

Supplemental Digital Content is available in the text.
Keywords: blood platelets, COVID-19, cytokines, hemostasis, inflammation, thrombosis
Abstract
Rationale:
In addition to the overwhelming lung inflammation that prevails in coronavirus disease 2019 (COVID-19), hypercoagulation and thrombosis contribute to the lethality of subjects infected with severe acute respiratory syndrome coronavirus 2. Platelets are chiefly implicated in thrombosis. Moreover, they can interact with viruses and are an important source of inflammatory mediators. While a lower platelet count is associated with severity and mortality, little is known about platelet function during COVID-19.
Objective:
To evaluate the contribution of platelets to inflammation and thrombosis in patients with COVID-19.
Methods and Results:
Blood was collected from 115 consecutive patients with COVID-19 presenting nonsevere (n=71) and severe (n=44) respiratory symptoms. We document the presence of severe acute respiratory syndrome coronavirus 2 RNA associated with platelets of patients with COVID-19. Exhaustive assessment of cytokines in plasma and in platelets revealed the modulation of platelet-associated cytokine levels in both patients with nonsevere and severe COVID-19, pointing to a direct contribution of platelets to the plasmatic cytokine load. Moreover, we demonstrate that platelets release their alpha- and dense-granule contents in both nonsevere and severe forms of COVID-19. In comparison to concentrations measured in healthy volunteers, phosphatidylserine-exposing platelet extracellular vesicles were increased in nonsevere, but not in severe cases of COVID-19. Levels of D-dimers, a marker of thrombosis, failed to correlate with any measured indicators of platelet activation. Functionally, platelets were hyperactivated in COVID-19 subjects presenting nonsevere and severe symptoms, with aggregation occurring at suboptimal thrombin concentrations. Furthermore, platelets adhered more efficiently onto collagen-coated surfaces under flow conditions.
Conclusions:
Taken together, the data suggest that platelets are at the frontline of COVID-19 pathogenesis, as they release various sets of molecules through the different stages of the disease. Platelets may thus have the potential to contribute to the overwhelming thrombo-inflammation in COVID-19, and the inhibition of pathways related to platelet activation may improve the outcomes during COVID-19.
Editorial, see p 1419
Meet the First Author, see p 1345
The outbreak of coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, emerged in Wuhan of Hubei province in China in December 2019 and rapidly spread to more than 196 countries worldwide.1,2 The World Health Organization declared the outbreak a serious public health emergency of international concern and by August 25, 2020, the virus had infected over 23.5 million and killed >810 492 individuals worldwide.2,3
SARS-CoV-2 is a positive-sense single stranded RNA virus.4,5 The main SARS-CoV-2 counter-receptor on human cells is the ACE2 (angiotensin-converting enzyme 2), highly expressed by nasopharyngeal airway epithelial cells as well as alveolar epithelial cells, vascular endothelial cells, and lung macrophages. The virus tropism likely explains the notorious respiratory symptoms observed during infection.6,7 An uncontrolled systemic inflammatory response, known as the cytokine storm, results from immune effector cell release of substantial amounts of proinflammatory cytokines, such as TNF (tumor necrosis factor), IL (interleukin)-1, IL-6, IL-7, and G-CSF (granulocyte-colony stimulating factor),8,9 which are suggested to contribute to SARS-CoV-2 lethality.1,10,11 While outstanding research aimed at understanding SARS-CoV-2 pathogenesis initially focused on lung inflammation, COVID-19 also implicates multi-organ damage, leading to multiple organ failure, notably of the respiratory, cardiac, renal, and hepatic systems.1,12
Thrombotic complications that manifest as microvascular thrombosis, venous, or arterial thrombosis are features detected in most patients with multi-organ failure.13–18 Disseminated intravascular coagulation is a condition in which blood clots form throughout the body, blocking small blood vessels. Although disseminated intravascular coagulation appeared rarely and was associated with bleeding manifestations in a multicenter retrospective study involving 400 patients with COVID-19 (144 critically ill),17 disseminated intravascular coagulation occurred in the majority (71%) of patients who died of COVID-19 according to a study of 183 consecutive patients.19 What drives disseminated intravascular coagulation during COVID-19 is currently unknown. It is important to note that accumulating evidence shows that the blood of patients with COVID-19 is hypercoagulable.14,17,19–21 Higher D-dimer and fibrin degradation product levels, a longer prothrombin time and activated partial thromboplastin time are observed in SARS-CoV-2-infected patients with hospitalization complications or in those who died, relative to survivors.17–19,21
Hematologic manifestation in patients with symptomatic COVID-19 include severe leukopenia and lymphopenia.14,19,22 Lower platelet counts are associated with increased risk of in-hospital mortality in patients with COVID-19, although the platelet levels are generally not considered as clinically relevant as patients typically do not require platelet transfusions.17,23–28 It is suggested that SARS-CoV-2 may reduce platelet production, increase platelet destruction, or more likely that platelet activation and thrombosis in patients may increase platelet consumption.26 As thrombosis and blood coagulation are chiefly controlled by platelets,29 it is critical to define whether platelets are activated in COVID-19.
Platelets are small (2–4 µm in diameter) anucleated cells derived from megakaryocytes in bone marrow and lungs.30,31 Nearly one trillion platelets patrol the blood vessels to maintain the integrity of the vasculature. Damage to blood vessels triggers the formation of a hemostatic plug to stop subsequent bleeding.32 Thrombus formation mediated by platelets can also implicate extracellular vesicles (EV; microparticles or microvesicles), which provide anionic phospholipids, such as phosphatidylserine, which can support the coagulation cascade.33
In addition to their role in hemostasis and thrombosis, platelets also contribute to immunity and inflammation.34–37 The extravasation of neutrophils and their invasion of inflamed tissues require their interaction with activated platelets.38,39 Moreover, the liberation of neutrophil extracellular trap, a process by which neutrophils release extracellular DNA, is observed in COVID-19.40–42 Neutrophil extracellular trap requires platelets and may thus contribute to thrombosis.43,44 Platelets express immune and inflammatory molecules such as IL-1,45 and a set of immune receptors including CD40L, TLR (Toll-like receptors),34 and the Fc receptor for IgG FcγRIIA (Fc gamma receptor IIa).46
Dengue virus can infect megakaryocytes,47 and megakaryocytes can upregulate type-I interferon genes in response to dengue and influenza viruses.48 While dengue virus can replicate in platelets,49 influenza virus can be internalized by platelets and is transported through the circulatory system in humans.50,51 Influenza virus-induced lung injury in mice can be prevented by targeting of platelet αIIbβ3 by an antagonist or other antiplatelet compounds such as clopidogrel, an antagonist of ADP (adenosine diphosphate) receptors, or blockers of protease-activated receptor-4, thus pointing to an active role by platelets in influenza pathogenesis.52 Influenza and herpes simplex virus-1 also activate platelet aggregation and thrombosis due to the presence of prevalent opsonizing antibodies and their interaction with FcγRIIA.53,54 Thus, activated platelets may contribute to both the overwhelming inflammation and thrombosis that prevail in COVID-19.
While thrombosis and coagulation abnormalities predict worse outcomes in COVID-19, the role of platelet function has yet to be investigated. Here, we examined platelet activation, secretion, adhesion, and aggregation in patients with severe and nonsevere COVID-19.
Methods
Supplement contains the detailed description of methods.
All data, analytic methods, and study materials supporting the findings of this study are provided in the article and supplemental material and are available from the corresponding author upon reasonable request.
Results
Patient Screening and Characterization
A total of 1544 patients with suspected SARS-CoV-2 infection (travelers, proximity to infected patients, and presence of symptoms) were screened according to the flow chart presented in Figure I in the Data Supplement. The clinical symptoms included fever, cough, dyspnea, fatigue, headache, chest pain, and pharyngalgia. Initial screening in addition to laboratory blood testing and high-resolution chest computed tomography imaging (CT scans; Figure II in the Data Supplement) led to the selection of 196 patients, who were then screened for the presence of 2019-nCoV RdRp and E genes using RT-PCR (reverse transcription polymerase chain reaction) (Figure III in the Data Supplement; see Table 1 and Figure IV in the Data Supplement for clinical data). The degree of COVID-19 severity (severe versus nonsevere) was defined at the time of patient admission using the American Thoracic Society guidelines for community-acquired pneumonia.55 There were 71 cases in the nonsevere COVID-19 (COVID-nonsevere) group and 44 cases in the severe COVID-19 group (COVID-severe), while 81 individuals were declared negative by both throat swab RT-PCR amplification of 2019-nCoV RdRp and E genes and chest CT scans. The latter individuals were excluded from the study given that they still could present symptoms of unidentified etiology. Furthermore, known comorbidities, medication (independent of COVID-19 treatment) and self-identified ethnicity/race are reported in the Data Supplement (Tables I through III in the Data Supplement). The median (interquartile range, IQR) age of severe cases (64.5 [72.75–41.25] years) was significantly higher than for nonsevere cases (48 [62–37] years). The hospitalization duration (median [IQR]) was also significantly higher for severe patients (20 [28.75–14] days) compared with nonsevere patients (14 [16–13] days) days, with 8.5% of total patients critically ill and admitted to the intensive care unit. There was no significant difference in body weight (median [IQR]) between nonsevere patients (75 [87–62] kg) and severe patients (82 [90–65.5] kg). Five COVID-severe patient deaths occurred in the intensive care unit, while all nonsevere patients survived.
Table 1.
