


Abstract
Diverse classes of silencing small (s)RNAs operate via ARGONAUTE-family proteins within RNA-induced-silencing-complexes (RISCs). Here, we have streamlined various embodiments of a Q-sepharose-based RISC-purification method that relies on conserved biochemical properties of all ARGONAUTEs. We show, in multiple benchmarking assays, that the resulting 15-min benchtop extraction procedure allows simultaneous purification of all known classes of RISC-associated sRNAs without prior knowledge of the samples-intrinsic ARGONAUTE repertoires. Optimized under a user-friendly format, the method – coined ‘TraPR’ for Trans-kingdom, rapid, affordable Purification of RISCs – operates irrespectively of the organism, tissue, cell type or bio-fluid of interest, and scales to minute amounts of input material. The method is highly suited for direct profiling of silencing sRNAs, with TraPR-generated sequencing libraries outperforming those obtained via gold-standard procedures that require immunoprecipitations and/or lengthy polyacrylamide gel-selection. TraPR considerably improves the quality and consistency of silencing sRNA sample preparation including from notoriously difficult-to-handle tissues/bio-fluids such as starchy storage roots or mammalian plasma, and regardless of RNA contaminants or RNA degradation status of samples.
INTRODUCTION
Silencing small (s)RNAs guide ARGONAUTE (AGO)-like proteins to complementary RNA/DNA to promote enhanced mRNA turnover, translational repression, chromatin compaction or DNA elimination (1–3). AGO-bound sRNAs are part of often larger RNA-induced-silencing-complexes (RISCs) in vivo, of which they are the minimal, functional cores. Understanding the biology underpinned by silencing sRNAs requires their characterization, which is most readily achieved by their en masse sequencing from total RNA preparations. The gold-standard in this respect entails the size-separation and selection of silencing sRNAs from polyacrylamide gels prior to cloning and sequencing (4–6). However, this tedious procedure also leads to the selection/cloning of degradation products from unrelated RNAs invariably contaminating total RNA samples to various extents. Additionally, gel-selection procedures result in RNA losses, making it challenging for samples with limited input quantity (4–6).
As an alternative to total RNA sequencing, various AGO-purification techniques enable the specific isolation of functional i.e. AGO-loaded sRNA classes suitable for subsequent northern/RT-qPCR analyses or their global exploration via cloning and deep-sequencing (7,8). Purifying sRNA-loaded AGOs largely circumvents the issue of contaminating RNAs (e.g. abundant breakdown products of tRNAs, rRNAs or mRNAs). It requires, however, the availability of specific IP-graded antibodies against individual members of often extended AGO families. Even so, disparate antibody efficacies affect the comparability of IPs conducted between distinct AGOs. Furthermore, IP-based approaches require prior knowledge about which AGO(s) accumulate(s) in a given sample, inherently biasing de novo exploration of silencing sRNAs. Finally, AGO antibodies rarely cross-react, even in related species, ultimately confining IP usage mostly to a small number of established model organisms (7,8). ‘AGO-protein-Affinity-Purification-by-Peptides’ (AGO-APP) solves some of these caveats by exploiting GW182’s high affinity for mammalian AGO2 (9). GW182 typifies so called ‘AGO-hook’ proteins that convey specialized functions to some –albeit not all– GW-repeat-interacting AGOs (10,11). Nonetheless, the efficacy of AGO-APP, in which a short GW182-derived peptide is used to isolate AGO:sRNA complexes (9), remains variable in different organisms. This presumably reflects AGO-intrinsic features influencing AGO:GW interactions (11). For example, AGO-APP does only efficiently purify two out of nine functional AGOs in Arabidopsis (9). Due to their multi-step nature, AGO-IP and AGO-APP are lengthy and remain mostly specialist-geared, nor are they optimally adapted to the high-throughput of clinical investigation involving samples from large patient cohorts. This is particularly pressing for analyses conducted, for instance, on body fluids, which contain minute amounts of bona fide silencing sRNAs amongst larger quantities of other RNA contaminants (reviewed in (12)).
A fast, simple and reliable method is thus desirable to purify functional silencing sRNAs without prior knowledge of the sample's AGO composition and irrespectively of the organism, tissue, cell type or bio-fluid of interest. Ideally, such a method should enable consistent, high-performance purification of AGO:sRNA complexes (i.e. minimal RISCs) including from recalcitrant tissues or starting material of substandard quality or quantity. In conceiving such a method, we considered an anion-exchange chromatography process originally used to separate, from contaminating free nucleic acids, germline-enriched silencing sRNAs (piRNAs) in association with their AGO-like effector proteins called PIWI (13). This method and its various subsequent embodiments (13–16) use the property of Q-sepharose, a positively-charged anion-exchange matrix, to strongly adsorb RNPs with surface-exposed RNA or naked RNAs (such as degradation fragments) under the ionic conditions of a standard sample's lysate. This bound material can be only eluted via high salt washes. By contrast, silencing sRNAs are shielded from affinity with Q-sepharose due to their embedding into AGOs. Hence, RISCs typically flow through, or are eluted under mild salt concentrations (13). This approach was successfully used to isolate in vivo-extracted (13–16) or to clean in vitro-assembled minimal RISCs (17). The underlying methods, however, were not established as purification procedures displaying the simplicity and universality envisioned here. Post-chromatography sRNA isolation/analysis, in particular, still relied on (i) laborious polyacrylamide gel-selection using radiolabeling (13–16), among additional caveats. These include (ii) the tediousness and technicality of AGO fractionation over linear or stepwise salt gradients (13,15), which, despite simplification (16), remains cumbersome to non-specialists; (iii) the uncontrolled purity-level of AGOs versus potentially co-eluting and unrelated RNPs; (iv) the unknown universality of the method, i.e. are all AGOs isolated and should the non-elution of possible AGO-free sRNAs be of concern?, (v) the approach's as-yet-undetermined applicability to a broad range of organisms and to the full spectrum of AGO:sRNA classes and functions across organisms and kingdoms of life; (vi) the crucial lack of side-by-side, extensive benchmarking against gold-standard procedures for optimal silencing sRNA profiling, including from (vii) difficult-to-handle tissues or (viii) sub-quality material exhibiting various degrees of RNA degradation.
Here, we present a universal method, based on generic lysis- and AGO elution- buffers, which, combined with a single-elution anion-exchange Microspin™ column, fulfils all the above requirements (i–viii). Streamlined to operate under a benchtop user-friendly format, the method enables universal, ultra-fast minimal RISC purification directly amenable to silencing sRNA-analytical methods including acrylamide gel-free library preparation for downstream deep-sequencing.
MATERIALS AND METHODS
Experimental models
Arabidopsis thaliana ecotype Columbia-0 (Col-0) was used as the genetic background for the transgenic plants expressing pAGO1::FLAG::AGO1 (18), pAGO3::FLAG-AGO3 (19), pAGO7::FLAG-AGO7 (20), as well as the ago1-27 (21), ago2-1 (22), ago4-5 (23), ago5-1 (24), ago6-2 (25), ago9-1 (24) and ago10-1 (26) mutants. Inflorescences and 1–5 days after pollination (DAP) siliques tissues were collected on plants grown in soil at 21°C and under a 16-h-light/8-h-dark regimen in growth chambers equipped with 36-Watt fluorescent lights at a 2:1 ratio of 840 Cool White:Grolux, with an average light intensity of 160 μmol m−2 .s−1 and 60% relative humidity. Seedling were grown on 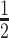 MS medium (pH5.7) without sucrose and selection was done in cabinets displaying 50% relative humidity at 21°C in a 12-h-light/12-h-dark regimen of 120 μmol m−2 s−1. Collected Arabidopsis tissues were flash-frozen in liquid nitrogen, ground using Silamat S6 (Ivoclar Vivadent) and stored at –80°C.
MS medium (pH5.7) without sucrose and selection was done in cabinets displaying 50% relative humidity at 21°C in a 12-h-light/12-h-dark regimen of 120 μmol m−2 s−1. Collected Arabidopsis tissues were flash-frozen in liquid nitrogen, ground using Silamat S6 (Ivoclar Vivadent) and stored at –80°C.
Paramecium tetraurelia wild type strain 51 mating type 7 was cultivated at 27°C in wheat grass powder (WGP, Pines international, USA) infusion-medium bacterized, the day before use, with Klebsiella pneumoniae and supplemented with 0.8 mg/l β–sitosterol (Merck). Autogamy was carried out at 27°C as described (27). 400 mL cultures of exponentially growing cells at 1000 cells/ml or of autogamous cells at 2000 cells/ml were centrifuged and flash-frozen in liquid nitrogen. Cell pellets were stored at –80°C.
Oryza sativa ssp. Japonica cv. Taipei 30 was grown in greenhouse in soil (Klasmann-Deilmann, Germany) in a 12-h-light, 30°C, 80% relative humidity and 12-h-dark, 22°C, 60% relative humidity regimen. Collected leaves were flash-frozen in liquid nitrogen and ground using Silamat S6. Powder was stored at –80°C.
