

Article, see p 1404
The global pandemic of coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), continues to spread, but the fundamental mechanisms behind the pathogenicity are poorly understood. Cardiovascular comorbidities are frequent in patients presenting with COVID-19 as is cardiac damage. In patients presenting to the hospital with severe SARS-CoV-2 infection, one-third have underlying cardiovascular disease and over a quarter exhibit myocardial injury as indicated by elevated TnT (troponin) levels.1 Plasma high-sensitivity CRP (C-reactive protein) and NT-proBNP (N-terminal pro-B-type natriuretic peptide) levels significantly associate with fatal outcome from COVID-19, suggesting that inflammation may be a potent mechanism for myocardial injury and a critical mediator of mortality.2 The disease appears to progress in 3 phases: initial illness caused by active infection, a second pulmonary phase, and, when severe, a third phase characterized by hyperinflammation, cytokine storm, high cardiac injury biomarker levels, and significant morbidity and mortality. In some COVID-19 patients, the host inflammatory response continues to amplify and results in systemic inflammation3 with a higher proportion of nonsurvivors exhibiting progressive thrombocytopenia. Consistent with this noted drop in platelet count, clots have been detected in small vessels of the lungs, heart, and liver of COVID-19 patients,4 and the prevalence of deep vein thrombosis in hospitalized patients with active infection is increased.5
The mechanisms underlining thrombosis in COVID-19 patients are not known and likely due to multiple processes including inflammation, oxygen demand injury, and plaque rupture triggered by the infection. Platelets mediate thrombotic vascular occlusion but are also increasingly recognized to have immunomodulatory activity. Several of our recent studies have characterized the role of viral infections in cardiac disease.6–8 Although robust data on the scope of acute myocardial infarction in COVID-19 are not yet available, myocardial infarction contributed to in-hospital mortality during previous severe acute respiratory syndrome/coronavirus epidemics. A recent study has also demonstrated that influenza and other respiratory viruses increase the incidence of acute myocardial infarction particularly in the first 7 days post-infection,9 suggesting that platelets may be directly involved in mediating an immune response but, when dysregulated, can also lead to thrombotic vascular occlusion.
Growing mechanistic and clinical data have shown that, in addition to thrombosis, platelets perform various immune functions during infection. Platelets can form heterotypic aggregates with cells mediating innate immunity including neutrophils, monocytes, eosinophils, and dendritic cells.7 Platelets also influence adaptive immunity and modulate B- and T-cell activity by secreting various granule proteins such as serotonin (5-HT [5-hydroxytryptamine]) and PF4 (platelet factor 4).7 Specific to ssRNA viral infections, we and others have shown that platelets act as immune cells by binding and internalizing viruses including influenza, HIV, and encephalomyocarditis.6,8,10 Platelet viral internalization leads to lysosomal degradation of the viral coat and activation of the pathogen-associated molecular pattern receptor, TLR7 (Toll-like receptor 7). TLR7 signals through AKT (protein kinase B),8,10 p38-MAPK (mitogen-activated protein kinase),8 and IRAK4-IKKb-SNAP23 (synaptosomal-associated protein 23)10 causing P-selectin8,10 and CD40L (CD40 ligand)8 surface expression that mediates interaction with neutrophils. Activation of TLR7 by influenza also results in C3 (complement 3)6 release from platelets that leads to complement cascade activation and release of neutrophil DNA. Platelets control the size of netting aggregates by further releasing GM-CSF (granulocyte-macrophage colony-stimulating factor).6 Dysregulation of this process by means of damage related to tissue factor and inflammatory cytokine release can cause uncontrolled netosis and potential immunothrombosis.11 Additionally, although direct platelet–SARS-CoV-2 interactions may be important in the prothrombotic response, platelet reactions to signals arising from the injured and infected lung may further contribute to thrombosis. As infection progresses and platelets become entangled in the nets and microthrombi, the overall ability of platelets to remove circulating virus is reduced and immune function may be compromised. These observations coupled with our population-based studies12 suggest that platelet-mediated immunity may be an important regulator of the ssRNA viral response and, specifically, could influence COVID-19–mediated thrombosis.