Clinical Analysis and Blood Parameters
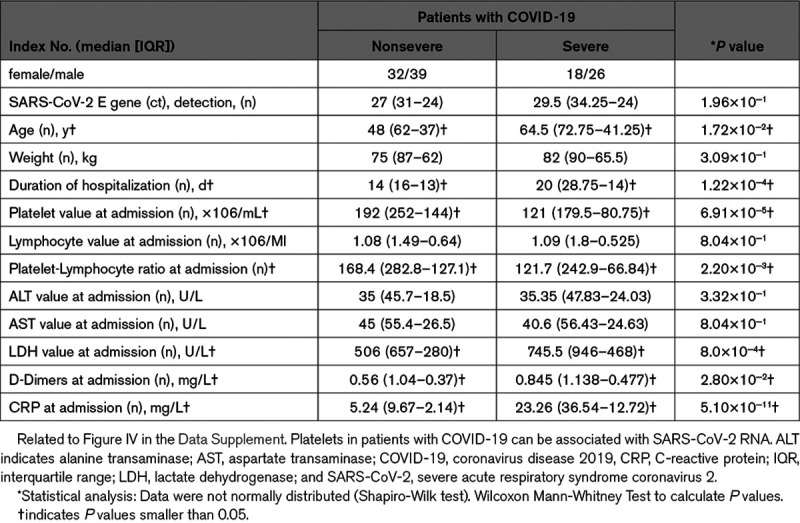
Blood Parameters and Thrombocytopenia
The severity of disease was established, and routine blood parameters were monitored at the time of admission for both the nonsevere and severe cases of COVID-19 (Table 1 and Figure IV in the Data Supplement).2 Lymphocyte counts, ALT (alanine aminotransferase) and AST (aspartate aminotransferase) data were similar between the 2 groups of infected patients. In contrast, the LDH (lactate dehydrogenase) and CRP (C-reactive protein) inflammation markers, which were higher than the expected normal range in both groups, were significantly increased in patients with severe COVID-19 in comparison with nonsevere patients, pointing to a possible cytokine storm and respiratory failure associated with disease severity (Table 1 and Figure IV in the Data Supplement).56 Furthermore, D-dimers were higher than the expected normal range in both groups and were significantly increased in patients with severe COVID-19 in comparison with nonsevere patients (Table 1 and Figure IV in the Data Supplement). The significant difference (P=0.028, Mann-Whitney) between the 2 groups of patients, however, appeared to be due to the particularly high levels of D-dimers in the 5 nonsurvivors, as the significance was lost (P=0.205, Mann-Whitney) when calculations were performed in their absence, indicating a procoagulant state in the blood, notably in nonsurvivors (Figure V in the Data Supplement).
Thus, we evaluated platelet numbers and platelet-lymphocyte ratios measured at the time of admission in the 2 groups. Platelet counts (median [IQR]) were in the lower range in both patients with COVID-19 severe (121 [179.5–80.75]×106/mL) and nonsevere (192 [252–144]×106/mL) in comparison to the expected count in healthy volunteers (130–400×106/mL). Moreover, we observed a modest, but statistically significant reduction in platelet counts between the 2 patient groups. The platelet-lymphocyte ratio was lower in severe patients compared with nonsevere patients, arguing in favor of the thrombosis mainly reported in more severe cases (Table 1 and Figure IV in the Data Supplement).
SARS-CoV-2 RNA has been identified in urine and stool samples in addition to semen, consistent with ACE2 expression outside the respiratory tract, such as in endothelial cells that line blood vessels, heart, kidney, and spermatozoa.57–60 We evaluated whether platelets are associated with SARS-CoV-2 RNA in COVID-19. Platelets were isolated from recruited healthy volunteers (n=17) and from patients with COVID-19 (38 nonsevere and 11 severe) at the time of admission, and we tested for the presence of SARS-CoV-2 RNA using primers designed to detect the E Gene. While no SARS-CoV-2 RNA was detected in healthy controls (0%, 0 out of 17), we detected SARS-CoV-2 RNA in platelets from patients with nonsevere (23.7%, 9 patients out of 38) and severe (18.2%, 2 patients out of 11) COVID-19 (Table IV and Figure VI in the Data Supplement). An intriguing observation was that individuals with positive platelets for SARS-CoV-2 RNA were significantly older (71 [74.50–57.00] years) than those who were negative (41 [53.00–34.50] years; P<0.0001), with no other parameters being different between the 2 groups (Table V in the Data Supplement). The data suggest that platelets may associate with SARS-CoV-2 RNA molecules, and that this event may be more likely to occur in older patients.
Circulating Cytokines
Elevated inflammatory cytokine levels are reported in the blood of patients with COVID-19 8,9 and may contribute to the overwhelming inflammation and to coagulation through the activation of endothelial cells.61,62 We used a multiplex assay to monitor 48 cytokines in plasma prepared from healthy volunteers (n=10) and from patients with COVID-19 (10 nonsevere and 9 severe) at the time of admission. In patients with nonsevere disease, the concentrations of 13 cytokines or growth-factors were significantly increased (epidermal growth factor [EGF], Eotaxin, FGF-2 [fibroblast growth factor-2], Fractalkine, IFN-γ [interferon gamma], IL-2, IL-12p40, IL-22, IL-27, MCP-3 [monocyte chemotactic protein-3], macrophage-derived chemokine [MDC], PDGF-AB/BB [platelet-derived growth factor-AA/BB], TGFβ, [transforming growth factor β]), the concentrations of 2 cytokines (IL-9 and IL-15) were significantly decreased, while that of 33 cytokines or growth-factors were similar to healthy controls (Figure 1 and Figure VII in the Data Supplement). In patients with severe disease, 27 cytokines or growth-factors (EGF, Eotaxin, FGF-2, FLT-3 L [Fms-like tyrosine kinase-3 L], Fractalkine, GM-CSF, GROα [growth-related oncogene α], IFNγ, IL-1α, IL-1β, IL-1-ra, IL-4, IL-6, IL-7, IL-8, IL-10, IL-13, IL-17A, IL-17E/IL-25, M-CSF [macrophage colony-stimulating factor], MDC, MIP-1β [macrophage inflammatory protein-1β], PDGF-AA, PDGF-AB/BB, TGF-α, TGF-β, VEGF-A [vascular endothelial growth factor-A]) were significantly increased, 2 cytokines (IL-3 and IL-18) were significantly decreased, and levels of 19 cytokines or growth-factors were similar to healthy controls (Figure 1 and Figure VII in the Data Supplement). The larger number of upregulated cytokines and growth-factors in severe as compared with nonsevere disease, reflects the notion that the inflammatory response may contribute to disease severity.
Figure 1.
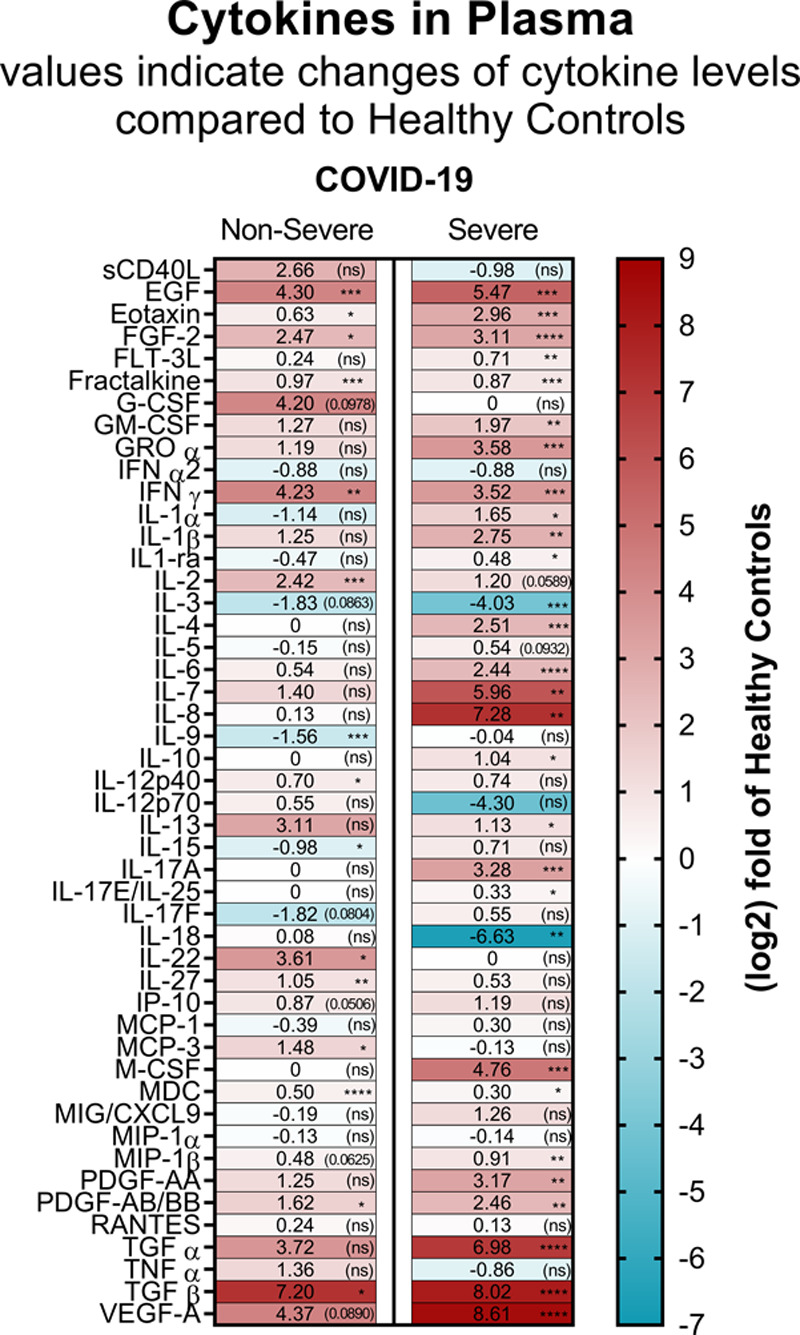
Cytokine and chemokine levels in plasma from patients with coronavirus disease 2019 (COVID-19). Heat map visualization of 48 cytokine/chemokine expression profiles in plasma of patients with COVID-19 nonsevere (median of n=10) and severe (median of n=9) relatively to the healthy controls (median of n=10). Cytokine/chemokine expression is represented as a (log2) fold change relative to healthy controls. The numbers in each part represent the change and statistical significance (in brackets), and the color codes refer to red for increased expression and blue for decreased expression. Absolute cytokine/chemokine values (pg/mL) and details on the statistical analysis are shown in Figure VII in the Data Supplement. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Platelets Produce Inflammatory Cytokines in COVID-19
We then determined the ability of patient with COVID-19 platelets to produce inflammatory mediators. Platelets derived from healthy volunteers and from severely and nonseverely affected patients were stimulated with low doses of α-thrombin (0.025 and 0.05 U/mL). Platelets were then lysed, and the amounts of IL-1β, IL-18, sCD40L, and the eicosanoid thromboxane B2 were determined. While IL-18 secretion and thromboxane production were unaffected in the disease, we found that COVID-19 platelets were more prone to release IL-1β and soluble CD40L upon exposure to 0.05 U/mL of thrombin in comparison with healthy volunteers (Figure 2A). Platelets from all subjects were similarly efficient at releasing the inflammatory mediators when higher concentrations of thrombin were used, suggesting that platelets are a source of inflammatory mediators and may be prone to release certain inflammatory molecules during SARS-CoV-2 infection.