Manihot esculenta Crantz cv. TMS60444 was used as wild type cassava plants, grown on soil (40% Klasmann Substrate 2, 10% Perlite, 50% Ricoter lawn soil, fertilized with Scotts Osmocote), in a greenhouse with a minimum of 14 hours of light, 60% relative humidity and day/night temperatures of 24°C/17°C respectively. Storage roots were harvested after a total of 6 months growth, washed and the epidermis peeled. Roots were flash-frozen and pulverized in liquid nitrogen using analytical mill (IKA). Powder was stored at –80°C.
Schizosaccharomyces pombe wild type strain SpYB 97 was cultured in 300 ml liquid Edinburgh minimal medium number 2 (EMM2) until OD 0,2, at 30°C in under 170 rpm agitation. Cells were arrested in early S-phase by adding 12 mM final hydroxyurea (HU) for 4 hours at 30°C under 170 rpm agitation. Cells were centrifuged at 1800g. for 5 min at room temperature and washed twice with pre-warmed medium and released during 70 min at 30°C under 170 rpm agitation in pre-warmed fresh medium. Cells were then centrifuged at 1800g. for 4 min at room temperature, washed with ice-cold 1× PBS and pelleted again. Dry pellets were flash-frozen in liquid nitrogen and stored at –80°C.
Drosophila melanogaster strain W1118 were grown at 25°C on medium supplemented with yeast. Ovary dissection was conducted on flies aged of 3 days, into cold PBS on ice. Dissected tissues were pelleted, flash-frozen in liquid nitrogen and stored at –80°C.
Caenorhabditis elegans strain N2 was grown at 23°C on NGM medium-containing Petri dishes freshly plated with E. coli OP50 (www.wormbook.org). Worm plates were washed with M9 buffer (22 mM KH2PO4, 42 mM Na2HPO4, 86 mM NaCl, 1 mM MgSO4), the suspension incubated on a wheel for 10 min, then centrifuged at 1600g for 1 min at room temperature. Pellets were washed with M9 until supernatant was clear then flash-frozen in liquid nitrogen and stored at –80°C.
Mus musculus line C57BL/6 aged of 6–8 weeks were maintained at the ETH Phenomics center (https://epic.ethz.ch). Dissected organs were flash-frozen in liquid nitrogen and stored at –80°C. Terminal blood collection was conducted in vena cava with 10% 0.5M EDTA, centrifuged at 2000g for 5 min at 4°C, then the plasma was collected, flash frozen in liquid nitrogen and stored at -80°C.
Mouse neuroblastoma cell line N2a (ATCC®CCL-131) was grown in DMEM medium (Sigma, D6429) supplemented with 10% fetal bovine serum (FBS, PanBiotech, P30-1502) and Penicillin-Streptomycin (Sigma, P0781) in T75 flasks (TPP, 90076) in an incubator at 37°C, infused with 5% CO2. Cells were washed with 1X PBS (Gibco, 10010), detached using 1 mL 0.25% Trypsin–EDTA (Gibco, 25200) and re-suspended in 10 ml DMEM 10% FBS medium. Cells were centrifuged at 500g for 5 min, then washed three times in 1× PBS. Dry pellets were flash-frozen in liquid nitrogen and stored at –80°C.
TraPR purification
TraPR operates robustly with the indicated buffer volumes as far as the resin is not saturated by an excessive amount of starting material. This, in our experience, is the primary cause for suboptimal purification or failure. Recommended amounts of starting material for the different organisms/organs used in this study (lysis volume of 400 μl) are provided in Supplementary Table S1. A simple, empirical method to ensure that a given amount of starting material does not exceed the TraPR column capacity is to prepare a clarified lysate from twice the chosen amount of sample and split it into two halves. The first half is subjected to total RNA extraction (control) and the second half is subjected to TraPR purification according to the procedure described below, to extract the E-fraction RNA. Total RNA and E-fraction RNA is then subjected to migration in 1% agarose (1× TBE) and stained with ethidium bromide. Under UV exposure, the strong rRNA signal observed in the high molecular weight (HMW) fraction of the total RNA preparation should be undetectable in the TraPR-isolated E-fraction RNA, indicating that the chosen amount of starting material for the organism/tissue under consideration has not exceeded column capacity.
The Q Sepharose HP resin (GE healthcare, GE17-1014-01) is equilibrated with 5 volume of equilibration buffer (20 mM HEPES–KOH pH 7.9, 10% (v/v) glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, conductivity adjusted at 8 mS cm−2 with potassium acetate at an indicative concentration of ∼0.1 M)) under gentle shaking. The resin is then re-suspended in storage buffer (20 mM HEPES–KOH, pH 7.9, 10% (v/v) glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 0.2 mM sodium azide, conductivity adjusted at 8 mS cm−2 with potassium acetate (KoAc) at an indicative concentration of ∼0.1 M), with a ratio of storage buffer:resin of 3:5 (v:v). Under constant gentle agitation, 800 μl of the mix (containing 500 μl of resin) is packed into Microspin™ columns (GE healthcare, GE27-35-650), which are then tightly closed. TraPR columns are stored at 4°C.
The main steps of the TraPR procedure are depicted in Figure 1A. To start the TraPR purification procedure, dry pellets of flash-frozen material are typically transferred into a Dounce homogenizer, and supplemented with 400 μl of lysis buffer (20 mM HEPES–KOH, pH 7.9, 10% (v/v) glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 0.1% (v/v) Triton X100, conductivity adjusted at 8 mS cm−2 with KoAc at an indicative concentration of ∼0.1 M). Lysis is performed on ice, using pestle A 50 times, then pestle B 50 times. Lysates are then transferred into 1.5 ml microcentrifuge tubes. For plants, the tissue powder is directly homogenized in 400 μl lysis buffer into 1.5 ml microcentrifuge tubes. Plasma samples are homogenized by pipetting up and down with an equal volume of Lysis buffer. All lysates are clarified at 10 000g for 5 min at 4°C and transferred into a fresh 1.5 ml microcentrifuge tube. 100 μl of clarified lysates are stored on ice as inputs (I-) fraction.
Figure 1.

(A) Schematics of the three-steps, 15 min Microspin™ anion-exchange chromatography procedure. (B) Left: immunoblot analysis of native AGOs extracted from Arabidopsis inflorescences using the procedure in (A). Right: anti-Flag immunoblot analysis of genomic FLAG-AGO3 expressed under the AGO3 promoter extracted from 1–5 days after pollination (DAP) siliques (top) or genomic FLAG-AGO7 expressed under the AGO7 promoter extracted from 2 weeks-old seedlings (bottom) using the procedure in (A). (C) RNA extracted from fractions purified in (B) was radiolabeled with T4 Polynucleotide Kinase (PNK) prior to migration on 17% denaturing polyacrylamide and transfer onto a nylon membrane. (D) Low molecular weight (LMW) RNA blot analysis of RNA extracted from fractions purified in (B). Radiolabeled oligonucleotides were used as probes to detect known Arabidopsis silencing sRNAs. Nomenclature for panels B–D. I: clarified lysate, E: AGO-eluted fraction, HS: High-salt resin wash after AGO elution. Clarified lysate used in lane I represents 20% of the material used for the TraPR purification; Ambion® DECADE™: RNA ruler, in nucleotides. EtBr: total RNA ethidium bromide staining. RNA spike: synthetic 22-nt RNA added in fractions post-TraPR purification as a control for RNA extraction. U6 RNA hybridization provides an internal control for successful RISCs isolation in the E-fraction. Prot. spike: FLAG-tagged protein added post-purification as a control for protein extraction, detected with an anti-Flag antibody. Coom.: total protein Coomassie blue staining.
TraPR columns are vortexed a few seconds to re-suspend the resin, their bottom is then opened and the columns centrifuged 15 seconds in a 2 mL collecting microcentrifuge tube on a benchtop mini-centrifuge (or 1000g on a regular centrifuge). This removes the storage buffer. The bottoms of the columns are then closed, their caps opened and 300 μl of clarified lysate is added onto the resin. Caps are closed and the resin and lysates are homogenized together by manual shaking. The bottoms of the columns are then re-opened and the columns transferred into a new 2 ml collecting microcentrifuge tube labelled ‘E-fraction’, centrifuged 15 s on a benchtop mini-centrifuge (or, alternatively, at 1000g on a regular centrifuge) to collect the flow-through. Column caps are then opened and 300 μl of TraPR elution buffer (20 mM HEPES–KOH, pH 7.9, 10% (v/v) glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, conductivity adjusted at 40 mS cm−2 with KoAc at an indicative concentration of ∼0.44M) added onto the resin, then the columns are centrifuged 15 seconds on a benchtop mini-centrifuge (or, alternatively, at 1000g on a regular centrifuge) with the flow-through collected into the 2 ml microcentrifuge tube labelled ‘E-fraction’. The elution step is repeated once with 300 μl of TraPR elution buffer collected in the 2 ml microcentrifuge tube labelled ‘E-fraction’. E-fraction tubes (containing purified RISC-associated silencing sRNAs) are closed and stored on ice. Columns are then transferred onto new collecting tubes and the resin washed three times with 300 μl of TraPR elution buffer, then the flow-through is discarded. Columns are then transferred into new 2 ml microcentrifuge tubes labelled ‘HS-fraction’, and non-RISC-associated RNAs demobilized by mixing the resin with 300 μl TraPR HS buffer (20 mM HEPES–KOH, pH 7.9, 10% (v/v) glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, conductivity of the HS buffer is adjusted at 110 mS cm−2 with KoAc at the indicative concentration of ∼4 M).
