


Abstract
R-loops are dynamic, co-transcriptional nucleic acid structures that facilitate physiological processes but can also cause DNA damage in certain contexts. Perturbations of transcription or R-loop resolution are expected to change their genomic distribution. Next-generation sequencing approaches to map RNA–DNA hybrids, a component of R-loops, have so far not allowed quantitative comparisons between such conditions. Here, we describe quantitative differential DNA–RNA immunoprecipitation (qDRIP), a method combining synthetic RNA–DNA-hybrid internal standards with high-resolution, strand-specific sequencing. We show that qDRIP avoids biases inherent to read-count normalization by accurately profiling signal in regions unaffected by transcription inhibition in human cells, and by facilitating accurate differential peak calling between conditions. We also use these quantitative comparisons to make the first estimates of the absolute count of RNA–DNA hybrids per cell and their half-lives genome-wide. Finally, we identify a subset of RNA–DNA hybrids with high GC skew which are partially resistant to RNase H. Overall, qDRIP allows for accurate normalization in conditions where R-loops are perturbed and for quantitative measurements that provide previously unattainable biological insights.
INTRODUCTION
R-loops are three-stranded nucleic acid structures consisting of a Watson–Crick RNA–DNA hybrid and a displaced single strand of DNA. They typically form during transcription, when nascent RNA hybridizes to its DNA template, and they appear to facilitate certain biological processes while provoking DNA damage in other contexts (1,2). R-loops are highly dynamic structures that can form in different locations depending on cell type (3,4) and growth conditions (5). Thus, it is expected that perturbations in growth conditions or depletion of R-loop resolving factors (6,7) would change their genomic levels and distribution. However, truly quantitative comparisons of R-loop levels between such conditions have so far proven elusive.
A number of recent studies have mapped where R-loops form genome-wide through next-generation sequencing approaches. In the most-widely adopted mapping technique, DNA:RNA immunoprecipitation sequencing (DRIP-seq) (8), the hybrid component of R-loops is directly immunoprecipitated using the S9.6 RNA–DNA hybrid antibody from restriction-digested genomic DNA (9). Various subsequent methods have modified DRIP-seq to increase the resolution by sonication (10–12), to map hybrids to a specific strand (4,10,11), or to capture R-loops in a more native context within permeabilized cells (12). Increasingly, hybrid mapping has been used to detect regions of change when growth conditions (5) or R-loop processing factors are altered (13–16). In all of these studies, comparisons are made after hybrid signal has been normalized to the total mapped reads from each sample. However, normalizing to total read counts makes a key assumption: that the RNA–DNA hybrid content remains constant between conditions. While this assumption may be appropriate for small perturbations, it is likely inaccurate when the R-loop content significantly changes between samples.
This issue is not specific to RNA–DNA hybrid mapping, and in a range of other next-generation sequencing approaches this type of normalization has been shown to introduce biases, obscure real changes between conditions and even lead to misinterpretations (17–19). Well-defined internal standards have proven to be a reliable tool to facilitate the accurate comparison of sequencing signal between experimental conditions (18–23). These spiked-in standards or ‘spike-ins’ are introduced during sample preparation, carried through the experimental workflow, and ultimately sequenced to provide an internal normalization factor independent of total mapped reads. In ChIP-seq, Drosophila chromatin spike-ins have been used to quantitatively normalize sequencing signal from histone marks on the human genome, revealing a genome-wide depletion of these marks that was not apparent with normalization using total read counts (21). In RNA-seq, synthetic RNA spike-ins reduced bias in quantifying short genes (20) and changed data interpretations compared to standard normalization, thereby reconciling apparently contradictory experimental data (18). Recently, Drosophila cells were added at the beginning of the DRIP-seq workflow and the Drosophila RNA–DNA hybrids used to normalize between conditions (24). As these hybrids are derived from cells, they cannot be quantified, and therefore this spike-in approach does not allow for absolute quantitation of cellular hybrid content, which is possible with a pure internal standard. Moreover, the benefit of including a spike-in was not evaluated or explored in this study. Synthetic RNA–DNA hybrids have also been used to confirm that RNA–DNA hybrids and adjacent dsDNA structures are compatible with a modified DRIP-seq protocol (11), but these spike-ins were not used for normalization.
Because RNA–DNA hybrids can be created in vitro, pure, synthetic RNA–DNA hybrids are a potential alternative that can be used to normalize hybrid signal and provide absolute quantification of the cellular hybrid content. Here, we describe quantitative differential DNA–RNA immunoprecipitation sequencing (qDRIP-seq), a modified, high-resolution form of strand-specific ssDRIP-seq (11) that is compatible with the use of synthetic RNA–DNA hybrids and Drosophila cell-based hybrids as internal standards. We show that qDRIP-seq allows for sensitive, high-resolution, strand-specific mapping of RNA–DNA hybrids, and facilitates comparisons in hybrid content between different biological conditions.
MATERIALS AND METHODS
Cell culture
HeLa cells were obtained from ATCC (Manassas, VA, USA), where they were tested for mycoplasma and verified by STR profiling. These cells were grown in DMEM (Gibco, Dublin, Ireland) supplemented with 10% FBS and 1% penicillin/streptomycin/glutamine, and grown in 5% CO2 at 37°C. DRB (Cayman Chemical Company, Ann Arbor, MI, USA) was mixed into pre-warmed media at 100 μM and added to cells for 40 min, or the indicated time, prior to cell harvest. Drosophila Schneider 2 (S2) cells were obtained from ThermoFisher Scientific and grown according to manufacturer's instructions at 27°C in Schneider's Drosophila Medium (Gibco) containing 10% heat-inactivated FBS, 50 units penicillin G and 50 μg streptomycin sulphate per milliliter of medium.
Preparation of spike-in hybrids
Escherichia coli genomic DNA was prepared as in (https://bio-protocol.org/bio101/e97). Briefly, 1.5 ml of an overnight culture was lysed in 0.6% SDS for 1 h, and genomic DNA was extracted using a standard phenol–chloroform isolation. The sequences used for spike-in hybrids were then amplified by PCR using Phusion DNA polymerase, and using primers containing the T7 polymerase promoter sequence. RNA was produced from these PCR templates using in vitro transcription from 120 ng of DNA template per spike-in, and purified using an RNeasy kit (#74104, Qiagen, Hilden, Germany), with DNase treatment performed on the columns for 15 min (#79254, Qiagen). The complementary DNA sequence was ordered directly as single-stranded ultramers or megamers (Integrated DNA Technologies, Coralville, IA, USA) (Supplementary Table S1). Annealing was performed between 400 ng of RNA and 100 ng of DNA in 45 μl of 1× buffer 2.1 (#B7202S, New England Biolabs, Ipswich, MA, USA), beginning with a denaturation step for 10 min at 95°C, and followed by seventy cycles of 90 s holds at decreasing intervals of 1°C until the temperature reached 25°C. Hybrids were then purified by 1 h of electrophoresis on a 0.9% agarose gel, followed by excision of the hybrid band and purification by centrifugation for 10 min at 5000 rpm as in (25). Ethanol precipitation was found to cause some dissociation of synthetic RNA-DNA hybrids, and was therefore not used for purification. The final concentrations of nucleic acids after cleanup were determined by fluorimetry using the Qubit HS RNA assay kit (#Q32852, Thermo Fisher Scientific, Waltham, MA, USA). To make the dsDNA spike-ins, sequences were amplified from the E. coli genome by PCR using Phusion DNA polymerase, purified by electrophoresis on a 0.9% agarose gel, excised and isolated by gel extraction. The ssDNA spike-in was purchased directly as an ultramer (Integrated DNA Technologies) (Supplementary Table S1). All seven spike-ins were diluted and combined to produce a stock solution of 2.5 pM for each spike-in. Five microliters of the spike-ins were added to individual experimental samples just prior to cell lysis, giving a final ratio of 3.76:1 compared to the cell count. This ratio was chosen to put the spike-in copy number roughly on parity with the average HeLa genome copy number, as HeLa cells are approximately hypertriploid.
Considerations for spike-in design
In addition to the two synthetic RNA–DNA spike-ins described (∼280 bp long), two additional RNA–DNA spike-ins were designed, each ∼130 bp long. These sequences were successfully synthesized, purified and recovered in DRIP (Figure 1B, C). However, standard size-selection with AMPure XP beads (Beckman Coulter, Brea, CA, USA) depleted these smaller spike-ins disproportionately during library preparation (Supplementary Figure S2B). Although altering the size-selection parameters improved the recovery of the shorter spike-ins, this unacceptably compromised library quality (Supplementary Figure S2B). Therefore, care must be taken when using spike-ins <150 bp in length, as these may not be compatible with all library synthesis procedures.
Figure 1.

Preparing and evaluating synthetic RNA–DNA hybrids as spike-ins for DRIP. (A) Experimental scheme showing how hybrids were synthesized. Briefly, target regions were amplified from E. coli genomic DNA with a flanking T7 promoter. RNA was prepared from these templates by in vitro transcription, then hybridized to a synthetic ssDNA oligo. Hybrids were purified by agarose gel electrophoresis. (B) Gel image showing RNase H reversible size-shifts after hybridization of RNA and DNA. Unlabeled samples were separated on a 2.5% agarose gel which was then stained with RedSafe nucleic acid staining solution. (C) qPCR of genomic (left) and spike-in (right) hybrids following transcription inhibition with DRB. RNase H (RH) treatment demonstrates antibody specificity. Error bars represent 95% confidence interval (CI) of the mean. Results are significantly different as determined by non-overlapping 95% CIs. In primer name, GB indicates gene body.
As a significantly cheaper alternative to the use of long DNA oligomers, hybrids were also made using PCR-generated dsDNA templates for annealing reactions together with purified RNA. While these hybrids could be used as spike-ins, both DNA strands were present in sequencing libraries, indicating that some hybrid molecules were formed containing the non-template strand. To generate a more homogeneous hybrid population, the synthetic hybrids used in this study were made by annealing RNA to commercially-sourced pure ssDNA templates as described above.