Figure 2.
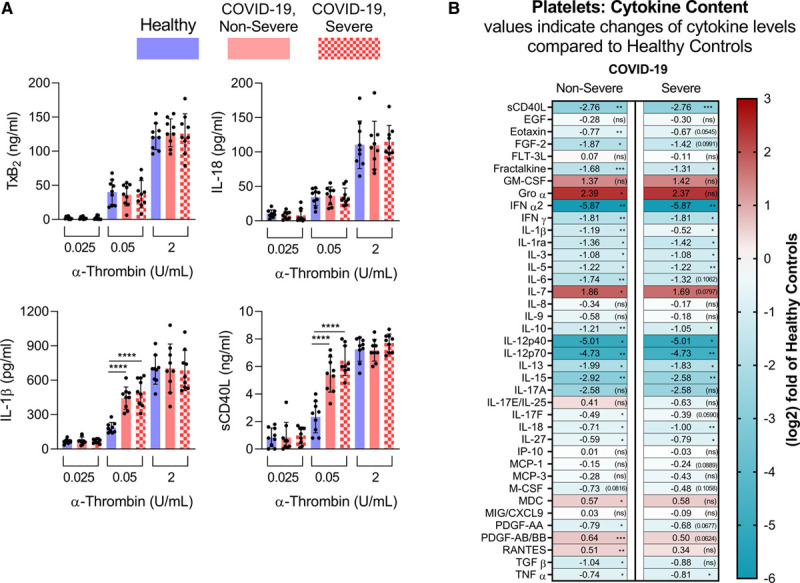
Platelets are prone to produce and release inflammatory molecules in patients with coronavirus disease 2019 (COVID-19). A, Platelets from healthy controls (n=9), COVID-19 nonsevere (n=9), and COVID-19 severe (n=9) patients were stimulated for 5 min at room-temperature with 0.025, 0.05, or 2 U/mL of α-thrombin. Thromboxane B2 (TxB2), IL (interleukin)-18, IL-1β, and soluble CD40 ligand (sCD40L) production was evaluated. Data are represented as mean±SD. Statistical analysis: Data were normally distributed (Shapiro-Wilk test). One-way ANOVA with subsequent Sidak multiple comparisons test. ***P<0.001 and ****P<0.0001. B, Heat map visualization of 39 cytokine/chemokine expression profiles in plasma of patients with COVID-19 nonsevere (median of n=10) and severe (median of n=9) relatively to the heathy controls (median of n=10). Cytokine/chemokine expression is represented as a (log2) fold change relative to healthy controls. The numbers in each part represent the change and statistical significance (in brackets) and the color codes refer to red for increased expression and blue for decreased expression. Absolute cytokine/chemokine values (pg/mL) and details on the statistical analysis are shown in Figure VII in the Data Supplement. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Because they are anucleated, the platelet cytokine content can mirror that of megakaryocytes, or may be regulated by translation upon platelet activation.63,64 However, nothing is known regarding the platelet cytokine content in COVID-19. Thus, we monitored cytokines in lysates prepared using platelets from patients with COVID-19 and included healthy volunteers as controls. While a few cytokines were below the detection limit (G-CSF, IL-1α, IL-2, IL-4, IL-5, IL-6, IL-10, IL-22, TGF-α, and VEGF), we detected 39 cytokines/growth-factors in platelets from healthy volunteers, including sCD40L, IFN α and γ, and IL-1β (Figure 2B and Figure VII in the Data Supplement), in agreement with their important cytokine content. Of note, we found significantly reduced levels for 22 cytokines/growth-factors (nonsevere disease) and 16 cytokines/growth-factors (severe disease), including cytokines relevant to virus and inflammatory responses (eg, sCD40L, eotaxin, IFN α and γ, IL-1β, TNF α and β; Figure 2B and Figure VII in the Data Supplement) in COVID-19. Together, the data show that platelets are sensitized to release certain cytokine cargo in COVID-19.
Platelet Degranulation in COVID-19
Platelet alpha- and dense-granule content, determined by the assessment of PF4 (platelet factor 4) and serotonin respectively, was examined in platelets. We found a reduction in both PF4 and serotonin in platelets from patients with nonsevere and severe COVID-19 in comparison with healthy volunteers (Figure 3A). Moreover, robust increases in PF4 and serotonin levels were measured in plasma from patients with COVID-19, independent of disease severity (Figure 3B), which might point to platelets degranulation during SARS-CoV-2 infection.
Figure 3.
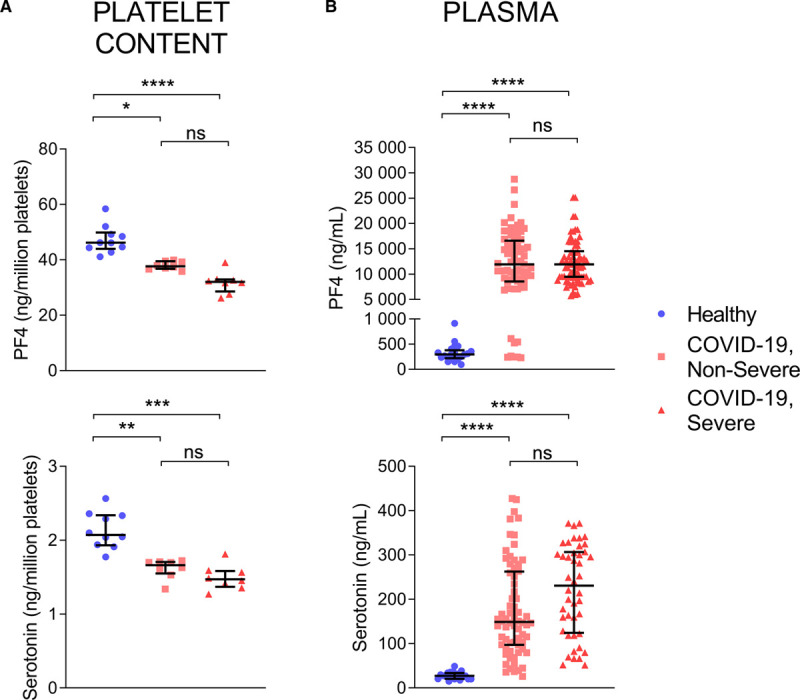
Platelets are degranulated in patients with coronavirus disease 2019 (COVID-19). Markers of platelet degranulation (PF4 [platelet factor 4] for alpha granules and serotonin for dense granules) were evaluated in plasma of patients with COVID-19. Concentrations of PF4 (upper) and serotonin (lower) were measured in platelets (A) and plasma (B) from healthy controls, patients with COVID-19 nonsevere and COVID-19 severe. For platelet content, values were expressed as ng per million platelets. Data are represented as median with interquartile range (IQR). Statistical analysis: ROUT method identified three outliers for PF4 and serotonin (platelet content), which were thus excluded from the analysis. Data were not normally distributed (Shapiro-Wilk test). Kruskal-Wallis test with subsequent Dunn multiple comparisons test. **P<0.01, ***P<0.001 ****P<0.0001, ns (nonsignificant). PF4: healthy controls (n=18 for plasma, n=10 for platelet content), COVID-19 nonsevere (n=71 for plasma, n=10 for platelet content), COVID-19 severe (plasma n=44, platelet content n=9). Serotonin: healthy controls (n=10 for plasma, n=10 for platelet content), COVID-19 nonsevere (n=18 for plasma, n=9 for platelet content), COVID-19 severe (plasma n=14, platelet content n=8).