Columns are centrifuged 15 s on a benchtop mini-centrifuge (or, alternatively, at 1000g on a regular centrifuge) and eluates collected. HS elution buffer is applied twice more, and the collected HS-fractions are stored on ice. The TraPR purification process thus generates 3 tubes labelled: I (Input) containing the clarified lysate; E for: AGO-Eluted fraction and HS, collecting the high-salt resin wash after AGO elution.
Note that the KoAc concentrations provided here for buffers' preparations are only indicative, not definitive. We strongly recommend conductivity measurement (using, for instance the UPC900 conductometer, GE healthcare) as the final criterion to ensure standardization of reagents and, hence, reproducibility of the purification procedure.
Protein analysis
Protein analysis detailed procedures are provided in Supplementary method 1. Commercial antibodies used in this study are listed in Supplementary Table S3. Peptides used for the production of antibodies directed against Arabidopsis AGO proteins are listed in Supplementary Table S4.
RNA analysis
RNA extraction (precipitation)
Total RNA extraction was performed using 1 mL of TRI Reagent® (Sigma, T9424), following the manufacturer's instructions. After overnight precipitation at -20°C, RNA pellets were recovered by centrifugation at 20 000g for 30 min at 4°C, washed three times with cold 80% ethanol and re-suspended in the appropriate solvent. TraPR-purified RNA was extracted from I-, E- and HS- fractions using one volume of 50:49:1 acidic Roti-Aqua-P/C/I pH 4.5–5 (Carl Roth, X985.1). Once homogenized, aqueous phases were recovered after centrifugation at 10 000 g for 10 min at 4°C, and transferred in tubes containing 10% (v/v) 3 M sodium acetate, pH 5.2, and 1 μl of Glycogen (Thermo Fisher Scientific, R0551), before mixing with 100% (v/v) cold isopropanol. After overnight incubation at –20°C, RNA pellets were recovered by centrifugation at 20 000g for 30 min at 4°C, washed three times with cold 80% ethanol and re-suspended in the appropriate solvent.
RNA extraction (silica columns)
Aqueous phases obtained after phenol treatment were mixed with 2 volume of RNA MAX buffer (Zymo research, R1070-2) and vortexed. 800 μl of the mix was transferred onto Zymo spin Ic columns (Zymo research, R1004) and centrifuged at 12 000g for 30 s whereupon the flow-through was discarded. This step was repeated until all the mix was passed through the columns. 400 μl of RNA Prep. buffer (Zymo research, R1060-2) was added onto the columns, which were then centrifuged at 12 000g for 1 min, whereupon the flow-through was discarded. 800 μl of RNA Wash buffer (Zymo research, R1003-3) was added to the column and centrifuged at 12 000g for 30 s, whereupon the flow-through was discarded. Washing was repeated once with 400 μl of RNA Wash buffer. Columns were then transferred into new 1.5 ml microcentrifuge tubes and 10 μl of water was pipetted directly onto the resin. After 1 min incubation at room temperature, RNA was recovered by centrifuging the columns at 12 000g for 1 min.
T4 polynucleotide kinase (PNK) RNA labelling
The clarified lysate used as input (I-fraction) represents 25% of the material used for the TraPR purification. The remaining 75% of clarified lysate are purified through the TraPR column (E-and HS-fractions).PNK labeling involved 10% of RNA extracted from the above mentioned I-, E- and HS- TraPR fractions. 100% of the labeling of E-fraction and 10% of the I- and HS-fractions were resolved on gel. RNA re-suspended in water was mixed in 1× PNK buffer B (0.5 M imidazole–HCl pH 6.4, 180 mM MgCl2, 50 mM DTT, 1 mM spermidine and 1 mM ADP, Thermo Fisher Scientific) with 10 μCurie [32P-γ]-ATP (Hartmann Analytic, SRP401) and 10 U of T4 Polynucleotide kinase (T4 PNK, Thermo Fisher Scientific, EK0031) in 20 μl total volume. Exchange reactions were conducted at 37°C during 45 min whereupon reactions were stopped by thermic denaturation at 95°C for 5 min. 10 μl of 4× RNA loading buffer (50% (v/v) glycerol, 50 mM Tris pH 7.7, 5mM EDTA, 0.03% Bromophenol Blue) were then added to the reaction tubes. DECADE™ Marker (Ambion, AM7778) was labelled in parallel using 1× PNK buffer A (50 mM Tris–HCl pH 7.6, 10 mM MgCl2, 5 mM DTT, 0.1 mM spermidine, Thermo Fisher Scientific) with 10 μCurie [32P-γ]-ATP and 10 U of T4 PNK in 20 μl total volume. An equimolar mix of 21- and 24-nt-long synthetic RNA oligonucleotides (Supplementary Table S2), used as ladder in Figure 2B, was labelled under the same conditions. Forward reactions were conducted at 37°C for 1 h, upon which reactions were stopped by thermic denaturation at 95°C for 5 min. Samples and molecular weight marker were resolved on 6 M urea, 17% poly-acrylamide (Euromedex, EU0061-B), 0.5X TBE gels before electro-transfer on Hybond®-NX membrane (GE Healthcare, RPN203T) using the Biorad MiniProtean II system. Membranes were exposed overnight on Imaging plate (Fujifilm, BAS-IP2025), scanned on a Typhoon™ FLA9000 phosphor-imager (GE Healthcare) and analyzed with the Typhoon™ FLA9000 V1.1 software (GE Healthcare).
Figure 2.

(A) PNK radiolabeling (as in Figure 1C) of total RNAs from the I-, E- and HS-fractions extracted via the procedure schematized in Figure 1A as applied to various organisms. (B) Top: as in (A) but with exponentially growing Paramecium cells (Veg.) and Paramecium cells at the onset of sexual events (T 0 arbitrarily defined as the time when 50% of the cells begin maternal macronucleus fragmentation, as evaluated cytologically). Two synthetic radiolabeled RNAs were used as rulers. Bottom: LMW RNA blot analysis of sRNAs co-extracted in (B, top). Radiolabeled oligonucleotides were used as probes to detect known Paramecium silencing sRNAs (Cl22, mtA, Sardine) accumulating under the indicated condition/stage. (C) Top: immunoblot analysis of native ALG1 extracted from C. elegans mixed (eggs+larvae+adults) populations using the procedure schematized in Figure 1A. Bottom: LMW RNA blot analysis of sRNAs co-extracted in (C, top). Radiolabeled oligonucleotides were used as probes to detect known C. elegans silencing sRNAs. (D) Top: immunoblot analysis of native AGO1 and AGO2 from mouse neuroblastoma cells (N2a; left) or dissected brain (right), extracted via the procedure schematized in Figure 1A. Bottom: LMW RNA blot analysis of sRNAs co-extracted in (D, top). Radiolabeled oligonucleotides were used as probes to detect known mouse miRNAs (let-7a, miR-16). Nomenclature for panels A–D. I: clarified lysate, E: AGO-eluted fraction, HS: High-salt resin wash after AGO elution. Clarified lysate used in lane I represent 20% of the material used for the TraPR purification; Ambion® DECADE™: RNA ruler, in nucleotides. EtBr: total RNA ethidium bromide staining. RNA spike: synthetic 22-nt RNA added in fractions post-TraPR purification as a control for RNA extraction. U6 RNA hybridization provides an internal control for successful RISCs isolation in E-fraction and equal RNA loading. Prot. spike: FLAG-tagged protein added post-purification as a control for protein extraction, detected with an anti-Flag antibody. Coom: total protein Coomassie blue staining.
PCR quantification procedures for DNA, mRNA and mature miRNA are provided in Supplementary method 2.
RNAse T1 treatment
Mouse liver lysates in TraPR lysis buffer supplemented with 100 U of RNAse T1 (Thermo Fischer scientific, EN0541) were incubated 30 min at room temperature on a rotating wheel. Half of the treated lysates was subjected to TRI Reagent® (Sigma, T9424), following the manufacturer's instructions to purify total RNA, the second half was clarified prior to being subjected to the TraPR procedure.
