

Abstract:
Antiarrhythmic drugs remain the mainstay therapy for patients with atrial fibrillation (AF). A major disadvantage of the currently available anti-AF agents is the risk of induction of ventricular proarrhythmias. Aiming to reduce this risk, several atrial-specific or -selective ion channel block approaches have been introduced for AF suppression, but only the atrial-selective inhibition of the sodium channel has been demonstrated to be valid in both experimental and clinical studies. Among the other pharmacological anti-AF approaches, “upstream therapy” has been prominent but largely disappointing, and pulmonary delivery of anti-AF drugs seems to be promising. Major contradictions exist in the literature about the electrophysiological mechanisms of AF (ie, reentry or focal?) and the mechanisms by which anti-AF drugs terminate AF, making the search for novel anti-AF approaches largely empirical. Drug-induced termination of AF may or may not be associated with prolongation of the atrial effective refractory period. Anti-AF drug research has been largely based on the “suppress reentry” ideology; however, results of the AF mapping studies increasingly indicate that nonreentrant mechanism(s) plays an important role in the maintenance of AF. Also, the analysis of anti-AF drug-induced electrophysiological alterations during AF, conducted in the current study, leans toward the focal source as the prime mechanism of AF maintenance. More effort should be placed on the investigation of pharmacological suppression of the focal mechanisms.
Key Words: atrial fibrillation, antiarrhythmics, reentry, arrhythmias, sodium channel
INTRODUCTION
Atrial fibrillation (AF) is a major clinical problem, and AF prevalence is predicted to significantly increase in the future. Despite marked progress in the use of catheter ablation in the past 2 decades, anti-AF drug approach remains the first-line therapy in patients with AF. A major disadvantage of the currently available anti-AF agents is the risk of induction of ventricular proarrhythmias. It has been reasoned that drug-induced atrial-specific or -selective prolongation of atrial effective refractory period (ERP) could provide an anti-AF effect with no or little risk of ventricular proarrhythmias. Most of the pharmacological research for suppression of AF during the past 25 years has been focused on the development of atrial-specific or -selective anti-AF agents. Several atrial-specific or -selective targets for AF have been suggested, with the most prominent being the ultrarapid delayed rectifier potassium (IKur), the small-conductance calcium-activated potassium (SK) channel, and sodium channel (INa) currents.1–3 The suggested potassium channels are present exclusively or largely in the atrium (so they are atrial-specific).1,3 The sodium channel is well expressed in both the atrium and ventricle, but the inhibition of INa can produce prominent atrial-selective electrophysiological alterations.2 Another prominent anti-AF investigational approach has been “upstream therapy” (ie, targeting nonelectrical parameters).1 Among the recent novel investigational anti-AF directions, pulmonary delivery of anti-AF agents for rapid cardioversion of AF seems to be rather promising.4,5
Electrophysiological mechanisms of AF generation and the mechanisms by which anti-AF drugs suppress AF are more debatable at the present time than they were 1 to 4 decades ago. Better understanding of these mechanisms may empower the search for superior anti-AF agents. This review is an attempt to analyze the prime investigational anti-AF pharmacological approaches and mechanisms by which drugs terminate AF, with the purpose of identifying valid research approaches and postulating future directions.
IKur INHIBITION FOR AF: NOT EFFECTIVE
The channel carrying IKur is found specifically in the atrium of mammals.3 Based on numerous supporting publications, “IKur inhibition for AF” was the most advanced and believed to be the most promising anti-AF pharmacological approach during the first decade of the 2000s.1 Later, it was recognized that this current is too small (or even absent) in the mammalian atrium,6–8 and inhibition of IKur abbreviates action potential duration (APD) in “healthy” atria and produces no or only a minor APD prolongation in electrically remodeled atrial cells.7,9 Specific inhibition of IKur does not significantly affect the occurrence of AF either in experimental or in clinical settings.7,10 As it turned out, all prominent IKur blockers that are effective against AF (vernakalant, AVE0118, AZD1305, etc.) concomitantly inhibit INa in an atrial-selective manner, and their atrial-selective ERP prolongation and anti-AF effects are largely or exclusively due to inhibition of INa.11–15 IKur blockers cause atrial-specific ERP prolongation exclusively or largely due to induction of postrepolarization refractoriness (PRR), a sodium channel–mediated parameter.11–13,15 Among the numerous investigational IKur blockers, only vernakalant, a prominent atrial-selective INa blocker,12,14,15 has been approved for the acute termination of recent-onset AF in the clinic (in Europe). Vernakalant has been shown to be an effective and safe anti-AF agent.16–18
SK CHANNEL INHIBITION FOR AF: ANOTHER “IKur STORY”?
SK channels are exclusively or largely located in the atrium.3,19,20 It has been suggested that specific inhibition of SK channel produces atrial-specific APD prolongation (and, thus, equivalent ERP prolongation), causing anti-AF effect without adverse consequences in the ventricles.3,8,20 Indeed, a number of agents that block the SK channels (eg, NS8593, UCL1684, N-(pyridin-2-yl)-4-(pyridin-2-yl)thiazol-2-amine [ICA], N-(4-methylpyridin-2-yl)-4-(pyridin-2-yl)thiazol-2-amine [ICAGEN], and AP14145) have been shown to prolong atrial ERP and suppress AF without prolongation of QT in experimental settings.3,8,20–22
Recently, it was demonstrated that NS8593 and UCL1684 cause no effect on atrial and ventricular APD but produce an atrial-selective prolongation of ERP due to induction of PRR, indicating atrial-selective inhibition of the sodium channel.23 Consistent with that, both NS8593 and UCL1684 potently inhibit peak INa in HEK cells at a holding potential −90 mV, and NS8593 blocks peak INa in an atrial-selective manner in canine cardiac myocytes (at holding potential −80 mV but not at −120 mV).23 Both agents effectively prevent the induction of AF due to rate-dependent depression of atrial excitability (ie, due to atrial-selective inhibition of the sodium channel).23
ICA and ICAGEN, prominent SK channel blockers that are effective against AF, also depress sodium channel–mediated parameters in an atrial-selective manner [such as Vmax, conduction velocity (CV)],20,24 indicating that these agents are atrial-selective INa blockers. It is yet to be determined whether a specific inhibition of SK channel may suppress AF.
High concentrations of some SK channel blockers may depolarize atrial resting membrane potential (RMP; by 1–3 mV), indirectly promoting INa inhibition.20,24 Mechanisms of this depolarization of RMP are unlikely to be directly related to SK channel inhibition because depolarization of RMP has not been observed with normal concentrations of SK channel blockers.8,25 At the same time, it is well known that the use of a toxic concentration of INa blockers is associated with a depolarization of RMP.26 Thus, available data indicate that “SK channel inhibition for AF” seems to be a repetition of the “IKur for AF” story described above.
TASK-1 Inhibition for AF
TASK-1, a two-pore domain potassium (K2P) channel, has been suggested as an atrial-specific target for suppression of AF.27 Interestingly, although AVE0118 is a more potent TASK-1 than IKur inhibitor,28 it causes little to no prolongation of atrial APD70-90,9,29 indicating that a potent inhibition of the TASK-1 channel does not significantly prolong atrial ERP. At the same time, AVE0118 is known to produce a significant atrial-specific prolongation of atrial ERP30 due to atrial-selective INa inhibition (causing PRR).29 The potential of TASK-1 channel blockers to inhibit INa should be investigated under physiologically relevant settings, considering our experience with IKur and SK channel blockers (described above).
IK1 Inhibition for AF
Persistent AF consistently augments IK1 that acts to shorten atrial APD and promote AF.31 Multichannel blockers that inhibit IK1 can suppress AF.31 Specific inhibition of IK1 (with PA-6) effectively cardioverts persistent AF in a goat AF model,32 but does not stop chronic, naturally occurring AF in dogs.33 Specific inhibition of IK1 did not promote proarrhythmias in these studies. Further research is needed for evaluation of specific IK1 inhibition for AF suppression.
ATRIAL-SELECTIVE SODIUM CHANNEL BLOCK FOR AF
Atrial-Selective INa Inhibition
The sodium channel is well expressed in the atria and ventricles, and INa inhibition is widely used for suppression of both atrial and ventricular arrhythmias.34 However, as it was first recognized in 2007,2 some INa blockers may cause a significant atrial-selective depression of the sodium channel–mediated parameters that may be useful for safe suppression of AF (Fig. 1).12–15,29,35–39 Among the prominent atrial-selective sodium channel blockers are ranolazine2 and vernakalant.12,14 The principal factors underlying the atrial-selective effects of INa blockers include a more positive RMP, a more negative steady-state inactivation relationship of the sodium channel, and a more gradual phase 3 of the action potential in atrial versus ventricular cells (Fig. 2).2,35,40 The more negative half-inactivation voltage and more positive RMP importantly reduce the fraction of resting sodium channels in atria versus ventricles. Because recovery from sodium channel block occurs predominantly during the resting state of the channel, accumulation of sodium channel blockade is expected to be greater in atria versus ventricles.35 INa blockers possessing rapid versus slow unbinding kinetics tend to be atrial-selective (eg, ranolazine vs. propafenone).37,40,41
FIGURE 1.
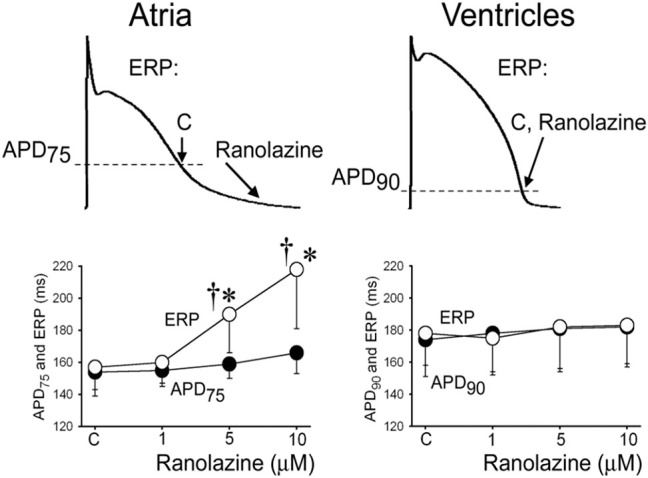
Ranolazine induces atrial-selective prolongation of ERP largely due to PRR. The arrows in upper panel illustrate the position on the action potential corresponding to the end of the ERP in atria and ventricles and the effect of ranolazine to shift the end of the ERP in atria but not ventricles. *P < 0.05 versus control. †P < 0.05 versus (n = 5–18). Data were obtained from canine hearts. CL = 500 ms. C = control. From Burashnikov et al,2 with permission.
FIGURE 2.