DRIP sonication
DRIP was adapted from the previously published procedure (8,26). 2 × 106 HeLa cells were resuspended in 1.6 ml TE and lysed with 50 μl 20% SDS and 5 μl Proteinase K 20 mg/ml (Thermo Fisher Scientific) for 3 h at 37°C. Synthetic RNA–DNA hybrid spike-ins (5 μl of a stock solution at 2.5 pM) were added to cells in TE buffer prior to lysis. For spike-in with Drosophila S2 cells, 1.3 × 106 S2 cells were mixed with 2 × 106 HeLa cells in TE prior to cell lysis to give a 1:1.5 S2:HeLa cell ratio, with the aim of obtaining ∼10% of total sequencing reads from the Drosophila genome.
DNA was extracted by phenol–chloroform extraction using phase lock tubes and ethanol precipitated. Precipitated DNA was gently spooled and washed with 70% ethanol without centrifugation. DNA was allowed to air dry for 20 min and resuspended on ice in 130 μl TE buffer. DNA was sonicated in 6 × 16 mm microtubes (Covaris, Woburn, MA, USA) to a peak fragment size of 300 bp, performed on a Covaris machine (E220 evolution) using SonoLab 7.3 software, with 10% Duty Factor, 200 cycles/burst, 140 peak incident power and for 60 s per tube. Where relevant, samples were treated with RNase H (5000 units/ml, New England Biolabs) prior to immunoprecipitation. For low RNase H treatment, 10 μg of sonicated DNA was digested with 6 μl RNase H in 1× RNase H digestion buffer in 80 μl total volume overnight at 37°C and purified using standard methods described above. For high RNase H treatment, 4 μg of sonicated DNA per reaction was digested with 18 μL RNase H in 180 μl total volume overnight at 37°C. Following RNase H treatment, DNA was purified with phenol-chloroform extraction and ethanol precipitation. For each immunoprecipitation, 4 μg of sonicated DNA (with or without pre-treatment with RNase H treatment) was bound with 20 μg of S9.6 antibody (Antibodies Incorporated, Davis, CA, USA) in 1× binding buffer (10 mM NaPO4 pH 7, 140 mM NaCl, 0.05% Triton X-100) overnight at 4°C. Dynabeads Protein G beads (Thermo Fisher Scientific) were added for 2 h. The use of magnetic beads compared to agarose beads increased our fold enrichment ∼2-fold relative to hybrid-negative genomic loci. Bound beads were washed three times in binding buffer and elution was performed in 250 μl elution buffer (50 mM Tris pH 8, 10 mM EDTA, 0.5% SDS, 8 μl Proteinase K 20 mg/ml) for 45 min with rotation at 55°C. DNA was purified with phenol–chloroform extraction and ethanol precipitation.
DRIP digestion
This was performed as described previously (8,26) and detailed above for DRIP sonication but with the following differences: ethanol precipitated, spooled and dried genomic DNA was resuspended in 200 μl water and fragmented with a cocktail of restriction enzymes (Bsrg1, EcoR1, HindIII, SspI, XbaI, 2 μl each) in NEB 2.1 buffer and 1 mM spermidine for 10 h at 37°C. DNA was extracted by phenol–chloroform extraction using phase lock tubes and ethanol precipitated. Immunoprecipitation and subsequent steps were performed as described above for DRIP sonication.
Library preparation and sequencing for qDRIP-seq
DNA libraries were prepared from three pooled technical replicate DRIPs per sample. While the DNA material from one IP (or less) is sufficient for successful library preparation, we found pooling IPs effective in minimizing technical variability due to sample handling in pilot experiments. DNA libraries were synthesized from ssDNA using the Accel-NGS 1S DNA library kit (Swift Biosciences, Ann Arbor, MI, USA), according to the manufacturer's protocol. Using multiplexing adapters from the 1S Plus Set A Indexing Kit (Swift Biosciences), adapter-ligated DNA was amplified by PCR. For inputs 12 PCR cycles were used to amplify 1 ng of DNA, and for IP samples 13 PCR cycles were used on the equivalent amount of DNA from one IP reaction. Following PCR, DNA fragments 200–600 bp were retained by size selection using a 0.6× volume ratio of AMPure XP beads (Beckman Coulter)/sample followed by 1.0× ratio. Library DNA was analyzed on a Bioanalyzer DNA HS (Agilent, Santa Clara, CA, USA) at the Stanford Protein and Nucleic Acid Facility, quantified by qPCR using NEBNext Library Quant Kit for Illumina (New England Biolabs), then pooled and sequenced on a HiSeq 4000 (Illumina, San Diego, CA, USA) at the Stanford Genome Sequencing Service Center, using 2 × 151 bp sequencing.
qPCR
qPCR was performed on a Roche LightCycler 480 Instrument II using SYBR-Green master mix (Bio-Rad Laboratories, Hercules, CA, USA). Primers used for qPCR are listed in Supplementary Table S2.
EU nascent transcription assay
Cells were pulsed for 1 h with 100 μM 5-ethynyl uridine (EU) from the Click-iT RNA Alexa Fluor 488 imaging kit (Thermo Fisher Scientific). Cells were fixed in 4% PFA/PBS for 15 min, and permeabilized with 0.25% Triton/PBS for 15 min. The Click-iT reaction was performed according to manufacturer's instructions. Cells were then incubated in DAPI for 15 min, and the slides mounted with Pro-Long Gold Antifade (Thermo Fisher Scientific) and imaged on a Zeiss Axioscope with a 20× objective (Zeiss, Oberkochen Germany). EU signal intensity from ≥1200 cells per condition was calculated using CellProfiler (version 3.1.8).
qDRIP analysis
Prior to alignment, Skewer (27) was used to remove adapter sequences, and Cutadapt (28) was used to remove low complexity G-rich tails from the beginning of R2 with ‘cutadapt -j $N_CORES –minimum-length 30 -U 12’. Trimmed reads were aligned to a custom genome combined from hg38 and the sequences of the synthetic spike-ins with bwa-mem (29). Reads were separated into positive and negative stranded files using SAMtools (30) and unix text-processing utilities. BEDTools (31) was used to convert these SAM files into BEDPE format, with subsequent sorting, filtering for concordant reads, and duplicate alignment removal performed using unix text-processing utilities. Genome browser tracks were produced with the BEDTools genomecov utility, and visualized using IGV (32). Reads from the spike-in did not have duplicate alignments removed, as we expect multiple coincidental fragments to derive from the spike-ins.
Peak calling and differential peak calling
Peaks were called directly from aligned reads from both strands using MACS2 (33) with default broad peak settings. BEDTools was then used to obtain coverage in each experiment over these consensus peaks. Using these read counts, we filtered out only those regions that showed strong (2-fold) reduction after RNase H treatment as measured by reads per million. We further filtered these RNase H-sensitive peaks to those deriving at least two-thirds of their reads from a single strand. For peak calling on the Drosophila genome, we required peaks to be highly statistically enriched above input (FDR cutoff of 1e–14) and to derive at least 70% of their reads from the expected strand.
For differential peak calling, we separately called peaks for the DMSO and DRB samples using the protocol described above. We then used BEDTools to merge these regions in combination with a comparable number of random 5000 bp peaks to include some unchanged background regions to reduce bias in the differential analysis. The DESeq2 R package (34) was then used to obtain fold change estimates and FDR-corrected P-values for differential expression across peaks.
Metaplots
Metaplots around genes, transcription start sites and other genome features were produced from genome browser tracks with deepTools (35). For gene centered analyses, we used Gencode V29 canonical genes filtered to only include those that had at least one sense read in a publicly available GRO-seq dataset obtained in HeLa cells (36). Tracks for GC and AT-skew were produced using the BEDTools nuc tool with further processing from unix text processing tools. To calculate G-quadruplex density around peak sites, the locations of G-quadruplexes as determined biochemically in (37) were turned into a track using the bedtools genomecov utility (where the track was 1 to indicate the presence of a G-quadruplex, or 0 to indicate the absence), and these values were used to produce metaplots with deepTools.
R-loop absolute quantitation and lifetime analysis
For absolute quantitation, we used BEDTools to calculate maximum read depths over each called peak. We additionally estimated the gene copy number over 10 kb intervals on the genome using our sequenced input samples, finding that most intervals fell into three populations of read-counts with modes having integral ratios of approximately 2:3:4. We thus assigned each 10 kb region as having 2N, 3N or 4N DNA content. Each hybrid peak was assigned a copy number from the modal copy number call across the peak. We then normalized the maximum read depths for peaks in each sample to the estimated genomic copy number at their respective sites, and divided it by the mean spike-in read count obtained for that sample. Interpreting these allelic fractions as the probability that a given site contained a hybrid, we carried out a numerical simulation of the number of expected hybrids in a cell. To do this, we first assumed that hybrid formation was completely uncorrelated between sites. We further made the simplifying assumption that every site either fully contained a hybrid, or did not contain a hybrid. In each simulated haploid genome, each site was randomly assigned as either hybrid-containing, or hybrid-absent using the allelic fraction at that site as a probability. We counted the number of peaks simulated to be hybrid-containing in 200 000 haploid genomes, and then summed pairs of these counts to obtain estimates for 100 000 diploid cells.
For the lifetime analysis, we first used BEDTools to select only those genes with RNase H reversible R-loop peaks, and binned each gene into 500bp regions. Gene specific rates of transcription were obtained from (38). For each bin, we calculated the time without new transcription as  , where ttreatment is the treatment time with DRB (40 minutes), vgene is the gene specific rate of transcription (in kb per minute) Xbin is the start position of the bin within the gene (in kb). We then calculated spike-in-normalized qDRIP read densities for DRP (ρDRB) and DMSO (ρDMSO) over these bins by normalizing the qDRIP read counts over these bins to their associated mean spike-in read count. Using these densities, we calculated the fraction remaining as the ratio of these read densities
, where ttreatment is the treatment time with DRB (40 minutes), vgene is the gene specific rate of transcription (in kb per minute) Xbin is the start position of the bin within the gene (in kb). We then calculated spike-in-normalized qDRIP read densities for DRP (ρDRB) and DMSO (ρDMSO) over these bins by normalizing the qDRIP read counts over these bins to their associated mean spike-in read count. Using these densities, we calculated the fraction remaining as the ratio of these read densities  . For each gene with at least 3 estimated data points, we then built two linear regression models to estimate the intercept a and decay constant β under models of zeroth order decay
. For each gene with at least 3 estimated data points, we then built two linear regression models to estimate the intercept a and decay constant β under models of zeroth order decay 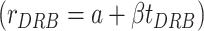 or first order decay
or first order decay  . To avoid high leverage points from skewing the estimated regression line, the logarithmic regression model was weighted by the square root of rDRB. For genes well fit by first order decay models (Pearson's R2 > 0.8), half-lives were calculated as log 2 over the estimated decay constant (ln(2)/β). To calculate the total number of resolution events per day in a cell, we made use of the differential equation for first order decay which relates the rate of decay (dN/dt) to the time constant (k) and the amount of material to decay (N) : dN/dt = –kN. As we found a mean resolution time of 11 minutes, we calculated a mean time constant of ln(2)/(11 min) or 0.063/min. Multiplying this by the estimated count of R-loops in a cell at steady state (300), we find that 18.9 R-loops would be resolved per minute, which is ∼27 000 per day.