Northern analysis
The clarified lysate used as input (I-fraction) represents 25% of the material used for the TraPR purification. The remaining 75% of clarified lysate are purified through the TraPR column (E-and HS-fractions). Depending on the biological model, a variable amount of the purification is resolved on gel, but the deposited amount of E- and HS-fractions is always identical. RNA re-suspended in 1X RNA loading buffer was denatured at 95°C for 5 min, followed by immediate incubation on ice. RNA was resolved on 6 M urea, 17% poly-acrylamide (Euromedex, EU0061-B), 0.5× TBE gels before electro-transfer on Hybond®-NX membrane (GE Healthcare, RPN203T) using Biorad MiniProtean II system. Chemical crosslinking was performed using the EDC (Sigma, E7750) method at 60°C for 90 min according to (28), then membranes were washed three times in water and pre-hybridized in PerfectHyb™plus buffer (Sigma, H7033) at 42°C for 30 min in rotating cylinders. Synthetic DNA oligonucleotide probes (Supplementary Table S2) were labeled in 1× PNK buffer A with 25 μCurie [32P-γ]-ATP and 10 U T4 PNK in a total volume of 20 μl. For P. tetraurelia mtA and Cl. 22 probes, an equimolar mix of the indicated DNA oligonucleotides was labeled. Radiolabeling reactions were conducted at 37°C during 1 h, then 30 μl of water were added and probes purified from free [32P-γ]-ATP using Illustra™ Microspin G-25 columns (GE Healthcare, 27-5325-01). Long DNA probes were amplified by PCR using oligonucleotide primers of which the sequences are listed in Supplementary Table S2. Agarose gel-purified PCR products were labelled by random priming using the Prime-a-gene® kit (Promega) without dCTP and in presence of 25 μCurie of [32P-α]-dCTP (Hartmann Analytic, SRP305) for 1 h at 37°C, purified on Illustra™ Microspin G-50 columns (GE Healthcare, 27-5330-01) and denatured at 95°C for 5 min. Membrane hybridizations were conducted overnight at 42°C in PerfectHyb™plus hybridization buffer, in rotating cylinders, then the membranes were washed 3 times for 20 min at 50°C with 2× SSC, 2% SDS buffer. Membranes were dried and exposed at least overnight on Imaging plate (Fujifilm, BAS-IP2025), scanned on a Typhoon FLA9000 phosphor-imager (GE Healthcare) and analyzed with the Typhoon™ FLA9000 V1.1 software (GE Healthcare). Membranes were stripped three times for 10 min in boiling 0.1% SDS, then washed once in water and pre-hybridized 30 min at 42°C in PerfectHyb™plus hybridization buffer prior to re-probing over-night.
sRNA library preparation
sRNA libraries presented in Figures 3A and 4 (replicate 1) and Supplementary Figure S5B were generated using the TruSeq Small RNA library preparation kit (Illumina) and sequenced via the Hiseq 2500 system (Illumina) at the Functional Genomic Center Zürich (www.fgcz.ch). For the plant sRNA samples, TRI Reagent®-extracted total RNA was resolved on 6 M urea, 17% poly-acrylamide, 1X TBE gels in parallel with radiolabeled 21- and 24-nt synthetic RNA oligonucleotides (Supplementary Table S2). After migration, gels were exposed for 1 h on Imaging plates (Fujifilm, BAS-IP2025), scanned on a Typhoon FLA9000 phosphor-imager (GE Healthcare) and the image generated was used for samples’ gel-selection at a size comprised between 20- and ∼25-nt. sRNAs were recovered from the gel using the ZR smallRNA™ PAGE Recovery kit (Zymo, R1070) following the provider's instructions, and eluted in 10 μl of water prior to library preparation. sRNA libraries presented in Figure 4 (replicates 2 and 3) and Figure 5 were generated in the Voinnet laboratory using the LEXOGEN small-RNA Seq Library Prep. Kit (052) following the provider's instructions. After PCR amplification, libraries were gel-selected on 10% poly-acrylamide (Euromedex, EU0061-B), 1× TBE gels stained with ethidium bromide. Excised gel portions were crushed in 500 mM NaCl, incubated overnight at 4°C on rotating wells and gel pieces removed by centrifugation at 5000g. for 5 min at room temperature on Costar® Spin-X 0.45 μm centrifuge filters (Corning Inc., 8162). DNA was precipitated by adding 1 volume of isopropanol, incubated at –20°C for 1h and centrifuged at 20 000g for 30 min at 4°C. Libraries were re-suspended in 10 μl water prior to quality control and quantification using the Agilent TapeStation 2200 system. Libraries were sequenced on the NextSeq 500 and HiSeq 2500 (Illumina) at the Functional Genomic Center Zürich (www.fgcz.ch). Drosophila sRNA libraries presented in Figure 3C and Supplementary Figure S3C-E were generated in the J. Brennecke laboratory at IMBA (Vienna) using a custom-made procedure in which gel-selection, ribo-depletion and library preparation were conducted according to (29) and (30). Library sequencing was conducted on a HiSeq2500 system (Illumina) at the Vienna Biocenter sequencing facility (www.viennabiocenter.org).
Figure 3.

(A) Size distribution and annotation of Arabidopsis inflorescence-derived sRNAs following TRUseq-based deep-sequencing of TRI®-Reagent-extracted total RNA (Total-RNA, top), polyacrylamide gel-selected sRNA (Gel-selected, middle) or sRNAs directly cloned after TraPR purification (bottom). n = 3 technical replicates for each condition, bars indicate the standard deviation. (B) 5′-terminal nucleotide composition of deep-sequenced sRNAs prepared by the methods indicated in (A). Nucleotide identity is displayed for 21-nt-long (left) and 24-nt-long (right) sRNAs. Bars indicate the standard deviation. (C) Size distribution and annotation of Drosophila ovary sRNAs following deep-sequencing, via an in-house protocol, of Gel-selected+ribo-depleted sRNAs (top), Gel-selected+ribo-depleted+oxidized sRNAs (middle), or sRNAs directly cloned after TraPR purification (bottom). 10 μg of total RNA were used for the first two methods whereas TraPR-based purification involved either 5 (TraPR 5) or 50 (TraPR 50) ovary pairs. n = 2 technical replicates for each preparation method, bars indicate the two independent datapoints.
Figure 4.

(A) Northern LMW RNA blot analysis of sRNAs found in the I-, E-, and HS-fractions following TraPR-based extraction conducted in mouse liver lysates treated for 30 min at room temperature with 100 U RNase T1 or not (Intact). TRI®-Reagent-extracted total RNA (Total-RNA) from identically treated lysates was analyzed in parallel. Radiolabeled oligonucleotides were used as probes to detect known mouse liver miRNAs (let-7a; miR-122). I: clarified lysate, E: AGO-eluted fraction, HS: High-salt resin wash after AGO elution. Clarified lysate used in lane I represent 20% of the material used for the TraPR purification; EtBr: total RNA ethidium bromide staining. U6 RNA hybridization provides an internal control for successful RISCs isolation in E- fraction and equal RNA loading. (B) Size distribution and genomic origin of mouse liver sRNAs following deep-sequencing of TRI®-Reagent-extracted total RNA (Total-RNA, left) or sRNAs directly purified by TraPR (right) from intact (top) or RNAse T1-treated (bottom) mouse liver lysates. n = 3 biological replicates for each condition, bars indicate the standard deviation. (C) Extended Pearson correlation matrix of rRNAs (black), miRNAs (green) and other sRNAs (gray) based on the sRNA reads count over each considered annotated loci in the libraries prepared in (B).
Figure 5.

(A) Size distribution and genomic origin of mouse plasma sRNA following Lexogen-based sequencing of TRI®-Reagent-extracted total RNA (Total-RNA, left) or sRNAs directly purified by TraPR (right). n = 4 biological replicates for each condition, bars indicate the standard deviation. (B) miRNA-dispersion analysis, based on the standard deviation of miRNA counts, in the libraries prepared in (A) as depicted per quartile reflecting miRNA abundance (C) Proportion of plasmatic tRNA-derived reads according to their sizes (<20-nt; 20-to-24-nt; > 24-nt) in the libraries prepared in (A). (D) 5′-terminal nucleotide composition of plasmatic tRNA-derived RNA in the 20-to-24-nt size range in the libraries prepared in (A). For panels B, C and D Wilcoxon rank sum test, n.s.: non-significant, *P < 10−3. (E) Proportion of plasmatic tRNA Val-AAC-derived sRNA reads according to their sizes (<20-nt; 20-to-24-nt; >24-nt) in the libraries prepared in (A). (F) 5′-terminal nucleotide composition of plasmatic tRNA Val-AAC-derived sRNAs in the 20-to-24-nt size range in the libraries prepared in (A). Bars indicate the standard deviation. (G) Single nucleotide sRNA profiles over tRNA Val-AAC using the libraries prepared in (A), displayed by size range (grey <20-nt; green 20-to-24-nt; black >24-nt). RPM: reads per million. For panels B–D, boxes indicate the first to third quartiles and whiskers show the highest and lowest values within 1.5 times the interquartile range (IQR).
Bioinformatic analyses
Sequencing libraries were pre-processed and mapped using SUSHI analysis (31). The quality of raw reads was controlled with FastQC to detect sequencing abnormalities. Adapter sequences were trimmed using Trimmomatic 0.36 with parameters ILLUMINACLIP:1:20:7:5:true AVGQUAL:10 MINLEN:15 (32). Trimmed reads were then mapped using Bowtie 1.2.1.1 using default parameters on the respective genomes with the following genome builds: Arabidopsis genome (TAIR10), Drosophila genome (BDGP6, release 92) or mouse genome (GRCm38, release 92). Reads were counted with Rsubread 1.26.1 (allowMultiOverlap = T, strandSpecific = 0, countMultiMappingReads = T, fraction = T) where reads mapping to multiple genomic loci were accounted fractionally (33). Reads counts were partitioned into different categories obtained from these annotations using as references Arabidopsis (TAIR10), Drosophila (BDGP6, repeatmasker dm6) or mouse (GRCm38, miRBase 22, GtRNAdb mm10). Raw reads counts were subsequently library-normalized and log-transformed with the rlog function of DEseq2 (34). Visualizations of library overview characteristics such as heatmaps of distance matrix and extended Pearson correlation matrix were plotted using R cran with the packages pheatmap and GGally. All other representations including histograms and tRNA read coverage profiles were generated using in house scripts in R using the packages ggplot2, rtracklayer (35) and GenomicAlignments (36).