In this issue of Circulation Research, Zaid et al13 provide further evidence that platelets contribute to the inflammatory cytokine milieu during COVID-19 and are potentially hyperactivated. Using a COVID-19 cohort of 115 patients of Moroccan ethnicity, Zaid et al showed that levels of ILs (interleukins) related to neutrophil and monocyte recruitment, and activation and T-cell, B-cell, and natural killer (NK) cells growth and proliferation were reduced in COVID-19 patients (Figure). In certain cases, the reduced levels of inflammatory cytokines in platelets were associated with increased plasma levels of the same cytokines. Additionally, platelet-specific granule content, including PF4/CXCL4 (C-X-C motif) and serotonin/5-HT was significantly increased in the plasma of COVID-19 patients, suggesting platelet activation. Platelet α-granule protein PF4 is a key regulator of T-cell development, necessary to limit Th17 (T helper 17 cell) differentiation.14 Outside of the nervous system, serotonin is stored in platelets and, once released, can lead to increased vascular permeability,15 in addition to neutrophil recruitment and T-cell activation. Serotonin also acts as a weak platelet agonist, which in synergy with ADP would facilitate platelet aggregation. Consistently the authors show that platelets derived from patients with active COVID-19 infection have an increased adhesion and aggregation potential, suggesting that serotonin and other platelet content can also potentially feed into the described microthrombosis that underlines COVID-19. Zaid et al also showed that platelets from COVID-19 patients released more IL1β and soluble CD40L at low levels of thrombin, further suggesting that platelets have an increased hyperactive potential and may be contributing to the overall inflammatory surge often observed during infection. Finally, blood from COVID-19 patients contained extracellular vesicles of platelet origin that may contribute to the coagulopathy observed in these patients due to the increased phosphatidylserine levels on their surface. Despite the exact mechanism of activation, we can conclude that platelets are hyperactivated, have an increased prothrombotic potential, and contribute to the elevated inflammatory cytokine pool that underlines the fatal course of COVID-19.
Figure.
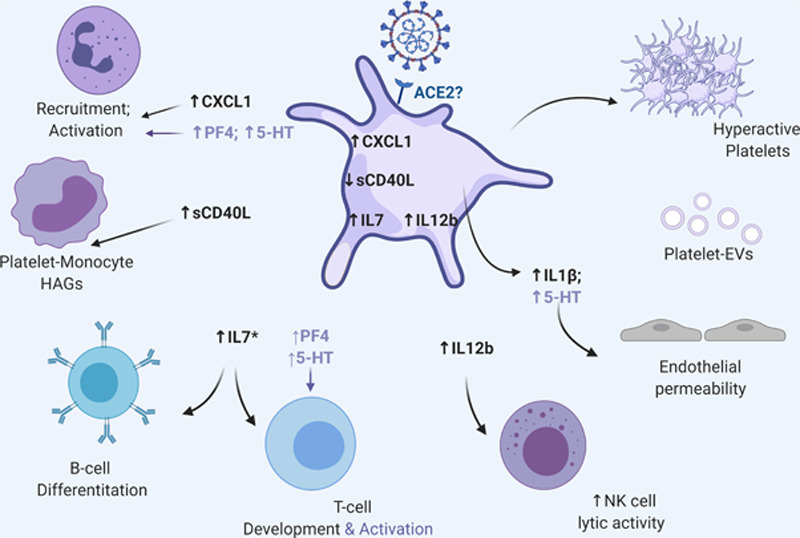
Coronavirus disease 2019 (COVID-19), platelets, and immunity. Severe acute respiratory syndrome coronavirus 2 RNA can be found in human platelets. COVID-19 changes platelet inflammatory cytokine profile important for interaction and immune cell activation. In most cases, changes in platelet cytokine level are reflected in the plasma by an undefined mechanism, and plasma levels of platelet-derived PS (phosphatidylserine)-extracellular vesicles (EVs) are also increased. Platelets from COVID-19 patients also exhibit increased baseline activation potential suggesting that they can be hyperactivated with disease severity. Hyperactivation is further reflected in the increased levels of platelet-specific granule proteins such as PF4 (platelet factor 4) and serotonin (5-HT [5-hydroxytryptamine]). PF4 and serotonin can further recruit and activate immune cells. Additionally, serotonin can increase platelet aggregation particularly in the presence of ADP, which is highly elevated during infection due to inflammasome activation and consequent cell death. Of note, in addition to the platelet-virus interactions, other signals coming from the damaged, infected, and ischemic endothelium (not depicted in this figure) can further contribute to the increased prothrombotic pathology that underlines COVID-19. This figure was generated using BioRender. ACE2 indicates angiotensin-converting enzyme 2; IL, interleukin; and sCD40L, soluble CD40 ligand.
