2-mediated pulmonary allergy68–72. |
| iNOS | Inducible nitric oxide synthase | M1-macrophage marker. Synthesizes nitric oxide54–56. |
| IL-6 | Interleukin-6 | Inflammatory cytokine highly expressed in acute and chronic inflammation73, 74. |
| TNF-α | Tumor necrosis factor α | Pro-inflammatory cytokine, produced predominantly by macrophages75, 76. |
| IL-12β | Interleukin-12β | Pro-inflammatory cytokine produced by antigen-presenting cells, including macrophages77, 78. |
| TGF-β1 | Transforming growth factor β1 | Cytokine that controls of growth, proliferation, differentiation and apoptosis of cells. Regulates the expression of genes involved in M2 polarization79, 80. |
| IL-1β | Interleukin-1β | Pro-inflammatory cytokine, signature marker of the host response to implants81–83. |
| MCP-1 | Monocyte chemoattractant protein-1 | Cytokine that regulates migration and infiltration of monocytes and macrophages84. |
| PPARγ | Peroxisome proliferator activated receptor γ | Transcriptional factor that regulates energy storage. Anti-inflammatory mediator with response elements in Arg-1 gene85–89. |
| MMP-9 | Gelatinase-B | Metalloproteinase secreted by monocyte-derived macrophages. Upregulated in M1 macrophages90–92. |
| MMP-10 | Stromelysin 2 | Moderates the inflammatory response of resident and infiltrating macrophages and regulates their tissue remodeling activity by promoting the expression of MMP-1393, |
| MMP-13 | Collagenase 3 | Regulates the resolution of scarring processes. Signature marker of the host response to implants81–83. |
| Col1A1 | Collagen type I alpha 1 | Matrix deposition of collagen type I, shown to be degraded by M2-like macrophages95. |
| CTGF | Connective tissue growth factor | Matricellular protein, major downstream mediator of skin fibrosis96. |
| VEGFα | Vascular endothelial growth factor α | Predominantly secreted by macrophages to induce angiogenesis and vascular development97. |
| PDGFα | Platelet-derived growth factor α | Regulates blood vessel formation and proliferation of cells of mesenchymal origin, including fibroblasts. Expression in monocyte-derived macrophages is regulated by IFN-γ98. |
| B2M | β-2 microglobulin | Most appropriate reference gene for macrophages99. |
Statistical analysis.
Comparisons of means were performed by either one-way or two-way analysis of variance (ANOVA), using at least p < 0.05 as statistical significance criteria followed by Tukey’s test to compare groups and Sidak’s test to compare time points. Shapiro-Wilk was used to test normality. All statistical tests were performed on GraphPad Prism V7 (La Jolla California, USA).
Results and Discussion
Effects of IL-4 and components of the coating in multiple macrophage populations.
To determine the phenotypic changes promoted by IL-4 on different macrophage populations, inducible nitric oxide synthase (iNOS, M1 marker) and arginase-1 (Arg-1, M2 marker) were used in co-labeling with F4/80, CD68 and CD11b. Immunolabeling tissue sections provide more accurate information regarding the localization and distribution of macrophage populations within the peri-implant tissue, compared to flow cytometry characterization. F4/80 is a cell surface glycoprotein and widely used marker for labelling of tissue-resident macrophages in mice, which is highly expressed in macrophages of numerous tissues including connective tissue and skin (Langerhans cells)58, 59, but also shown to be expressed with lower intensity in dendritic cells100. CD68 (Cluster of Differentiation 68) is a protein highly expressed by cells in the monocyte lineage (e.g., monocytic phagocytes, osteoclasts), including both circulating macrophages and tissue-resident macrophages (e.g., Kupffer cells, microglia)60–62. Consequently, this marker has been equivocally used as a general “pan-macrophage” marker, rather than a macrophage subpopulation. CD11b is expressed on the surface of many leukocytes including monocytes, neutrophils, natural killer cells, granulocytes and macrophages, as well as on 8% of spleen cells and 44% of bone marrow cells54, 63. Functionally, CD11b regulates leukocyte adhesion and migration to mediate the inflammatory response, herein considered predominantly as a marker for recruited macrophages63.
Quantification of Arg-1 and iNOS positive cells in the peri-implant tissue at 3, 7 and 14 days post-implantation (Figure 1a) showed higher expression of Arg-1 in the IL-4 eluting implant, consistent with a regulatory M2-like host response, while the pristine implanted group presented higher expression of iNOS, characteristic of an inflammatory M1 host response. Also, both markers are generally more expressed at 7 days post-implantation, while no expression of either Arg-1 and iNOS were seen at 90 days (not shown). All of these expression patterns are consistent with previous studies25, 31, and therefore 7 days was chosen to perform further comparisons between macrophage populations.
Figure 1.