RESULTS
Rapid and easy purification of functional silencing sRNAs in Arabidopsis
To implement a rapid, single-step procedure for anion-exchange-based AGO purification, we first studied the model plant Arabidopsis thaliana, which expresses nine functional out of ten AtAGOs encompassing three phylogenetic clades (37). In preliminary high-resolution anion-exchange chromatography, AtAGOs and their cognate silencing sRNAs were co-eluted at moderate salt concentrations, as opposed to other cellular RNAs remaining adsorbed on the resin. Building on this, systematic fraction bulk-analyses were conducted to optimize lysis and elution procedures, upon which the approach was adapted to a workflow based upon Q Sepharose-resin-packed Microspin™ columns. This allowed isolation of all nine functional AtAGOs from crude inflorescence extracts within 15 min on a benchtop microfuge (Figure 1A-B; Supplementary Figure S1A). AtAGO-specific signals were detected in input (I-) and eluate (E-) fractions, but were at, or below, detection in the high-salt (HS-) fraction, which contains the bulk of cellular nucleic acids (Figure 1B, C; Supplementary Figure S1B). Radioactive 5′ end-labelling, via polynucleotide kinase (PNK), of total RNA from E-fraction yielded discrete 21-nt and 24-nt signals diagnostic of miRNAs and transposon(TE)/repeat-derived siRNAs, respectively (38). Specific elution of AGO-loaded silencing sRNAs in the E-fraction was confirmed by northern-blot analysis and RT-qPCR measurement of cognate si/miRNAs as opposed to DNA and other cellular RNAs (U6, rRNA, mRNA, snoRNA), which all eluted in the HS-fraction (Figure 1D; Supplementary Figure S1B). Confirming purification of native minimal RISC(s), the E-fraction was amenable to AGO immunoprecipitation (IP) without dialysis/desalting, yielding considerably less RNA contaminants than IP conducted from crude lysates (Supplementary Figure S1C). Directly applying phenol-extracted RNA from the E-fraction onto silicate-based columns bypassed the overnight precipitation used in Figure 1C-D, granting complete extraction of northern/PCR/sequencing-ready silencing sRNAs within ∼30 min (Supplementary Figure S1D; see below).
TraPR universally accesses all known classes of RISC-associated silencing sRNAs
Given the conserved structural and biochemical properties of AGO/AGO-like proteins (38,39), we systematically tested the procedure described above across a wide range of organisms. E-fraction labelling in ciliates (P. tetraurelia) showed a discrete 25-nt signal during conjugation (stage-T0; Figure 2A). Northern analysis of E-fraction RNA obtained from samples in vegetative growth versus conjugation detected 23-nt-long Ptiwi12-dependent endogenous siRNAs (e.g. Cl22 (40)) in vegetative samples, and 25-nt-long Ptiwi01/09-dependent scanRNAs (e.g. mtA, Sardine (41,42); Figure 2B) in conjugation samples. Cassava (M. esculenta) storage roots accumulate high starch levels, usually complicating efficient RNA extraction by standard methods (43). Nevertheless, PNK labelling and northern analysis indicated robust isolation of 21-nt/24-nt miRNAs/siRNAs in the E-fraction (Figure 2A, Supplementary Figure S2A). Similar results were obtained using rice (O. sativa) samples in which a stronger signal centered on 24-nt siRNAs presumably reflects the higher TE load of cereals' genomes (44) (Figure 2A, Supplementary Figure S2B). End-labelling of E-fraction total RNA obtained from hydroxyurea-synchronized fission yeast (S. pombe) yielded a discrete yet fuzzy 21-to-25-nt signal typical of the pericentromeric-repeat siRNAs produced by the PAZ-domain-deficient SpDcr1 (Figure 2A) (45). Their sole effector in heterochromatin formation, SpAgo1, was purified in the E-fraction while it was undetectable in the HS-fraction (Supplementary Figure S2C). In C. elegans mixed stages (eggs, larvae, adults), somatic ALG-1 and associated 22-nt miRNAs were present specifically in the E-fraction (Figure 2C), as were 15 out-of-the 22 AGO/PIWI proteins predicted to be functional in this organism (46) (Supplementary Figure S2F), including the main PRG effectors of the germline-specific 21-U piRNA-like RNAs (Figure 2C). A PNK-labelled signal at ∼22-nt also reflected efficient miRNA purification from adult mouse (M. musculus) brain and N2a neuroblastoma cells (Figure 2A, D). Accordingly, mmAGO1/AGO2, dominating in these cells/tissues (47), were co-purified and enriched with cognate miRNAs in the E- versus HS-fraction, as were miR-16/miR-21 versus snoR202 upon RT-qPCR analysis of mouse liver RNAs (Supplementary Figure S2D). Finally, PNK labelling of mouse testis-derived RNA —also eluted in the E-fraction— yielded a strong, 27–30-nt signal typical of germline-specific mammalian piRNAs mediating TE silencing (48) (Supplementary Figure S2E). These results in mice are in line with the original Q-sepharose-based multi-step separation of miRNA-loaded RISC and piRNA-laoded RISCs at respectively ∼ 0.1 and ∼0.4 M salt concentrations (13).
In every organism tested, PNK labelling yielded no significant signal other than silencing sRNAs in the E-fraction, in stark contrast to the corresponding input and HS-fractions (Figures 1C and 2). Likewise, in polyacrylamide gel analyses of E-fractions, neither total RNA ethidium bromide staining nor northern blotting for the abundant U6 RNA showed residual signal within the 15-to-150-nt range. Importantly, all HS-fractions tested lacked AGO/AGO-like proteins, even upon mass spectrometry analyses in C. elegans (Supplementary Figure S2F), the model organism producing one of the most extensive and diverse suites of these proteins (46). Therefore, the fast and easy procedure depicted in Figure 1A functions universally, without any adjustment, by allowing purification of all classes of RISC-associated silencing sRNAs known to date. Accordingly, it was coined ‘TraPR’, for Trans-kingdom, rapid, affordable Purification of RISCs.
TraPR enables direct silencing sRNA cloning for optimal deep-sequencing analyses
Abundant t/r/snRNAs and their fragments often contaminate sRNA libraries prepared from total RNA. We therefore benchmarked TraPR against existing library preparation methods, i.e. direct TRI®-reagent-extraction (Total-RNA: sub-standard, rapid) or gel-selection (Gel-selected: gold-standard, long, tedious) in Arabidopsis, where contaminations in libraries are substantial. RNA prepared in triplicates via each method was ligated, amplified using standard TRUseq® library preparation, and deep-sequenced (Figure 3A). Clustering analysis revealed comparable performances for the TraPR-based and the days-long gel-selection procedures, both yielding the typical 21-nt and 24-nt size distribution (Figure 3A, middle, bottom, Supplementary Figure S3A). With the Total-RNA approach, however, the defined size distribution of canonical silencing sRNAs was substantially masked by other RNAs (predominantly tRNA fragments) spanning the 16-to-37-nt size range (Figure 3A, top). Individual miRNA- and TE-derived sRNA- reads counts display a higher Pearson correlation (r² = 0.803 and r² = 0.892, respectively) between Gel-selected libraries and TraPR libraries, as opposed to Total-RNA libraries compared with gel-selected libraries (Supplementary Figure S3B). The high similarity observed in TraPR and gel-selected libraries for Arabidopsis miRNAs and TE-derived sRNAs indicates that TraPR is technically on par with the gold-standard procedure for plant silencing sRNA library preparation and does not introduce bias in the sequenced sRNAs. Further supporting high-quality purification of cognate minimal RISCs, the TraPR-isolated 21-nt and 24-nt silencing sRNAs displayed ∼90% 5′ terminal- Uridine and Adenosine, respectively. These are the established cargo preferences of AGO1 and AGO4, which serve as main effectors of the most abundant Arabidopsis silencing sRNAs, i.e. 21-nt-long miRNAs and 24-nt-long TE/repeat-derived siRNAs, respectively (8) (Figure 3B).