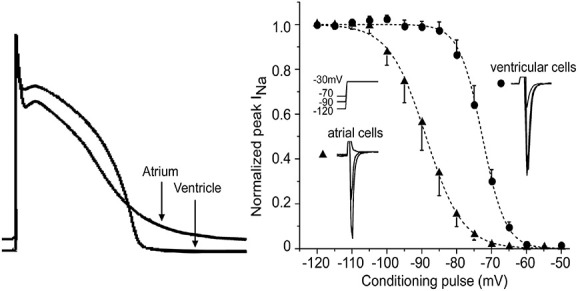
The principal factors underlying the atrial-selective effects of INa blockers are (1) a more positive RMP, (2) a more gradual phase 3 of the action potential in atrial versus ventricular cells resulting in a shorter DI in the atrium, and (3) a more negative steady-state inactivation relationship of the sodium channel. Right panel reproduced from Burashnikov et al,2 with permission.
Concomitant inhibition of potassium channels (particularly IKr) can strongly enhance inhibition of the sodium channel.11,37,38,42 This augmentation is due to the atrial predominant shortening of the diastolic interval (DI) by IKr blockers secondary to atrial predominant prolongation of APD. Specific block of IKr more greatly prolongs the atrial versus ventricular ERP and APD at normal activation rates.43–46 All prominent atrial-selective INa blockers (eg, ranolazine, vernakalant, and AZD1305) inhibit IKr, which directly and/or indirectly contributes to their atrial selectivity and anti-AF efficacy.11
The atrial selectivity of INa blockers has been demonstrated largely in “healthy” atria and ventricles.2,13,35,36,40 Electrical abnormalities altering APD and RMP either in the atrium or in the ventricle may significantly modify the atrial selectivity of INa. Abbreviation of atrial APD, a feature of electrically remodeled atria,47,48 significantly reduces the efficacy of INa blockers,42,49 which may reduce their atrial-selective effect.
Anti-AF Efficacy and Safety of Atrial-Selective INa Blockers
Atrial-selective INa blockers (eg, ranolazine and vernakalant) have been shown to be effective and safe in preventing and terminating new-onset and paroxysmal AF in both experimental and clinical studies.2,35–37,40,50,51 In the experimental settings, anti-AF efficacy of atrial-selective agents is associated with a prominent use-dependent depression of atrial excitability, not permitting the occurrence of rapid atrial activation. This is likely to be the underlying mechanism of prevention and termination of paroxysmal AF by atrial-selective INa blockers in clinical settings.40
Various combinations of drugs may produce a potent synergistic atrial-selective depression of sodium channel–mediated parameters and effectively prevent and terminate AF, with the most prominent being the combination of ranolazine and dronedarone.39,52 The atrial selectivity and anti-AF efficacy and safety of this combination was first demonstrated in canine cardiac preparations.52 Later, the combination of ranolazine and dronedarone has been reported to be reasonably effective against AF, safe and tolerable in patients with paroxysmal AF in the HARMONY clinical trial.39
Ventricular proarrhythmias do not or very rarely occur with the use of atrial-selective INa blockers, and it is believed due to 2 principal factors. First, these drugs have rapid or relatively rapid unbinding kinetics from the sodium channel and, therefore, do not cause severe conduction disturbances in the ventricles, which are strongly associated with ventricular proarrhythmias.11,35,37 The second factor is inhibition of late INa. All atrial-selective INa blockers inhibit late INa (more potently than peak INa)53 that commonly prevents excessive potential prolongation of QT (caused by any reason), largely eliminating the risk of TdP.37,53 In fact, atrial-selective INa blockers can effectively prevent ventricular arrhythmias associated with acute coronary disease, heart failure, and long QT syndromes, an effect largely attributed to their action to block late INa.37,53,54 Also, at rapid ventricular activation rates, atrial-selective INa blockers can potently inhibit peak INa, which may contribute to their antiarrhythmic effect in the ventricle.53,55
The above data on the anti-AF efficacy of atrial-selective INa blockers are related to paroxysmal AF. Atrial-selective INa blockers are poorly effective against persistent AF (Box 1).
BOX 1. Investigational Atrial-Selective Anti-AF Pharmacology.
For the past 30 years, anti-AF pharmacological research has been largely focused on the development of atrial-selective agents, mainly aiming at the reduction of proarrhythmia in ventricles. However, by now, among these approaches, the atrial-selective sodium channel inhibition has been the only experimentally and clinically proven method in terms of efficacy and safety. The other prominent atrial-selective anti-AF approaches (ie, IKur and SK channel inhibition) do not seem to be valid. The available data indicate that anti-AF efficacy and safety of the IKur and SK channel blockers are largely or exclusively due to atrial-selective inhibition of the sodium channel.
IK-ACh INHIBITION FOR AF
The channels underlying the acetylcholine-regulated inward-rectifying potassium current (IK-ACh) and the constitutively active IK-ACh are found exclusively in atria and have been suggested to be an atrial-specific target for AF treatment.56 Although block of IK-ACh may prolong atrial APD and effectively suppress AF in experimental studies,57 anti-AF efficacy of IK-ACh inhibitors in clinical studies has been disappointing.58
UPSTREAM THERAPY FOR AF
A possible limitation of the ion channel block approach for AF treatment is that nonelectrical factors (largely structural remodeling) may contribute to the generation of AF,59 so that interventions reducing/preventing these factors (referred to as “upstream therapies”) may be required for effective AF suppression.60 It is believed that atrial structural remodeling promotes AF by causing conduction disturbances, promoting reentry.60 Generally, although numerous experimental and clinical studies were very promising in 2000–2005, revealing a significant AF reduction with various upstream therapy agents (ie, angiotensin converting enzyme inhibitors [ACEIs], angiotensin receptor blockers [ARBs], statins, and polyunsaturated fatty acids [PUFAs]) in a number of AF pathologies, results from large randomized clinical trials have been quite disappointing.11,61
The contribution of structural remodeling in the development of AF remains poorly understood.59,60,62 The atrium develops structural remodeling to a greater degree than the ventricle in response to pressure and volume overload (the atrium is much thinner and smaller than the ventricle).62 Advanced atrial remodeling is associated with a significant depression of atrial excitability that acts to reduce the capability of atria to maintain AF.62,63 Recently, it was suggested that the burden of AF may be reduced in advanced versus mild/median atrial structural remodeling.62
PULMONARY DELIVERY OF ANTI-AF AGENTS
Pulmonary delivery of anti-AF agents seems to be capable of effectively and safely cardioverting AF.5 This innovative investigational method for acute cardioversion of AF has been recently validated in preclinical studies.4,5,64 Among the advantages of the pulmonary delivery are the potential of rapid increase and decrease of the plasma drug level and atrial-predominant electrophysiological effects.5,64 Both are useful for reduction of ventricular proarrhythmia risk.5 The clinical trial testing this novel approach is underway (“INhalation of Flecainide to Convert Recent Onset SympTomatic AF to siNus rhyThm” [INSTANT], NCT03539302).
ANY OTHER DIRECTIONS?
Few advancements have been made in the development of novel, effective rhythm control pharmacological agents in the past 30 years. The current anti-AF pharmacological research has been largely conceived and developed for suppression of reentrant activity, consistent with the thinking: “AF is largely reentry.”1,34 However, at the present time, the mechanisms of AF maintenance (reentry or focal?) are more debatable than in previous decades, and the mechanisms by which anti-AF drugs suppress AF remain rather speculative (the latter at least in part due to the former). Understanding these mechanisms may empower the search for the development of novel anti-AF treatment strategies.
ELECTROPHYSIOLOGICAL MECHANISMS OF AF: REENTRY OR FOCAL?
For decades, it has been accepted that AF is largely maintained by a reentrant mechanism(s).65–70 There are functional and anatomical reentries, and the former is commonly considered to be the prevailing type of reentry for the maintenance of AF.69,70 Functional reentry types are the “leading circuit” and “spiral wave” (rotor). In the 1970s and 1980s, the “leading circuit” reentry was believed to be the dominant type of reentry responsible for AF.65–67 According to the theory, the “leading circuit” is largely determined by wavelength (WL) and characterized by the absence of the fully excitable gap (EG) and the presence of a refractory core.71 The WL is the product of atrial ERP and CV, estimating the minimal reentrant pathway (the shorter the WL, the greater the probability for reentry).71 In 1990–2000, the “leading circuit” theory was largely replaced with the “spiral wave” as the prime mechanism of rapid arrhythmias.69,70,72 Critical characteristics of the rotor are that the curved wavefront tip meets with wavetail in a single spot (called the phase singularity point), the rotor core is not refractory (it is excitable but not excited), and the EG is present.69,70,72 The spiral wave activity is determined by the sink-source relationship of the inner-curved wavefront tip with the tissue.69,70,72
During the late 20th century, researchers had little doubt that AF was largely maintained by multiple simultaneous reentrant wavelets.65–67,73–75 In the 21st century, with more advanced mapping technologies and increased numbers of groups investigating AF with mapping techniques, multiple simultaneous reentries during AF were not observed,76–85 with rare exceptions.86 Nowadays, although some groups consistently observe relatively stable or unstable rotors during AF,87–89 the other groups either do not record rotors at all or record them rarely and only short-lived.76,77,79,80,82,83,85,90–93 Using simultaneous epicardial and endocardial surface mapping, some researches consistently observed an intramural, anatomically determined reentrant circuit during AF (without any reentry on the surfaces),84 while other groups did not detect reentry at all (intramurally or on the surface).78,81 Importantly, most of those who consistently demonstrated sustained or frequent short-lived rotors during AF used the phase mapping approach for rotor detection87–89 that was recently reported to have a low specificity, usually interpreting conduction blocks as rotors.93 This may explain mutually exclusive mapping AF data, ie, “consistent reentry”87–89 and “no or rare reentry.”76–83,85,90–93
A heterogeneous propagation of activation (ie, fibrillatory conduction) commonly occurs during AF, often producing half- or three-quarter-loops of activations. These loops are often interpreted as evidence for reentry (incomplete reentry).94 Such three-quarter-loops of activations, however, could be either true reentry or would-be-reentry.82,83 A single, rapidly firing source readily produces would-be-reentries.82,83 It seems that many of the reported reentries in reality are would-be-reentries.
The double-layer hypothesis for the maintenance of persistent AF has been recently introduced.78,81 It is based on the experimental and clinical mapping studies demonstrating (1) the presence of a substantial endoepicardial electrical dissociation in the setting of persistent AF (creating electrical double layers), (2) absence of evidence for reentry (on the surface and intramurally), and (3) consistent presence of sporadic wavelets and short-lived focal breakthroughs on the surfaces.77,78,81 It has been postulated that during persistent AF, the electrical double layers constantly “feed” each other with electrical activation, thereby being the prime mechanism of persistent AF maintenance.78,81 However, sustained AF can readily occur without endoepicardial dissociation, indicating that such dissociation is not required for the maintenance of long-lasting AF.