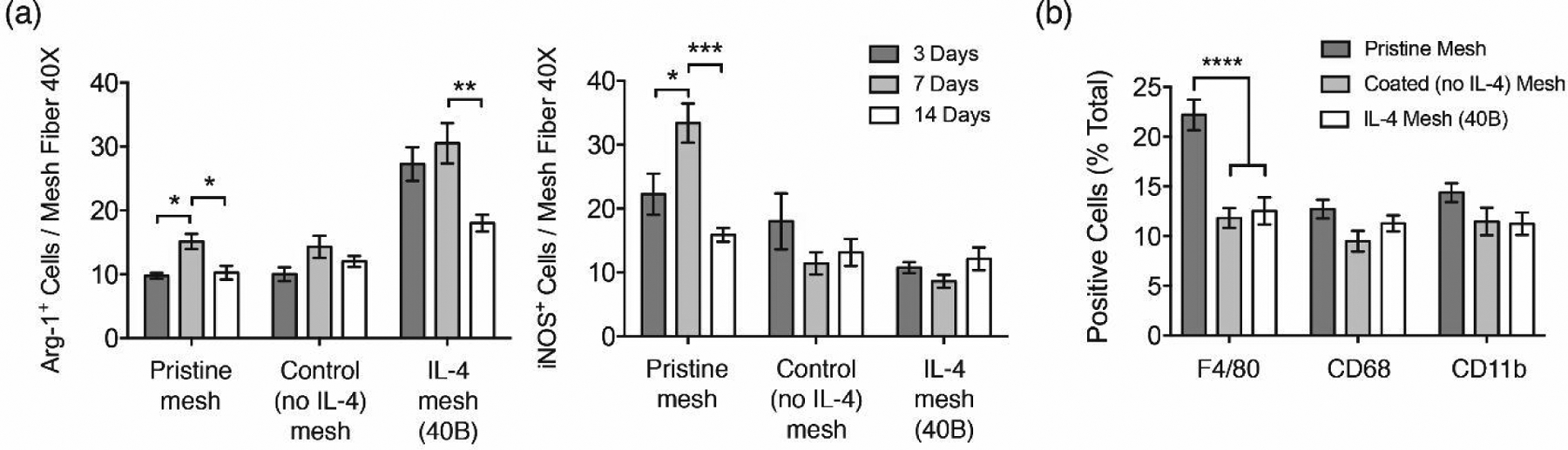
(a) Image analysis quantification of Arg-1+ and iNOS+ cells surrounding single mesh fibers in tissue cross sections of mice implanted with a 1 cm2 of pristine, coated (no IL-4) and IL-4 loaded (40B) mesh for 3, 7 and 14 days. Representative images of these samples can be found in Figures S1 and S2. (b) Image analysis quantification of F4/80+, CD68+ and CD11b+ macrophages as percentages from total cells (DAPI) surrounding single mesh fibers in tissue cross sections of mice implanted with a 1 cm2 piece of pristine, coated (no IL-4) and IL-4 loaded (40B) mesh for 7 days. Bars represent the mean ± SEM (N = 6).Statistical significance as (*) p < 0.05 and (****) p < 0.0001, using two-way ANOVA with Tukey’s test. All other differences are non-significant.
Co-labelling of Arg-1 with F4/80+, CD68+ and CD11b+ macrophage populations was performed seven days post-implantation to identify and quantify M2-like macrophages. Quantification of cells expressing each of these pan-macrophage markers revealed that mice implanted with coated (no IL-4) and IL-4 eluting implants were associated with a reduction in F4/80+ macrophages, compared to the pristine implant group (Figure 1b). There were no differences in the percentage of cells for both CD68 and CD11b in any of the implanted groups. Therefore, these results suggest that the coating may prevent recruitment and/or proliferation of the F4/80+ macrophage population, likely through interactions of the microenvironment with the surface of the coated implant. Alternatively, one or more components of the coating may exert distinct effects on the recruitment of the F4/80+ macrophage population but not the CD68 and CD11b populations. Also, the higher percentage of F4/80+ macrophages in the pristine group seems to suggest that the response to the bare polypropylene biomaterial includes delayed or inhibited recruitment of other macrophage subsets, which may be a consequence of the low degree of vascularization in the subcutaneous tissue.
The percentage of M2-like macrophages on each of these populations was also quantified, revealing that mice implanted with IL-4 eluting mesh presented a consistently higher percentage of Arg-1+ macrophages in all populations, compared to all other groups (Figure 2). The percentage of Arg-1+ macrophages in both coated (no-IL4) mesh and pristine groups was similar, which is consistent with previous studies demonstrating that the observed shift towards an M2 phenotype is due to local delivery of IL-423–27.
Figure 2.
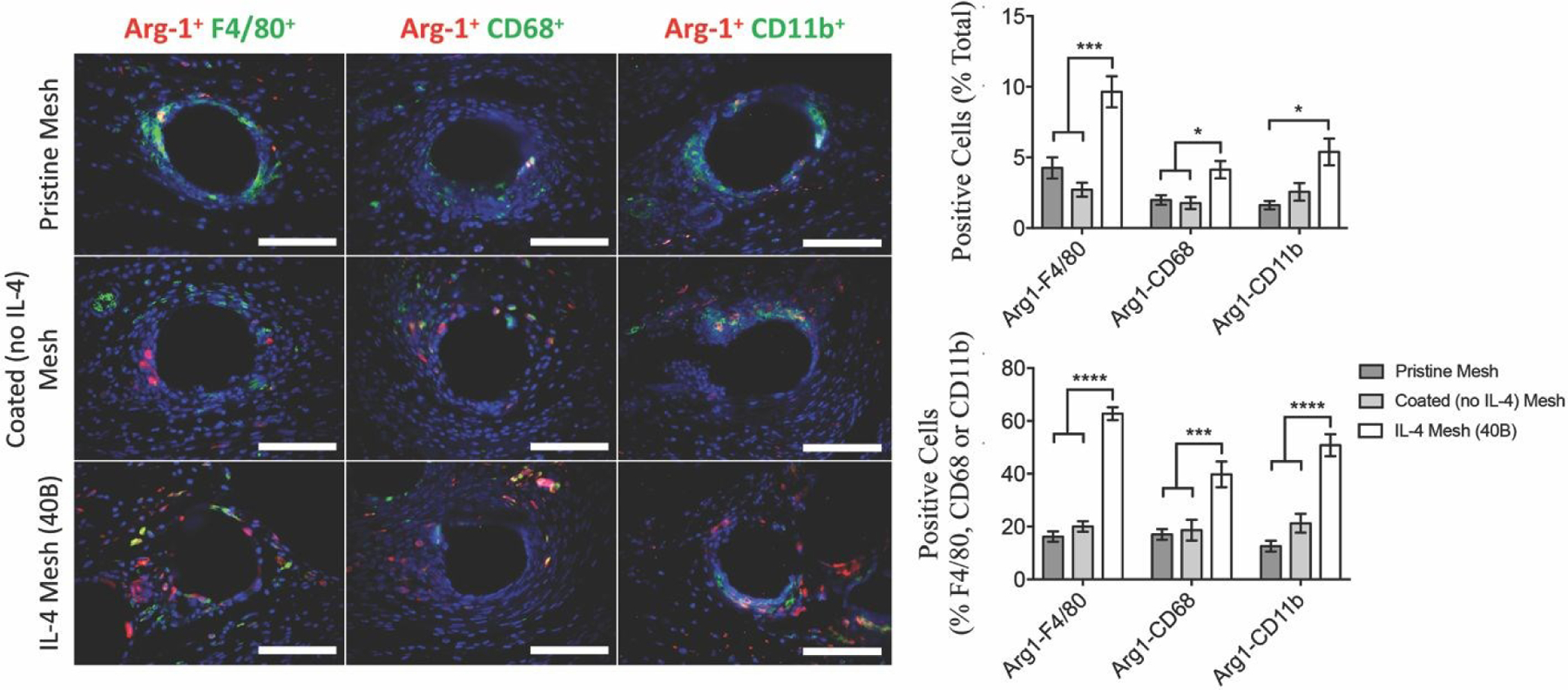
Image analysis of F4/80+, CD68+ and CD11b+ macrophages co-labeled with Arg-1 (M2 macrophage marker), as percentages of total cells (DAPI) surrounding single mesh fibers of tissue cross sections of mice implanted with a 1 cm2 piece of pristine, coated (no IL-4) and IL-4 loaded (40B) mesh for 7 days. Scale bars represent 50 μm. Bars represent the mean ± SEM (N = 6). Statistical significance as (*) p < 0.05 and (****) p < 0.0001, using two-way ANOVA with Tukey’s test. All other differences are non-significant.