For an independent comparative silencing sRNA deep-sequencing analysis, we turned to Drosophila ovaries, which harbor 21-nt siRNAs, ∼22-nt miRNAs and 23-to-29-nt piRNAs, respectively in complex with Ago2, Ago1 and PIWI proteins (Piwi, Aub, Ago3). piRNA cloning in flies is severely complicated by the high abundance of the 30-nt long 2S rRNA, which, based on a previous study, is efficiently removed via anion-exchange chromatography (14). Typical piRNA library generation from fly gonads entails the gel-based selection of 18-to-29-nt sRNAs from total RNA, and/or the removal of 2S rRNA as well as other abundant ncRNA fragments by antisense-oligo based depletion or oxidation of RNA samples, which renders only the 2′-O-methylated siRNA and piRNA populations compatible for cloning (29). We prepared sequencing libraries in duplicates from three different experimental conditions using optimized library preparation procedures with randomized adapters (30,49). Prior to adapter ligation, sRNAs were either gel-selected and 2S rRNA-depleted (Gel-selected+ribo-depleted), gel-selected, 2S rRNA-depleted and oxidized (Gel-selected+ribo-depleted+oxidized) or TraPR-extracted (Figure 3C). 2S rRNA, while still abundant in the Gel-selected+ribo-depleted libraries, was eliminated in TraPR-based libraries, qualitatively on-par with the Gel-selected+ribo-depleted+oxidized libraries. While effective in removing contaminants, a major drawback of the oxidization procedure is that it eliminates the majority of miRNAs (29), which lack the 2′-O-methyl modification. Yet, and beside their biological interest per se, miRNAs are most commonly used as effective normalization means when comparing piRNA populations between different experimental conditions in gonads. TraPR libraries fully preserved the miRNA complement while displaying essentially no silencing-unrelated sRNA contaminants (Figure 3C). Furthermore, sRNA profiles were near-identical in TraPR-based libraries prepared from 50 or from as little as two ovary pairs (Figure 3C; Supplementary Figure S3C), unravelling a wide scalability of the method down to minute amounts of dissected material (Supplementary Figure S3D, left). As expected, in a miRNA-focused analysis, the Gel-selected+ribo-depleted and TraPR libraries clustered together, while the Gel-selected+ribo-depleted+oxidized libraries were formed a clear outlier group due to oxidization-induced loss-of-miRNAs (Supplementary Figure S3D, right). Overall, miRNA reads counts were highly correlated (r² = 0.99) between Gel-selected+ribo-depleted and TraPR-based libraries, as opposed to the Gel-selected+ribo-depleted+oxidized libraries (Supplementary Figure S3E). Silencing sRNA reads derived from TEs (constituted of the 2′-O-methylated siRNA and piRNA populations) showed a high Pearson correlation (r2 = 0.99) between all three library types. The highest Pearson correlations, observed for all silencing sRNA classes, were found between technically distinct TraPR-generated libraries independently of the amount of ovary pairs, demonstrating the method's robustness and consistency over a broad range of input quantities. We conclude that minimal RISC purification by TraPR enables fast, comprehensive and unbiased generation of silencing sRNA libraries from highly variable amounts of input material.
TraPR is resilient to RNA degradation
While silencing sRNAs are protected by AGO/AGO-like proteins, degradation of abundant cellular RNAs is a major complicating factor when sequencing libraries are prepared from samples of suboptimal RNA quality. To test whether TraPR also alleviates this problem, we deliberately treated a non-clarified mouse liver lysate for 30 minutes with RNase T1, and subsequently extracted RNA via TraPR or via TRI®-Reagent. We compared the corresponding I-/E-/HS-fractions to those of an untreated (i.e. intact) lysate. As expected, widespread RNA degradation was observed in the Total-RNA and TraPR-input (I)-fractions, as attested by nonspecific ethidium bromide staining and U6 RNA hybridization (Figure 4A). By contrast, the RNase treatment did not prevent the strong TraPR-based enrichment, in the E- versus I-fraction, of mmAGO1/AGO2 dominating in the liver (Supplementary Figure S4A). Accordingly, the RNA degradation pattern was observed only in the I- and HS-fractions but not in the RISC-containing E-fraction, in which abundant let-7a and liver-specific miR-122 were readily detectable by northern analysis (Figure 4A).
Encouraged by these results, we prepared sequencing libraries in independent biological triplicates of Total-RNA- and TraPR-purified sRNAs from intact versus RNase-treated mouse liver lysates. Total-RNA libraries from the RNAse-treated lysate were heavily contaminated, causing strongly reduced silencing sRNA levels. In contrast, the TraPR-based libraries prepared from the RNAse-treated lysate were on-par with the Total-RNA libraries prepared from intact lysates (Supplementary Figure S4B). Remarkably, the sRNA length distributions were near-identical in TraPR libraries prepared from RNase-treated and intact lysates, both displaying the dominant ∼22-nt mammalian miRNA peak. Moreover, both libraries were nearly devoid of tRNA contaminants, which were more abundant in Total-RNA libraries prepared even from intact lysates (Figure 4B). Total-RNA libraries from RNase-treated lysates instead displayed mostly tRNA-derived reads at the expense of miRNA reads (Figure 4B). Consistently, in both global sRNA count analyses (Supplementary Figure S4C, left) and miRNA-focused analyses (Supplementary Figure S4C, right), the Total-RNA libraries from intact lysates clustered together with TraPR-based libraries independently of RNAse treatments for the latter. In contrast, Total-RNA libraries from intact lysates strongly diverged from RNase-treated lysates. Furthermore, all TraPR libraries clustered in this analysis, suggesting that the method robustly allows comparison of miRNA species regardless of sample RNA quality (Supplementary Figure S4C, right). Total-RNA and TraPR datasets from intact lysates were highly similar interns of both miRNA and tRNA species (Figure 4C). Similarly, TraPR libraries were highly comparable even when cloning sRNAs from RNAse T1 digested lysates. This was confirmed by correlating miRNA read counts, which revealed no overt miRNA bias between Total-RNA intact lysate and intact lysates' TraPR libraries (r2 = 0.970) or between RNase-treated and intact lysates’ TraPR libraries (r2 = 0.978) (Figure 4C). Therefore, TraPR allows the consistent isolation of bona fide silencing sRNAs, granting highly reproducible sequencing results even from RNA-degradation-prone samples or samples with vastly suboptimal RNA quality.
TraPR enables reproducible, high-yield silencing sRNA isolation from mammalian plasma
Despite its major biomedical importance as a putative source of biomarkers, reproducible silencing sRNA profiling from mammalian plasma remains a challenge (12). This is mainly due to widespread RNA-degradation, minute silencing sRNA content and the diversity of silencing sRNA carriers in plasma (12). These may include microparticles such as exosomes, micro-vesicles or apoptotic bodies as well as high-density lipoprotein (reviewed in (50)). RNA-binding protein carriers also exist, among which AGO2 is found either freely in the plasma or within cell-derived vesicles including exosomes (51). This likely reflects AGO2’s recycling and degradation in multivesicular bodies and autophagosomes, respectively (52,53). TraPR’s ability in isolating AGO:sRNA complexes, even under adverse conditions, prompted us to test its performances with murine plasma samples, of which 150 μl were subjected to TRI®-Reagent-based (Total-RNA) or TraPR-based extraction prior to library preparation via the Lexogen Small RNA-Seq Library Prep kit. In read size-distribution analyses (n = 4 samples each), Total-RNA libraries contained up to 80% tRNA-derived reads in the 29-to-33-nt range and only ∼7% miRNA reads. TraPR-based libraries, by contrast, were nearly entirely constituted of miRNA reads peaking, expectedly, at 22-nt (Figure 5A). In miRNA-focused analyses, the four TraPR-based libraries consistently showed the highest Pearson correlations compared to the four Total-RNA libraries (0.959 < r2 < 0.989 versus 0.943 < r2 < 0.976; Supplementary Figure S5A). This most likely reflects the robust and consistent detection of low abundant miRNAs granted by TraPR, as revealed by analyses of reads count dispersion conducted on abundance-based quartiles (Figure 5B; Wilcoxon rank sum test, quartile1: P < 4.5 × 10–5, quartile2: P<5 × 10–4). The substantially higher reliability of TraPR in detecting low-abundance species agrees with the miRNA content of TraPR-based libraries being at least one order-of-magnitude higher than in Total-RNA libraries (Figure 5A). Similar conclusions were reached in comparing biological triplicates prepared with TRUseq™ from Illumina (Supplementary Figure S5B, C). Noteworthy, Lexogen versus Illumina libraries prepared downstream of TraPR-based sRNA isolation showed a consistently higher Pearson correlation in miRNA reads counts (r2 = 0.903) than their Total-RNA counterparts (r2 = 0.879). Collectively, these results identify sRNA sample preparation, besides library cloning, as a key bottleneck in silencing sRNA sequencing from plasma. Our results also suggest that a substantial fraction of plasma-borne miRNAs—which the TRI®-Reagent-based Total-RNA approach isolates in a carrier-independent manner—are indeed bound to AGO(s) (51) and, hence, highly amenable to TraPR-based purification. Beside exploration via sequencing, known circulating silencing sRNAs such as miRNAs may be quantified as biomarkers in mammalian plasma, a setting under which we decided to additionally benchmark TraPR. We found that RT-qPCR based detection of miR-16 and miR-21 was enhanced by > two orders-of-magnitude in the TraPR-purified E-fraction compared to Total-RNA extracted from the same volume (150 μl) of mouse plasma I-fraction (Supplementary Figure S5D).
tRNA-derived reads dominate mammalian biofluid sRNA libraries such as plasma. While most of these molecules likely represent degradation products, some tRNA-derived sRNAs, ∼22-nt in length, are in fact loaded into mammalian AGOs upon their processing by RNaseZ or Dicer (54). These AGO:tRNA complexes are RNAi-competent and have attracted attention as possible global regulators of RNA silencing in mammals (54). Consistent with this previous work (54), the proportion of 20-to-24-nt reads mapping to tRNAs is strongly increased in TraPR versus Total-RNA-based plasma libraries, whereas the inverse is true for longer (>24-nt) reads (Figure 5C). Furthermore, the 20-to-24-nt tRNA reads display a clear trend towards a 5′ terminal Uridine residue (signature of preponderant Ago cargoes in mammals) only in TraPR- but not in Total-RNA libraries (Figure 5D). This was even more pronounced when exclusively examining tRNA Val-AAC and tRNA His-GTG, the two most TraPR-enriched 5′ U/A tRNA fragments in the 20-to-24-nt size class (Figure 5E-G and Supplementary Figure S5E-G). Therefore, TraPR allows the unbiased detection of bona fide Ago:sRNA complexes from as little as 150 μl plasma.