Platelet Extracellular Vesicles in COVID-19
EV released by platelets can participate in both inflammation and the coagulation process due to the exposure of phosphatidylserine.33,65,66 However, it is currently unknown whether EV are produced in COVID-19. EV smaller than 1 µm and of platelet origin identified by the surface expression of CD41 were detected by high sensitivity flow cytometry. To determine whether EV expressed surface phosphatidylserine, we included annexin V conjugated with a fluorescent probe in the analyses. EV were sensitive to detergent treatment, which confirmed their membrane moiety. Moreover, no annexin V+ EV were detected when Ca2+ ions were chelated by ethylenediaminetetraacetic acid, which further ensured the specificity of the detection approach (Figure VIII in the Data Supplement). While the total number of platelet EV (exposing- and nonexposing phosphatidylserine) were significantly increased in both groups of patients with COVID-19 in comparison to healthy volunteers, the levels of phosphatidylserine-exposing platelet EV were only significantly increased in nonsevere forms of the disease (Figure 4A and 4B). Moreover, the levels of both types of EV were significantly lower in patients with severe compared with nonsevere COVID-19 (Figure 4A and 4B).
Figure 4.
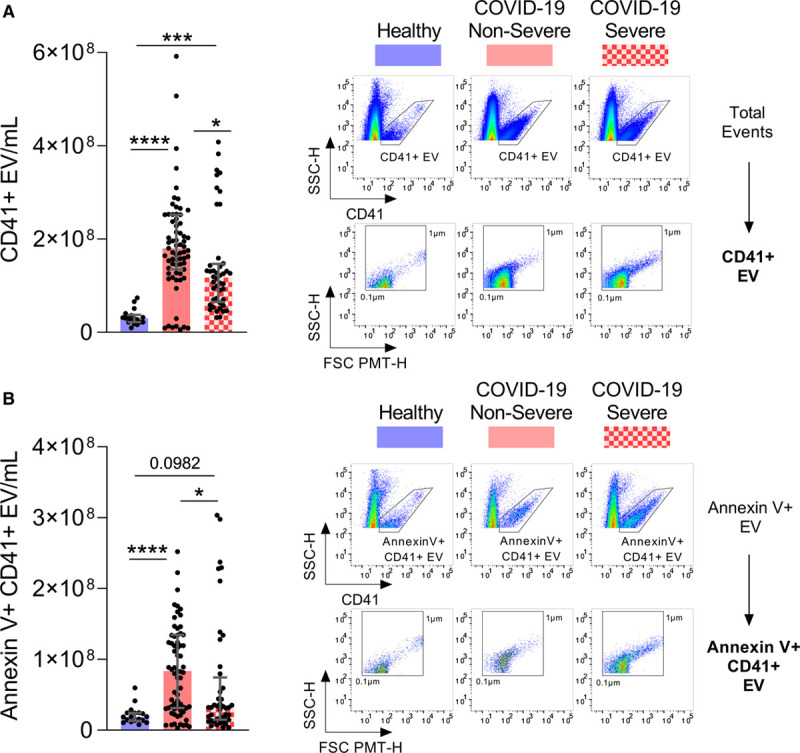
Platelet extracellular vesicles are released in patients with coronavirus disease 2019 (COVID-19). Circulating platelet extracellular vesicles (CD41+ extracellular vesicle [EV]) expressing phosphatidylserine or not were analyzed in plasma from healthy controls (n=18), patients with nonsevere (n=71) and severe COVID-19 (n=44). A, Total CD41+ EV were quantified (left) and representative scatter plots of CD41+ EV relative size and inner complexity are illustrated (right). B, Annexin V+ CD41+ EV were quantified (left) and representative scatter plots of AnV+CD41+ EV relative size are illustrated (right). The gating strategy is illustrated in Figure VIII in the Data Supplement. Samples with EV-concentrations close to the median of the whole population for each group were selected for representation. Data are represented as median with interquartile range (IQR). Statistical analysis: Data were not normally distributed (Shapiro-Wilk test). Kruskal-Wallis test with subsequent Dunn multiple comparisons test. FSC indicates forward scatter; and SSC, sideward scatter. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
Normalization of EV-numbers to platelet concentrations showed significantly increased concentrations of both types of EV (CD41+EV and annexin V+ CD41+ EV) per platelet in both patients with nonsevere and severe COVID-19 compared with healthy controls (Figure IX in the Data Supplement), suggesting increased production of EV in this disease. The concentrations of platelet EV, however, could not be solely explained by the number of circulating platelets. There was a small but significant positive correlation between CD41+ EV and the number of circulating platelets (r=0.19, P=0.0420), but we observed the absence of a correlation between the number of circulating platelets and annexin V+CD41+ EV (r=0.09, P=0.3397; Table VI in the Data Supplement). The data might suggest that the number of circulating phosphatidylserine-exposing platelet EV may be reduced in patients with the most severe inflammatory status, potentially due to their reduced production or their accumulation in certain organs or tissues.
Correlative Studies Implicating the Measured Blood and Platelet Parameters
Given that concentrations of D-dimers, CRP, platelets, lymphocytes, platelet/lymphocyte ratio, ALT, AST, LDH, plasma PF4 and serotonin, platelet EV (CD41+ EV), and phosphatidylserine-exposing platelet EV (annexin V+CD41+ EV), were measured for all patients, we evaluated potential correlations between these variables and disease outcomes. As measured outcomes, we took in account (1) the duration of hospitalization, (2) time before patients were tested negative for the presence of SARS-CoV-2 RdRp or E genes RNA, and (3) mortality. For these analyses, we confirmed the absence of confounding effects due to race or self-identified ethnicity, and sex (Tables VII and VIII in the Data Supplement).
We observed a significant positive correlation for both CRP and LDH levels measured on admission with the hospitalization duration (CRP: r=0.35, P=0.0002; LDH: r=0.40, P≤0.0001), and the time until the patient was tested negative for the presence of virus (CRP: r=0.39, P≤0.0001; LDH: r=0.37, P≤0.0001; Table 2). Moreover, D-dimers, CRP, and LDH levels measured on admission were significantly associated with mortality (Table IX in the Data Supplement), supporting notions of levels of D-dimers,17,19,21,67 CRP,56,68 and LDH56,67 as predictors of critical clinical outcome in COVID-19. Furthermore, age was found to be significantly associated with mortality (Table IX in the Data Supplement), but did not correlate with the duration of hospitalization (r=0.10, P=0.2856; Table 2).
Table 2.
Pearson Correlation of Clinical Parameters (on Admission) and Outcome (Duration of Hospitalization; Time Until Tested Negative for SARS-CoV-2)
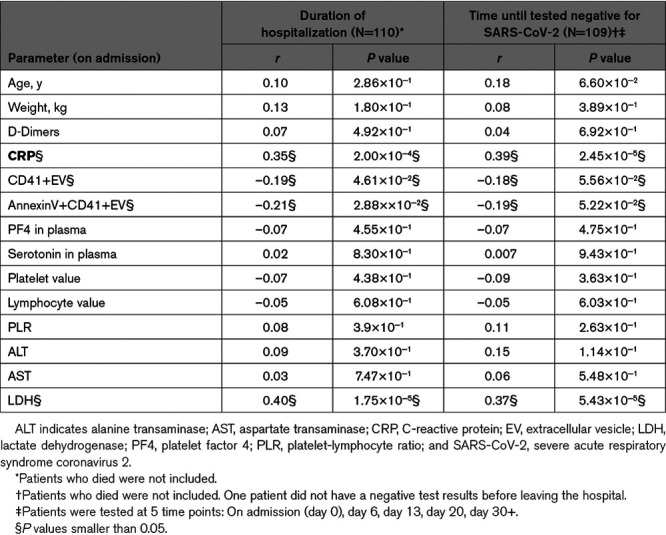
Surprisingly, D-dimers did not correlate with any of the measured platelet parameters (Table X in the Data Supplement). In contrast, we found modest but negative correlations between levels of platelet EV and CRP (CD41+EV: r=−0.19, P=0.0401; annexin V+CD41+EV r=−0.17, P=0.0620) and LDH (CD41+EV: r=−0.27, P=0.0040; annexin V+CD41+EV r=−0.21, P=0.0256; Table X in the Data Supplement). Moreover, we found modest but significant negative correlations between levels of CD41+ EV and annexinV+CD41+ EV with hospitalization duration (CD41+EV: r=−0.19, P=0.0461; annexin V+CD41+EV r=−0.21, P=0.0288; Table 2).
Platelets Are Hyperactivated in COVID-19
Platelet activation results from a complex balance of stimulatory and inhibitory signaling pathways involving several PKC (protein kinase C) isoforms expressed in human platelets. Among these kinases, PKCδ is a key regulator of platelet granule secretion, activation, and aggregation activity.69,70 To determine whether PKCδ is involved in platelet activation during this infection, platelets collected from severe and nonsevere patients were stimulated (or not) with a low dose of α-thrombin (0.05 U/mL), lysed and then analyzed by immunoblotting for the presence of phosphorylated-PKCδ Tyr311, a phosphorylation site that potentiates the activity of the enzyme.71 We found that PKCδ phosphorylation on Tyr311 residue was increased in response to a low dose of α-thrombin in patients with severe (P<0.05) and nonsevere (P=0.0638) COVID-19, while phosphorylation was undetectable in controls (Figure 5), thus suggesting that platelet activation signaling pathways may be sensitized in the disease.