Similarly, the percentage of M1-like macrophages was quantified using iNOS co-labelling with F4/80, CD68 or CD11b. Results show that both coated (no IL-4) and IL-4 mesh groups presented diminished percentages of iNOS+ macrophages in the F4/80+ and CD68+ populations, compared to the pristine mesh group (Figure 3). There were no differences among groups in the CD11b population. These results suggest that the presence of the coating prevents the interactions of the polypropylene surface with F4/80+ and CD68+ macrophages as well as polarization to M1 phenotype, while it does not exert effects on CD11b+ macrophages. This seems to be in agreement with previous studies that have shown diminished M1 macrophage response in presence of coated implants38, 101–103.
Figure 3.
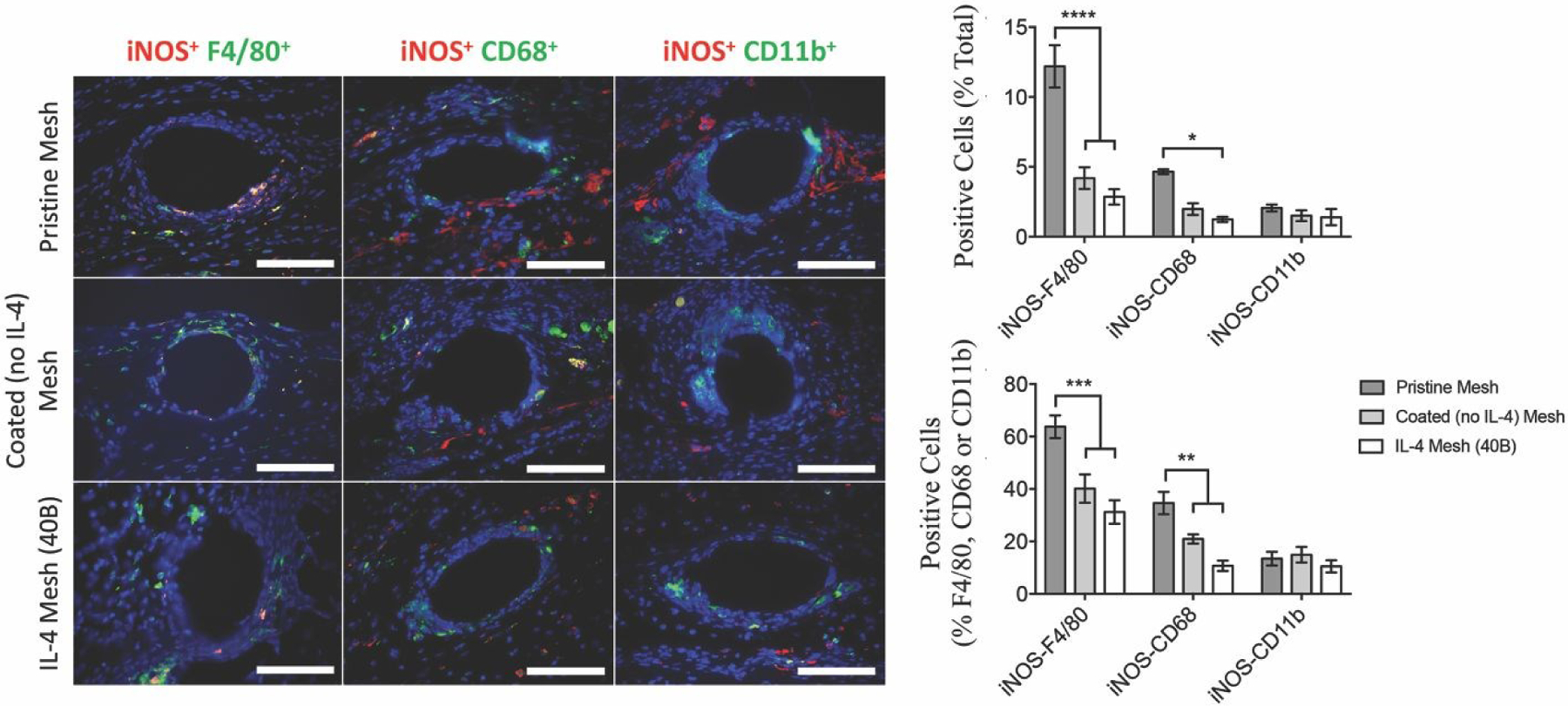
Image analysis of F4/80+, CD68+ and CD11b+ macrophages co-labeled with iNOS (M1 macrophage marker), as percentages of total cells (DAPI) surrounding single mesh fibers of tissue cross-sections of mice implanted with a 1 cm2 piece of pristine, coated (no IL-4) and IL-4 loaded (40B) mesh for 7 days. Scale bars represent 50 μm. Bars represent the mean ± SEM (N = 6). Statistical significance as (*) p < 0.05 and (****) p < 0.0001, using two-way ANOVA with Tukey’s test. All other differences are non-significant.