Focal activations during AF are observed by practically all groups (short-lived, sustained, or both), and these activations can be real focal source(s) or breakthrough(s) from any remote sources (reentry or focal).80–85,87,89,95–97 It was long ago postulated that AF can be maintained by focal sources,98,99 and there has been an increasing amount data supporting this hypothesis, particularly during the past 10 years.76,79,80,82,83,90,97,100 Interestingly, although the data from 1960 to 2000 almost universally supported reentry as the only mechanism of AF maintenance,65–67 starting from about the beginning of the 21st century, an increasing number of studies have not or rarely observed reentry77,78,81 and/or have consistently detected focal activations as either predominant or the only mechanism of AF maintenance.76,79,80,82,83,85,90,100 In short, the prime mechanism of AF maintenance is rather debatable in 2020, and as a result (at least partly), the search for novel anti-AF pharmacological drugs/approaches remains largely empirical.
MECHANISMS OF AF TERMINATION BY ANTI-AF DRUGS
The drug-induced alterations in atrial electrophysiological parameters associated with AF cardioversion may shed some light on electrophysiological mechanisms of AF maintenance, which, in turn, may help in developing better anti-AF drugs.
Prolongation of Atrial ERP?
As it has been for decades, the current investigational anti-AF pharmacological approaches are mostly focused on prolonging atrial ERP.1,3,12,15,35,36,38,40,41 This concept has largely stemmed from the doctrine that “AF is reentry,” supported by the fact that prolongation of ERP generally suppresses reentry. However, it should be recognized that a significantly prolonged ERP does not permit the occurrence of any rapid activation, independently of the underlying mechanism of AF (reentry or focal).11,40
In the experimental settings, it seems that any intervention that shortens atrial ERP promotes AF occurrence (supporting the causality between these 2).7,47,48,67,95,101,102 AF itself shortens atrial ERP due to electrical remodeling, and this shortening is likely to be critical for the maintenance and recurrence of AF (“AF begets AF”).47,48 By contrast, the initiation of new-onset AF in the clinic seems to be usually accompanied with prolongation of atrial ERP. In the diseases that are associated with AF (eg, heart failure, hypertension, and valvular heart disease), atrial ERP is commonly prolonged in the absence of current AF in both experimental and clinical studies.62,63,103–105 Moreover, available data indicate that atrial baseline ERP is longer in patients who will develop new-onset AF versus those who will remain in sinus rhythm.106 Prolongation of atrial ERP generally acts to suppress AF initiation.1,11 Perhaps, there are critical pro-AF factors that promote AF in these diseases despite prolongation of atrial ERP (such as intracellular calcium abnormalities).107 Still, even in the setting of prolonged atrial ERP in diseased atria, further prolongation of atrial ERP with pharmacological agents suppresses the induction of AF.86,108
The efficacy of anti-AF drugs to prolong atrial ERP critically depends on APD duration and rate of activation (Fig. 3). When achievable with anti-AF drugs, a significant prolongation of atrial ERP is well associated with an efficient prevention of AF (Fig. 3). In nonelectrically or moderately electrically remodeled atria (“paroxysmal AF”), commonly having normal or moderately altered action potential waveform at sinus rhythm, both IKr and INa blockers significantly prolong atrial ERP at the resting heart rate, and both are effective in preventing AF initiation.1,11 In electrically remodeled atria having short atrial APD (“persistent AF”), the ability of IKr and INa blockers to prolong atrial ERP is reduced,86,109,110 and that is associated with a considerable decrease in the efficacy of these blockers to prevent the recurrence of AF in remodeled atria (Fig. 3).61,111
FIGURE 3.
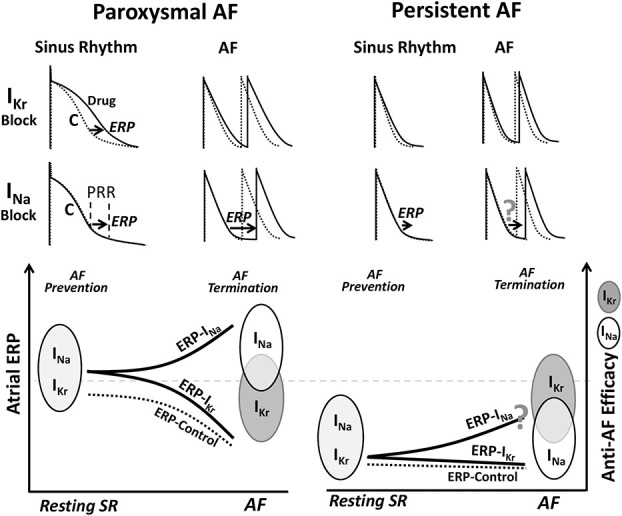
Schematic relations of anti-AF drug-induced ERP prolongation with anti-AF efficacy of the agents. The capability of anti-AF drugs to prolong atrial ERP seems to correlate well with their efficacy to prevent both the paroxysmal and persistent AF and terminate paroxysmal but not persistent AF. IKr blockers prolong ERP due to prolongation of APD and INa blockers due to depression of excitability, causing PRR. C, control. Please see text for details.
The capability of anti-AF drugs to prolong atrial ERP during AF seems to correlate with the efficiency of these drugs to terminate paroxysmal (particularly new-onset AF) but not or less so with persistent AF (Fig. 3). Few data exist on drug-induced ERP alterations during AF.112 In nonremodeled atria, INa blockers cause a significant prolongation of ERP at rapid pacing rates (due to use dependency) that is associated with a high rate of AF termination in experimental studies.2,52,67,113 INa blockers are quite effective in acute cardioversion of paroxysmal AF in the clinic (particularly those lasting <48 hours),17,61 and rate-dependent ERP prolongation seems to account for this high anti-AF efficacy.1,11 Specific IKr inhibitors typically cause little prolongation of atrial ERP at very rapid activation rates (due to reverse use dependency),86,114 which may explain their generally weaker efficiency to cardiovert paroxysmal AF than INa blockers (Fig. 3).16,17,61
Anti-AF drugs are commonly less effective in cardioversion of persistent versus paroxysmal AF,61 and the reason for that is poorly understood. It seems that the capability of anti-AF drugs to prolong atrial ERP is reduced during persistent versus paroxysmal AF (Fig. 3).110,112,115 Still, INa blockers should prolong atrial ERP during AF to a greater degree than that of IKr blockers (Fig. 3; as measured during persistent AF116) due to their use and reverse use dependencies, respectively, and therefore should be more effective in terminating persistent AF. Contrary to this reasoning, IKr blockers (dofetilide and ibutilide) are generally more efficient than INa blockers (flecainide and propafenone) in termination of persistent AF (Fig. 3).61 Acute intravenous dofetilide or ibutilide have been reported to stop persistent AF quite effectively in clinical studies, reaching 44%–88% success rates.117–119 These IKr blockers seem to cardiovert persistent AF with minor or without prolongation of atrial ERP (IKr blockers do not or only mildly prolong atrial APD/ERP at very rapid activation rates).86,112,114 In a goat model of persistent AF, a high dose of d-sotalol acutely terminates persistent AF at 100% without prolongation of atrial ERP and slowing CV.111,112 Thus, prolongation of atrial ERP per se does not seem to be a required condition for termination of persistent AF. Our understanding of mechanisms by which anti-AF drugs cardiovert AF (ie, “ERP prolongation”) and/or AF mechanisms (ie, AF is largely reentry) cannot seem to reasonably explain the termination of persistent AF by anti-AF agents.
Drug-Induced Termination of AF: Widening of EG
The understanding of mechanisms underlying AF termination by anti-AF drugs has undergone substantial changes during the past 3 decades. In 1980–2000, the dominant theory was that both INa and IKr blockers terminated AF by prolonging WL.44,67,71,74,75,113,120 Cardioversion of AF by drug-induced prolongation of WL was explained with termination of reentry due to elimination of the fully EG leading to conduction block, enlargement of the reentry core(s) size, and decrease of the reentrant circuit numbers.44,67,71,74,75,86,113,120 Of note, drug-induced prolongation of WL can realistically occur only due to lengthening of ERP (without CV slowing with IKr blockers and despite it with INa blockers; CV slowing causes WL shortening). So, lengthening of ERP alone might account for the anti-AF actions of antiarrhythmics.
During the first decade of the 2000s, it became increasingly obvious that termination of AF by anti-AF drugs does not correlate well with WL (that can be prolonged, not altered, or shortened), but it is consistently associated with widening of the EG, improvement in AF organization, and prolongation of AF cycle length (AFCL), while ERP may or may not be prolonged (Fig. 4).112,115,121–131 When measured or estimated, a fully excitable temporal EG (the time difference between ERP and AFCL at any given location) is commonly present during sustained AF without drugs (accounting for 10%–40% of AFCL)132 and is significantly prolonged by the drugs.112,123,125 Drug-induced lengthening of the temporal EG (ie, “DI”) during AF provides a longer period for recovery of electrical activity, resulting in less complexity in propagation of excitation during AF (Fig. 4). The spatial EG during AF, estimated by deducting WL from pathlength (which are ERP × CV and AFCL × CV, respectively), is also increased by anti-AF agents (by 30%–190%).112 The spatiotemporal EG is highly difficult to quantify, and it is likely to be heterogeneous during the most types and stages of AF. The consistent drug-induced improvement in AF organization112,115,122–125,127–130 indicates that the “average” spatiotemporal EG is widened by anti-AF agents. In the following analysis, the term “EG” will be used to reflect the spatiotemporal EG. Of note, when schematically illustrating reentry with EG, the latter is commonly not specified.69
FIGURE 4.