Figure 5.
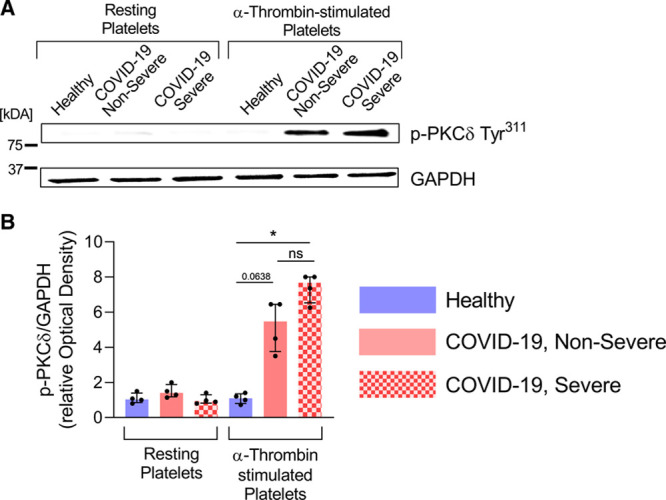
PKCδ (protein kinase C) phosphorylation is increased in platelets of patients with coronavirus disease 2019 (COVID-19). PKCδ phosphorylation on Tyr311 residue is increased in response to α-thrombin in patients with severe and nonsevere COVID-19. Platelets were stimulated (or not) with 0.05 U/mL of α-thrombin for 5 min at room temperature. Platelet lysates were analyzed by SDS-PAGE for P-PKCδ Tyr311. GAPDH was evaluated in each condition. A, Immunoblot representative of 4 donors. B, Densitometric analysis of P-PKCδ Tyr311 normalized to GAPDH was performed, data were expressed as relative Optical Density (n=4). Data are represented as median with interquartile range (IQR). Statistical analysis: Kruskal-Wallis test with subsequent Dunn multiple comparisons test. *P<0.05.
The relationship between platelet activation and the severity of COVID-19 was then investigated by functional assays. Platelets from patients with severe and nonsevere COVID-19 were collected and analyzed by optical aggregometry under arterial flow conditions using various doses of α-thrombin. Platelets exposed to suboptimal concentrations of α-thrombin (0.05 U/mL) were highly activated (≥2.5-fold increase) in both severe and nonsevere patients compared with controls (Figure 6A and 6B). When platelets from all groups were stimulated with a higher concentration of α-thrombin (2 U/mL), no significant differences were observed, pointing to a lower platelet stimulation threshold in patients with COVID-19 (Figure 6A and 6B). Moreover, the formation of platelet aggregates was visualized by rhodamine-based immunofluorescence on a collagen-coated surface under flow conditions, and the ratio of adherent platelets to total platelets (activated versus nonactivated platelets) was calculated. The number of adherent platelets was significantly higher in severe patients compared with nonsevere patients and controls (Figure 6C), suggesting that platelets are more prone to clotting in severe disease.
Figure 6.
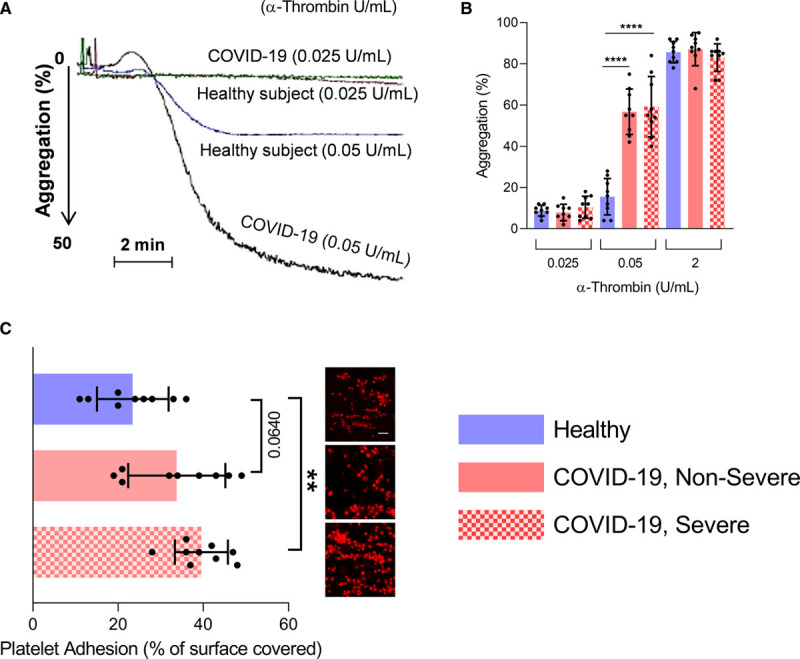
Platelets are prone to aggregation in patients with coronavirus disease 2019 (COVID-19). Platelet aggregation and adhesion were evaluated in healthy donors, patients with nonsevere and severe COVID-19 (n=9). A and B, Platelets were stimulated with 0.025, 0.05, or 2 U/mL α-thrombin. A, Representation of light transmission curve of platelets aggregation from healthy controls and patients with COVID-19. B, Quantification (%) of maximal platelet aggregation in healthy controls (n=9), patients with COVID-19 nonsevere (n=9) or COVID-19 severe (n=9). C, Platelet adhesion on collagen was evaluated under flow condition after 5 min. Data are presented as percentage of surface covered by platelets. A representative image for each condition is illustrated (Scale bar=50 µm). Data points close to the mean of each population for each group were selected for representative images. Data are represented as mean±SD. Statistical analysis: Data were normally distributed (Shapiro-Wilk test). One-way ANOVA with subsequent Sidak multiple comparisons test. **P<0.001, ****P<0.0001
The results are visualized in the graphical abstract.
Discussion
Both inflammation and thrombosis are clinical manifestations observed during SARS-CoV-2 infection. They can be lethal, and the understanding of cellular and molecular effectors in COVID-19 may reveal novel therapeutic approaches, critical for the treatment of patients while vaccines are still lacking. Platelets can interact with viruses and can participate in both inflammation and thrombosis. Thus, for this study, we examined platelets in a cohort of patients with nonsevere and severe COVID-19 with confirmed lower range (but not thrombocytopenic) platelet counts, in agreement with the current literature.23–27 Our findings identify SARS-CoV-2 RNA associated with patient platelets and reveal that platelets express proinflammatory molecules and are hyperactivated in COVID-19.
Of interest, was the detection of SARS-CoV-2 RNA associated with platelets from some patients with COVID-19. The presence of viral RNA in platelet endosomal compartments may activate platelet TLR-7, a process that occurs in cases of influenza and encephalomyocarditis virus infections.50,72 However, it remains to be established whether platelets represent bona fide target cells susceptible to infection, or whether platelets might have captured circulating SARS-CoV-2 RNA molecules. Spike glycoprotein S mediates SARS-CoV-2 attachment to the cellular receptor ACE2 and entry by fusion of the viral envelope with cell membranes.73 Studies have previously highlighted that conflictual results can be obtained depending on the choice of primers used in the assessment of ACE2 mRNA by quantitative PCR.74 Attempts to detect ACE2 mRNA using intron-spanning primers in nucleic acid preparations isolated from enriched platelets (97.04±1.29% pure) yielded negative results (cycle threshold value 38.16±3.00), suggesting the platelets either express ACE2 mRNA below the assay detection limit or express no ACE2 mRNA (Figure XA in the Data Supplement), in agreement with findings from others.75 These findings contrast with other attempts to detect ACE2 using commercially available nonintron spanning primers in the same platelet preparations, and which led to successful detection of ACE2 in platelets from both healthy and COVID-19 individuals (Figure XB in the Data Supplement). However, genomic DNA was identified in these samples despite the fact that platelets are anucleated (Figure XC in the Data Supplement), which might explain the detection of ACE2 in these experimental conditions. Given the high purity of the platelet preparations examined, we thus speculate that cell free genomic DNA (eg, in extracellular vesicles, such as apoptotic bodies, or DNA in complexes with proteins) might interact with platelets. However, the platelet proteome does not necessarily mirror the platelet transcriptome. For instance, platelets can adsorb or internalize plasma proteins, such as fibrinogen, which is mainly produced by the liver and is captured and stored in platelet alpha granules.76 Although ACE2 protein can be shed from cells77 and might thus be transferred to platelets, we did not detect any significant ACE2 expression on platelets, as assessed using immunofluorescence (Figure XD in the Data Supplement), consistent with its absence in documented platelet granule proteome.78 We nonetheless do not completely rule out the potential involvement of ACE2 in platelet and SARS-CoV-2 interactions in vivo. Indeed, we found that age was a variable associated with presence of the SARS-CoV-2 RNA in platelets (Table V in the Data Supplement), which was only detected in ≈20% of the tested infected individuals. Future studies specifically designed to thoroughly evaluate ACE2 expression in platelets in larger samples of individuals, taking in consideration, for instance, age, sex, race, comorbidities, and the presence of SARS-Cov-2 in platelets may definitively reveal whether ACE2 expression by platelets may be relevant to COVID-19 pathogenesis.