. To avoid high leverage points from skewing the estimated regression line, the logarithmic regression model was weighted by the square root of rDRB. For genes well fit by first order decay models (Pearson's R2 > 0.8), half-lives were calculated as log 2 over the estimated decay constant (ln(2)/β). To calculate the total number of resolution events per day in a cell, we made use of the differential equation for first order decay which relates the rate of decay (dN/dt) to the time constant (k) and the amount of material to decay (N) : dN/dt = –kN. As we found a mean resolution time of 11 minutes, we calculated a mean time constant of ln(2)/(11 min) or 0.063/min. Multiplying this by the estimated count of R-loops in a cell at steady state (300), we find that 18.9 R-loops would be resolved per minute, which is ∼27 000 per day.
To analyze biological correlates of lifetimes, 500 bp windows were scored as ‘more stable,’ ‘average’ or ‘less stable’ based on whether their remaining quantity at a given time fell over one standard deviation above the mean, within a standard deviation of the mean, or more than one standard deviation below the mean. Bedtools was used to overlap tracks of total transcription (GRO-seq from (36)), nucleotide content (determined with bedtools and unix utilities), and G-quadruplex counts (as determined biochemically in (37)), and position in gene (using the Gencode V29 canonical annotations.) To determine the relative collision orientation across each bin, we used the replication fork direction across each 500 bp window (as determined by Repli-Seq in (39)) and compared it to the annotated direction of transcription in each bin. Thus, a region with an RFD of 1 (indicating that the leading strand is found to fully coincide with the Watson strand) would be scored as ‘fully co-directional’ in a Watson gene, and ‘fully head-on’ in a Crick gene.
Data processing, data visualization and statistical analysis
Data processing for metaplots, differential peak calls, lifetime analysis, and absolute quantitation was performed with a standard Python scientific stack (Python 3.6.8, NumPy 1.14.0, Pandas 0.22.0). Data were visualized with the Python packages Matplotlib (version 2.1.2) and Seaborn (version 0.8.1). Statistical analysis was primarily performed in Python using the Scipy stats package (1.0.0) for statistical tests and statsmodels (0.8.0) for regression analysis. R (Version 3.1.0) was used for Negative Binomial regression, and for differential peak calling statistics with the Bioconductor DESeq2 package (Love 2014) (version 1.6.3).
RESULTS
Design and preparation of RNA–DNA hybrid spike-ins
To carry out quantitative RNA–DNA hybrid mapping, we wanted to generate internal standards compatible with a hybrid mapping approach. To test the ability of synthetic RNA–DNA hybrids to serve as internal standards or ‘spike-ins’, we began by selecting E.coli sequences with little homology to the human genome which could therefore be clearly distinguished from the human genome in sequencing and by qPCR. We designed four RNA–DNA hybrid spike-ins (L132, H136, L286, H281), although only the two longer spike-ins (L286 and H281) were ultimately used to normalize the sequencing signal. We also designed two double-stranded DNA (dsDNA) spike-ins (LDNA, HDNA) and one ssDNA spike-in (ssDNA) as negative internal controls (Supplementary Table S1). To generate the spike-ins, we amplified target regions from purified E. coli genomic DNA using primers containing a flanking T7 promoter. RNA was then prepared by in vitro transcription using T7 polymerase, purified, and annealed to synthetic complementary ssDNA (Figure 1A). To account for potential biases in sequence recognition with the S9.6 antibody (40), our hybrid and dsDNA spike-ins were designed both with low and high GC-content (Supplementary Table S1). After confirming an RNase-H-reversible size shift in our hybridization product by gel electrophoresis (Figure 1B), we excised the shifted hybrid band and isolated pure hybrids with a gentle gel crushing procedure (25). Ethanol precipitation caused some hybrid dissociation, whereas our purification approach was faster and produced cleaner intact hybrids, as we confirmed by subsequent visualization on a gel (Supplementary Figure S1A).
We next determined how efficiently our synthetic RNA–DNA hybrids were isolated by immunoprecipitation as compared to genomic hybrids. To best control for technical variation between samples, we introduced spike-ins during the initial cell lysis. Genomic DNA was then extracted from 2 million HeLa cells, fragmented by sonication and DRIP-qPCR was performed. We fragmented genomic DNA by sonication rather than restriction enzyme digestion as this has been shown to improve resolution and reduce certain biases in hybrid mapping near promoters (41). We first confirmed that spike-ins were efficiently recovered (Figure 1C), that our antibody concentration was not limiting for genomic R-loops (Supplementary Figure S1B), and that spike-ins did not compete for available antibody with genomic sites (Supplementary Figure S1C). We additionally confirmed that our spike-ins remained stable even during extended periods of immunoprecipitation (Supplementary Figure S1D).
Next, we tested whether the spike-ins would remain unchanged after global hybrid perturbation using 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), a potent inhibitor of RNA Pol II transcription that reduces hybrids at many genomic sites (3,4). After confirming that DRB reduced transcription outside of the nucleolus by imaging nascent RNA incorporating 5-ethynyl-uridine (EU) (Supplementary Figure S1E, F), we compared changes in levels of genomic hybrids and spike-ins by qPCR. DRB treatment dramatically reduced hybrids at genomic loci, but left the yield of spike-in hybrids unchanged (Figure 1C), indicating that the spike-ins can serve as effective standards for qPCR under strong perturbations of genomic R-loops. Although the spike-ins were purified (Supplementary Figure S1A), we did not obtain 100% recovery from the IP. As sonication is proposed to disrupt RNA–DNA hybrids (42), we tested recovery of both genomic and spike-in hybrids following fragmentation with either sonication or restriction digestion. Sonication consistently decreased the yield of recovery compared to enzyme digestion. However, we still did not obtain 100% yield for spike-in constructs with restriction digestion (Supplementary Figure S1G), indicating that there are additional losses or dissolution of hybrids during the DRIP procedure which cannot fully be attributed to sonication. This further highlights the need for spike-ins, which could prevent misinterpretations of technical variation in this loss for biological variation. In all cases, signal both from genomic and spike-in sites was highly RNase H sensitive (Figure 1C), confirming the purity of the synthetic hybrids and the S9.6 antibody's specificity for hybrid structures.
High resolution, strand-specific sequencing of genomic and spike-in hybrids
To sequence both genomic and spike-in hybrids, we adopted an approach similar to ssDRIP-seq which directly sequences the template ssDNA strand of the hybrid (11). We chose this approach because the library preparation method used in DRIP-seq (8) requires double-stranded DNA, and we did not efficiently capture the single-stranded DNA used in the hybrid spike-ins. Sequencing RNA as in RDIP (10) or DRIPc (4) would work in principle, but we sought to avoid RNA-based techniques due to the S9.6 antibody's off-target affinity for double-stranded RNA (43,44). We also modified the ssDRIP-seq protocol to fragment the genome by sonication, rather than enzymatic digestion (Figure 2A, Supplementary Figure S2A). We found that this workflow, using a highly sensitive sequencing library preparation procedure, reduced the necessary number of cells from 10 million to 1–2 million, and is even possible with <0.5 million cells. After processing we found that only the longer spike-ins (L286, H281) could be used for analysis, as sequencing experiments showed that our shorter length hybrid spike-ins (L132, H136) were removed during the size selection step of library preparation (Supplementary Figure S2B), along with most DNA fragments under 150 bp (Supplementary Figure S2C). Because very little of the genomic DNA was fragmented smaller than 150 bp during sonication (Supplementary Figure S2A), we do not expect this size selection to substantially bias sequencing results on the genome.
Figure 2.

qDRIP provides strand-specific, high resolution RNA–DNA hybrid mapping. (A) Schematic of qDRIP experimental process. (B) Representative genome browser view of qDRIP-seq signal. From top to bottom: two qDRIP-seq biological replicates, RNase H digested sample pooled prior to IP, and input pooled from replicates. All tracks normalized by reads per million mapped. Negative strand signal in red, positive in blue. Bent arrows represent TSS, while large triangular arrows represent TES (transcription end site). (C) Read counts from template strand (TS) and non-template strand (NTS) of hybrids, as well as from ssDNA and dsDNA negative controls. (D) GC (green) and AT (red) skew across coding strand of qDRIP peaks, including 600 bp flanking 5’- and 3’-ends. (E) Fractions of qDRIP peaks overlapping noted genomic features (P = 2.5e–2798, chi-square test). (F) Scaled metaplot of sense hybrids between TSS and first-intron exon boundary, as well as 1 kb upstream of TSS and 1 kb downstream of first intron-exon boundary. Tracks shown are mean IP (blue) and pooled input (grey). Bands represent 95% CI of mean read signal.
We sequenced IPs from two biological replicates, a matched input and a control IP treated with RNase H prior to pulldown. After adapter trimming, reads were aligned to human genome build hg38, with duplicate alignments removed prior to further analysis. We found qDRIP-seq signal to derive primarily from the template strand of transcribed areas within the genome, consistent with isolation of co-transcriptional RNA–DNA hybrids (Figure 2B). From our spike-ins, we detected significant hybrid signal only from the DNA strand of the synthetic hybrids, but not from the ssDNA or dsDNA controls (Figure 2C). Only a small number of reads were detected in the input, consistent with the expected coverage for regions with no enrichment. The strand bias indicates that the template DNA strand must be enriched on the genome. Although the reasons for this are not clear, it is possible that sonication disrupts the non-template DNA strand (42) or that this strand is removed during the wash steps of the immunoprecipitation (11) (Supplementary Figure S2D).