Overwhelmingly, studies have shown ACE2 (angiotensin-converting enzyme 2)-TMPRSS2 (transmembrane protease, serine 2) as the predominant receptor-protease axis for SARS-CoV-2 cellular uptake. Interestingly, Zaid et al13 detected-CoV-2 RNA in platelets but were unable to detect ACE2 RNA or protein expression. These observations are consistent with a previous study using similar methods of CD45 (receptor linked protein tyrosine phosphatase)-depleted platelets.16 Due to the limitations of this type of platelet isolation and challenges with detecting low abundant transcripts by SYBR™ Green qPCR13 or RNA sequencing,16 lack of ACE2 RNA expression does not appear definitive. Resolution limitations also exist for the detection of low abundant proteins using epifluorescence microscopy.13 In our experience, confocal microscopy coupled with a 100X lens6 is necessary for proper imaging of low expressed proteins such as TLR7.6 Differences in antibody-epitopes against ACE2 may also contribute to the limitations of the detection of this protein. Lastly, lack of ACE2 expression in platelets is in conflict with a study claiming its presence.17 Taken together, the question of how SARS-CoV-2 is taken up by platelets continues to be without an answer. Regardless of the method of detection, low ACE2 levels would not explain the profound hyperactive platelet phenotype that has been described.13,16 These observations raise the possibility that ACE2 may not be the only receptor for SARS-CoV-2 in platelets. Similarly, although virus-receptor recognition is necessary for proper infection, other paths for SARS-CoV-2 internalization may also be utilized by platelets. Insights gained from other cells suggest that viral internalization can occur with micropinocytosis or phagocytosis of fragments from infected cells undergoing apoptosis, uptake of virus-containing microparticles, or uptake of IgG antibodies containing scaffolded viral particles.11 Non-ACE2 mechanisms may lead to viral internalization independent of the endosomal/lysosomal compartments of platelets. Consequently, lack of endosomal-viral localization may lead to failed TLR7 response and dysregulated platelet-immune cell activation manifested by the increased risk for immunothrombosis in COVID-19 patients. Lastly, studies of platelets incubated with SARS-CoV-2 collected directly from Vero cells17 could be misleading by causing nonvirus-related coagulation. Unpurified viral stock contains cell fragments, high quantities of ADP, ATP, fibrinogen, and other procoagulants. Further studies using purified virus are necessary to establish and delineate the proper virus-receptor interaction and direct effect between SARS-CoV-2 and platelets.
As we gain a greater understanding of the broad clinical implications of SARS-CoV-2 infection, we continue to recognize the disease complexity. However, the pressing need for new discoveries related to the COVID-19 complexity also requires a cautious balance between careful observation and rapid data release. Thrombosis, due to platelets and coagulation, clearly plays a role in multiple pathobiological processes triggered by COVID-19. The data by Zaid et al13 add to our understanding and should lead to further studies clarifying the interactions between platelets and SARS-CoV-2 during infection and how the interaction is reflected in platelet-mediated immunity and thrombosis. The importance of these future discoveries is necessary to guide the timing and type of therapies needed to mitigate thrombosis and coagulation in patients with SARS-CoV-2 infection.