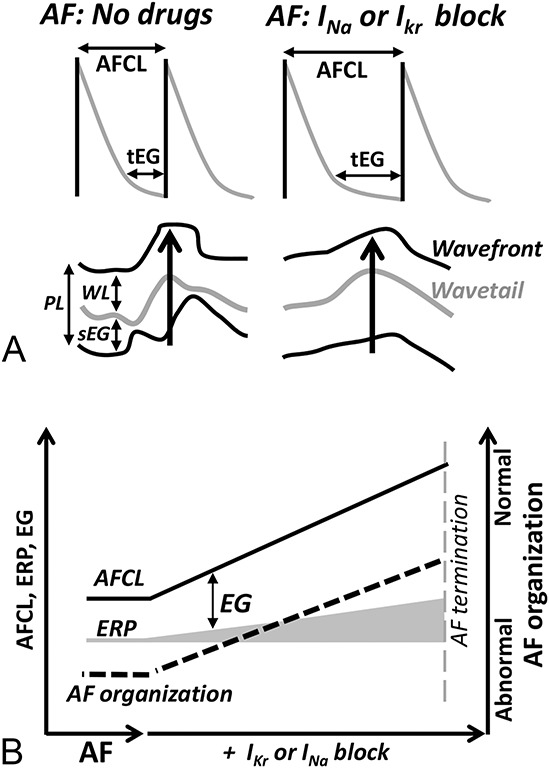
Cardioversion of AF by anti-AF drugs is associated with prolongation of the EG, improvement of AF organization, and lengthening AFCL, while ERP, CV, and WL may or may not be altered. A, Schematic illustrations of drug-induced prolongation of the temporal EG (tEG) and the local “AFCL” (upper panel) as well as lengthening of the spatial EG (sEG) and improvement of AF organization (bottom panel). B, Schematic summary of anti-AF agent-induced changes in the EG, ERP, AFCL, and AF organization. PL, pathlength (AFCL × CV). WL, wavelength (ERP × CV). Please see the text for details.
During AF, CV is not significantly affected by IKr blockers and considerably slowed by INa blockers.112 A paradoxical improvement in AF organization despite CV slowing with INa blockers can be explained with a prominent prolongation of the EG.112,123 Anti-AF drugs consistently induce prolongation of AFCL, but how the degree of this prolongation is related to cardioversion efficacy is not clear. D-sotalol less significantly prolongs AFCL but more effectively cardioverts persistent AF in an experimental study.112
AF MAINTENANCE: REENTRY OR FOCAL? INSIGHT FROM PHARMACOLOGICAL DATA
Thus, termination of AF by anti-AF agents is consistently associated with (1) prolongation of the EG, (2) improvement in AF organization, and (3) lengthening of AFCL (ERP, WL, and CV may or may not be altered; Fig. 4).112,115,121–125,127–130,133 Is this pharmacological pattern more consistent with reentrant or focal source activities as the prime underlying mechanism of AF maintenance?
Among these 3 drug-induced consistently altered parameters, widening of the EG seems to be the most important for the analyses because the EG is directly related to reentrant mechanisms and because the improvement in AF organization is a consequence of the EG widening. Drug-induced prolongation of AFCL seems to be less specific for identification of electrophysiological mechanisms underlying AF. The prime cause of drug-induced prolongation of AFCL is reduction of the activation rate of the AF-driving source. The EG does not regulate AFCL directly. In case of “mother” reentry, AFCL is determined by reentrant circulating time (EG shortening may indirectly slow CV around the reentry core), and in case of a focal source, the EG is largely a consequence of activation rate of the source (Fig. 5, insert).
FIGURE 5.
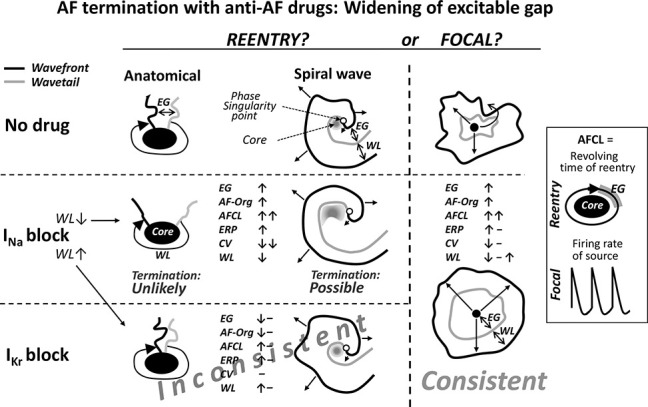
Simplified schematic illustrations of drug-induced alterations of AF-related electrophysiological parameters during AF and the consistency of these alterations with reentrant and focal sources as the underlying mechanisms of AF maintenance. The pattern of the anti-AF agent-induced alterations [ie, prolongation of EG and AFCL, improvement in AF organization (AF-org), etc.] seems to be more consistent with focal source than with reentry as the prime mechanism of AF maintenance. Please see text for details.
Reentry?
Consistent termination of the anatomical reentry with INa blockers associated with a significant widening of the EG around the reentry core cannot seem to be reasonably rationalized, that is, anatomical reentry should become more stable than less stable (Fig. 5). Theoretically, despite widening of the “general” EG, conduction block may occur in some critical part of reentrant anatomical circuit, which may interrupt the circuit. This critical reentrant point, however, should be either rather narrow (“1-dimensional”) or the block should be 3-D spanning from the core to nonexcitable area; otherwise, the conduction should proceed around the block, continuing the circulatory movement. If such reentry termination scenarios occur, they may account for some but unlikely for most of the cases of anatomical reentry termination with widening of the EG.
INa block–induced termination of functional reentry can be associated with prolongation of the EG but only with a critical reservation (Fig. 5). If AF is maintained by functional reentry, INa inhibition can prolong the EG around the reentry core (and in the rest of the atrium, thereby improving AF organization) due to greater slowing of CV than prolongation of ERP (Fig. 5).112 However, this should be accompanied with shortening of WL112,121,126,134 that, according to ample evidence, promotes—not suppresses—functional reentry.44,67,71,74,75,113,120 Yet, several studies reported that INa inhibition could shorten WL but still terminate AF.112,121,126,134 The mechanism underlying INa block–induced termination of AF, despite shortening WL, was studied using mathematical modeling (investigation of the mechanisms in the real cardiac tissue was not possible because recorded spiral waves were short-lived).126,134 Only the rotor(s) was considered as the underlying mechanism of AF. According to the mathematical modeling, INa inhibition could cardiovert AF by (1) enlargement of the spiral wave core leading to destabilization of the prime spiral wave (Figs. 5), (2) decreased probability of anchoring to functional obstacles, and (3) reduction in the number of secondary reentrant wavelets.126,134 No EG was reported in these 2 studies.126,134 However, INa inhibition should increase the EG in the spiral wave (Fig. 5).70
The only mechanism that has been suggested to accommodate prolongation of the EG with INa block–induced cardioversion of rotor is an increased probability of invasion and distraction of the spiral wave core by wandering wavelets.121 This suggestion is based on experimental study in which (1) sustained spiral waves were consistently observed in isolated canine atria in the presence of acetylcholine, (2) INa inhibition with pilsicainide shortened WL and prolonged the EG near the reentry core, and (3) wandering wavelets were able to invade and destroy the spiral core.121 These experimental data need to be verified because the opposite mechanism of reentrant AF termination by pilsicainide was shown in another canine vagally mediated AF model in which pilsicainide prolongs WL and eliminates EG, thereby stopping reentry.135 Also, the core of the spiral wave is not readily accessible to the intruder wavelets because the wavefront curvature is high near the spiral core.69,70,72
No experimental or theoretical data rationally explain the termination of reentrant AF with IKr blockers associated with prolongation of the EG. If AF is maintained by reentry, inhibition of IKr should act to shorten, not prolong, the EG around the reentrant core(s)70 because it acts to lengthen ERP without CV slowing (available data indicate that ERP may or may not be prolonged during AF112,116) (Fig. 5). Thus, if AF is maintained by reentry, specific IKr blockers, acting to reduce the EG, should either not affect or aggravate AF organization, contradicting the improvement of AF organization consistently caused by IKr blockers.112,125,129 These reasonings seem to speak against reentry as the prime underlying mechanism of AF.
The above speculations are based on the limited available data and/or theoretical considerations of pharmacological termination of reentrant AF associated with widening of the EG (Fig. 5). There seem to be other possibilities of such AF cardioversion, considering that the ERP, WL, temporal EG, and CV may vary significantly along the reentrant pathway. When selecting and maneuvering these and other parameters or factors in mathematical modeling, there well may be some explanations of drug-induced termination of reentry with EG widening. Such explanations should rationally validate the termination of reentry (1) by both the INa and IKr blockers, and (2) in most, rather than specific, cases.
When not considering the mechanism of reentry termination in the setting of the EG widening, prolongation of AFCL by INa blockers can be naturally explained with any type of reentry due to slowing of CV and increasing of the core size, leading to lengthening of the circulating time of reentry (ie, “AFCL”). If AF is maintained by reentry, IKr inhibition cannot seem to reasonably account for a significant prolongation of AFCL because IKr inhibition should not substantially affect the revolving time of the reentry (ie, “AFCL”) in the presence of an extended EG (Fig. 5).
One may explain the association of drug-induced AF cardioversion and improvement of AF organization with a reduced appearance of new reentrant wavelets. This seems to be a reverse causality logic because normalization of AF organization induced by anti-AF agents is likely to be a consequence of their direct anti-AF effect (ie, due to slowing the rate of activation of the driving source) rather than the anti-AF mechanism itself. Also, AF does not seem to be often (or ever) caused by continuously appearing and disappearing unstable multiple reentries (as discussed above).76–85,91
Thus, consistent termination of reentry associated with prolongation of the EG cannot seem to be reasonably explained with IKr inhibition and can be explained with INa inhibition but with some critical reservations (eg, there should be shortening not lengthening of WL). If AF is maintained by reentry, a significant prolongation of AFCL itself can be readily explained with INa but not with IKr blockers. Because cardioversion of AF is consistently associated with prolongation of the EG and AFCL as well as improvement of AF organization with both the INa and IKr blockers,112,115,122,123,125,127–129 consistent AF cardioversion should be rationally explained with reentry with both the INa and IKr blockers. That does not seem to be the case (Fig. 5). The alterations of electrophysiological parameters during AF by anti-AF agents do not seem to be consistent with reentry as the prime mechanism of AF maintenance.
Focal?
Mechanistically, anti-AF drug-induced pharmacological alterations in the specific electrophysiological parameters during AF (ie, prolongation of AFCL and EG, and an improvement of AF organization, with or without altering ERP, CV, and WL)112,115,121–125,127–130 are consistent with focal source as the mechanism of AF maintenance (Fig. 5). Spontaneous or drug-induced termination of automaticity and triggered activity is typically associated with prolongation of both the cycle length and DI (ie, EG), with or without prolongation of APD.136,137
There is an important caveat with the “focal source” hypothesis. Although there are data showing that INa blockers can suppress rapid focal activity,138 there are no such data for IKr blockers. According to our current understanding, IKr inhibition should not suppress rapid focal sources. However, our understanding cannot also reasonably explain (1) how reentry can be consistently terminated by IKr blockers without alterations of atrial ERP and CV with a significant widening of the EG112 and (2) why IKr blockers are generally more efficient in termination of persistent AF than INa blockers, despite that the latter more greatly prolong atrial ERP than the former at rapid activation rates (as discussed above).