Next we sought to determine how our results compared to those obtained from existing methods for sequencing R-loops. To provide an appropriate basis of comparison, we re-analyzed two previously published R-loop sequencing datasets obtained in HeLa cells: DRIP-seq data (45), and a recent RDIP-seq dataset (46). DRIP-seq remains the most popular sequencing method for R-loops, although it has limited resolution and no strand-specificity. By contrast, RDIP-seq incorporates a sonication step, but it also derives strand-specific signal by sequencing the RNA, raising the possibility of non-specific signal. At the human beta-Actin gene (ACTB), a well-validated site of R-loop enrichment, all three methods detected robust R-loops. However, the extent and enrichment of the signal varied between samples, with DRIP-seq identifying regions lying far upstream of the ACTB transcription start site (TSS) as R-loop forming, and RDIP-seq finding R-loops across the gene body but not at the previously studied R-loop-forming pause site (47,48) and only weakly at the TSS (Supplementary Figure S3A). By contrast, qDRIP-seq only detected signal downstream of the ACTB gene TSS, and this signal extended across the gene body and into the 3′ pause site. When taking a broader view of the genome, we found that RDIP-seq showed strand biases consistent with those observed by qDRIP-seq, and both methods showed good enrichment at TSS sites and within gene bodies (Supplementary Figure S3B). DRIP-seq and qDRIP-seq both showed strong association to genic regions, although the enrichment tended to be stronger in DRIP-seq than qDRIP-seq. This difference in enrichment between qDRIP-seq and DRIP-seq may derive from the use of sonication rather than restriction digest to fragment the genome (Supplementary Figure S2A), which we found to decrease percent recovery by qPCR (Supplementary Figure S1G).
To confirm that these observations held more generally, we examined the distribution of signal in all three sequencing methods around the TSS of the top 10,000 expressed genes in HeLa cells by GRO-seq (Supplementary Figure S3C) (36). Gene promoters have been shown in a variety of R-loop mapping methods to form R-loops robustly (3,4,8,10–12,42). Both qDRIP-seq and RDIP-seq detect R-loops only within gene bodies, and find no signal upstream of the TSS. By contrast, DRIP-seq finds a broad profile that extends approximately 2 kb upstream of the TSS. This pattern may reflect either the lower resolution obtained from fragmenting the genome by restriction digestion, or antisense R-loops upstream of the promoter that cannot be distinguished from sense signal downstream of the promoter. In terms of signal enrichment, qDRIP-seq does not detect as robust of a signal at the TSS compared to DRIP-seq, consistent with our observations at individual sites. Both qDRIP-seq and RDIP-seq show high signal enrichment within the gene body. Overall, we conclude that qDRIP-seq and RDIP-seq have higher resolution than DRIP-seq at the cost of slightly reduced sensitivity. RDIP-seq and qDRIP-seq both offer strand-specific and high-resolution R-loop mapping, but in qDRIP-seq the DNA moiety of the RNA–DNA hybrid is sequenced. This avoids the potential issues of non-specific S9.6 binding to dsRNA (43,44).
We then turned our attention to analysis of the data we obtained using qDRIP-seq. We performed peak calling against a matched input sample and, to facilitate downstream analysis, selected 16,895 peaks with strong strand bias and RNase H sensitivity, representing 191 Mb (6.3%) of genome space. These values are similar to previous reports of the hybrid-containing fraction of the genome, which have found between five and ten percent of the genome to form hybrids (4,11). We found a broad distribution of peak sizes (Supplementary Figure S4A), with a median of 3.7 kb and an interquartile range between 1.1 and 14 kb. The size distribution of reads contained within these peaks was comparable to that found in the control IP sample as a whole (Supplementary Figure S2C). As most R-loops are thought to be 100–2,000 bp in length (6,8,49), most peak sites likely represent a population average of several individual R-loops, as recently suggested by single-molecule experiments (50). Across qDRIP-seq peaks, we found patterns of GC-skew (asymmetry in G content between strands) and AT-skew (asymmetry in A content between strands) as previously reported (Figure 2D) (8,11). We additionally found qDRIP-seq peaks to be highly and significantly underrepresented in intergenic regions and particularly over-represented at the 3′- and 5′-ends of genes when compared to the total fraction of the genome within these compartments (Figure 2E), consistent with other reports (4,10). Within intergenic regions, R-loops were highly enriched over repetitive regions as identified by RepeatMasker on the human genome (Supplementary Figure S4B).
We next asked how qDRIP-seq hybrid signal correlated with known features of the genome. Across promoters, sense hybrid signal was precisely bounded by the TSS, and increased strikingly across the first exon to peak downstream of the first intron-exon boundary, consistent with previous reports of R-loops forming robustly across the first exon (12) (Figure 2F). This may reflect a role of the splicing machinery in preventing R-loop formation (51) or increased RNA Pol II pausing at the intron-exon boundary (52). These patterns are consistent with co-transcriptional R-loop formation and demonstrate the high resolution and strand selectivity of our mapping protocol. We also found that transcription correlated with R-loop density at the TSS (Supplementary Figure S4C), and no R-loop signal was detected around the TSS of non-expressed genes (Supplementary Figure S4D). Around promoters positive for hybrids, histone modifications associated with active transcription were over-represented compared to an expression-matched set of non-hybrid-containing promoters (Supplementary Figure S4E), as previously reported for hybrids (4,10,11).
Having shown that qDRIP-seq can detect hybrids on synthetic RNA–DNA templates and on the human genome, we next ascertained whether qDRIP-seq would also be compatible with a Drosophila cell-based spike-in that has previously been used to normalize RNA–DNA hybrids in sequencing (24). We detected robust signal across a number of loci in the Drosophila genome (Supplementary Figure S5A), with signal generally derived from the template strand as it is on the human genome. After performing peak calling to find robust, highly enriched sites of hybrid formation (see methods), we examined the pattern of hybrids at the TSS of genes containing these peaks. As on the human genome, hybrids were significantly enriched above background only in gene bodies, and were precisely bounded by the TSS (Supplementary Figure S5B). Comparing two biological replicates of control cells, we found that synthetic hybrid spike-ins and the cell-based spike-ins predict similar degrees of normalization between the replicates (Supplementary Figure S5C), establishing consistency between these spike-in approaches.
Synthetic spike-ins allow for absolute quantitation of genomic hybrid fractions
Our hybrid spike-ins were prepared in vitro and purified, added at levels similar to the genome copy number in HeLa cells, and carried through the entire experimental workflow. Spike-in read counts should therefore be similar to a genomic site that forms R-loops 100% of the time. We thus reasoned that spike-ins could be used to estimate the absolute hybrid frequency at each peak site: that is to say that a site forming R-loops at approximately 5% of all copies in a population of cells would be expected to have ∼5% of the spike-in read depth. Summing these frequencies throughout the genome, we can therefore estimate the number of hybrids per cell.
To accurately quantify the percent of hybrid-containing molecules at each site, we first estimated the genome copy number at every hybrid peak site by examining input DNA read counts over 10 kb bins. We found a mean DNA content at each locus slightly greater than 3N (Supplementary Figure S6A), consistent with measurements and known copy number alterations in HeLa cells (Supplementary Figure S6B) (53). After normalizing the read depth over each qDRIP peak site to its respective genome copy number, we then compared these read depths to the read depth measured over the spike-ins.
We find that the mean qDRIP peak region contains a hybrid at ∼0.8% of copies (Figure 3A), although some hybrids at housekeeping genes were found to form at rates between 1 and 10%. These percentages are approximately consistent with previous site-by-site measurements obtained by qPCR (54). As comparisons to a pure internal standard also account for dissolution of hybrids during the experimental workflow, these values represent an independent estimate of genomic fractions from qPCR.
Figure 3.

Absolute quantitation of genomic hybrids using the spike-in. (A) Histogram of estimated maximum genomic hybrid frequencies across consensus qDRIP peaks. Data is pooled from mean of two biological replicates. (B) Histogram of distribution of R-loop frequencies in diploid cells obtained from numerical simulation on genome-frequency data.
We next used these data to simulate the count of hybrids expected in the genome of cells. To do this, we first assumed that hybrid formation was uncorrelated between different sites in the genome, and that hybrid formation was a binary choice (either fully present, or fully absent.) These assumptions allowed us to use the allelic fraction of hybrids at each peak as the probability that each peak formed an R-loop in any given cell. To simulate the hybrid count in a single cell, we used these probabilities to assign each peak site to either contain a hybrid or not, and then counted the number of sites assigned to contain hybrids. Using this simulation procedure for 100,000 cells, we found the mean unperturbed cell to have ∼300 R-loops at steady state (Figure 3B). This estimate likely represents a lower bound due to incomplete recovery of hybrids in the sequencing protocol, and the strong possibility that more than one distinct R-loop structure may form within a single genomic peak site.
Synthetic RNA–DNA spike-ins allow for accurate hybrid signal normalization across R-loop perturbations
Having established a method to sequence genomic and synthetic RNA–DNA hybrids, we next evaluated the spike-ins under experimental conditions where R-loop formation is strongly perturbed. As a proof of principle, we chose to examine hybrid levels in the presence and absence of the RNA Pol II transcription elongation inhibitor DRB, which should acutely perturb R-loop formation at predictable sites. In particular, we expected that DRB treatment would reduce hybrids in places where DRB inhibits transcription, such as downstream of the pause site in Pol-II-transcribed genes, but not within non-pol-II genes such as ribosomal DNA. As DRB would reduce the total number of hybrid-containing sites on the genome, we also expected that raw read counts across remaining areas would be correspondingly increased. If normalizing by total read counts, this would lead to an artifactually high signal at these regions after DRB treatment relative to controls. A similar pattern of overestimation has previously been observed for nascent transcription by GRO-seq after DRB treatment (36).