Gene expression analysis of the host response to IL-4 eluting meshes
Gene expression analyses were done on the peri-implant tissue from explanted IL-4 eluting, coated and pristine polypropylene implants, to assess changes in the early (3, 7 and 14 days) and late (90 days) stages of the host response against biomaterials. SHAM controls were done where the surgery was performed without polypropylene mesh implantation. The study included representative genes for M1 and M2 macrophage responses, inflammation, tissue remodeling and angiogenesis.
Gene expression analysis showed that most genes involving inflammation, M1 or M2 macrophage activation were upregulated in all groups, most likely due to the surgical procedure (Figure 4). Inflammatory gene expression showed prolonged upregulation in all polypropylene groups while gene expression was resolved by 14 days in SHAM controls. As expected, the expression of genes representative of tissue remodeling was higher at later stages (14 and 90 days), suggesting both tissue remodeling and capsule deposition. The significant and persistent upregulation of IL-1b and MMP-13 as indicated by the heat map is in agreement with previous studies that have shown similar responses to alternative non-degradable biomaterials81–83. Taken together, the upregulation of these genes in response to the biomaterials tested here supports the notion that this is a key signature of the host response to non-degradable biomaterials. Of note, both Fizz-1 and PPAR-γ are strongly downregulated at early stages in all groups implanted with polypropylene mesh, even with delivery of IL-4. PPAR-γ has been shown to promote M2 macrophage polarization via PPAR-γ response elements in the promoter region of Arg-185–87. It also has been shown to be a major mediator of the anti-inflammatory effects of porcine acellular bladder matrix scaffolds88. However, not all macrophages respond to PPAR-γ activation in the same way89, in which mechanisms mediating these differences have not been elucidated yet. Similarly, Fizz-1 is shown to be highly upregulated in bone marrow-derived and peritoneal macrophages in response to IL-4 or extracellular matrix scaffolds68–72. Therefore, these results seem to suggest that different macrophage populations, likely of the F4/80+ subset, have distinct susceptibility to IL-4 and hence, a distinct phenotypic profile. Of note, there were no significant changes in the expression of genes tested and associated to angiogenesis, suggesting that the delivered IL-4 does not exert any effects with angiogenesis.
Figure 4.

Taqman gene expression heat map assessing the expression of gene targets at early (3, 7 and 14 days) and late (90 days) stages of the host response against biomaterials from peri-implant tissue isolated from polypropylene explants. Healthy tissue was used to normalize gene expression levels, using B2M as housekeeping gene. Significant differences in upregulated or downregulated genes are shown in Figure 5. Each square represent the mean of the samples in the group (N = 6).
In addition, it can be seen that the gene expression profile of pristine and coated implants is similar, while the profile of the IL-4 eluting implant group is different in numerous genes. Analysis of these differentially expressed genes is shown in Figure 5. Results from the genes tested revealed that mice with IL-4 eluting implants were associated with early-stage upregulation of M2-like macrophage genes such as Arg-1 (3 and 7 days), IL-10 (14 days) and YM-1 (3, 7 and 14 days), compared to both coated (no IL-4) and pristine implants. However, a downregulation in Fizz-1 (3 and 7 days) was observed in both IL-4 and coated (no IL-4) groups. The IL-4 eluting implant group was also associated with downregulation of M1-like macrophage genes such as IL-6 (7 days) and IL-12β (7 days), compared to both coated (no IL-4) and pristine implant groups. It has been widely shown that macrophages exposed to IL-4 have reduced expression of iNOS as a consequence of M2 polarization54. Interestingly, iNOS was highly upregulated in the IL-4 eluting implant group at 3 days only, compared to all other groups. This may be due to compensatory mechanisms on macrophages, or that the effects of IL-4 may trigger higher expression of iNOS in other cells such as T-cells and dendritic cells56. The IL-4 eluting implant group has also shown decreased expression of other genes at early stages of the host response, which include TGF-β1 (3 days), PPARγ (7 days) and PDGFα (3 days). TGF-β1 has been shown to promote collagen synthesis104–106, but this early downregulation is unlikely to be responsible for reduced capsule deposition.
Figure 5.

Significantly upregulated and downregulated genes at early (3, 7 and 14 days) and late (90 days) stages of the host response against biomaterials from peri-implant tissue isolated from polypropylene explants. Healthy tissue was used to normalize gene expression levels, using B2M as housekeeping gene. Bars represent the mean ± SEM (N = 6). Statistical significance as (*) p < 0.05, (**) p < 0.01 and (****) p < 0.0001, using two-way ANOVA with Tukey’s test. All other differences are non-significant.
Even though MMP-10 (Stromelysin 2) is highly expressed at later stages, the IL-4 eluting implant group showed higher expression at 7 days post-implantation when compared to both coated (no-IL4) and pristine implants. At later stages of the host response, MMP-13 (collagenase-3) expression in the IL-4 group was higher than all other groups at 14 and 90 days post-implantation. These proteolytic enzymes have been shown to contribute to the resolution of scarring processes. Specifically, MMP-10 has been shown to suppress the inflammatory response of resident and infiltrating macrophages and regulate their tissue remodelling activity by promoting the expression of MMP-13, mitigating the formation of scarring tissue93, 94. In addition, tissue matrix deposition was evaluated by the expression of both Col1A1 and CTGF (connective tissue growth factor). While Col1A1 was similarly expressed in all groups at 7, 14 and 90 days post-implantation, expression at 3 days in the IL-4 eluting implant group was found to be diminished compared to all other groups. While the expression of CTGF is diminished in both IL-4 eluting and coated (no IL-4) implant groups at 14 days, lower expression is maintained only in the IL-4 eluting implant group, suggesting that the components of the coating may have inhibitory effects on the expression of CTGF, but further inhibited by the presence of IL-4. These changes in proteolytic and matrix deposition activity promoted by the delivery of IL-4 further explain the decrease in fibrotic capsule deposition and improved peri-implant tissue quality observed in previous studies using IL-4 eluting implants to modulate the host macrophage response23–27, 107.