Influenza virus and herpes simplex virus-1 can trigger platelet degranulation when antibodies opsonize viral particles, and thereby activate platelet FcyRIIA.53,54 While antibodies specifically directed at SARS-CoV-2 are not expected in most individuals at an early stage of the pathogenesis, they might be produced at a later stage of infection, or prevailing cross-reacting antibodies against other more common coronaviruses that generate minor cold symptoms in humans (229E, NL63, OC43, and HKU1) may suffice to form immune complexes and activate platelet FcyRIIA.4,5 Thus, differences in antibody profiles, especially in neutralizing antibodies, among patients with severe and nonsevere COVID-19 may contribute to platelet interaction with the virus, and to the overwhelming inflammation. However, with the dissemination of the SARS-CoV-2 virus in the population, possible reinfections and vaccination, the activation of platelet FcyRIIA due to the presence of SARS-CoV-2 antibodies may be even more relevant in subsequent years.
SARS-CoV-2 entry into endothelial cells, which express ACE2, and the loss of endothelium integrity may favor recruitment of circulating platelets to sites of infection, their activation and degranulation, suggesting that the inhibition of endothelial cell activation may improve disease outcome.79 Megakaryocytes in lungs bear an immune profile distinct from the megakaryocytes found in bone marrow.31 While this remains speculative, megakaryocytes in lungs, a privileged location that might support their infection by respiratory viruses, might be susceptible to SARS-CoV-2 infection and might transfer viral RNA or viral particles as proplatelets are produced. Moreover, dengue virus47 can infect megakaryocytes, which impacts the megakaryocyte transcriptome.48 Hence, platelets produced by infected megakaryocytes, and by megakaryocytes in the inflamed environment, may present a different transcriptome, which may explain their potency at producing inflammatory cytokines, such as IL-1, when activated by α-thrombin.
From platelet degranulation and cytokine release analyses, we propose that platelets can contribute to the cytokine storm reported in COVID-19. A limited number of cytokines (Groα, IL-7, MDC, PDGF-AB/BB, RANTES [regulated upon activation normal T cell expressed and secreted]) was found to be increased in platelets in COVID-19, which may be either due to (1) overexpression by megakaryocytes and subsequent transfer to newly formed platelets, (2) overexpression by platelets through increased translation, or (3) increased capture of these cytokines from blood. On the contrary, some cytokines or growth-factors (eg, eotaxin, IFNγ, IL-1β, TGFβ) related to viral responses were significantly decreased in platelets, but increased in the blood, suggesting that platelets may contribute to the release of these molecules in the disease. While molecules such as CD40L, PF4, and serotonin are abundant in platelet granules and are released upon degranulation, cytokines such as IL-1 may be produced de novo following platelet activation.63 Therefore, the presence of CD40L, PF4, and serotonin are clear indicators of platelet degranulation in vivo. These molecules secreted by platelets can have effects on numerous cell types as well as the vasculature. Serotonin for instance, which we found to be abundantly released in this disease, can directly impact blood vessel integrity and promote leukocyte recruitment and cytokine release.53,80 Of potential relevance, soluble CD40L, which we found to be released by platelets in COVID-19, can contribute to both activation of CD40 bearing cells and thrombosis by stabilizing glycoprotein αIIbβ3.81 This may not be unique to SARS-CoV-2 as dengue virus also induces the release of CD40L from human platelets.82
Platelet EV convey molecules from the mother platelets.65 They can transport platelet-derived cytokines, as well as other proinflammatory molecules such as damage associated molecular patterns.65,83,84 We made the original observation that platelet EV are significantly increased in the circulation in this disease. This increase could be due to an increased production by megakaryocytes or platelets, the reduction of their clearance, or a combination of both effects. Surprisingly, levels of platelet EV were significantly lower in severe disease as compared with nonsevere disease. Moreover, phosphatidylserine-exposing platelet EV were increased in nonsevere patients, but they were not significantly elevated in comparison to nonsevere COVID-19 nor healthy volunteers in severe COVID-19. We observed that the reduced platelet number in more severe patients could not fully explain the reduced levels of phosphatidylserine-exposing platelet EV (Table VI in the Data Supplement), which might point to the consumption or sequestration of these particular subtypes of EV. Given that both CRP and LDH, but not D-dimers, were inversely associated with concentrations of platelet-derived EV, the data further suggest that inflammation, rather than thrombosis, might affect production, consumption or sequestration of platelet-derived EV.
At the time of revising this article, 2 independent studies reported that platelets are hyperactivated and form aggregates with leukocytes in COVID-19.75,85 Manne et al75 further observed gene expression changes in pathways associated with protein ubiquitination, antigen presentation and mitochondrial dysfunction in COVID-19, which added to our observations regarding the altered cytokine and growth factor contents, supports the notion that platelets may respond to SARS-CoV-2 infection by the modulation of its transcriptome and the secretion of inflammatory molecules. Moreover, mRNA from the SARS-CoV-2 N1 gene could be identified in platelets of a subset of patients, an observation we also made in our cohort.75
Hottz et al,85 on the contrary, demonstrated that activated platelets could associate with monocytes, which induced tissue factor expression by the latter. Intriguingly, they found that an unidentified plasma component present in COVID-19 could activate platelets, and that monocyte activation by platelets implicates P-selectin and αIIbβ3.85 Platelet activation, assessed by surface expression of P-selectin and CD63, was, however, only evident in patients with severe COVID-19 and strongly correlated with levels of D-dimers, which contrasts with our findings. In our cohort, D-dimers, as well as CRP and LDH, were indicators of more severe disease, consistent with the observations made by others.17,19,21,56,67,68 With the exception of platelet EV, which levels inversely correlated with CRP and LDH, none of the measured platelet-derived mediators correlated with D-dimers or the markers of inflammation. The different experimental approaches to assess activation and the relatively low number of nonsevere patients analyzed (4 outpatients with self-limiting COVID-19 syndrome and 2 asymptomatic subjects)85 possibly explain the apparent discrepancy between their study and ours. In our study, 71 patients with nonsevere disease were included, who all presented symptoms and were hospitalized. As platelets were not activated in patients with asymptomatic and self-limiting COVID-19,85 but were highly activated in our cohort of nonsevere and severe patients, it points to a potential role of platelets through the different stages of the pathogenesis. Moreover, that D-dimers failed to correlate with platelet counts or markers of platelet degranulation may further suggest that platelets, in the great majority of patients with symptomatic COVID-19, can be activated independently of thrombosis.
Using functional assays, we observed that platelets from patients with COVID-19 are hyperresponsive. Platelets were sensitized to release inflammatory cytokines, and they aggregated and adhered to a collagen surface more efficiently when originating from patients with COVID-19. As such, hyperactivation of platelets may contribute to the disease pathogenesis through both the release of inflammatory mediators and thrombosis. In summary, our study has revealed that platelets are hyperactivated in COVID-19 and may contribute to the observed systemic inflammatory response and thrombotic events observed in this disease. The blockade of platelet activation pathways may improve the outcomes in this disease.
Acknowledgments
We are grateful to the patients and the members of the coronavirus disease 2019 (COVID-19) laboratory of Cheikh Zaid Hospital in Rabat for their technical assistance with the RT-PCR tests, laboratory blood tests and the patients’ clinical data collection. We thank Anne-Sophie Julien from the Department of Mathematics and Statistics at Université Laval for support with the statistical analyses. Y. Zaid, F. Guessous, and E. Boilard conceived the initial concept and designed the study. F. Puhm, I. Allaeys, A. Naya, M. Oudghiri, L. Khalki, Y. Limami, N. Zaid, K. Sadki, R. Ben El Haj, B. Belefquih, A. Cheikh, L. Flamand, and Y. Cherrah participated to design the study, performed experiments and were involved in the data extraction. W. Mahir, L. Belayachi, A. Benouda, and M.-A. Langlois contributed critical reagents, biospecimens and instruments. E. Boilard, Y. Zaid, F. Puhm, I. Allaeys, L. Flamand, and F. Guessous wrote the article, and all authors read and approved the final article.
Sources of Funding
This study was supported by Cheikh Zaid Foundation and, a New Frontier Research Fund (NFRN-2019-00004) awarded to LF and EB and a Foundation grant from the Canadian Institutes of Health Research to EB. E. Boilard is recipient of senior award from the Fonds de Recherche du Québec en Santé (FRQS). F. Puhm is recipient of a postdoctoral fellowship from the FRQS. This study was approved by the Local Ethics Committee of Cheikh Zaid Hospital, Rabat, Morocco. Project: CEFCZ/PR/2020-PR04.
Disclosures
None.
Supplemental Materials
Expanded Materials & Methods
Online Figures I–X
Online Tables I–X
Supplementary Material
Footnotes
Nonstandard Abbreviations and Acronyms
- ACE2
- angiotensin-converting enzyme 2
- ALT
- aminotransferase
- AST
- aspartate aminotransferase
- COVID-19
- coronavirus disease 2019
- CRP
- C-reactive protein
- EV
- extracellular vesicle
- G-CSF
- granulocyte-colony stimulating factor
- IL
- interleukin
- IQR
- interquartile range
- LDH
- lactate dehydrogenase
- PF4
- platelet factor 4
- PKC
- protein kinase C
- SARS-CoV-2
- severe acute respiratory syndrome coronavirus 2
- TLR
- Toll-like receptor
- TNF
- tumor necrosis factor
Y.Z. and F.P. contributed equally to this article.
For Sources of Funding and Disclosures, see page 1416.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.120.317703.
Contributor Information
Younes Zaid, Email: y.zaid@um5s.net.ma.
Florian Puhm, Email: florian.puhm@crchudequebec.ulaval.ca.