We thus performed qDRIP-seq in HeLa cells treated with 100 μM DRB for 40 min, a time-point previously shown to reduce but not eliminate hybrids at a subset of human genes (4). As expected for inhibition of transcription elongation, treatment with DRB severely affected hybrid formation (Figure 4A, Supplementary Figure S7A, B) and nascent transcription (Supplementary Figure S7C–E) reducing hybrid signal within the 5’-ends of long genes while retaining hybrids lying far downstream from promoters. As short DRB treatment is not expected to affect transcription from non-RNA Pol-II-transcribed genes, these genomic regions effectively act as a natural internal standard that can be used to evaluate whether synthetic spike-ins are effective in allowing accurate normalization between samples (36). At the polymerase-I-transcribed 18S and 28S ribosomal DNA genes, we confirmed by qPCR that hybrid signal remained unchanged (Supplementary Figure S8A). Using read counts over annotated Pol I and Pol III genes, we found that synthetic spike-in hybrids significantly reduced the extent to which signal at these regions was overestimated (Figure 4B). Similarly, in metaplots of qDRIP-seq read density of Pol I genes, we found that normalizing with read counts overestimated the hybrid signal under DRB treatment. By contrast, normalizing using spike-ins approximately equalized signal between the control and DRB treatment conditions. Although differences in signal were not statistically significant, the trend of correction was within our expectation (Supplementary Figure S8B, C).
Figure 4.

qDRIP allows for effective normalization when R-loops are acutely perturbed by transcription inhibition. (A) Genome browser view taken from a long gene (AUH) showing effects of DRB on hybrid formation. In order from top to bottom, tracks shown are two biological replicate DRB-treated IP samples, two biological replicate control (DMSO) IP samples, and a track showing genes. All tracks are normalized by reads per million mapped. Red indicates negative strand signal, while blue indicates positive strand signal. Bent arrows represent TSS, while large triangular arrows represent TES. (B) Scatter plots showing read-counts over Pol I or Pol III-transcribed regions compared for DMSO and DRB treatment using read counts to normalize (left) and spike-ins to normalize (right). Individual regions are shown as grey dots, while the regression line and bootstrapped 95% CI are shown as a grey line and grey band, respectively. Blue diagonal line represents expected trend line if read counts are equal. The normalization factor calculated using read counts is 1.373 with a bootstrapped 95% CI of (1.247, 1.459), while the normalization factor calculated using spike-ins is 1.170 with a bootstrapped 95% CI of (1.063, 1.242). Altogether, spike-ins significantly reduced overestimation of non-pol II genes (P = 0.006, non-parametric bootstrap of the difference of means). (C) Metaplots of DMSO (blue) and DRB (red) IP signal over the first 200 kb of all genes longer than 200 kb expressed in HeLa cells by GRO-seq (36), normalized using total read counts (left) or spike-in read counts (right).
As a CDK9/PTEF-b inhibitor, DRB blocks transcription by preventing engaged RNA polymerases from transitioning to processive elongation after the 5′-pause site (55). However, DRB does not affect polymerases that have cleared this stage of the transcription cycle. We thus expected that transcription at the 3′-end of long genes should be largely unaffected by DRB treatment. Using publicly available GRO-seq data collected from DRB-treated and control samples (36), we confirmed that nascent transcription remained mostly unchanged at these sites after 30 min of treatment using the same dose of DRB (Supplementary Figure S8D). We reasoned that R-loop formation should also remain largely unchanged in these regions after 40 min of DRB treatment, which we confirmed by qPCR measurements at the 3′ ends of a selection of long genes (Supplementary Figure S8E). As hypothesized, normalization of hybrids with mapped read counts showed a marked increase at the 3′-end of genes (Figure 4C). In contrast, the signal was equalized at these regions using spike-in read counts, consistent with nascent transcript levels and the known biology of DRB (Figure 4C). Interestingly, the DRB hybrid signal was equivalent to that in the control ∼130 kb downstream of the TSS. This is consistent with measurements of transcriptional elongation rates in HeLa cells which suggest that an average polymerase would have traveled 135 kb in 40 min (38). Thus, we find that normalization by total read counts consistently overestimates hybrid levels in regions that should be unaffected by DRB, whereas normalizing with spike-ins brings these regions close to parity.
Hybrid spike-ins facilitate accurate differential peak calling
Having shown that synthetic spike-in hybrids could effectively normalize qDRIP-seq signal across genome features that are unaffected by DRB treatment, we next asked whether spike-ins could facilitate unbiased discovery of differential hybrid-containing regions. In most sequencing experiments, regions where IP signal changes between conditions are not known a priori, and are generally discovered through differential peak calling. We thus performed differential peak calling between our DMSO and DRB samples over 81,151 preliminary peaks from the union of the two conditions. We selected differential regions between the samples at a 1% false discovery rate, and further filtered these calls to include only strand-specific and RNase H-sensitive qDRIP-seq peaks in the final differential peak set.
With spike-ins, we found 94 hybrid sites that were significantly induced and 4812 sites that were significantly repressed after DRB treatment (Figure 5A). Using qPCR, we confirmed that regions called as significantly down (Supplementary Figure S9A) and up (Supplementary Figure S9B) in DRB were indeed significantly altered. Without spike-in normalization, we found that 754 (4.5%) peaks were called as increased in hybrid content under transcription inhibition, an increase of 660 peaks (Figure 5A). We additionally found 394 fewer peaks called as significantly down with read-count normalization compared to spike-in normalization. Given the co-transcriptional nature of R-loops, we expect that most regions should decrease in hybrid formation after DRB addition. Therefore, we suspected that many regions called as increased only in the read-count normalization were likely false positives, and regions that failed to be called as down in the read-count normalization were likely false negatives.
Figure 5.

Spike-ins facilitate accurate differential peak calling. (A) Comparison of DESeq2 calls normalized using total read counts (left) or DESeq2 calls normalized using spike-in read counts (right). Using a cutoff of 0.01 for the FDR corrected p-value, sites significantly increased in the DRB are highlighted in red, and sites significantly decreased in the DRB are highlighted in blue, for both normalization methods. (B) qPCR measurements over genes called as significantly down by spike-in normalization, but non-significant by total read counts. For all qPCR measurements, error bars represent 95% CI of mean value. Results are significantly different (marked as *) as determined by non-overlapping 95% CIs. In primer name, GB indicates gene body, 3’ indicates TES proximal regions, 5’ indicates TSS proximal regions. (C) Same as for (B), but for regions significantly up by read-count normalization.
To test whether these classes of peaks actually represented false negative and false positive calls, we examined changes at a few representative peaks from each class by qPCR. Regions were selected to represent a variety of positions within genes (5′, 3′ and gene body). At the peaks that were significantly decreased after DRB treatment only with spike-in normalization, qPCR measurements confirmed a significant decrease (Figure 5B). Thus, normalizing our differential peak calls with spike-ins strikingly increased the sensitivity of these calls. We additionally tested the specificity of our calls by examining regions called as increased by read-count normalization, but unchanged by spike-in normalization. By qPCR, these regions had no significant differences (Figure 5C), again confirming that spike-ins properly normalized differential calls. Finally, we selected regions that were predicted to have almost no change between our conditions as called by spike-in normalization, but that would be predicted as slightly (1.2-fold) increased in the DRB sample by read-count normalization. Measuring these regions by qPCR, we found that levels did not show any consistent bias towards greater signal in DRB (Supplementary Figure S9C). Thus, regions that are predicted to have identical signal using spike-in normalization, but that show consistent (if small) biases by read-count normalization, show no bias when measured by qPCR. Taken together, these results show that normalization with spike-ins substantially increases the sensitivity and specificity of our differential peak calls, and confirms the importance of using internal standards to discover regions that change in hybrid content between biological conditions.
Observation and analysis of RNase H-resistant signal
Throughout these sequencing experiments, we included a control sample that had been pre-treated with RNase H prior to pulldown with the S9.6 hybrid antibody. RNase H specifically degrades RNA contained within an RNA–DNA hybrid; therefore, treatment with RNase H would be expected to reduce measured hybrid levels to those observed in the input sample. RNase H treatment substantially reduced signal across genomic regions (Figure 2B) and spike-in constructs (Figure 2C). However, we were surprised to observe that substantial signal remained in some regions after RNase H treatment. Deeming this remaining signal as RNase H-resistant (RHR), we sought to better understand its characteristics.
Among known genome features, we found RHR signal to be particularly high immediately downstream of promoters (Figure 6A), although RHR signal was detectable above input across entire gene bodies (Supplementary Figure S10A). High positive GC-skew immediately downstream of the TSS has previously been identified as a major contributing factor to R-loop formation in these regions (8). Given that promoters had high RHR signal, we asked whether the degree of RHR signal at promoters correlated with GC-skew downstream of the TSS. Indeed, RHR signal correlated to GC-skew downstream of the TSS even more strongly than total qDRIP-seq signal, and RHR signal was highly restricted to these regions of high GC-skew (Figure 6B).
Figure 6.

RNase H resistant signal. (A) Aggregate plot of qDRIP-seq signal around the TSS of top 10,000 expressed genes as determined by GRO-seq (36). Tracks are IP (blue), RHR (red) and input (grey). Error bands represent 95% CI of mean. (B) Heatmaps of mean IP signal, RNase H-resistant signal and GC-skew around top 10,000 promoters ranked by GC-skew immediately (0–500 bp) downstream of the TSS. Correlation coefficient between IP signal and GC-skew was 0.06, whereas correlation coefficient for RHR signal was 0.22 (Spearman's rho). (C) GC-skew around RNase H resistant regions within the full (unfiltered) qDRIP-seq peak set. qDRIP peaks (red) compared to regions of equal lengths randomly selected from non-resistant qDRIP-peaks (blue). As before, bands represent 95% CI of mean read signal. (D) Same as (C), but showing biochemically determined G-quadruplex density (37) over these regions. (E) RNase H-resistant signal around RH-resistant peak calls. Peaks lying 5’ in genes (which DRB should affect) are in blue, while peaks lying 3’ in genes (which DRB should not affect) are in red. Left panel is RNase H treatment in control cells, while right panel is RNase H treatment in DRB treated cells. As before, error bands represent 95% CI of the mean.