Therefore, gene expression results are consistent with surface marker expression and demonstrate that delivery of IL-4 promotes an M2-like macrophage response, upregulating M2-macrophage genes such as Arg-1, IL-10, and YM-1; increasing MMP activity, but downregulating M1-macrophage genes IL-6 and IL-12β. Downregulation in Fizz-1 and CTGF in both IL-4 eluting and coated (no-IL4) implant groups suggests that downregulation in these genes are mediated by the effects of the components of the coating.
Glycosaminoglycan production and correlation with the macrophage host response against biomaterials.
Glycosaminoglycans (GAGs) are important components of the extracellular matrix which regulate the release and bioactivity of growth factors and cytokines108–110. Therefore, they are capable of regulating the infiltration and response of macrophages in the host response. The relative amount and distribution of GAGs at the peri-implant tissue was detected using image Alcian Blue staining and image analysis. Results show that GAGs are mainly located in the immediate peri-implant tissue, immediately after the first layers of cells interfacing the implant surface. Quantification though image analysis revealed that the production of GAGs is higher at early stages of the host response (7 days), and diminishes gradually as the host response evolves towards later stages (Figure 6). The IL-4 eluting implant group presented a significantly higher deposition of GAGs than both pristine and coated implant groups at 7 and 14 days post-implantation, then similar at 90 days. Quantification of GAGs at 3 days was not included due to absent or aberrant staining due to the presence of platelet aggregation, low cell count and tissue swelling during hemostasis.
Figure 6.

Representative images of Alcian Blue stained peri-implant tissue cross-sections showing single mesh fibers of mice implanted with (a) pristine, (b) coated (no IL-4) and (c) IL-4 eluting implants at 7 days post-implantation. Scale bars represent 100 μm. Image analysis of glycosaminoglycan production (top right) at the peri-implant tissue surrounding single mesh fibers of mice implanted with pristine, coated (no IL-4) and IL-4 eluting implants for 7, 14 and 90 days. Bars represent the mean ± SEM (N = 6). Statistical significance as (*) p < 0.05, using two-way ANOVA with Tukey’s test. All other differences are non-significant.
The pattern of GAG production seems to overlap with the pattern of cell markers and genes expressed at early stages of the host response, especially for those involving M2-macrophage response in the IL-4 eluting implant group. These results suggest that delivery of IL-4 promotes the production of GAGs, directly or indirectly though M2 macrophage polarization, which may aid in the regulation of delivery and bioactivity of other anti-inflammatory cytokines and growth factors. This is in agreement with previous literature describing that GAGs can bind M2-like proteins such as IL-4 and IL-10111–113.
Limitations and future directions.
To evaluate the macrophage host response against non-degradable implants, we performed subcutaneous implantation of polypropylene surgical mesh in a mouse model, focusing our analyses mostly on the tissue-implant interface. A murine model was chosen due to the robustness and reliability of both surgical procedure and host response characterization. However, this model presents limited clinical relevance for two major reasons. First, both innate and adaptive immunity in mice and human present significant discrepancies in surface marker expression, signalling pathways and balance of leukocyte subsets114. Secondly, subcutaneous implantation may present a distinct macrophage response compared to other tissues. For example, surgical mesh implants such as those investigated in the present are used intraabdominally for hernia and transabdominally or transvaginally for pelvic organ prolapse21, 22, 115.
Study of macrophage populations was limited to the use of three common markers - F4/80, CD68 and CD11b. Similarly, the identification of M1 and M2 macrophages was limited to the use of iNOS and Arg-1, respectively. While characterization of multiple markers simultaneously using flow cytometry is possible, immunolabelling of macrophages in tissue cross-sections has the advantage of providing information regarding the localization and distribution of macrophage populations directly adjacent to the implant surface or within the peri-implant tissue. Regardless, flow cytometry analyses using of several markers for additional characterization of macrophages isolated from peri-implant tissue explants will be performed in future studies. Similarly, while previous studies have suggested that the markers presented herein can be used to differentiate tissue-resident versus circulation derived macrophage populations, no lineage tracing experiments have been performed in the present study. Therefore, additional work is needed to specifically link the markers studied here to specific macrophage origins.
Gene expression assays were performed to evaluate more exhaustively the early phenotypic changes promoted by IL-4 and the coating components. However, it must be noted that these assays are performed in samples from tissue surrounding the implant and hence contribution of multiple cell types to the gene expression profile may occur. Further protein studies will be performed in future studies to reveal the downstream activation cascades of these genes, as the expression of some genes may not be necessarily correlated with protein expression.
Cell quantification through image analysis to study the host response and macrophage characterization was performed in the area of single fibers rather than knots, in order to normalize the host response to the same surface area of mesh in our analysis. Surface area normalization in mesh knots is difficult due to the complex variety of shapes, including but not limited to number of fibers, distance between fibers and fused fibers. Recent published studies have demonstrated that density (and hence area) of the mesh is a local driver of the host macrophage response, where increased mesh density is associated with a denser cellular response21, 22. While investigation of the differences in the host response between fibers and knots was beyond the scope of the present study, it will be important to evaluate the effects of the IL-4 eluting implants in knots to illustrate cytokine effects in the presence of high density and complex geometry of the implant.