DISCUSSION
Here, we have shown how a simplified Q-sepharose-based procedure allows purification of all known classes of silencing sRNAs identified to date. In most instances, we have provided western-based or mass spectrometry evidence that silencing sRNAs co-purify with their cognate AGO/PIWI effector protein(s), thereby validating the notion that, at the very least, TraPR isolates minimal RISCs across kingdoms. This universality is most likely granted by the high conservation of AGOs’ structural and biochemical properties among organisms, including, chiefly, their isoelectric point. These properties are accommodated by a generic, mild AGO-elution buffer ensuring that free nucleic acids, including tRNA, rRNA and RNA degradation products, remain strongly adsorbed on the Q-sepharose matrix, being only eluted at much higher salt concentrations. Although the TraPR embodiment described here entails the use of a single, universal E-buffer, we have implemented TraPR variants allowing further empirical deconvolution of the E-fraction with buffers spanning a range of mild salt concentration. This, in theory, should enable sub-fractionation of distinct AGO:sRNA complexes, as was done with the original Q-sepharose-based method applied to the separation of miRNA- versus piRNA-loaded RISCs in rat (13). Such a TraPR-based sub-fractionation approach conducted in Arabidopsis indeed revealed the existence of separate pools of AtAGO1 and enabled the isolation of distinct AtAGO4-clade members (i.e. AtAGO4, AtAGO6, AtAGO9) together with sub-populations of discrete heterochromatic siRNA cargoes (data not shown).
In its most straightforward embodiment, TraPR provides a universal, simple and rapid solution to silencing sRNA isolation and downstream analyses in virtually any organism including non-model species, which are largely neglected in silencing sRNA-exploration studies. Importantly, deep-sequencing conducted in plants, flies and mouse revealed no overt silencing sRNA bias incurred by TraPR compared to methods relying on total, instead of AGO-bound, sRNA isolation. Thus, putative AGO-unloaded silencing sRNA species are not a cause for concern regarding the exhaustiveness of silencing sRNA repertoires generated by the method. This incidentally strongly suggests that silencing sRNAs commonly sequenced after total RNA-based extraction and ligation-based cloning are mostly loaded entities. Noteworthy, TraPR-isolated AGO:sRNA complexes remain compatible with more targeted, downstream analyses because we have shown that E-fractions are directly amenable to subsequent AGO-IPs.
In all cases tested, TraPR outperforms tedious gold-standard methods, granting isolation of silencing sRNAs via a standardized, three-step procedure requiring no adjustment. Due to its robustness, resilience to RNA degradation, ease-of-use, complete operation at room-temperature, and compatibility with minute amounts of starting material, TraPR should be ideally suited for biomarker identification and clinical diagnosis/prognosis in a variety of tissues and bio-fluids.
DATA AVAILABILITY
Sequencing data have been deposited into the Gene Expression Omnibus (GEO) under the accession number GSE134516. Original images have been deposited in Mendeley and are available under doi: 10.17632/xgfhrtbw48.1. Mass spectrometry data are deposited on PRIDE (http://www.ebi.ac.uk/pride; Project: PXD015395).
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Laure C. David, Mattia Valentini and Bogdan Mateescu for providing M. esculenta, S. pombe and M. musculus samples, respectively. The authors thank Arturo Mari-Ordoñez and the Voinnet team for fruitful discussions and critical reading of the manuscript. The FGCZ is deeply acknowledged for its support, especially at the initial stages of technology development.
Notes
Present address: Stefan Oberlin, Department of Microbiology and Immunology, UCSF Diabetes center, University of California, San Fransisco, USA.
Present address: Julia Svozil, Lonza Ltd, PCP, Visp 3930, Switzerland.
Present address: Veronica Barragan-Borrero, Gregor Mendel Institute (GMI), Vienna, 1030, Austria.
Contributor Information
Thomas Grentzinger, Department of Biology, Swiss Federal Institute of Technology (ETH), Zürich, 8092, Switzerland.
Stefan Oberlin, Department of Biology, Swiss Federal Institute of Technology (ETH), Zürich, 8092, Switzerland.
Gregory Schott, Department of Biology, Swiss Federal Institute of Technology (ETH), Zürich, 8092, Switzerland.
Dominik Handler, Institute of Molecular Biotechnology of the Austrian Academy of Sciences (IMBA), Vienna, 1030, Austria.
Julia Svozil, Department of Biology, Swiss Federal Institute of Technology (ETH), Zürich, 8092, Switzerland.
Veronica Barragan-Borrero, Department of Biology, Swiss Federal Institute of Technology (ETH), Zürich, 8092, Switzerland.
Adeline Humbert, Institut Jacques Monod, Université de Paris, CNRS, Paris, 75013, France.
Sandra Duharcourt, Institut Jacques Monod, Université de Paris, CNRS, Paris, 75013, France.
Julius Brennecke, Institute of Molecular Biotechnology of the Austrian Academy of Sciences (IMBA), Vienna, 1030, Austria.
Olivier Voinnet, Department of Biology, Swiss Federal Institute of Technology (ETH), Zürich, 8092, Switzerland.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
TraPR development in O.V.'s laboratory was supported by an ETH core grant attributed to his team. Work in S.D.'s laboratory was supported by intramural funding from the CNRS; Fondation de la Recherche Médicale (Equipe FRM [DEQ20160334868]); Agence Nationale de la Recherche (ANR POLYCHROME). Funding for open access charge is through core support from ETH attributed to O.V.
Conflict of interest statement. A patent application has been filed with regard to the TraPR technology, listing G.S., O.V and T.G. as inventors. The technology has been exclusively licensed to Lexogen GmbH (Vienna, Austria) from which several TraPR-based kits are now available.