To generalize these patterns, we performed peak calling on the RHR signal from our control qDRIP-seq experiment, obtaining 125,187 peaks over 106.8 Mb. These peaks were considerably smaller on average than those obtained for qDRIP without RNase H with a median peak size of 0.4 kb and an interquartile range between 0.29 and 0.58 kb (Supplementary Figure S10B compared to Supplementary Figure S4A). The mean read lengths within the control and RHR peaks were 283 and 237 bp, respectively (Supplementary Figure S10C). Peak calling revealed that RHR regions were not restricted to promoter regions, and were found at terminators, gene bodies and intergenic regions (Supplementary Figure S10D). While the majority of qDRIP-seq peak regions were not contained within an RHR peak, a number of RHR peaks were not contained within a called qDRIP-seq peak (Supplementary Figure S10E). To better understand the source of these apparently ‘new’ RHR regions, we examined the distribution of qDRIP-seq signal before RNase H treatment around these peaks. We found a small but significant accumulation of this signal above input at RHR peaks (Supplementary Figure S10F). We therefore conclude that these apparently new peaks consist of low levels of signal that may not have met the threshold for peak calling in the original qDRIP-seq sample, but that are highly selected for after RH treatment.
Focusing on genic RHR peaks where there was an annotated direction of transcription, we asked whether GC-skew was elevated over these regions. We found that GC-skew over RHR peaks was elevated compared to nearby regions of the genome and regions selected from DRIP-seq peaks, confirming that these patterns in nucleotide content generalize outside of promoter regions (Figure 6C). High concentrations of guanine on one strand are also associated with secondary structures such as G-quadruplexes (56). We therefore also examined the correlation to biochemically-determined G-quadruplex forming sequences (37), likewise finding a strong association of these regions with RNase H-resistant peaks (Figure 6D). Thus, RHR signal displays general correlations to these sequence features.
There are two major possibilities that might explain widespread RHR signal in qDRIP. As certain secondary structures (57,58) have been shown in specific contexts to be resistant to RNase H, this signal could represent regions that are not efficiently digested by the enzyme. Alternatively, if RNase H does completely and uniformly digest all hybrids on the genome, this enrichment could be an off-target effect of the S9.6 antibody for particular DNA sequences or secondary structures. As S9.6 and RNase H are the two key reagents for studying RNA–DNA hybrids, it is critical to understand which of these possibilities contributes to this unexpected RHR signal.
To address this issue, we first asked whether increasing RNase H treatment could affect this signal. If some regions of the genome are partially resistant to RNase H, increasing the enzyme concentration and treatment time might be expected to reduce this RHR signal. Conversely, if the signal derives from off-target S9.6 binding, increases in enzyme concentration would not reduce this signal, and might in fact further purify the resistant component of the signal. We thus examined the degree of RHR signal under these increased treatment conditions, finding that increased treatment reduces signal across the genome (Supplementary Figure S11A) and systematically brings down signal at peaks of different levels of GC-skew (Supplementary Figure S11B). This indicates that RHR signal is affected by more stringent digestion conditions, implying that this signal comes from partial rather than complete resistance to digestion. Second, transcription should induce R-loops, but it would not be expected to affect the off-target propensity of S9.6 to recognize particular DNA sequences or structures. DRB treatment therefore provides a means to test whether RHR signal represents bona fide R-loops. If the RHR signal is reduced after DRB treatment, this is consistent with reduced digestion by RNase H, but not with off-target binding. As previously discussed, short treatments with DRB are not expected to affect transcription far from the TSS, but should affect peaks towards the 5′ ends of genes. Without DRB treatment, we find that these two regions have identical RHR signal; however, DRB treatment substantially reduces RHR signal at peaks where DRB is expected to act (Figure 6E), indicating that the observed RHR signal does indeed come from partially resistant RNA–DNA hybrids on the genome.
We finally asked whether RHR signal could be detected with other R-loop mapping methods performed in HeLa cells. Using publicly available datasets, we found accumulation of RDIP-seq signal around the RHR sites, both in samples from control cells and those endogenously over-expressing RNase H1 (Supplementary Figure S11C), indicating that some RNA–DNA hybrids are resistant to human RNase H1 acting in vivo, and not only to ex vivo treatment as we have performed. We also analyzed data from RR-ChIP-seq, a method which does not use S9.6 but in which RNA is sequenced following immunoprecipitation of GFP-tagged human RNase H1 in cells (46). We did not detect signal at the RHR sites from samples immunoprecipitated with either catalytically inactive RNase H1-D210N or wild-type RNase H1 (Supplementary Figure S11D).
Determination of hybrid lifetimes
Having shown that synthetic spike-in hybrids could accurately normalize between control and DRB-treated samples, we leveraged the spike-ins as a tool to carry out a natural experiment to estimate intrinsic R-loop lifetimes genome-wide. Because DRB inhibits new transcription but does not halt actively elongating RNA Pol II, it does not instantaneously halt transcription across the entire gene body. Instead, polymerases that began elongation before DRB treatment continue to transcribe. At the time of cell harvest, regions lying increasingly upstream from the last initiated polymerases (or the ‘front’ of transcription) will have spent increasingly more time without new transcription, and therefore without the formation of new R-loops. Thus, every gene effectively contains a natural timecourse of transcription inhibition (Figure 7A).
Figure 7.

R-loop lifetimes. (A) Schematic of transcription with and without DRB. (B) Ratio of DRB to control signal in RNase H-sensitive peaks, compared to estimated time without transcription. Error bands are 95% CI of the mean. Horizontal dotted line indicates a 2-fold decrease in DRB signal. (C) qPCR measurements during a DRB timecourse at regions predicted to be unstable (top) or stable (bottom) by pseudo-timecourse obtained from sequencing data. Error bars represent 95% CI of the mean. In primer name, GB indicates gene body. (D) GC content across 500 bp regions with shorter, longer or close to average (NS) lifetimes (P = 2.5e–143, Kruskal–Wallis test). (E) Biochemically determined G-quadruplex counts (37) over the same regions as (D) (P = 2.7e–7, ANOVA on Negative Binomial regression, likelihood ratio test). (F) Relative replication fork directionality (RFD) (39) to transcription over the same regions as (D), where 1 represents fully co-directional and –1 represents fully head-on (P = 3.5e–12, Kruskal–Wallis test). (G) Distribution of half-lives assuming first-order decay.
Using gene-specific rates of transcription previously determined by 4sUDRB-Seq (38) and the length of DRB treatment (40 minutes), we can estimate the position of the last front of transcription. We can additionally work backwards from the position of this front, using the rate of transcription to estimate the time without new transcription at each position upstream from the front. By comparing this time to the remaining fraction of hybrid signal after DRB treatment, we can find areas that are longer or shorter lived than the average, and make some quantitative estimates of hybrid half lives.
To determine R-loop lifetimes genome-wide, we compared the inferred time without transcription (calculated using transcription rates) to the fraction of remaining hybrid signal without transcription in our experiment. At early time points, we observed parity between the hybrid content in DMSO and DRB (Figure 7B), consistent with our previous findings at the ends of long genes and with the idea that hybrids are not instantaneously resolved (Figure 4C, Supplementary Figure S8E). At progressively longer time points without transcription, we found a striking reduction in signal. Hybrid levels declined to ∼20% of what is obtained in control samples, with a half-life of ∼15 min (Figure 7B). These results are in line with qPCR timecourses at a limited selection of genes that found promoter hybrid levels diminish with a half-life of ∼10 min during a DRB treatment timecourse (4).
We noticed that there was wide variability in the quantity of remaining signal (as indicated by the 95% confidence interval), with some regions being resolved much faster or slower than the average. After validating these apparent differences in kinetics with a DRB treatment time course (Figure 7C, Supplementary Table S3), we asked whether there were any genomic factors that could predict whether regions had increased or decreased hybrid stability. Strikingly, hybrid lifetimes strongly correlated with GC content, with shorter-lived hybrids having lower average GC-content and more stable hybrids having higher GC content (Figure 7D). We additionally found that an increase in G-quadruplex-forming sequences significantly correlated with longer hybrid lifetimes by negative binomial regression (Figure 7E). Using Repli-seq data obtained in HeLa cells (39), we also found that sites predicted to participate in head-on replication-transcription collisions had longer lifetimes than co-directional collisions (Figure 7F), consistent with measurements of higher hybrid content in these regions (45). We did not observe any consistent trends for GC-skew or AT-skew (Supplementary Figure S12A, B). Low stability and high stability hybrids were both somewhat over-represented at the 5′ and 3′-ends of genes, but there was no consistent trend across categories (Supplementary Figure S12C). Lifetimes also did not consistently correlate with sense transcription, suggesting that total transcription may influence hybrid levels (Supplementary Figure S4C) through formation but not resolution (Supplementary Figure S12D).
We finally sought to quantify the half-lives of specific hybrids. Previous measurements from DRB timecourses using qPCR (4) have found hybrids to be resolved in a manner approximately consistent with first order (exponential) decay. To generalize this pattern, we determined whether logarithmic fits of the remaining proportion of reads after DRB added any explanatory value beyond a simple linear model. As we only had one true time-point of DRB inhibition, we fit each curve to pseudo time-points collected across single genes, reasoning that sites within a single gene would likely be resolved by similar co-transcriptional mechanisms. In 224 (85%) of these genes, we found that exponential models fit the data better as measured by Pearson's R2 (Supplementary Figure S12E). Furthermore, the generally high quality of these fits confirmed that site-to-site variation in lifetimes was fairly low across these gene bodies. Among the 107 genes fitted extremely well (R2 > 0.95) by an exponential model, we found that 88% had hybrids with mean half-lives between 7 and 15 min, with a mean of 11.0 min (Figure 7G). These values suggest that previous measurements at selected promoters (4) generalize across the genome.
With genome-wide estimates of both the hybrid count per cell and the mean lifetimes of these hybrids, we next estimated the rate at which cells resolve hybrids. A half-life of 11 minutes with first order decay implies that 6.3% of cellular hybrids are turned over every minute. Using our previous result that cells contain ∼300 hybrids at steady state, we calculated that 19 hybrids are resolved every minute, for a total of 27,000 per day. As a frame of reference, this is approximately double the rate of depurination in human cells (59). Altogether, these results underscore the power of absolute quantitative measurements as provided by qDRIP.
DISCUSSION
A persistent challenge for the R-loop field has been accurate comparison of hybrid levels between conditions where R-loops are perturbed. More generally, normalization using the conventional approach of total read count assumes that total signal remains unchanged between samples in next-generation sequencing experiments. Sequencing experiments under conditions that strongly decrease or increase signal at a subset of genomic sites will therefore inherently suffer from over- or under-estimation of signal at unchanged sites, respectively. Spiked-in standards have been shown to correct these biases, revealing changes between conditions that were otherwise obscured, and preventing misinterpretations (17–21), but this approach has not been widely recognized as needed for R-loop mapping.