Thus, as judging from limited available data, mechanistically, the alterations of electrophysiological parameters during AF caused by anti-AF drugs seem to be more consistent with the focal source than with reentry as the prime underlying mechanism of AF maintenance (Fig. 5). This assumption should be considered in conjunction with continuously increasing evidence, indicating that the focal source can be an important or even the prime mechanism of AF maintenance.76,79,80,82,83,85,90,97–99,139
Pharmacological Suppression of Focal AF
It seems that anti-AF pharmacological research should pay more attention to suppression of the rapid focal mechanisms. The cellular and ionic mechanisms of focal sources underlying AF maintenance, as well as mechanisms of pharmacological suppression of the focal sources (beyond “ERP prolongation”), are essentially unknown. A critical problem is that, although at least some AFs are focal, atrial single cells and thin atrial superfused preparations do not seem to be capable of generating rapid sustained activity. That impedes the investigation of ionic and cellular mechanisms of focal AF and, therefore, specific pharmacological inhibition of these focal sources. Spontaneous activity in atrial isolated single cells and thin tissue slices, when it occurs, is much slower than AF (in fact, it is commonly slower than the resting sinus rhythm), nonsustained, and usually artificially induced by rapid pacing in the presence of isoproterenol, high Ca2+, acetylcholine, etc.52,136,140–143 Such focal activity can hardly explain even atrial tachycardia, and the relevance of its pharmacological suppression of AF is unclear. Some rapid repetitive activity in atrial single cells and thin superfused preparations reported in several studies144,145 is not reproducible.52,136,140,143,146
It seems that atrial sustained rapid focal sources exist only in in vivo, in situ, and coronary-perfused cardiac preparations, and thus, pharmacological suppression of focal AF should be largely studied in these conditions (ie, largely empirically). An obstacle is the absence of indisputable and reproducible sustained focal AF models (without resorting to arguments such as “it may be microreentry,” or “reentry is intramural”). Focal AF models should be established or may be recognized among the existing AF models. Some that are believed to be well-proven reentrant AF models65,94 may be, in fact, focal AF models.76,85
Little is known about potential specific targets for pharmacological suppression of focal AF. Abnormalities in intracellular calcium handling are a common finding in the atrial cells isolated from patients with AF, and these abnormalities may be involved in the generation of focal AF.107,142 However, the role of intracellular calcium abnormalities in the generation of sustained AF is disputable. Long-lasting AF may both promote107 and suppress147 focal intracellular calcium-mediated arrhythmogenic mechanisms (the latter by silencing calcium signaling).148 There has been some preclinical research aimed at the development of anti-AF agents normalizing intracellular calcium activity107 but that has not been translated to clinical practice yet.
Among the current anti-AF drugs, specific IKr blockers seem to be the most efficient in cardioverting persistent AF, and this seems to be associated with little or no prolongation of atrial ERP (as discussed above).112 It is important to understand if IKr blockers can suppress rapid focal sources and, if they can, what is the underlying mechanism.
Thus, focal arrhythmogenic mechanism(s) capable of sustaining AF and mechanisms of pharmacological suppression of such source(s) are very poorly defined. Understanding these mechanisms could be helpful for the development of novel anti-AF agents. A significant drug-induced prolongation of atrial ERP is likely to prevent and terminate any rapid arrhythmic mechanisms (or at least slow them down), and this essentially empirical approach seems to be the best currently feasible option in the search for novel anti-AF agents (Box 2).
BOX 2. The Prime Strategy for Anti-AF Pharmacological Research for the Future: Suppression of the Rapid Focal Mechanisms.
The current understanding of the prime electrophysiological mechanism of AF, as well as mechanisms by which anti-AF agents suppress AF, is debatable. Results of the mapping studies seem to cast doubt on the dogma that AF is primarily maintained by reentry. The analysis of electrophysiological alterations induced by anti-AF agents during AF, conducted in the current study, leans toward focal sources as the prime mechanism of AF maintenance. Until now, investigational pharmacological anti-AF strategies have largely stemmed from the “AF is reentry” ideology. It seems that suppression of the rapid focal mechanisms should be an important or the prime strategy for anti-AF pharmacological research.
IK1 + INa Inhibition for Suppression of Persistent AF?
Separate inhibition of INa and IK1 prolongs atrial ERP and may suppress AF.1,32 A combination of ik1 and atrial-selective INa inhibition should synergistically prolong atrial ERP through enhancement of INa block by IK1 reduction (the latter depolarizes RMP and shortens DI). This approach may be particularly relevant for suppression of persistent AF. The efficacy of INa blockers to inhibit INa (and therefore prolong atrial ERP) seems to be reduced in persistent versus paroxysmal AF, which may, in turn, contribute to a reduced effectiveness of INa blockers in suppressing persistent versus paroxysmal AF. Several factors may decrease the ability of INa blockers to inhibit INa in long-lasting versus short-lasting AF, ie, (1) 2–5 mV more negative RMP14,149 (consistent with augmentation of IK131), (2) shorter atrial APD,47,48 (3) longer DI (including during AF),150 and (4) a right-ward shift of the steady-state inactivation curve for the sodium channel (increasing the availability of the sodium channel).149 In patients with persistent AF, inhibition of IK1 should act to normalize atrial RMP (shifting RMP to a more positive potential) and shorten DI (secondary to prolongation of late repolarization), enhancing the inhibition of the sodium channel. This should be translated to a greater ERP prolongation and thus to a more efficient cardioversion and prevention of persistent AF.
CONCLUSIONS
Among the novel anti-AF pharmacological approaches suggested during the past 30 years, to date, only atrial-selective inhibition of INa has been shown to be valid in terms of efficacy and safety in both experimental and clinical studies. Noteworthy, concomitant atrial-selective inhibition of INa is likely to be the prime anti-AF mechanism of IKur and SK channel blockers.
Prolongation of atrial ERP is the mainstay of the current anti-AF pharmacological strategy, and, when achievable, anti-AF drug-induced prolongation of ERP seems to well correlate with the anti-AF efficacy of the drugs. However, significant lengthening of ERP with antiarrhythmics may not be attainable during persistent AF and does not seem to be required for the cardioversion of persistent AF.
Pharmacological research for anti-AF drugs has been largely conceived and intended for suppression of reentry. However, mapping studies increasingly indicate that nonreentrant mechanism(s) plays an important role in the maintenance of AF. Also, electrophysiological alterations caused by anti-AF agents during AF seem to be more consistent with focal rather than reentrant mechanisms. Pharmacological research should place more effort for the development of drugs suppressing focal sources.
ACKNOWLEDGMENTS
The author is grateful to Donna Loyle, MS, for editorial assistance.
Footnotes
Supported by grants from W.W. Smith Charitable Trust and NHLBI (HL47678, HL138103, and HL152201).
The author reports no conflicts of interest.
REFERENCES
- 1.Nattel S, Carlsson L. Innovative approaches to anti-arrhythmic drug therapy. Natrevdrug Discov. 2006;5:1034–1049. [DOI] [PubMed] [Google Scholar]
- 2.Burashnikov A, Di Diego JM, Zygmunt AC, et al. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diness JG, Sorensen US, Nissen JD, et al. Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:380–390. [DOI] [PubMed] [Google Scholar]
- 4.Verrier RL, Bortolotto AL, Silva BA, et al. Accelerated conversion of atrial fibrillation to normal sinus rhythm by pulmonary delivery of flecainide acetate in a porcine model. Heart Rhythm. 2018;15:1882–1888. [DOI] [PubMed] [Google Scholar]
- 5.Verrier RL, Belardinelli L. Pulmonary delivery of antiarrhythmic drugs for rapid conversion of new-onset atrial fibrillation. J Cardiovasc Pharmacol. 2020;75:276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavri BB, Greenberg HE, Kraft WK, et al. MK-0448, a specific Kv1.5 inhibitor: safety, pharmacokinetics and pharmacodynamic electrophysiology in experimental animal models and in humans. Circ Arrhythm Electrophysiol. 2012;5:1193–1201. [DOI] [PubMed] [Google Scholar]
- 7.Burashnikov A. Are there atrial selective/predominant targets for “upstream” atrial fibrillation therapy? Heart Rhythm. 2008;5:1294–1295. [DOI] [PubMed] [Google Scholar]
- 8.Qi XY, Diness JG, Brundel B, et al. Role of small conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation. 2014;129:430–440. [DOI] [PubMed] [Google Scholar]
- 9.Wettwer E, Hala O, Christ T, et al. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110:2299–2306. [DOI] [PubMed] [Google Scholar]
- 10.Camm AJ, Dorian P, Hohnloser SH, et al. A randomized, double-blind, placebo-controlled trial assessing the efficacy of S66913 in patients with paroxysmal atrial fibrillation. Eur Heart J Cardiovasc Pharmacother. 2019;5:21–28. [DOI] [PubMed] [Google Scholar]
- 11.Burashnikov A, Antzelevitch C. Novel pharmacological targets for the rhythm control management of atrial fibrillation. PharmacolTher. 2011;132:300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burashnikov A, Pourrier M, Gibson JK, et al. Rate-dependent effects of vernakalant in the isolated non-remodeled canine left atria are primarily due to block of the sodium channel. Comparison with ranolazine and dl-sotaol. Circ Arrhythm Electrophysiol. 2012;5:400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burashnikov A, Zygmunt AC, Di Diego JM, et al. AZD1305 exerts atrial-predominant electrophysiological actions and is effective in suppressing atrial fibrillation and preventing its re-induction in the dog. J Cardiovasc Pharmacol. 2010;56:80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wettwer E, Christ T, Endig S, et al. The new antiarrhythmic drug Vernakalant: ex-vivo study of human atrial tissue from sinus rhythm and chronic atrial fibrillation. Cardiovasc Res. 2013;98:145–154. [DOI] [PubMed] [Google Scholar]