Isabelle Allaeys, Email: Isabelle.Allaeys@crchudequebec.ulaval.ca.
Abdallah Naya, Email: anaya@mabiotech.com.
Mounia Oudghiri, Email: mouniaoudghiri@gmail.com.
Loubna Khalki, Email: lkhalki@um6ss.ma.
Youness Limami, Email: limami@hotmail.com.
Nabil Zaid, Email: zaidnabil@hotmail.com.
Khalid Sadki, Email: ksadki@fsr.ac.ma.
Rafiqua Ben El Haj, Email: b.rafiqua@gmail.com.
Wissal Mahir, Email: wissal.maher@gmail.com.
Lamiae Belayachi, Email: belayachi.lamiae@gmail.com.
Bouchra Belefquih, Email: b.belefquih@biolife.ma.
Amina Benouda, Email: benoudaamina@yahoo.fr.
Amine Cheikh, Email: cheikh.amine@gmail.com.
Marc-André Langlois, Email: langlois@uottawa.ca.
Yahia Cherrah, Email: cherrahy@yahoo.fr.
Louis Flamand, Email: louis.flamand@crchudequebec.ulaval.ca.
Novelty and Significance
What Is Known?
Inflammation, hypercoagulation, and thrombosis are hallmarks of severe coronavirus disease 2019 (COVID-19).
Platelets are chiefly implicated in thrombosis.
Platelets can interact with different microbes, including viruses.
Platelets are a major source of inflammatory mediators.
What New Information Does This Article Contribute?
Severe acute respiratory syndrome coronavirus 2 RNA molecules can be associated with human platelets.
Platelets are hyperactivated in COVID-19, both in nonsevere and severe forms of the disease.
Platelets have enhanced adhesion properties, molecules from their alpha and dense granule are elevated in blood, and they are a source of inflammatory cytokines.
Platelet extracellular vesicles are elevated in blood of patients with COVID-19.
None of the measured markers of platelet activation were associated with levels of D-dimers.
In addition to the overwhelming inflammation that occurs in the setting of severe acute respiratory syndrome coronavirus 2, hypercoagulation and thrombosis are now recognized hallmark of COVID-19 and contribute to the lethality of disease. Platelets are implicated in thrombosis and can interact with different microbes including viruses. Moreover, platelets are a major source of inflammatory mediators. In this study, we found that severe acute respiratory syndrome coronavirus 2 RNA molecules are associated with human platelets. Moreover, platelets are hyperactivated in COVID-19, both in nonsevere and severe forms of the disease with enhanced adhesion properties and enhanced inflammatory cytokines. Thus, these data suggest that platelets participate in COVID-19 infection and may mediate thrombotic disease.
References
- 1.Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20:363–374. doi: 10.1038/s41577-020-0311-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, Liu L, Shan H, Lei CL, Hui DSC, et al. ; China Medical Treatment Expert Group for Covid-19. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. 2020https://covid19.who.int CdC-WhOpoM. Accessed September 14, 2020
- 4.Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu H, Zhong L, Deng J, Peng J, Dan H, Zeng X, Li T, Chen Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12:8 doi: 10.1038/s41368-020-0074-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pedersen SF, Ho YC. SARS-CoV-2: a storm is raging. J Clin Invest. 2020;130:2202–2205. doi: 10.1172/JCI137647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–1069. doi: 10.1001/jama.2020.1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ; HLH Across Speciality Collaboration, UK. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meredith Wadman JC-F, Jocelyn K, Catherine M. How does coronavirus kill? Clinicians trace a ferocious rampage through the body, from brain to toes. Science Mag. In press
- 13.Vulliamy P, Jacob S, Davenport RA. Acute aorto-iliac and mesenteric arterial thromboses as presenting features of COVID-19. Br J Haematol. 2020;189:1053–1054. doi: 10.1111/bjh.16760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135:2033–2040. doi: 10.1182/blood.2020006000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Llitjos JF, Leclerc M, Chochois C, Monsallier JM, Ramakers M, Auvray M, Merouani K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost. 2020;18:1743–1746. doi: 10.1111/jth.14869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, Kucher N, Studt JD, Sacco C, Alexia B, et al. ; Humanitas COVID-19 Task Force. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res. 2020;191:9–14. doi: 10.1016/j.thromres.2020.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JCT, Fogerty AE, Waheed A, Goodarzi K, Bendapudi PK, Bornikova L, Gupta S, et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood. 2020;136:489–500. doi: 10.1182/blood.2020006520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McFadyen JD, Stevens H, Peter K. The emerging threat of (Micro) thrombosis in COVID-19 and its therapeutic implications. Circ Res. 2020;127:571–587. doi: 10.1161/CIRCRESAHA.120.317447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844–847. doi: 10.1111/jth.14768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, Der Nigoghossian C, Ageno W, Madjid M, Guo Y, et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up. J Am Coll Cardiol. 2020;75:2950–2973. doi: 10.1016/j.jacc.2020.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7:e438–e444. doi: 10.1016/S2352-3026(20)30145-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arentz M, Yim E, Klaff L, Lokhandwala S, Riedo FX, Chong M, Lee M. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington State. JAMA. 2020;323:1612–1614. doi: 10.1001/jama.2020.4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, Shang Y. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost. 2020;18:1469–1472. doi: 10.1111/jth.14848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. 2020;506:145–148. doi: 10.1016/j.cca.2020.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lippi G, Favaloro EJ. D-dimer is associated with severity of coronavirus disease 2019: a pooled analysis. Thromb Haemost. 2020;120:876–878. doi: 10.1055/s-0040-1709650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu P, Zhou Q, Xu J. Mechanism of thrombocytopenia in COVID-19 patients. Ann Hematol. 2020;99:1205–1208. doi: 10.1007/s00277-020-04019-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Sun W, Guo Y, Chen L, Zhang L, Zhao S, Long D, Yu L. Association between platelet parameters and mortality in coronavirus disease 2019: retrospective cohort study. Platelets. 2020;31:490–496. doi: 10.1080/09537104.2020.1754383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao D, Zhou F, Luo L, Xu M, Wang H, Xia J, Gao Y, Cai L, Wang Z, Yin P, et al. Haematological characteristics and risk factors in the classification and prognosis evaluation of COVID-19: a retrospective cohort study. Lancet Haematol. 2020;7:e671–e678. doi: 10.1016/S2352-3026(20)30217-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts HR, Hoffman M, Monroe DM. A cell-based model of thrombin generation. Semin Thromb Hemost. 2006;32suppl 132–38. doi: 10.1055/s-2006-939552 [DOI] [PubMed] [Google Scholar]
- 30.Machlus KR, Italiano JE., Jr. The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol. 2013;201:785–796. doi: 10.1083/jcb.201304054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544:105–109. doi: 10.1038/nature21706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–2494. doi: 10.1056/NEJMra071014 [DOI] [PubMed] [Google Scholar]
- 33.Ridger VC, Boulanger CM, Angelillo-Scherrer A, Badimon L, Blanc-Brude O, Bochaton-Piallat ML, Boilard E, Buzas EI, Caporali A, Dignat-George F, et al. Microvesicles in vascular homeostasis and diseases. Position paper of the European Society of Cardiology (ESC) working group on atherosclerosis and vascular biology. Thromb Haemost. 2017;117:1296–1316. doi: 10.1160/TH16-12-0943 [DOI] [PubMed] [Google Scholar]
- 34.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264–274. doi: 10.1038/nri2956 [DOI] [PubMed] [Google Scholar]
- 35.Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle cuisine: platelets served with inflammation. J Immunol. 2015;194:5579–5587. doi: 10.4049/jimmunol.1500259 [DOI] [PubMed] [Google Scholar]
- 36.Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123:2759–2767. doi: 10.1182/blood-2013-11-462432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li C, Li J, Li Y, Lang S, Yougbare I, Zhu G, Chen P, Ni H. Crosstalk between platelets and the immune system: old systems with new discoveries. Adv Hematol. 2012;2012:384685 doi: 10.1155/2012/384685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, Nácher M, Pitaval C, Radovanovic I, Fukui Y, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346:1234–1238. doi: 10.1126/science.1256478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imhof BA, Jemelin S, Ballet R, Vesin C, Schapira M, Karaca M, Emre Y. CCN1/CYR61-mediated meticulous patrolling by Ly6Clow monocytes fuels vascular inflammation. Proc Natl Acad Sci U S A. 2016;113:E4847–E4856. doi: 10.1073/pnas.1607710113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5:e138999 doi: 10.1172/jci.insight.138999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, Daßler-Plenker J, Guerci P, Huynh C, Knight JS, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. 2020;217:e20200652 doi: 10.1084/jem.20200652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, Mostyka M, Baxter-Stoltzfus A, Borczuk AC, Loda M, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136:1169–1179. doi: 10.1182/blood.2020007008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768–2776. doi: 10.1182/blood-2013-10-463646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Constantinescu-Bercu A, Grassi L, Frontini M, Salles C, II, Woollard K, Crawley JT. Activated alphaIIbbeta3 on platelets mediates flow-dependent NETosis via SLC44A2. Elife. 2020;9:e53353 doi: 10.7554/eLife.53353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, Weyrich AS. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154:485–490. doi: 10.1083/jcb.200105058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karas SP, Rosse WF, Kurlander RJ. Characterization of the IgG-Fc receptor on human platelets. Blood. 1982;60:1277–1282 [PubMed] [Google Scholar]