Here, we describe qDRIP-seq, a method that combines stranded, high-resolution hybrid sequencing with synthetic RNA–DNA hybrid spike-ins for cross-condition normalization. We first show that our sequencing procedure recognizes hybrid-containing sites generally consistent with known biology (Figure 2). We additionally use the spike-ins to make absolute estimates of the genomic R-loop fraction at different genomic sites, and used these estimates to model the count of hybrids in an average cell (Figure 3). In cells treated with the Pol II transcription elongation inhibitor DRB, we also show that normalization using total read count overestimates hybrid signal at non-pol II transcribed genes and the ends of long genes. By contrast, normalization using synthetic RNA–DNA hybrid standards corrects these biases (Figure 4). Finally, we find that the use of hybrid spike-ins reduces the false-positive and false-negative rates of differential hybrid peak calling between control and DRB-inhibited conditions (Figure 5), and we identify a set of RNA–DNA hybrids that show partial resistance to RNase H (Figure 6). Collectively, these data demonstrate the need for and the potential utility of synthetic RNA–DNA hybrid spike-ins in hybrid mapping experiments.
Here, we also provide the first data of hybrid lifetimes at a genomic scale (Figure 7). Measuring kinetic off-rates as opposed to thermodynamic steady state levels could prove to be a powerful new approach to study R-loop biology. For example, hybrid lifetimes would be expected to increase after depletion of R-loop processing factors. Thus, identifying the specific sites where hybrid lifetimes increase could reveal where these factors act. As our lifetime estimates critically rely on the ratio of DRB treated to control R-loop signal, spike-in normalization was crucial for accurate results because normalizing by total read count overestimates hybrid signal at the ends of genes. Interrogating lifetimes genome-wide allowed us to discover that high-GC content and G-quadruplex formation, but not high transcription or nucleotide skew, correlate with longer R-loop lifetimes on the genome. Where sufficient data were available across genes to make lifetime estimates, we also found hybrid levels at most genes diminish exponentially with a half-life of ∼11 min. While many factors may influence R-loop lifetimes, the observed exponential decay implies that R-loop resolution does not increase when R-loops are depleted across the genome. This implies that R-loop resolution is not rate-limiting at steady state.
Our estimate that mammalian cells must resolve on the order of 27,000 R-loops per day brings into clear focus the substantial resources that cells must invest to turn over R-loops. Even if 99% of R-loops occur in contexts where they are benign, this would leave 260 potential detrimental events per day, a rate comparable to serious events such as DNA base damage (59). If R-loops only cause damage in specific contexts as some recent studies suggest (60,61), it would be interesting to understand what necessitates this speedy resolution even outside of these contexts. Additionally, this high rate of resolution may partially explain the large and diverse set of pathways required for efficient R-loop processing and resolution (6,7).
Beyond these insights into cellular hybrid content and lifetimes, our sequencing results also open up some interesting questions regarding the nature of the signal obtained by hybrid pulldown and sequencing. We are the first to report substantial RNase H-resistant regions on the genome (Figure 6), and define these regions as having distinct nucleotide and sequence characteristics. Although the source of this signal is not precisely defined, our data suggest that this signal likely represents bona-fide co-transcriptional R-loops that show decreased sensitivity, rather than complete resistance to RNase H. Intriguingly, RDIP-seq also exhibits RNase H1-resistant hybrid signal at these sites, indicating that this phenomenon can be detected using multiple R-loop mapping methods. DRIP-seq results including an RNase H control do not detect these resistant regions (5,26), possibly because resistant regions are relatively small compared to the size of the restriction fragments used in DRIP-seq. While the S9.6 antibody is known to show biases in sequence recognition (40), these results indicate that RNase H may show a possibly distinct set of biases, either in recognition or catalytic activity. RR-ChIP-seq, which uses RNase H1 itself as a tool for R-loop detection, does not exhibit hybrid signal at the same RHR sites as qDRIP and RDIP-seq. Although there are alternative interpretations, this could indicate that RNase H binds poorly to these resistant sites. This would be consistent with our observation of RNase H resistance at these sites. Overall these results indicate that the use of RNase H as a tool for R-loop detection (16,62) requires careful evaluation, since RNase H does not seem to bind or act on all R-loops uniformly. Furthermore, our work highlights the need to optimize RNase H digestion conditions in hybrid mapping experiments, as restricting the genomic regions analyzed to those that are deemed RNase H sensitive could lead to certain biases depending on the extent of digestion, and hence in the regions analyzed.
Additionally, our work implies that the choice of sonication or restriction digest poses an important trade-off in sequencing RNA–DNA hybrids. While sonication substantially improves the resolution of hybrid signal, it also reduces sensitivity in qDRIP-seq. Although the decrease in sensitivity is relatively minor for qDRIP-seq when compared to the gains in resolution and strand-specificity, DRIP-seq may still be the more appropriate technique for experiments requiring extreme sensitivity. Nevertheless, use of spike-ins allowed us to quantify the loss of hybrid material throughout the experimental procedure, as even with gentle fragmentation, pure hybrids can only be isolated with ∼50% yield (Supplementary Figure S1G). While our method inherently assumes that genomic hybrids are recovered with the same efficiency as the synthetic hybrids, these losses highlight the importance of including standards that can account for any variation in pulldown efficiency.
We have additionally shown our approach to be compatible with cell-based hybrid spike-ins such as those recently used in DRIP-seq (24). As an important technical note, we found that the Drosophila genome tended to have weaker hybrid signal within peaks when compared to the human genome. This necessitates careful selection of peak regions within the Drosophila genome to avoid counting background signal in the normalization, which was also the approach taken for the previous study using this cell-based spike-in for DRIP-seq (24). In a head-to-head comparison, we found a cell-based spike-in to behave similarly to the synthetic hybrids used in this study (Supplementary Figure S5), although it will be useful to further evaluate their relative behaviour under conditions in which R-loops are perturbed. Cell-based spike-ins provide some advantages over synthetic spike-ins, as they provide greater sequence diversity, and are easier to prepare than biochemically pure hybrids. On the other hand, cell-based spike-ins could vary between biological replicates due to different growth conditions affecting the hybrid content of cells, and the impure nature of the material does not allow for quantitative comparisons in yield (as we do in Supplementary Figure S1G) or absolute quantification of hybrids in the sequencing experiment (Figure 3). Overall, we believe that there are trade-offs between these two normalization standards, and that there may be contexts in which a cell-based spike-in or a synthetic spike-in might be more appropriate.
While this study provides valuable quantitative insights, there is still room for improvement in the method as described. In particular, our conclusions are based on counts from only two spike-in sequences, as we found that shorter spike-ins were not compatible with size selection during library preparation. Our data suggest that hybrids shorter than 150 bp are unlikely to be useful for normalization (Supplementary Figure S2), which is an important consideration in designing additional spike-ins. Two spike-ins were sufficient to correctly normalize between DMSO and DRB, but additional spike-ins might provide more statistical certainty that could help to normalize conditions with more subtle R-loop perturbations. Additionally, a larger panel of spike-ins could be used for further characterization of the size and sequence bias and dynamic range of DRIP. Making such a library of RNA–DNA hybrids would require a substantial concerted and likely collaborative effort, such as that required for the spike-in library developed for RNA-seq by the External RNA Controls Consortium (ERCC) (20). Nevertheless, we have demonstrated in principle the value of using spike-ins for R-loop mapping genome-wide and set the stage for further development of a more extensive set of controls.
In summary, qDRIP-seq provides high-resolution, strand-specific maps of RNA–DNA hybrids, and allows for quantitative comparisons to be made between conditions where R-loop levels are perturbed. There is increasing interest in factors purported to resolve R-loops in cells (2,6,63), and as many of these factors alter cellular R-loop content, the use of spike-in standards will be particularly important for these studies moving forward.
DATA AVAILABILITY
Analysis notebooks for these experiments have been deposited on github (https://github.com/cimprichlab/crossley_et_al_2019). Next generation sequencing data from this study have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE134084. Public datasets were downloaded from GEO with the accession numbers GSE87607 and GSE120371 for RDIP-seq and GSE87607 for RR-ChIP-seq.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lionel Sanz and Frédéric Chédin for sharing unpublished protocols and important discussions regarding combining sonication and DRIP. We thank members of the Cimprich lab for critically reading this manuscript and also Dan Jarosz and Hannah Long for providing helpful feedback.
Notes
Present address: Stephan Hamperl, Institute of Epigenetics and Stem Cells, Helmholtz Zentrum München, München, Bayern D-81377, Germany.
Contributor Information
Magdalena P Crossley, Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA 94305, USA.
Michael J Bocek, Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA 94305, USA.
Stephan Hamperl, Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA 94305, USA.
Tomek Swigut, Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA 94305, USA.
Karlene A Cimprich, Department of Chemical and Systems Biology, Stanford University School of Medicine, Stanford, CA 94305, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Leukemia and Lymphoma Society [5455-17 to M.P.C.]; National Institutes of Health [GM119334 to K.A.C., S10OD018220 to the Stanford Functional Genomics Facility, T32-CA09302 to M.J.B.]; office of the Vice Provost for Graduate Education at Stanford [to M.J.B.]; V Foundation [D2018-017 to K.A.C.]; qDRIP methodology studies were supported by the V foundation and the full set of spike-ins and extended sequencing studies were supported by the NIH [GM119334]. K.A.C. is an ACS Research Professor. Funding for open access charge: NIH [GM119334].
Conflict of interest statement. None declared.