REFERENCES
- 1. Wilson R.C., Doudna J.A.. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013; 42:217–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chalker D.L., Yao M.C.. DNA elimination in ciliates: transposon domestication and genome surveillance. Annu. Rev. Genet. 2011; 45:227–246. [DOI] [PubMed] [Google Scholar]
- 3. Matzke M.A., Mosher R.A.. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014; 15:394–408. [DOI] [PubMed] [Google Scholar]
- 4. Ow M.C., Lau N.C., Hall S.E.. Small RNA library cloning procedure for deep sequencing of specific endogenous siRNA classes in Caenorhabditis elegans. Methods Mol. Biol. 2014; 1173:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McGinn J., Czech B.. Small RNA library construction for high-throughput sequencing. Methods Mol. Biol. 2014; 1093:195–208. [DOI] [PubMed] [Google Scholar]
- 6. Tsuzuki M., Watanabe Y.. Profiling new small RNA sequences. Methods Mol. Biol. 2017; 1456:177–188. [DOI] [PubMed] [Google Scholar]
- 7. Brennecke J., Aravin A.A., Stark A., Dus M., Kellis M., Sachidanandam R., Hannon G.J.. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007; 128:1089–1103. [DOI] [PubMed] [Google Scholar]
- 8. Mi S., Cai T., Hu Y., Chen Y., Hodges E., Ni F., Wu L., Li S., Zhou H., Long C. et al.. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell. 2008; 133:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hauptmann J., Schraivogel D., Bruckmann A., Manickavel S., Jakob L., Eichner N., Pfaff J., Urban M., Sprunck S., Hafner M. et al.. Biochemical isolation of Argonaute protein complexes by Ago-APP. Proc. Natl Acad. Sci. U.S.A. 2015; 112:11841–11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Behm-Ansmant I., Rehwinkel J., Doerks T., Stark A., Bork P., Izaurralde E.. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006; 20:1885–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Azevedo J., Cooke R., Lagrange T.. Taking RISCs with Ago hookers. Curr. Opin. Plant Biol. 2011; 14:594–600. [DOI] [PubMed] [Google Scholar]
- 12. Das S., Extracellular R.N.A.C.C., Ansel K.M., Bitzer M., Breakefield X.O., Charest A., Galas D.J., Gerstein M.B., Gupta M., Milosavljevic A. et al.. The extracellular RNA communication consortium: establishing foundational knowledge and technologies for extracellular RNA research. Cell. 2019; 177:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lau N.C., Seto A.G., Kim J., Kuramochi-Miyagawa S., Nakano T., Bartel D.P., Kingston R.E.. Characterization of the piRNA complex from rat testes. Science. 2006; 313:363–367. [DOI] [PubMed] [Google Scholar]
- 14. Lau N.C., Robine N., Martin R., Chung W.J., Niki Y., Berezikov E., Lai E.C.. Abundant primary piRNAs, endo-siRNAs, and microRNAs in a Drosophila ovary cell line. Genome Res. 2009; 19:1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grentzinger T., Armenise C., Pelisson A., Brun C., Mugat B., Chambeyron S.. A user-friendly chromatographic method to purify small regulatory RNAs. Methods. 2014; 67:91–101. [DOI] [PubMed] [Google Scholar]
- 16. Grentzinger T., Chambeyron S.. Fast and accurate method to purify small noncoding RNAs from Drosophila ovaries. Methods Mol. Biol. 2014; 1093:171–182. [DOI] [PubMed] [Google Scholar]
- 17. Salomon W.E., Jolly S.M., Moore M.J., Zamore P.D., Serebrov V.. Single-Molecule imaging reveals that argonaute reshapes the binding properties of its nucleic acid guides. Cell. 2015; 162:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bologna N.G., Iselin R., Abriata L.A., Sarazin A., Pumplin N., Jay F., Grentzinger T., Dal Peraro M., Voinnet O.. Nucleo-cytosolic shuttling of ARGONAUTE1 prompts a revised model of the plant MicroRNA pathway. Mol. Cell. 2018; 69:709–719. [DOI] [PubMed] [Google Scholar]
- 19. Jullien P.E., Grob S., Marchais A., Pumplin N., Chevalier C., Otto C., Schott G., Voinnet O.. Functional characterization of Arabidopsis ARGONAUTE 3 in reproductive tissue. 2018; bioRxiv doi:18 December 2018, preprint: not peer reviewed 10.1101/500769. [DOI] [PubMed]
- 20. Azevedo J., Garcia D., Pontier D., Ohnesorge S., Yu A., Garcia S., Braun L., Bergdoll M., Hakimi M.A., Lagrange T. et al.. Argonaute quenching and global changes in Dicer homeostasis caused by a pathogen-encoded GW repeat protein. Genes Dev. 2010; 24:904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morel J.B., Godon C., Mourrain P., Béclin C., Boutet S., Feuerbach F., Proux F., Vaucheret H.. Fertile hypomorphic ARGONAUTE (ago1) mutants impaired in post-transcriptional gene silencing and virus resistance. Plant Cell. 2002; 14:629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lobbes D., Rallapalli G., Schmidt D.D., Martin C., Clarke J.. SERRATE: a new player on the plant microRNA scene. EMBO Rep. 2006; 7:1052–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greenberg M.V., Ausin I., Chan S.W., Cokus S.J., Cuperus J.T., Feng S., Law J.A., Chu C., Pellegrini M., Carrington J.C. et al.. Identification of genes required for de novo DNA methylation in Arabidopsis. Epigenetics. 2011; 6:344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Katiyar-Agarwal S., Gao S., Vivian-Smith A., Jin H.. A novel class of bacteria-induced small RNAs in Arabidopsis. Genes Dev. 2007; 21:3123–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng X., Zhu J., Kapoor A., Zhu J.K.. Role of Arabidopsis AGO6 in siRNA accumulation, DNA methylation and transcriptional gene silencing. EMBO J. 2007; 26:1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takeda A., Iwasaki S., Watanabe T., Utsumi M., Watanabe Y.. The mechanism selecting the guide strand from small RNA duplexes is different among argonaute proteins. Plant Cell Physiol. 2008; 49:493–500. [DOI] [PubMed] [Google Scholar]
- 27. Guérin F., Arnaiz O., Boggetto N., Denby Wilkes C., Meyer E., Sperling L., Duharcourt S.. Flow cytometry sorting of nuclei enables the first global characterization of Paramecium germline DNA and transposable elements. BMC Genomics. 2017; 18:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pall G.S., Hamilton A.J.. Improved northern blot method for enhanced detection of small RNA. Nat. Protoc. 2008; 3:1077–1084. [DOI] [PubMed] [Google Scholar]
- 29. Hayashi R., Schnabl J., Handler D., Mohn F., Ameres S.L., Brennecke J.. Genetic and mechanistic diversity of piRNA 3′-end formation. Nature. 2016; 539:588–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jayaprakash A.D., Jabado O., Brown B.D., Sachidanandam R.. Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Res. 2011; 39:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hatakeyama M., Opitz L., Russo G., Qi W., Schlapbach R., Rehrauer H.. SUSHI: an exquisite recipe for fully documented, reproducible and reusable NGS data analysis. BMC Bioinformatics. 2016; 17:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 34. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lawrence M., Gentleman R., Carey V.. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics. 2009; 25:1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J.. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vaucheret H. Plant ARGONAUTES. Trends Plant Sci. 2008; 13:350–358. [DOI] [PubMed] [Google Scholar]
- 38. Xie Z., Johansen L.K., Gustafson A.M., Kasschau K.D., Lellis A.D., Zilberman D., Jacobsen S.E., Carrington J.C.. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004; 2:E104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Poulsen C., Vaucheret H., Brodersen P.. Lessons on RNA silencing mechanisms in plants from eukaryotic argonaute structures. Plant Cell. 2013; 25:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marker S., Le Mouël A., Meyer E., Simon M.. Distinct RNA-dependent RNA polymerases are required for RNAi triggered by double-stranded RNA versus truncated transgenes in Paramecium tetraurelia. Nucleic Acids Res. 2010; 38:4092–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arnaiz O., Mathy N., Baudry C., Malinsky S., Aury J.M., Denby Wilkes C., Garnier O., Labadie K., Lauderdale B.E., Le Mouël A. et al.. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLos Genet. 2012; 8:e1002984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Singh D.P., Saudemont B., Guglielmi G., Arnaiz O., Goût J.F., Prajer M., Potekhin A., Przybòs E., Aubusson-Fleury A., Bhullar S. et al.. Genome-defence small RNAs exapted for epigenetic mating-type inheritance. Nature. 2014; 509:447–452. [DOI] [PubMed] [Google Scholar]
- 43. Khatabi B., Arikit S., Xia R., Winter S., Oumar D., Mongomake K., Meyers B.C., Fondong V.N.. High-resolution identification and abundance profiling of cassava (Manihot esculenta Crantz) microRNAs. BMC Genomics. 2016; 17:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jeong D.H., Park S., Zhai J., Gurazada S.G., De Paoli E., Meyers B.C., Green P.J.. Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell. 2011; 23:4185–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Djupedal I., Kos-Braun I.C., Mosher R.A., Söderholm N., Simmer F., Hardcastle T.J., Fender A., Heidrich N., Kagansky A., Bayne E. et al.. Analysis of small RNA in fission yeast; centromeric siRNAs are potentially generated through a structured RNA. EMBO J. 2009; 28:3832–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Youngman E.M., Claycomb J.M.. From early lessons to new frontiers: the worm as a treasure trove of small RNA biology. Front Genet. 2014; 5:416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ringwald M., Eppig J.T., Kadin J.A., Richardson J.E.. GXD: a Gene Expression Database for the laboratory mouse: current status and recent enhancements. The Gene Expresison Database group. Nucleic Acids Res. 2000; 28:115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. O’Donnell K.A., Boeke J.D.. Mighty Piwis defend the germline against genome intruders. Cell. 2007; 129:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Andersen P.R., Tirian L., Vunjak M., Brennecke J.. A heterochromatin-dependent transcription machinery drives piRNA expression. Nature. 2017; 549:54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fritz J.V., Heintz-Buschart A., Ghosal A., Wampach L., Etheridge A., Galas D., Wilmes P.. Sources and functions of extracellular small RNAs in human circulation. Annu. Rev. Nutr. 2016; 36:301–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arroyo J.D., Chevillet J.R., Kroh E.M., Ruf I.K., Pritchard C.C., Gibson D.F., Mitchell P.S., Bennett C.F., Pogosova-Agadjanyan E.L., Stirewalt D.L. et al.. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:5003–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gibbings D.J., Ciaudo C., Erhardt M., Voinnet O.. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009; 11:1143–1149. [DOI] [PubMed] [Google Scholar]
- 53. Gibbings D., Mostowy S., Jay F., Schwab Y., Cossart P., Voinnet O.. Selective autophagy degrades DICER and AGO2 and regulates miRNA activity. Nat. Cell Biol. 2012; 14:1314–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haussecker D., Huang Y., Lau A., Parameswaran P., Fire A.Z., Kay M.A.. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA. 2010; 16:673–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been deposited into the Gene Expression Omnibus (GEO) under the accession number GSE134516. Original images have been deposited in Mendeley and are available under doi: 10.17632/xgfhrtbw48.1. Mass spectrometry data are deposited on PRIDE (http://www.ebi.ac.uk/pride; Project: PXD015395).