- 15.van Hunnik A, Lau DH, Zeemering S, et al. Antiarrhythmic effect of vernakalant in electrically remodeled goat atria is caused by slowing of conduction and prolongation of postrepolarization refractoriness. Heart Rhythm. 2016;13:964–972. [DOI] [PubMed] [Google Scholar]
- 16.deSouza IS, Tadrous M, Sexton T, et al. Pharmacologic cardioversion of recent-onset atrial fibrillation: a systematic review and network meta-analysis. Europace. 2020;22:854–869. [DOI] [PubMed] [Google Scholar]
- 17.Simon A, Niederdoeckl J, Skyllouriotis E, et al. Vernakalant is superior to ibutilide for achieving sinus rhythm in patients with recent-onset atrial fibrillation: a randomized controlled trial at the emergency department. Europace. 2017;19:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varkevisser R, van der Heyden MA, Tieland RG, et al. Vernakalant is devoid of proarrhythmic effects in the complete AV block dog model. Eur J Pharmacol. 2013;720:49–54. [DOI] [PubMed] [Google Scholar]
- 19.Tuteja D, Xu D, Timofeyev V, et al. Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol. 2005;289:H2714–H2723. [DOI] [PubMed] [Google Scholar]
- 20.Skibsbye L, Poulet C, Diness JG, et al. Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc Res. 2014;103:156–167. [DOI] [PubMed] [Google Scholar]
- 21.Skibsbye L, Diness JG, Sorensen US, et al. The duration of pacing-induced atrial fibrillation is reduced in vivo by inhibition of small conductance Ca(2+)-activated K(+) channels. J Cardiovasc Pharmacol. 2011;57:672–681. [DOI] [PubMed] [Google Scholar]
- 22.Diness JG, Skibsbye L, Simo-Vicens R, et al. Termination of vernakalant-resistant atrial fibrillation by inhibition of small-conductance Ca(2+)-activated K(+) channels in pigs. Circ Arrhythm Electrophysiol. 2017;10:e005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burashnikov A, Barajas-Martinez H, Hu D, et al. The SK channel inhibitors NS8593 and UCL1684 prevent the development of atrial fibrillation via atrial-selective inhibition of sodium channel activity. J Cardiovasc Pharmacol. 2020;76:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skibsbye L, Wang X, Axelsen LN, et al. Antiarrhythmic mechanisms of SK channel inhibition in the rat atrium. J Cardiovasc Pharmacol. 2015;66:165–176. [DOI] [PubMed] [Google Scholar]
- 25.Nagy N, Marton Z, Kiss L, et al. Role of Ca(2)+-sensitive K+ currents in controlling ventricular repolarization: possible implications for future antiarrhytmic drug therapy. Curr Med Chem. 2011;18:3622–3639. [DOI] [PubMed] [Google Scholar]
- 26.Bigger JT, Jr, Mandel WJ. Effect of lidocaine on the electrophysiological properties of ventricular muscle and purkinje fibers. J Clin Invest. 1970;49:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt C, Wiedmann F, Beyersdorf C, et al. Genetic ablation of TASK-1 (tandem of P domains in a weak inward rectifying K(+) channel-related acid-sensitive K(+) channel-1) (K2P3.1) K(+) channels suppresses atrial fibrillation and prevents electrical remodeling. Circ Arrhythm Electrophysiol. 2019;12:e007465. [DOI] [PubMed] [Google Scholar]
- 28.Kiper AK, Rinne S, Rolfes C, et al. Kv1.5 blockers preferentially inhibit TASK-1 channels: TASK-1 as a target against atrial fibrillation and obstructive sleep apnea? Pflugers Arch. 2015;467:1081–1090. [DOI] [PubMed] [Google Scholar]
- 29.Burashnikov A, Barajas-Martinez H, Hu D, et al. Atrial-selective prolongation of refractory period with AVE0118 is due principally to inhibition of sodium channel activity. J Cardiovasc Pharmacol. 2012;59:539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blaauw Y, Gogelein H, Tieleman RG, et al. “Early” class III drugs for the treatment of atrial fibrillation: efficacy and atrial selectivity of AVE0118 in remodeled atria of the goat. Circulation. 2004;110:1717–1724. [DOI] [PubMed] [Google Scholar]
- 31.Ehrlich JR. Inward rectifier potassium currents as a target for atrial fibrillation therapy. J Cardiovasc Pharmacol. 2008;52:129–135. [DOI] [PubMed] [Google Scholar]
- 32.Ji Y, Varkevisser R, Opacic D, et al. The inward rectifier current inhibitor PA-6 terminates atrial fibrillation and does not cause ventricular arrhythmias in goat and dog models. Br J Pharmacol. 2017;174:2576–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szatmári V, Ji Y, Herwijnen BV, et al. Efficacy of pentamidine analogue 6 in dogs with chronic atrial fibrillation. J Vet Intern Med. 2018;32:1549–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosen MR, Janse MJ. Concept of the vulnerable parameter: the Sicilian Gambit revisited. J Cardiovasc Pharmacol. 2010;55:428–437. [DOI] [PubMed] [Google Scholar]
- 35.Burashnikov A, Antzelevitch C. Atrial-selective sodium channel blockers: do they exist?. Jcardiovasc Pharmacol. 2008;52:121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carvas M, Nascimento BC, Acar M, et al. Intrapericardial ranolazine prolongs atrial refractory period and markedly reduces atrial fibrillation inducibility in the intact porcine heart. J Cardiovasc Pharmacol. 2010;55:286–291. [DOI] [PubMed] [Google Scholar]
- 37.Antzelevitch C, Burashnikov A, Sicouri S, et al. Electrophysiological basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2011;8:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguilar M, Xiong F, Qi XY, et al. potassium channel blockade enhances atrial fibrillation-selective antiarrhythmic effects of optimized state-dependent sodium channel blockade. Circulation. 2015;132:2203–2211. [DOI] [PubMed] [Google Scholar]
- 39.Reiffel JA, Camm AJ, Belardinelli L, et al. The HARMONY trial: combined ranolazine and dronedarone in the management of paroxysmal atrial fibrillation: mechanistic and therapeutic synergism. Circ Arrhythm Electrophysiol. 2015;8:1048–1056. [DOI] [PubMed] [Google Scholar]
- 40.Burashnikov A, Antzelevitch C. New development in atrial antiarrhythmic drug therapy. Nat Rev Cardiol. 2010;7:139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burashnikov A, Belardinelli L, Antzelevitch C. Atrial-selective sodium channel block strategy to suppress atrial fibrillation: ranolazine versus propafenone. J Pharmacol Exp Ther. 2012;340:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burashnikov A, Belardinelli L, Antzelevitch C. Inhibition of IKr potentiates development of atrial-selective INa block leading to effective suppression of atrial fibrillation. Heart Rhythm. 2015;12:836–844. [DOI] [PubMed] [Google Scholar]
- 43.Spinelli W, Parsons RW, Colatsky TJ. Effects of WAY-123,398, a new class III antiarrhythmic agent, on cardiac refractoriness and ventricular fibrillation threshold in anesthetized dogs: a comparison with UK-68798, E-4031, and dl-sotalol. J Cardiovasc Pharmacol. 1992;20:913–922. [DOI] [PubMed] [Google Scholar]
- 44.Wang J, Feng J, Nattel S. Class III antiarrhythmic drug action in experimental atrial fibrillation. Differences in reverse use dependence and effectiveness between d-sotalol and the new antiarrhythmic drug ambasilide. Circulation. 1994;90:2032–2040. [DOI] [PubMed] [Google Scholar]
- 45.Echt DS, Berte LE, Clusin WT, et al. Prolongation of the human monophasic action potential by sotalol. Am J Cardiol. 1982;50:1082–1086. [DOI] [PubMed] [Google Scholar]
- 46.Burashnikov A, Di Diego JM, Sicouri S, et al. Atrial-selective effects of chronic amiodarone in the management of atrial fibrillation. Heart Rhythm. 2008;5:1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morillo CA, Klein GJ, Jones DL, et al. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteeristics of a new model of sustained atrial fibrillation. Circulation. 1995;91:1588–1595. [DOI] [PubMed] [Google Scholar]
- 48.Wijffels MC, Kirchhof CJ, Dorland R, et al. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–1968. [DOI] [PubMed] [Google Scholar]
- 49.Le Grand B, Le Heuzey JY, Perier P, et al. Cellular Electrophysiologic effects of flecainide on human atrial fibers. Cardiovasc Res. 1990;24:232–238. [DOI] [PubMed] [Google Scholar]
- 50.Bechard J, Gibson JK, Killingsworth CR, et al. Vernakalant selectively prolongs atrial refractoriness with no effect on ventricular refractoriness or defibrillation threshold in pigs. J Cardiovasc Pharmacol. 2011;57:302–307. [DOI] [PubMed] [Google Scholar]
- 51.Guerra F, Romandini A, Barbarossa A, et al. Ranolazine for rhythm control in atrial fibrillation: a systematic review and meta-analysis. Int J Cardiol. 2017;227:284–291. [DOI] [PubMed] [Google Scholar]
- 52.Burashnikov A, Sicouri S, Di Diego JM, et al. Synergistic effect of the combination of dronedarone and ranolazine to suppress atrial fibrillation. J Am Coll Cardiol. 2010;56:1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burashnikov A. Late INa inhibition as an antiarrhythmic strategy. J Cardiovasc Pharmacol. 2017;70:159–167. [DOI] [PubMed] [Google Scholar]
- 54.Verrier RL, Kumar K, Nieminen T, et al. Mechanisms of ranolazine's dual protection against atrial and ventricular fibrillation. Europace. 2013;15:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burashnikov A, Antzelevitch C. Role of late sodium channel current block in the management of atrial fibrillation. Cardiovasc Drugs Ther. 2013;27:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobrev D, Friedrich A, Voigt N, et al. The G protein-gated potassium current Ik,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005;112:3697–3706. [DOI] [PubMed] [Google Scholar]
- 57.Hashimoto N, Yamashita T, Tsuruzoe N. Tertiapin, a selective Ik,ACh blocker, terminates atrial fibrillation with selective atrial effective refractory period prolongation. Pharmacol Res. 2006;54:136–141. [DOI] [PubMed] [Google Scholar]
- 58.Walfridsson H, Anfinsen OG, Berggren A, et al. Is the acetylcholine-regulated inwardly rectifying potassium current a viable antiarrhythmic target? Translational discrepancies of AZD2927 and A7071 in dogs and humans. Europace. 2015;17:473–482. [DOI] [PubMed] [Google Scholar]
- 59.Allessie MA, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002;54:230–246. [DOI] [PubMed] [Google Scholar]