- 47.Vogt MB, Lahon A, Arya RP, Spencer Clinton JL, Rico-Hesse R. Dengue viruses infect human megakaryocytes, with probable clinical consequences. PLoS Negl Trop Dis. 2019;13:e0007837 doi: 10.1371/journal.pntd.0007837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell RA, Schwertz H, Hottz ED, Rowley JW, Manne BK, Washington AV, Hunter-Mellado R, Tolley ND, Christensen M, Eustes AS, et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood. 2019;133:2013–2026. doi: 10.1182/blood-2018-09-873984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simon AY, Sutherland MR, Pryzdial EL. Dengue virus binding and replication by platelets. Blood. 2015;126:378–385. doi: 10.1182/blood-2014-09-598029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019;10:1780 doi: 10.1038/s41467-019-09607-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jansen AJG, Spaan T, Low HZ, Di Iorio D, van den Brand J, Tieke M, Barendrecht A, Rohn K, van Amerongen G, Stittelaar K, et al. Influenza-induced thrombocytopenia is dependent on the subtype and sialoglycan receptor and increases with virus pathogenicity. Blood Adv. 2020;4:2967–2978. doi: 10.1182/bloodadvances.2020001640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lê VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, Adrian I, Errazuriz-Cerda E, Frasquilho S, Antunes L, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med. 2015;191:804–819. doi: 10.1164/rccm.201406-1031OC [DOI] [PubMed] [Google Scholar]
- 53.Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, Levesque T, Becker Y, Tessandier N, Melki I, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S A. 2018;115:E1550–E1559. doi: 10.1073/pnas.1720553115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boilard E, Paré G, Rousseau M, Cloutier N, Dubuc I, Lévesque T, Borgeat P, Flamand L. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood. 2014;123:2854–2863. doi: 10.1182/blood-2013-07-515536 [DOI] [PubMed] [Google Scholar]
- 55.Metlay JP, Waterer GW, Long AC, Anzueto A, Brozek J, Crothers K, Cooley LA, Dean NC, Fine MJ, Flanders SA, et al. Diagnosis and treatment of adults with community-acquired pneumonia. An official clinical practice guideline of the American thoracic society and infectious diseases society of America. Am J Respir Crit Care Med. 2019;200:e45–e67. doi: 10.1164/rccm.201908-1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poggiali E, Zaino D, Immovilli P, Rovero L, Losi G, Dacrema A, Nuccetelli M, Vadacca GB, Guidetti D, Vercelli A, et al. Lactate dehydrogenase and C-reactive protein as predictors of respiratory failure in CoVID-19 patients. Clin Chim Acta. 2020;509:135–138. doi: 10.1016/j.cca.2020.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786 [DOI] [PubMed] [Google Scholar]
- 58.Danilczyk U, Penninger JM. Angiotensin-converting enzyme II in the heart and the kidney. Circ Res. 2006;98:463–471. doi: 10.1161/01.RES.0000205761.22353.5f [DOI] [PubMed] [Google Scholar]
- 59.Chen Y, Chen L, Deng Q, Zhang G, Wu K, Ni L, Yang Y, Liu B, Wang W, Wei C, et al. The presence of SARS-CoV-2 RNA in the feces of COVID-19 patients. J Med Virol. 2020;92:833–840. doi: 10.1002/jmv.25825 [DOI] [PubMed] [Google Scholar]
- 60.Perry MJ, Arrington S, Neumann LM, Carrell D, Mores CN. It is currently unknown whether SARS-CoV-2 is viable in semen or whether COVID-19 damages sperm. Andrology. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–3777. doi: 10.1182/blood-2002-06-1887 [DOI] [PubMed] [Google Scholar]
- 62.Dolmatova EV, Wang K, Mandavilli R, Griendling KK. The effects of sepsis on endothelium and clinical implications. Cardiovasc Res. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122:379–391. doi: 10.1016/j.cell.2005.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang H, Lang S, Zhai Z, Li L, Kahr WHA, Chen P, Brkić J, Spring CM, Flick MJ, Degen JL, et al. Fibrinogen is required for maintenance of platelet intracellular and cell-surface P-selectin expression. Blood. 2009;114:425–436. doi: 10.1182/blood-2008-03-145821 [DOI] [PubMed] [Google Scholar]
- 65.Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and disease. Platelets. 2017;28:214–221. doi: 10.1080/09537104.2016.1265924 [DOI] [PubMed] [Google Scholar]
- 66.Owens AP, III, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–1297. doi: 10.1161/CIRCRESAHA.110.233056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li X, Xu S, Yu M, Wang K, Tao Y, Zhou Y, Shi J, Zhou M, Wu B, Yang Z, et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol. 2020;146:110–118. doi: 10.1016/j.jaci.2020.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, Xie C, Ma K, Shang K, Wang W, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. 2020;71:762–768. doi: 10.1093/cid/ciaa248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zaid Y, Senhaji N, Darif Y, Kojok K, Oudghiri M, Naya A. Distinctive roles of PKC delta isozyme in platelet function. Curr Res Transl Med. 2016;64:135–139. doi: 10.1016/j.retram.2016.05.001 [DOI] [PubMed] [Google Scholar]
- 70.Murugappan S, Tuluc F, Dorsam RT, Shankar H, Kunapuli SP. Differential role of protein kinase C delta isoform in agonist-induced dense granule secretion in human platelets. J Biol Chem. 2004;279:2360–2367. doi: 10.1074/jbc.M306960200 [DOI] [PubMed] [Google Scholar]
- 71.Hall KJ, Jones ML, Poole AW. Coincident regulation of PKCdelta in human platelets by phosphorylation of Tyr311 and Tyr565 and phospholipase C signalling. Biochem J. 2007;406:501–509. doi: 10.1042/BJ20070244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. 2014;124:791–802. doi: 10.1182/blood-2013-11-536003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buchholz UJ, Bukreyev A, Yang L, Lamirande EW, Murphy BR, Subbarao K, Collins PL. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc Natl Acad Sci U S A. 2004;101:9804–9809. doi: 10.1073/pnas.0403492101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma D, Chen CB, Jhanji V, Xu C, Yuan XL, Liang JJ, Huang Y, Cen LP, Ng TK. Expression of SARS-CoV-2 receptor ACE2 and TMPRSS2 in human primary conjunctival and pterygium cell lines and in mouse cornea. Eye (Lond). 2020;34:1212–1219. doi: 10.1038/s41433-020-0939-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, Petrey AC, Tolley ND, Guo L, Cody M, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136:1317–1329. doi: 10.1182/blood.2020007214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Princen HM, Moshage HJ, Emeis JJ, de Haard HJ, Nieuwenhuizen W, Yap SH. Fibrinogen fragments X, Y, D and E increase levels of plasma fibrinogen and liver mRNAs coding for fibrinogen polypeptides in rats. Thromb Haemost. 1985;53:212–215 [PubMed] [Google Scholar]
- 77.Wysocki J, Garcia-Halpin L, Ye M, Maier C, Sowers K, Burns KD, Batlle D. Regulation of urinary ACE2 in diabetic mice. Am J Physiol Renal Physiol. 2013;305:F600–F611. doi: 10.1152/ajprenal.00600.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zufferey A, Schvartz D, Nolli S, Reny JL, Sanchez JC, Fontana P. Characterization of the platelet granule proteome: evidence of the presence of MHC1 in alpha-granules. J Proteomics. 2014;101:130–140. doi: 10.1016/j.jprot.2014.02.008 [DOI] [PubMed] [Google Scholar]
- 79.Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado Del Pozo C, Prosper F, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181:905–913.e7. doi: 10.1016/j.cell.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, Cifuni SM, Mauler M, Cicko S, Bader M, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. 2013;121:1008–1015. doi: 10.1182/blood-2012-06-437392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.André P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD. CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med. 2002;8:247–252. doi: 10.1038/nm0302-247 [DOI] [PubMed] [Google Scholar]
- 82.Nunez-Avellaneda D, Mosso-Pani MA, Sanchez-Torres LE, Castro-Mussot ME, Corona-de la Pena NA, Salazar MI. Dengue virus induces the release of sCD40L and changes in levels of membranal CD42b and CD40L molecules in human platelets. Viruses. 2018;10:357 doi: 10.3390/v10070357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Paré A, Rousseau M, Naika GS, Lévesque T, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124:2173–2183. doi: 10.1182/blood-2014-05-573543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Della Valle P, Monno A, D’Alberti V, Maria Gasparri A, Franchini S, et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci Transl Med. 2018;10:eaao3089 doi: 10.1126/scitranslmed.aao3089 [DOI] [PubMed] [Google Scholar]
- 85.Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, Righy C, Franco S, Souza TML, Kurtz P, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 2020;136:1330–1341. doi: 10.1182/blood.2020007252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bou Khzam L, Hachem A, Zaid Y, Boulahya R, Mourad W, Merhi Y. Soluble CD40 ligand impairs the anti-platelet function of peripheral blood angiogenic outgrowth cells via increased production of reactive oxygen species. Thromb Haemost. 2013;109:940–947. doi: 10.1160/TH12-09-0679 [DOI] [PubMed] [Google Scholar]
- 87.Hachem A, Yacoub D, Zaid Y, Mourad W, Merhi Y. Involvement of nuclear factor κB in platelet CD40 signaling. Biochem Biophys Res Commun. 2012;425:58–63. doi: 10.1016/j.bbrc.2012.07.049 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.