Conclusions
The polarization study on F4/80+, CD68+ and CD11b+ macrophage subpopulations at the peri-implant tissue demonstrated that each of these populations possess distinct susceptibility to polarize to M2-like phenotypes under the effects of delivered IL-4. These results are consistent with gene expression analyses, showing that delivery of IL-4 promotes an M2-like macrophage response. Increased proteolytic activity and diminished matrix deposition are possible mechanisms explaining the decreased fibrotic capsule deposition and improved peri-implant tissue quality shown in previous studies using IL-4 eluting implants to modulate the host macrophage response. The pattern of M2-like gene expression seems to be correlated with the production of GAGs at early stages of the host response, promoted by the delivery of IL-4, suggesting a major role in this response. However, the phenotypic profile described in this response differs slightly in a few signature genes when compared to the response generated by IL-4 on macrophages derived from the bone marrow. This suggests that the host response against subcutaneous polypropylene implants is predominated by F4/80+ macrophage populations, further supporting our hypothesis regarding distinct susceptibility to cytokines. Therefore, careful consideration to the biological context, including macrophage phenotype, implant location, pathophysiological conditions and type of implanted material is required to derive the best strategy for immunomodulatory biomaterials.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (NIH) grants K12HD043441 (BNB) and R01AG055564 (BNB).
Footnotes
Conflicts of Interest
There are no conflicts of interest to declare.
References
- 1.Brown BN, Sicari BM and Badylak SF, Frontiers in immunology, 2014, 5, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown BN, Ratner BD, Goodman SB, Amar S and Badylak SF, Biomaterials, 2012, 33, 3792–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mosser DM and Edwards JP, Nature reviews. Immunology, 2008, 8, 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon S and Taylor PR, Nature reviews. Immunology, 2005, 5, 953–964. [DOI] [PubMed] [Google Scholar]
- 5.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A and Locati M, Trends Immunol, 2004, 25, 677–686. [DOI] [PubMed] [Google Scholar]
- 6.Wynn TA, Chawla A and Pollard JW, Nature, 2013, 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mills CD, Critical reviews in immunology, 2012, 32, 463–488. [DOI] [PubMed] [Google Scholar]
- 8.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ and Mann DL, Immunity, 2014, 40, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, Pinto AR, Klapproth K, Henri S, Malissen B, Rodewald HR, Rosenthal NA, Bajenoff M, Prinz M, Jung S and Sieweke MH, J Exp Med, 2014, 211, 2151–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ginhoux F and Guilliams M, Immunity, 2016, 44, 439–449. [DOI] [PubMed] [Google Scholar]
- 11.Gordon S and Pluddemann A, BMC Biol, 2017, 15, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon S, Pluddemann A and Martinez Estrada F, Immunol Rev, 2014, 262, 36–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortiz AM, DiNapoli SR and Brenchley JM, J Virol, 2015, 89, 5883–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown BN, Valentin JE, Stewart-Akers AM, McCabe GP and Badylak SF, Biomaterials, 2009, 30, 1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown BN, Londono R, Tottey S, Zhang L, Kukla KA, Wolf MT, Daly KA, Reing JE and Badylak SF, Acta Biomater, 2012, 8, 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sussman EM, Halpin MC, Muster J, Moon RT and Ratner BD, Ann Biomed Eng, 2014, 42, 1508–1516. [DOI] [PubMed] [Google Scholar]
- 17.Guo R, Merkel AR, Sterling JA, Davidson JM and Guelcher SA, Biomaterials, 2015, 73, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, Cao H, Zhao Y, Cheng M, Qin H, Cheng T, Hu Y, Zhang X and Liu X, Sci Rep, 2017, 7, 42707. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Goodman SB, Biomaterials, 2007, 28, 5044–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin TH, Kao S, Sato T, Pajarinen J, Zhang R, Loi F, Goodman SB and Yao Z, J Biomed Mater Res A, 2015, 103, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nolfi AL, Brown BN, Liang R, Palcsey SL, Bonidie MJ, Abramowitch SD and Moalli PA, Am J Obstet Gynecol, 2016, DOI: 10.1016/j.ajog.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown BN, Mani D, Nolfi AL, Liang R, Abramowitch S and Moalli PA, Am J Obstet Gynecol, 2015, DOI: 10.1016/j.ajog.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato T, Pajarinen J, Behn A, Jiang X, Lin TH, Loi F, Yao Z, Egashira K, Yang F and Goodman SB, J Biomed Mater Res A, 2016, DOI: 10.1002/jbm.a.35759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pajarinen J, Tamaki Y, Antonios JK, Lin TH, Sato T, Yao Z, Takagi M, Konttinen YT and Goodman SB, J Biomed Mater Res A, 2015, 103, 1339–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hachim D, LoPresti ST, Yates CC and Brown BN, Biomaterials, 2016, 112, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spiller KL, Nassiri S, Witherel CE, Anfang RR, Ng J, Nakazawa KR, Yu T and Vunjak-Novakovic G, Biomaterials, 2015, 37, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeves AR, Spiller KL, Freytes DO, Vunjak-Novakovic G and Kaplan DL, Biomaterials, 2015, 73, 272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang X, Sato T, Yao Z, Keeney M, Pajarinen J, Lin TH, Loi F, Egashira K, Goodman S and Yang F, Journal of orthopaedic research : official publication of the Orthopaedic Research Society, 2016, 34, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keeney M, Waters H, Barcay K, Jiang X, Yao Z, Pajarinen J, Egashira K, Goodman SB and Yang F, Biomaterials, 2013, 34, 10287–10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodman SB, Gibon E, Pajarinen J, Lin TH, Keeney M, Ren PG, Nich C, Yao Z, Egashira K, Yang F and Konttinen YT, J R Soc Interface, 2014, 11, 20130962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hachim D, Wang N, Lopresti ST, Stahl EC, Umeda YU, Rege RD, Carey ST, Mani D and Brown BN, J Biomed Mater Res A, 2017, 105, 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linehan E and Fitzgerald DC, Eur J Microbiol Immunol (Bp), 2015, 5, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stout RD and Suttles J, Immunol Rev, 2005, 205, 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J and Sambhara S, Aging Cell, 2004, 3, 161–167. [DOI] [PubMed] [Google Scholar]