- 60.Goette A, Kalman JM, Aguinaga L, et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: definition, characterization, and clinical implication. Heart Rhythm. 2017;14:e3–e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fuster V, Ryden LE, Cannom DS, et al. 2011 ACCF/AHA/HRS focused updates incorporated into the ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation A report of the American college of cardiology foundation/American heart association task force on practice guidelines developed in partnership with the European society of cardiology and in collaboration with the European heart rhythm association and the heart rhythm society. J Am Coll Cardiol. 2011;57:e101–e198. [DOI] [PubMed] [Google Scholar]
- 62.Burashnikov A, Antzelevitch C. Is extensive atrial fibrosis in the setting of heart failure associated with a reduced atrial fibrillation burden? Pacing Clin Electrophysiol. 2018;41:1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burashnikov A, Di Diego JM, Sicouri S, et al. A temporal window of vulnerability for development of atrial fibrillation with advancing heart failure. Eur J Heart Fail. 2014;16:271–280. [DOI] [PubMed] [Google Scholar]
- 64.De Antonio VZ, Silva AC, Stocco FG, et al. Pulmonary delivery of flecainide causes a rate-dependent predominant effect on atrial compared to ventricular depolarization duration revealed by intracardiac recordings in an intact porcine model. J Cardiovasc Electrophysiol. 2018;29:1563–1569. [DOI] [PubMed] [Google Scholar]
- 65.Allessie MA, Lammers WJEP, Bonke FIM, et al. Experimental evaluation of Moe's multiple wavelet hypothesis of atrial fibrillation. In: Zipes DP, Jalife J, eds Cardiac Electrophysiology and Arrhythmias . Grune & Stratton; Philadelphia, PA: 1985:265–276. [Google Scholar]
- 66.Konings KT, Kirchhof CJ, Smeets JR, et al. High-density mapping of electrically induced atrial fibrillation in humans. Circulation. 1994;89:1665–1680. [DOI] [PubMed] [Google Scholar]
- 67.Wang Z, Page P, Nattel S. Mechanism of flecainide's antiarrhythmic action in experimental atrial fibrillation. Circ Res. 1992;71:271–287. [DOI] [PubMed] [Google Scholar]
- 68.Haissaguerre M, Shah AJ, Cochet H, et al. Intermittent drivers anchoring to structural heterogeneities as a major pathophysiological mechanism of human persistent atrial fibrillation. J Physiol. 2016;594:2387–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pandit SV, Jalife J. Rotors and the dynamics of cardiac fibrillation. Circ Res. 2013;112:849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nattel S, Xiong F, Aguilar M. Demystifying rotors and their place in clinical translation of atrial fibrillation mechanisms. Nat Rev Cardiol. 2017;14:509–520. [DOI] [PubMed] [Google Scholar]
- 71.Rensma PL, Allessie MA, Lammers WJEP, et al. Length of excitation wave and susceptibility to reentrant atrial arrhythmias in normal conscious dogs. Circ Res. 1988;62:395–410. [DOI] [PubMed] [Google Scholar]
- 72.Pertsov AM, Davidenko JM, Salomonsz R, et al. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res. 1993;72:631–650. [DOI] [PubMed] [Google Scholar]
- 73.Moe GK, Rheinboldt WC, Abildskov JA. A computer model of atrial fibrillation. Am Heart J. 1964;67:200–220. [DOI] [PubMed] [Google Scholar]
- 74.Nattel S, Bourne G, Talajic M. Insights into mechanisms of antiarrhythmic drug action from experimental models of atrial fibrillation. J Cardiovasc Electrophysiol. 1997;8:469–480. [DOI] [PubMed] [Google Scholar]
- 75.Asano Y, Saito J, Matsumoto K, et al. On the mechanism of termination and perpetuation of atrial fibrillation. Am J Cardiol. 1992;69:1033–1038. [DOI] [PubMed] [Google Scholar]
- 76.Lee S, Sahadevan J, Khrestian CM, et al. High density mapping of atrial fibrillation during vagal nerve stimulation in the canine heart: restudying the moe hypothesis. J Cardiovasc Electrophysiol. 2013;24:328–335. [DOI] [PubMed] [Google Scholar]
- 77.Allessie MA, de Groot NM, Houben RP, et al. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: longitudinal dissociation. Circ Arrhythm Electrophysiol. 2010;3:606–615. [DOI] [PubMed] [Google Scholar]
- 78.de Groot N, van der Does L, Yaksh A, et al. Direct proof of endo-epicardial asynchrony of the atrial wall during atrial fibrillation in humans. Circ Arrhythm Electrophysiol. 2016;9:e003648. [DOI] [PubMed] [Google Scholar]
- 79.Wolf M, Tavernier R, Zeidan Z, et al. Identification of repetitive atrial activation patterns in persistent atrial fibrillation by direct contact high-density electrogram mapping. J Cardiovasc Electrophysiol. 2019;30:2704–2712. [DOI] [PubMed] [Google Scholar]
- 80.Takahashi Y, Akiyoshi K, Sekigawa M, et al. Endocardial contact mapping of the left atrial appendage in persistent atrial fibrillation. J Cardiovasc Electrophysiol. 2020;31:112–118. [DOI] [PubMed] [Google Scholar]
- 81.de Groot NMS, Allessie MA. Pathophysiology of atrial fibrillation: focal patterns of activation. Pacing Clin Electrophysiol. 2019;42:1312–1319. [DOI] [PubMed] [Google Scholar]
- 82.Lee S, Sahadevan J, Khrestian CM, et al. Simultaneous biatrial high-density (510-512 electrodes) epicardial mapping of persistent and long-standing persistent atrial fibrillation in patients: new insights into the mechanism of its maintenance. Circulation. 2015;132:2108–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee S, Sahadevan J, Khrestian CM, et al. Characterization of foci and breakthrough sites during persistent and long-standing persistent atrial fibrillation in patients: studies using high-density (510-512 electrodes) biatrial epicardial mapping. J Am Heart Assoc. 2017;6:e005274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hansen BJ, Zhao J, Csepe TA, et al. Atrial fibrillation driven by micro-anatomic intramural re-entry revealed by simultaneous sub-epicardial and sub-endocardial optical mapping in explanted human hearts. Eur Heart J. 2015;36:2390–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee S, Khrestian CM, Sahadevan J, et al. Reconsidering the multiple wavelet hypothesis of atrial fibrillation. Heart Rhythm. 2020. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li D, Benardeau A, Nattel S. Contrasting efficacy of dofetilide in differing experimental models of atrial fibrillation. Circulation. 2000;102:104–112. [DOI] [PubMed] [Google Scholar]
- 87.Narayan SM, Krummen DE, Shivkumar K, et al. Treatment of atrial fibrillation by the ablation of localized sources: CONFIRM (conventional ablation for atrial fibrillation with or without focal impulse and rotor modulation) trial. J Am Coll Cardiol. 2012;60:628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zaman JAB, Sauer WH, Alhusseini MI, et al. Identification and characterization of sites where persistent atrial fibrillation is terminated by localized ablation. Circ Arrhythm Electrophysiol. 2018;11:e005258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Haissaguerre M, Hocini M, Denis A, et al. Driver domains in persistent atrial fibrillation. Circulation. 2014;130:530–538. [DOI] [PubMed] [Google Scholar]
- 90.Nitta T, Ishii Y, Miyagi Y, et al. Concurrent multiple left atrial focal activations with fibrillatory conduction and right atrial focal or reentrant activation as the mechanism in atrial fibrillation. J Thorac Cardiovasc Surg. 2004;127:770–778. [DOI] [PubMed] [Google Scholar]
- 91.de Groot NM, Houben RP, Smeets JL, et al. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: epicardial breakthrough. Circulation. 2010;122:1674–1682. [DOI] [PubMed] [Google Scholar]
- 92.Lee G, Kumar S, Teh A, et al. Epicardial wave mapping in human long-lasting persistent atrial fibrillation: transient rotational circuits, complex wavefronts, and disorganized activity. Eur Heart J. 2014;35:86–97. [DOI] [PubMed] [Google Scholar]
- 93.Podziemski P, Zeemering S, Kuklik P, et al. Rotors detected by phase Analysis of filtered, epicardial atrial fibrillation electrograms colocalize with regions of conduction block. Circ Arrhythm Electrophysiol. 2018;11:e005858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gray RA, Pertsov AM, Jalife J. Incomplete reentry and epicardail breakthrough patterns during atrial fibrillation in the sheep heart. Circulation. 1996;94:2649–2661. [DOI] [PubMed] [Google Scholar]
- 95.Schuessler RB, Grayson TM, Bromberg BI, et al. Cholinergically mediated tachyarrhythmias induced by a single extrastimulus in the isolated canine right atrium. Circ Res. 1992;71:1254–1267. [DOI] [PubMed] [Google Scholar]
- 96.Lee G, McLellan AJ, Hunter RJ, et al. Panoramic characterization of endocardial left atrial activation during human persistent AF: insights from non-contact mapping. Int J Cardiol. 2017;228:406–411. [DOI] [PubMed] [Google Scholar]
- 97.Yamabe H, Kanazawa H, Ito M, et al. Prevalence and mechanism of rotor activation identified during atrial fibrillation by noncontact mapping: lack of evidence for a role in the maintenance of atrial fibrillation. Heart Rhythm. 2016;13:2323–2330. [DOI] [PubMed] [Google Scholar]
- 98.Scherf D, Romano FJ, Terranova R. Experimental studies on auricular flutter and auricular fibrillation. Am Heart J. 1948;36:241–251. [DOI] [PubMed] [Google Scholar]
- 99.Prinzmetal M, Rakata L, Borduas JL, et al. The nuture of spontanous auricular fibrillation in man. JAMA. 1955;157:1175–1182. [DOI] [PubMed] [Google Scholar]
- 100.Zhou S, Chang CM, Wu TJ, et al. Nonreentrant focal activations in pulmonary veins in canine model of sustained atrial fibrillation. Am J Physiol Heart Circ Physiol. 2002;283:H1244–H1252. [DOI] [PubMed] [Google Scholar]
- 101.Allessie MA, Bonke FIM, Schopman JG. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. Circ Res. 1973;33:54–62. [PubMed] [Google Scholar]
- 102.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003;107:2355–2360. [DOI] [PubMed] [Google Scholar]
- 103.Sanders P, Morton JB, Davidson NC, et al. Electrical remodeling of the atria in congestive heart failure: electrophysiological and electroanatomic mapping in humans. Circulation. 2003;108:1461–1468. [DOI] [PubMed] [Google Scholar]
- 104.Stambler BS, Fenelon G, Shepard RK, et al. Characterization of sustained atrial tachycardia in dogs with rapid ventricular pacing-induced heart failure. J Cardiovasc Electrophysiol. 2003;14:499–507. [DOI] [PubMed] [Google Scholar]
- 105.Lau DH, Mackenzie L, Kelly DJ, et al. Hypertension and atrial fibrillation: evidence of progressive atrial remodeling with electrostructural correlate in a conscious chronically instrumented ovine model. Heart Rhythm. 2010;7:1282–1290. [DOI] [PubMed] [Google Scholar]