REFERENCES
- 1. Crossley M.P., Bocek M., Cimprich K.A.. R-loops as cellular regulators and genomic threats. Mol. Cell. 2019; 73:398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stirling P.C., Hieter P.. Canonical DNA repair pathways influence R-loop-driven genome instability. J. Mol. Biol. 2017; 429:3132–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen L., Chen J.Y., Zhang X., Gu Y., Xiao R., Shao C., Tang P., Qian H., Luo D., Li H. et al.. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Mol. Cell. 2017; 68:745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanz L.A., Hartono S.R., Lim Y.W., Steyaert S., Rajpurkar A., Ginno P.A., Xu X., Chedin F.. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell. 2016; 63:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stork C.T., Bocek M., Crossley M.P., Sollier J., Sanz L.A., Chedin F., Swigut T., Cimprich K.A.. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife. 2016; 5:e17548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Santos-Pereira J.M., Aguilera A.. R loops: new modulators of genome dynamics and function. Nat. Rev. Genet. 2015; 16:583–597. [DOI] [PubMed] [Google Scholar]
- 7. Costantino L., Koshland D.. The Yin and Yang of R-loop biology. Curr. Opin. Cell. Biol. 2015; 34:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ginno P.A., Lott P.L., Christensen H.C., Korf I., Chedin F.. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 2012; 45:814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boguslawski S.J., Smith D.E., Michalak M.A., Mickelson K.E., Yehle C.O., Patterson W.L., Carrico R.J.. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J. Immunol. Methods. 1986; 89:123–130. [DOI] [PubMed] [Google Scholar]
- 10. Nadel J., Athanasiadou R., Lemetre C., Wijetunga N.A., P O.B., Sato H., Zhang Z., Jeddeloh J., Montagna C., Golden A. et al.. RNA:DNA hybrids in the human genome have distinctive nucleotide characteristics, chromatin composition, and transcriptional relationships. Epigenet. Chromatin. 2015; 8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu W., Xu H., Li K., Fan Y., Liu Y., Yang X., Sun Q.. The R-loop is a common chromatin feature of the Arabidopsis genome. Nat. Plants. 2017; 3:704–714. [DOI] [PubMed] [Google Scholar]
- 12. Dumelie J.G., Jaffrey S.R.. Defining the location of promoter-associated R-loops at near-nucleotide resolution using bisDRIP-seq. Elife. 2017; 6:e28306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang X., Chiang H.C., Wang Y., Zhang C., Smith S., Zhao X., Nair S.J., Michalek J., Jatoi I., Lautner M. et al.. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017; 8:15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gorthi A., Romero J.C., Loranc E., Cao L., Lawrence L.A., Goodale E., Iniguez A.B., Bernard X., Masamsetti V.P., Roston S. et al.. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature. 2018; 555:387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Manzo S.G., Hartono S.R., Sanz L.A., Marinello J., De Biasi S., Cossarizza A., Capranico G., Chedin F.. DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome. Biol. 2018; 19:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen L., Chen J.Y., Huang Y.J., Gu Y., Qiu J., Qian H., Shao C., Zhang X., Hu J., Li H. et al.. The augmented R-loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol. Cell. 2018; 69:412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loven J., Orlando D.A., Sigova A.A., Lin C.Y., Rahl P.B., Burge C.B., Levens D.L., Lee T.I., Young R.A.. Revisiting global gene expression analysis. Cell. 2012; 151:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu Z., Chen K., Xia Z., Chavez M., Pal S., Seol J.H., Chen C.C., Li W., Tyler J.K.. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014; 28:396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen K., Hu Z., Xia Z., Zhao D., Li W., Tyler J.K.. The overlooked fact: fundamental need for spike-in control for virtually all genome-wide analyses. Mol. Cell. Biol. 2015; 36:662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang L., Schlesinger F., Davis C.A., Zhang Y., Li R., Salit M., Gingeras T.R., Oliver B.. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011; 21:1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Orlando D.A., Chen M.W., Brown V.E., Solanki S., Choi Y.J., Olson E.R., Fritz C.C., Bradner J.E., Guenther M.G.. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 2014; 9:1163–1170. [DOI] [PubMed] [Google Scholar]
- 22. Vale-Silva L.A., Markowitz T.E., Hochwagen A.. SNP-ChIP: a versatile and tag-free method to quantify changes in protein binding across the genome. BMC Genomics. 2019; 20:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guertin M.J., Cullen A.E., Markowetz F., Holding A.N.. Parallel factor ChIP provides essential internal control for quantitative differential ChIP-seq. Nucleic Acids Res. 2018; 46:e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Svikovic S., Crisp A., Tan-Wong S.M., Guilliam T.A., Doherty A.J., Proudfoot N.J., Guilbaud G., Sale J.E.. R-loop formation during S phase is restricted by PrimPol-mediated repriming. EMBO J. 2019; 38:e99793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun Y., Sriramajayam K., Luo D., Liao D.J.. A quick, cost-free method of purification of DNA fragments from agarose gel. J. Cancer. 2012; 3:93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanz L.A., Chedin F.. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2019; 14:1734–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang H., Lei R., Ding S.W., Zhu S.. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics. 2014; 15:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011; 17:10–12. [Google Scholar]
- 29. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laitem C., Zaborowska J., Isa N.F., Kufs J., Dienstbier M., Murphy S.. CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat. Struct. Mol. Biol. 2015; 22:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chambers V.S., Marsico G., Boutell J.M., Di Antonio M., Smith G.P., Balasubramanian S.. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015; 33:877–881. [DOI] [PubMed] [Google Scholar]
- 38. Fuchs G., Voichek Y., Benjamin S., Gilad S., Amit I., Oren M.. 4sUDRB-seq: measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome. Biol. 2014; 15:R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Petryk N., Kahli M., d’Aubenton-Carafa Y., Jaszczyszyn Y., Shen Y., Silvain M., Thermes C., Chen C.L., Hyrien O.. Replication landscape of the human genome. Nat. Commun. 2016; 7:10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Konig F., Schubert T., Langst G.. The monoclonal S9.6 antibody exhibits highly variable binding affinities towards different R-loop sequences. PLoS One. 2017; 12:e0178875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Halasz L., Karanyi Z., Boros-Olah B., Kuik-Rozsa T., Sipos E., Nagy E., Mosolygo L.A., Mazlo A., Rajnavolgyi E., Halmos G. et al.. RNA–DNA hybrid (R-loop) immunoprecipitation mapping: an analytical workflow to evaluate inherent biases. Genome Res. 2017; 27:1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wahba L., Costantino L., Tan F.J., Zimmer A., Koshland D.. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 2016; 30:1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Z.Z., Pannunzio N.R., Hsieh C.L., Yu K., Lieber M.R.. Complexities due to single-stranded RNA during antibody detection of genomic rna:dna hybrids. BMC Res. Notes. 2015; 8:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hartono S.R., Malapert A., Legros P., Bernard P., Chedin F., Vanoosthuyse V.. The affinity of the S9.6 antibody for double-stranded RNAs impacts the accurate mapping of R-loops in fission yeast. J. Mol. Biol. 2018; 430:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamperl S., Bocek M.J., Saldivar J.C., Swigut T., Cimprich K.A.. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 2017; 170:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tan-Wong S.M., Dhir S., Proudfoot N.J.. R-loops promote antisense transcription across the mammalian genome. Mol. Cell. 2019; 76:600–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Skourti-Stathaki K., Proudfoot N.J., Gromak N.. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 2011; 42:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hatchi E., Skourti-Stathaki K., Ventz S., Pinello L., Yen A., Kamieniarz-Gdula K., Dimitrov S., Pathania S., McKinney K.M., Eaton M.L et al.. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell. 2015; 57:636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li X., Manley J.L.. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005; 122:365–378. [DOI] [PubMed] [Google Scholar]
- 50. Malig M., Hartono S.R., Giafaglione J.M., Sanz L.A., Chedin F.. Ultra-deep coverage single-molecule R-loop footprinting reveals principles of R-loop formation. J. Mol. Biol. 2020; 432:2271–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bonnet A., Grosso A.R., Elkaoutari A., Coleno E., Presle A., Sridhara S.C., Janbon G., Geli V., de Almeida S.F., Palancade B.. Introns protect eukaryotic genomes from transcription-associated genetic instability. Mol. Cell. 2017; 67:608–621. [DOI] [PubMed] [Google Scholar]
- 52. Nojima T., Gomes T., Grosso A.R.F., Kimura H., Dye M.J., Dhir S., Carmo-Fonseca M., Proudfoot N.J.. Mammalian NET-Seq reveals genome-wide Nascent transcription coupled to RNA processing. Cell. 2015; 161:526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Landry J.J., Pyl P.T., Rausch T., Zichner T., Tekkedil M.M., Stutz A.M., Jauch A., Aiyar R.S., Pau G., Delhomme N. et al.. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda). 2013; 3:1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chedin F. Nascent connections: R-loops and chromatin patterning. Trends Genet. 2016; 32:828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yankulov K., Yamashita K., Roy R., Egly J.M., Bentley D.L.. The transcriptional elongation inhibitor 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription factor IIH-associated protein kinase. J. Biol. Chem. 1995; 270:23922–23925. [DOI] [PubMed] [Google Scholar]
- 56. Gellert M., Lipsett M.N., Davies D.R.. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. U.S.A. 1962; 48:2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Weldon C., Behm-Ansmant I., Hurley L.H., Burley G.A., Branlant C., Eperon I.C., Dominguez C.. Identification of G-quadruplexes in long functional RNAs using 7-deazaguanine RNA. Nat. Chem. Biol. 2017; 13:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wanrooij P.H., Uhler J.P., Shi Y., Westerlund F., Falkenberg M., Gustafsson C.M.. A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop. Nucleic Acids Res. 2012; 40:10334–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lindahl T., Barnes D.E.. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000; 65:127–133. [DOI] [PubMed] [Google Scholar]
- 60. Costantino L., Koshland D.. Genome-wide map of R-loop-induced damage reveals how a subset of R-loops contributes to genomic instability. Mol. Cell. 2018; 71:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Garcia-Pichardo D., Canas J.C., Garcia-Rubio M.L., Gomez-Gonzalez B., Rondon A.G., Aguilera A.. Histone mutants separate R loop formation from genome instability induction. Mol. Cell. 2017; 66:597–609. [DOI] [PubMed] [Google Scholar]
- 62. Yan Q., Shields E.J., Bonasio R., Sarma K.. Mapping native R-loops genome-wide using a targeted nuclease approach. Cell Rep. 2019; 29:1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bhatia V., Herrera-Moyano E., Aguilera A., Gomez-Gonzalez B.. The role of replication-associated repair factors on R-loops. Genes (Basel). 2017; 8:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Analysis notebooks for these experiments have been deposited on github (https://github.com/cimprichlab/crossley_et_al_2019). Next generation sequencing data from this study have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE134084. Public datasets were downloaded from GEO with the accession numbers GSE87607 and GSE120371 for RDIP-seq and GSE87607 for RR-ChIP-seq.