- 35.Garrido A, Cruces J, Ceprian N, Vara E and de la Fuente M, Int J Mol Sci, 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franceschi C and Campisi J, J Gerontol A Biol Sci Med Sci, 2014, 69 Suppl 1, S4–9. [DOI] [PubMed] [Google Scholar]
- 37.Arnardottir HH, Dalli J, Colas RA, Shinohara M and Serhan CN, J Immunol, 2014, 193, 4235–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hachim D, Iftikhar A, LoPresti ST, Nolfi AL, Ravichandar S, Skillen C and Brown BN, J Control Release, 2019, DOI: 10.1016/j.jconrel.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oviedo-Socarras T, Vasconcelos AC, Barbosa IX, Pereira NB, Campos PP and Andrade SP, Microvasc Res, 2014, 93, 23–29. [DOI] [PubMed] [Google Scholar]
- 40.Boniakowski AE, Kimball AS, Jacobs BN, Kunkel SL and Gallagher KA, J Immunol, 2017, 199, 17–24. [DOI] [PubMed] [Google Scholar]
- 41.Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, Li LO, Becker L, Yuan W, Chait A, Braun KR, Potter-Perigo S, Sanda S, Wight TN, Pennathur S, Serhan CN, Heinecke JW, Coleman RA and Bornfeldt KE, Proc Natl Acad Sci U S A, 2012, 109, E715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostuni R, Kratochvill F, Murray PJ and Natoli G, Trends Immunol, 2015, 36, 229–239. [DOI] [PubMed] [Google Scholar]
- 43.Rhee I, Arch Pharm Res, 2016, 39, 1588–1596. [DOI] [PubMed] [Google Scholar]
- 44.Oliva N, Carcole M, Beckerman M, Seliktar S, Hayward A, Stanley J, Parry NM, Edelman ER and Artzi N, Sci Transl Med, 2015, 7, 272ra211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campos PP, Vasconcelos AC, Ferreira MA and Andrade SP, Histol Histopathol, 2011, 26, 433–442. [DOI] [PubMed] [Google Scholar]
- 46.Labonte AC, Kegerreis B, Geraci NS, Bachali P, Madamanchi S, Robl R, Catalina MD, Lipsky PE and Grammer AC, PLoS One, 2018, 13, e0208132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mendes JB, Campos PP, Ferreira MA, Bakhle YS and Andrade SP, J Biomed Mater Res B Appl Biomater, 2007, 83, 408–415. [DOI] [PubMed] [Google Scholar]
- 48.Roszer T, Cells, 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown BN, Mani D, Nolfi AL, Liang R, Abramowitch SD and Moalli PA, Am J Obstet Gynecol, 2015, 213, 668 e661–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nolfi AL, Brown BN, Liang R, Palcsey SL, Bonidie MJ, Abramowitch SD and Moalli PA, Am J Obstet Gynecol, 2016, 215, 206 e201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hachim D and Brown BN, J Biomed Mater Res A, 2018, 106, 2078–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sacerdote P, Palliat Med, 2006, 20 Suppl 1, s9–15. [PubMed] [Google Scholar]
- 53.Martucci C, Panerai AE and Sacerdote P, Pain, 2004, 110, 385–392. [DOI] [PubMed] [Google Scholar]
- 54.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S and Guerau-de-Arellano M, PLoS One, 2015, 10, e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rath M, Muller I, Kropf P, Closs EI and Munder M, Frontiers in immunology, 2014, 5, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xue Q, Yan Y, Zhang R and Xiong H, Int J Mol Sci, 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raes G, Van den Bergh R, De Baetselier P, Ghassabeh GH, Scotton C, Locati M, Mantovani A and Sozzani S, J Immunol, 2005, 174, 6561; author reply 6561–6562. [DOI] [PubMed] [Google Scholar]
- 58.Gordon S, Lawson L, Rabinowitz S, Crocker PR, Morris L and Perry VH, Curr Top Microbiol Immunol, 1992, 181, 1–37. [DOI] [PubMed] [Google Scholar]
- 59.Lee SH, Starkey PM and Gordon S, J Exp Med, 1985, 161, 475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.da Silva RP and Gordon S, Biochem J, 1999, 338 ( Pt 3), 687–694. [PMC free article] [PubMed] [Google Scholar]
- 61.Holness CL, Da Silva R, Fawcett J, Gordon S and Simmons D, Journal of Biological Chemistry, 1993, 268, 9661–9666. [PubMed] [Google Scholar]
- 62.Smith MJ and Koch GL, J Cell Sci, 1987, 87 ( Pt 1), 113–119. [DOI] [PubMed] [Google Scholar]
- 63.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG and Gordonov S, Nature immunology, 2012, 13, 1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mosser DM and Zhang X, Immunol Rev, 2008, 226, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ip WKE, Hoshi N, Shouval DS, Snapper S and Medzhitov R, Science, 2017, 356, 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iyer SS and Cheng G, Critical reviews in immunology, 2012, 32, 23–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang NC, Hung SI, Hwa KY, Kato I, Chen JE, Liu CH and Chang AC, J Biol Chem, 2001, 276, 17497–17506. [DOI] [PubMed] [Google Scholar]
- 68.Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F and Hassanzadeh Gh G, Journal of leukocyte biology, 2002, 71, 597–602. [PubMed] [Google Scholar]
- 69.Loke P, Nair MG, Parkinson J, Guiliano D, Blaxter M and Allen JE, BMC Immunol, 2002, 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huleihel L, Dziki JL, Bartolacci JG, Rausch T, Scarritt ME, Cramer MC, Vorobyov T, LoPresti ST, Swineheart IT, White LJ, Brown BN and Badylak SF, Seminars in immunology, 2017, 29, 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.LoPresti ST and Brown BN, J Immunol Regen Med, 2018, 1, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dziki JL, Wang DS, Pineda C, Sicari BM, Rausch T and Badylak SF, J Biomed Mater Res A, 2017, 105, 138–147. [DOI] [PubMed] [Google Scholar]