- 106.Lee JM, Lee H, Janardhan AH, et al. Prolonged atrial refractoriness predicts the onset of atrial fibrillation: a 12-year follow-up study. Heart Rhythm. 2016;13:1575–1580. [DOI] [PubMed] [Google Scholar]
- 107.Dobrev D, Wehrens XHT. Calcium-mediated cellular triggered activity in atrial fibrillation. J Physiol. 2017;595:4001–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Burashnikov A, Di Diego JM, Barajas-Martinez H, et al. Ranolazine effectively suppresses atrial fibrillation in the setting of heart failure. Circ Heart Fail. 2014;7:627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tse HF, Lau CP. Electrophysiologic actions of dl-sotalol in patients with persistent atrial fibrillation. J Am Coll Cardiol. 2002;40:2150–2155. [DOI] [PubMed] [Google Scholar]
- 110.Sato T, Mitamura H, Kurita Y, et al. Electropharmacologic effects of pilsicainide, a pure sodium channel blocker, on the remodeled atrium subjected to chronic rapid pacing. J Cardiovasc Pharmacol. 2001;38:812–820. [DOI] [PubMed] [Google Scholar]
- 111.Wijffels MC, Dorland R, Allessie MA. Pharmacologic cardioversion of chronic atrial fibrillation in the goat by class IA, IC, and III drugs: a comparison between hydroquinidine, cibenzoline, flecainide, and d-sotalol. J Cardiovasc Electrophysiol. 1999;10:178–193. [DOI] [PubMed] [Google Scholar]
- 112.Wijffels MC, Dorland R, Mast F, et al. Widening of the excitable gap during pharmacological cardioversion of atrial fibrillation in the goat: effects of cibenzoline, hydroquinidine, flecainide, and d-sotalol. Circulation. 2000;102:260–267. [DOI] [PubMed] [Google Scholar]
- 113.Wang JJ, Bourne GW, Wang ZG, et al. Comparative mechanisms of antiarrhythmic drug action in experimental atrial fibrillation—importance of Use-Dependent effects on refractoriness. Circulation. 1993;88:1030–1044. [DOI] [PubMed] [Google Scholar]
- 114.Pallandi RT, Lovell NH, Campbell TJ. Class III antiarrhythmic effects of dofetilide in rabbit atrial myocardium. J Cardiovasc Pharmacol Ther. 1996;1:229–234. [DOI] [PubMed] [Google Scholar]
- 115.Fynn SP, Todd DM, Hobbs WJ, et al. Effect of amiodarone on dispersion of atrial refractoriness and cycle length in patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2003;14:485–491. [DOI] [PubMed] [Google Scholar]
- 116.Kirchhof P, Engelen M, Franz MR, et al. Electrophysiological effects of flecainide and sotalol in the human atrium during persistent atrial fibrillation. Basic Res Cardiol. 2005;100:112–121. [DOI] [PubMed] [Google Scholar]
- 117.Wolbrette DL, Hussain S, Maraj I, et al. A quarter of a century later: what is dofetilide's clinical role today?. J Cardiovasc Pharmacol Ther. 2019;24:3–10. [DOI] [PubMed] [Google Scholar]
- 118.Steinberg JS, Shah Y, Szepietowska B. Pharmacologic conversion during dofetilide treatment for persistent atrial fibrillation. Pacing Clin Electrophysiol. 2017;40:667–671. [DOI] [PubMed] [Google Scholar]
- 119.Vos MA, Golitsyn SR, Stangl K, et al. Superiority of ibutilide (a new class III agent) over DL-sotalol in converting atrial flutter and atrial fibrillation. The Ibutilide/Sotalol Comparator Study Group. Heart. 1998;79:568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kirchhof C, Wijffels M, Brugada J, et al. Mode of action of a new class IC drug (ORG 7797) against atrial fibrillation in conscious dogs. J Cardiovasc Pharmacol. 1991;17:116–124. [DOI] [PubMed] [Google Scholar]
- 121.Kawase A, Ikeda T, Nakazawa K, et al. Widening of the excitable gap and enlargement of the core of reentry during atrial fibrillation with a pure sodium channel blocker in canine atria. Circulation. 2003;107:905–910. [DOI] [PubMed] [Google Scholar]
- 122.Kakugawa H, Shimizu A, Yamagata T, et al. Decrease in the spatial dispersion at the termination of atrial fibrillation by intravenous cibenzoline. Circ J. 2003;67:810–815. [DOI] [PubMed] [Google Scholar]
- 123.Shan Z, van d V, Blaauw Y, et al. Fractionation of electrograms and linking of activation during pharmacologic cardioversion of persistent atrial fibrillation in the goat. J Cardiovasc Electrophysiol. 2004;15:572–580. [DOI] [PubMed] [Google Scholar]
- 124.Fujiki A, Sakabe M, Nishida K, et al. Drug-induced changes in fibrillation cycle length and organization index can predict chemical cardioversion of long-lasting atrial fibrillation with bepridil alone or in combination with aprindine. Circ J. 2004;68:1139–1145. [DOI] [PubMed] [Google Scholar]
- 125.Santos PE, Duytschaever M, Allessie MA. Low-frequency oscillations of atrial fibrillation cycle length in goats: characterization and potentiation by class III antiarrhythmic almokalant. J Electrocardiol. 2008;41:711–723. [DOI] [PubMed] [Google Scholar]
- 126.Kneller J, Kalifa J, Zou R, et al. Mechanisms of atrial fibrillation termination by pure sodium channel blockade in an ionically-realistic mathematical model. Circ Res. 2005;96:e35–e47. [DOI] [PubMed] [Google Scholar]
- 127.Aunes-Jansson M, Edvardsson N, Stridh M, et al. Decrease of the atrial fibrillatory rate, increased organization of the atrial rhythm and termination of atrial fibrillation by AZD7009. J Electrocardiol. 2013;46:29–35. [DOI] [PubMed] [Google Scholar]
- 128.Husser D, Stridh M, Sornmo L, et al. Time-frequency analysis of the surface electrocardiogram for monitoring antiarrhythmic drug effects in atrial fibrillation. Am J Cardiol. 2005;95:526–528. [DOI] [PubMed] [Google Scholar]
- 129.Singh SM, D'Avila A, Kim SJ, et al. Intraprocedural use of ibutilide to organize and guide ablation of complex fractionated atrial electrograms: preliminary assessment of a modified step-wise approach to ablation of persistent atrial fibrillation. J Cardiovasc Electrophysiol. 2010;21:608–616. [DOI] [PubMed] [Google Scholar]
- 130.Tuan J, Osman F, Jeilan M, et al. Increase in organization index predicts atrial fibrillation termination with flecainide post-ablation: spectral analysis of intracardiac electrograms. Europace. 2010;12:488–493. [DOI] [PubMed] [Google Scholar]
- 131.Blaauw Y, Schotten U, van HA, et al. Cardioversion of persistent atrial fibrillation by a combination of atrial specific and non-specific class III drugs in the goat. Cardiovasc Res. 2007;75:89–98. [DOI] [PubMed] [Google Scholar]
- 132.Duytschaever M, Mast F, Killian M, et al. Methods for determining the refractory period and excitable gap during persistent atrial fibrillation in the goat. Circulation. 2001;104:957–962. [DOI] [PubMed] [Google Scholar]
- 133.Gharaviri A, Verheule S, Eckstein J, et al. Effect of Na+-channel blockade on the three-dimensional substrate of atrial fibrillation in a model of endo-epicardial dissociation and transmural conduction. Europace. 2018;20(suppl_3):iii69–iii76. [DOI] [PubMed] [Google Scholar]
- 134.Comtois P, Sakabe M, Vigmond EJ, et al. Mechanisms of atrial fibrillation termination by rapidly unbinding Na+ channel blockers. Insights from mathematical models and experimental correlates. Am J Physiol Heart Circ Physiol. 2008;295:H1489–H1504. [DOI] [PubMed] [Google Scholar]
- 135.Shinagawa K, Mitamura H, Takeshita A, et al. Determination of refractory periods and conduction velocity during atrial fibrillation using atrial capture in dogs: direct assessment of the wavelength and its modulation by a sodium channel blocker, pilsicainide. J Am Coll Cardiol. 2000;35:246–253. [DOI] [PubMed] [Google Scholar]
- 136.Wang TM, Luk HN, Sheu JR, et al. Inducibility of abnormal automaticity and triggered activity in myocardial sleeves of canine pulmonary veins. Int J Cardiol. 2005;104:59–66. [DOI] [PubMed] [Google Scholar]
- 137.Tanaka Y, Obata K, Ohmori T, et al. Angiotensin II induces automatic activity of the isolated Guinea pig pulmonary vein myocardium through activation of the IP(3) receptor and the Na(+)-Ca(2+) exchanger. Int J Mol Sci. 2019;20:1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Takahara A, Sugiyama A, Hashimoto K. Effects of class I antiarrhythmic drugs on the digitalis-induced triggered activity arrhythmia model: a rationale for the short-term use of class I drugs against triggered arrhythmias. Heart Vessels. 2004;19:43–48. [DOI] [PubMed] [Google Scholar]
- 139.Chauhan VS, Verma A, Nayyar S, et al. Focal source and trigger mapping in atrial fibrillation: randomized controlled trial evaluating a novel adjunctive ablation strategy. Heart Rhythm. 2020;17:683–691. [DOI] [PubMed] [Google Scholar]
- 140.Wit AL, Boyden PA. Triggered activity and atrial fibrillation. Heart Rhythm. 2007;4(3 suppl):S17–S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Voigt N, Li N, Wang Q, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yeh YH, Wakili R, Qi XY, et al. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol. 2008;1:93–102. [DOI] [PubMed] [Google Scholar]
- 143.Sicouri S, Glass A, Belardinelli L, et al. Antiarrhythmic effects of ranolazine in canine pulmonary vein sleeve preparations. Heart Rhythm. 2008;5:1019–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen YJ, Chen SA, Chen YC, et al. Effects of rapid atrial pacing on the arrhythmogenic activity of single cardiomyocytes from pulmonary veins: implication in initiation of atrial fibrillation. Circulation. 2001;104:2849–2854. [DOI] [PubMed] [Google Scholar]
- 145.Patterson E, Lazzara R, Szabo B, et al. Sodium-calcium exchange initiated by the Ca2+ transient: an arrhythmia trigger within pulmonary veins. J Am Coll Cardiol. 2006;47:1196–1206. [DOI] [PubMed] [Google Scholar]
- 146.Cha TJ, Ehrlich JR, Zhang L, et al. Atrial tachycardia remodeling of pulmonary vein cardiomyocytes: comparison with left atrium and potential relation to arrhythmogenesis. Circulation. 2005;111:728–735. [DOI] [PubMed] [Google Scholar]
- 147.Christ T, Rozmaritsa N, Engel A, et al. Arrhythmias, elicited by catecholamines and serotonin, vanish in human chronic atrial fibrillation. Proc Natl Acad Sci U S A. 2014;111:11193–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Greiser M. Calcium signalling silencing in atrial fibrillation. J Physiol. 2017;595:4009–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bosch RF, Zeng X, Grammer JB, et al. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–131. [DOI] [PubMed] [Google Scholar]
- 150.Shan Z, Yan J, Zhou J, et al. Role of progressive widening of the temporal excitable gap for perpetuation of atrial fibrillation in the goat. Circ J. 2010;74:655–663. [DOI] [PubMed] [Google Scholar]