Acknowledgments
Support for this publication is provided by an American Heart Association coronavirus disease 2019 (COVID-19) Rapid Response Award to J.E. Freedman and The University of Massachusetts Center for Clinical and Translational Science (UMCCTS)-Clinical and Translational Science Awards (CTSA) grant to M. Koupenova (UMCCTS is funded by The National Center for Advancing Translational Sciences (NCATS): grants UL1TR001453, TL1TR00454, and KL2TR001455).
Disclosures
None.
Footnotes
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
For Disclosures, see page 1421.
References
- 1.Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, Wang H, Wan J, Wang X, Lu Z. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5:811–818. doi: 10.1001/jamacardio.2020.1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li B, Yang J, Zhao F, Zhi L, Wang X, Liu L, Bi Z, Zhao Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in china. Clin Res Cardiol. 2020;109:531–538. doi: 10.1007/s00392-020-01626-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, Law KI, Tang BS, Hon TY, Chan CS, et al. ; HKU/UCH SARS Study Group. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/s0140-6736(03)13412-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L, Feng X, Zhang D, Jiang C, Mei H, Wang J, Zhang C, Li H, Xia X, Kong S, et al. Deep vein thrombosis in hospitalized patients with COVID-19 in Wuhan, China: prevalence, risk factors, and outcome. Circulation. 2020;142:114–128. doi: 10.1161/CIRCULATIONAHA.120.046702 [DOI] [PubMed] [Google Scholar]
- 6.Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019;10:1780 doi: 10.1038/s41467-019-09607-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. 2018;122:337–351. doi: 10.1161/CIRCRESAHA.117.310795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. 2014;124:791–802. doi: 10.1182/blood-2013-11-536003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwong JC, Schwartz KL, Campitelli MA, Chung H, Crowcroft NS, Karnauchow T, Katz K, Ko DT, McGeer AJ, McNally D, et al. Acute myocardial infarction after laboratory-confirmed influenza infection. N Engl J Med. 2018;378:345–353. doi: 10.1056/NEJMoa1702090 [DOI] [PubMed] [Google Scholar]
- 10.Banerjee M, Huang Y, Joshi S, Popa GJ, Mendenhall MD, Wang QJ, Garvy BA, Myint T, Whiteheart SW. Platelets endocytose viral particles and are activated via TLR (toll-like receptor) signaling. Arterioscler Thromb Vasc Biol. 2020;40:1635–1650. doi: 10.1161/ATVBAHA.120.314180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koupenova M. Potential role of platelets in COVID-19: implications for thrombosis. Res Pract Thromb Haemost. 2020;4:737–740. doi: 10.1002/rth2.12397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koupenova M, Mick E, Mikhalev E, Benjamin EJ, Tanriverdi K, Freedman JE. Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors. Arterioscler Thromb Vasc Biol. 2015;35:1030–1037. doi: 10.1161/ATVBAHA.114.304954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, Limami Y, Zaid N, Haj RBE, Mahir W, et al. Platelets can associate with SARS-CoV-2 RNA and are hyperactivated in COVID-19. Circ Res. 2020;127:1404–1418. doi: 10.1161/CIRCRESAHA.120.317703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi G, Field DJ, Ko KA, Ture S, Srivastava K, Levy S, Kowalska MA, Poncz M, Fowell DJ, Morrell CN. Platelet factor 4 limits Th17 differentiation and cardiac allograft rejection. J Clin Invest. 2014;124:543–552. doi: 10.1172/JCI71858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cloutier N, Paré A, Farndale RW, Schumacher HR, Nigrovic PA, Lacroix S, Boilard E. Platelets can enhance vascular permeability. Blood. 2012;120:1334–1343. doi: 10.1182/blood-2012-02-413047 [DOI] [PubMed] [Google Scholar]
- 16.Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, Petrey AC, Tolley ND, Guo L, Cody M, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136:1317–1329. doi: 10.1182/blood.2020007214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, Liu M, Zhao X, Xie Y, Yang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020;13:120 doi: 10.1186/s13045-020-00954-7 [DOI] [PMC free article] [PubMed] [Google Scholar]