- 73.Gabay C, Arthritis Res Ther, 2006, 8 Suppl 2, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rodriguez A, Meyerson H and Anderson JM, J Biomed Mater Res A, 2009, 89, 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parameswaran N and Patial S, Crit Rev Eukaryot Gene Expr, 2010, 20, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Riches DW, Chan ED and Winston BW, Immunobiology, 1996, 195, 477–490. [DOI] [PubMed] [Google Scholar]
- 77.Liu J, Cao S, Kim S, Chung EY, Homma Y, Guan X, Jimenez V and Ma X, Curr Immunol Rev, 2005, 1, 119–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ma X, Yan W, Zheng H, Du Q, Zhang L, Ban Y, Li N and Wei F, F1000Res, 2015, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gong D, Shi W, Yi SJ, Chen H, Groffen J and Heisterkamp N, BMC Immunol, 2012, 13, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Worthington JJ, Fenton TM, Czajkowska BI, Klementowicz JE and Travis MA, Immunobiology, 2012, 217, 1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luttikhuizen DT, van Amerongen MJ, de Feijter PC, Petersen AH, Harmsen MC and van Luyn MJ, Biomaterials, 2006, 27, 5763–5770. [DOI] [PubMed] [Google Scholar]
- 82.Mesure L, De Visscher G, Vranken I, Lebacq A and Flameng W, PLoS One, 2010, 5, e12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boehler RM, Graham JG and Shea LD, Biotechniques, 2011, 51, 239–240, 242, 244 passim. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deshmane SL, Kremlev S, Amini S and Sawaya BE, J Interferon Cytokine Res, 2009, 29, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW and Chawla A, Nature, 2007, 447, 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yao Q, Liu J, Zhang Z, Li F, Zhang C, Lai B, Xiao L and Wang N, J Biol Chem, 2018, 293, 16572–16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gallardo-Soler A, Gomez-Nieto C, Campo ML, Marathe C, Tontonoz P, Castrillo A and Corraliza I, Mol Endocrinol, 2008, 22, 1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bullers SJ, Baker SC, Ingham E and Southgate J, Tissue Eng Part A, 2014, 20, 2390–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gautier EL, Chow A, Spanbroek R, Marcelin G, Greter M, Jakubzick C, Bogunovic M, Leboeuf M, van Rooijen N, Habenicht AJ, Merad M and Randolph GJ, J Immunol, 2012, 189, 2614–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gong Y, Hart E, Shchurin A and Hoover-Plow J, J Clin Invest, 2008, 118, 3012–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang WC, Sala-Newby GB, Susana A, Johnson JL and Newby AC, PLoS One, 2012, 7, e42507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hanania R, Sun HS, Xu K, Pustylnik S, Jeganathan S and Harrison RE, J Biol Chem, 2012, 287, 8468–8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McMahan RS, Birkland TP, Smigiel KS, Vandivort TC, Rohani MG, Manicone AM, McGuire JK, Gharib SA and Parks WC, J Immunol, 2016, 197, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rohani MG, McMahan RS, Razumova MV, Hertz AL, Cieslewicz M, Pun SH, Regnier M, Wang Y, Birkland TP and Parks WC, J Invest Dermatol, 2015, 135, 2377–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Madsen DH, Leonard D, Masedunskas A, Moyer A, Jurgensen HJ, Peters DE, Amornphimoltham P, Selvaraj A, Yamada SS, Brenner DA, Burgdorf S, Engelholm LH, Behrendt N, Holmbeck K, Weigert R and Bugge TH, J Cell Biol, 2013, 202, 951–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chujo S, Shirasaki F, Kondo-Miyazaki M, Ikawa Y and Takehara K, J Cell Physiol, 2009, 220, 189–195. [DOI] [PubMed] [Google Scholar]
- 97.Ramanathan M, Pinhal-Enfield G, Hao I and Leibovich SJ, Mol Biol Cell, 2007, 18, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morelli PI, Martinsson S, Ostergren-Lunden G, Friden V, Moses J, Bondjers G, Krettek A and Lustig F, Atherosclerosis, 2006, 184, 39–47. [DOI] [PubMed] [Google Scholar]
- 99.Stephens AS, Stephens SR and Morrison NA, BMC Res Notes, 2011, 4, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R and Yona S, Nature reviews. Immunology, 2014, 14, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wolf MT, Dearth CL, Ranallo CA, LoPresti ST, Carey LE, Daly KA, Brown BN and Badylak SF, Biomaterials, 2014, 35, 6838–6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Udpa N, Iyer SR, Rajoria R, Breyer KE, Valentine H, Singh B, McDonough SP, Brown BN, Bonassar LJ and Gao Y, Tissue Eng Part A, 2013, 19, 2713–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goodman SB, Yao Z, Keeney M and Yang F, Biomaterials, 2013, 34, 3174–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Horiguchi M, Ota M and Rifkin DB, J Biochem, 2012, 152, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bujak M and Frangogiannis NG, Cardiovasc Res, 2007, 74, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eickelberg O, Kohler E, Reichenberger F, Bertschin S, Woodtli T, Erne P, Perruchoud AP and Roth M, Am J Physiol, 1999, 276, L814–824. [DOI] [PubMed] [Google Scholar]
- 107.Mokarram N, Merchant A, Mukhatyar V, Patel G and Bellamkonda RV, Biomaterials, 2012, 33, 8793–8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taylor KR and Gallo RL, FASEB J, 2006, 20, 9–22. [DOI] [PubMed] [Google Scholar]
- 109.Schultz GS and Wysocki A, Wound Repair Regen, 2009, 17, 153–162. [DOI] [PubMed] [Google Scholar]
- 110.Schonherr E and Hausser HJ, Dev Immunol, 2000, 7, 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lortat-Jacob H, Garrone P, Banchereau J and Grimaud JA, Cytokine, 1997, 9, 101–105. [DOI] [PubMed] [Google Scholar]
- 112.Kunze G, Kohling S, Vogel A, Rademann J and Huster D, J Biol Chem, 2016, 291, 3100–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang L, Prog Mol Biol Transl Sci, 2010, 93, 1–17. [DOI] [PubMed] [Google Scholar]
- 114.Mestas J and Hughes CC, J Immunol, 2004, 172, 2731–2738. [DOI] [PubMed] [Google Scholar]
- 115.Liang R, Knight K, Abramowitch S and Moalli PA, Curr Opin Obstet Gynecol, 2016, 28, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.