


Abstract
tRNAs are the most highly modified RNAs in all cells, and formation of 5-methyluridine (m5U) at position 54 in the T arm is a common RNA modification found in all tRNAs. The m5U modification is generated by the methyltransferase TrmA. Here, we test and prove the hypothesis that Escherichia coli TrmA has dual functions, acting both as a methyltransferase and as a tRNA chaperone. We identify two conserved residues, F106 and H125, in the RNA-binding domain of TrmA, which interact with the tRNA elbow and are critical for tRNA binding. Co-culture competition assays reveal that the catalytic activity of TrmA is important for cellular fitness, and that substitutions of F106 or H125 impair cellular fitness. We directly show that TrmA enhances tRNA folding in vitro independent of its catalytic activity. In conclusion, our study suggests that F106 and H125 in the RNA-binding domain of TrmA act as a wedge disrupting tertiary interactions between tRNA’s D arm and T arm; this tRNA unfolding is the mechanistic basis for TrmA’s tRNA chaperone activity. TrmA is the second tRNA modifying enzyme next to the pseudouridine synthase TruB shown to act as a tRNA chaperone supporting a functional link between RNA modification and folding.
INTRODUCTION
All transfer RNAs (tRNAs) are post-transcriptionally modified, and these modifications occur at a higher frequency in comparison to other non-coding RNAs (e.g. rRNA, mRNA) with a median of eight modifications per tRNA (1,2). Generally, tRNA modifications are thought to contribute to tRNA structure, function, folding, and stability. However, the significance of these individual modifications remains poorly understood, particularly for modifications found in the elbow region of tRNA. Despite the fact that many of these modifications are highly conserved, they are typically not essential and are dispensable for growth of bacteria and yeast, which raises the questions of why tRNA modification enzymes are important for cells, what their biological functions are, and why they are conserved.
A prevalent hypothesis is that tRNA modification enzymes function as tRNA chaperones, facilitating the proper folding of tRNA (3–5) as many modification enzymes need to unfold their substrate RNA in order to access the target nucleotide for modification. Previously, Gutgsell et al. (3) demonstrated that TruB, a modification enzyme that catalyzes formation of the conserved pseudouridine (Ψ) at position 55 in the TΨC-loop in all Escherichia coli tRNAs, contributes to cellular fitness in bacterial co-culture competition assays between a wild-type E. coli strain and a ΔtruB E. coli strain. However, this fitness disadvantage was rescued by expressing a catalytically inactive variant of TruB, suggesting that loss of the TruB protein itself rather than the pseudouridine formed by TruB is responsible for imparting a cellular fitness disadvantage. Therefore, the authors hypothesized that TruB functions as a tRNA chaperone (3). Recent work in our lab has indeed proven the hypothesis that TruB is a tRNA chaperone (4); we identified a key amino acid residue, K64, critical for tRNA binding and demonstrate that loss of tRNA binding by TruB hinders tRNA folding in vitro and confers a cellular fitness disadvantage in in vivo co-culture competition. Moreover, we dissected the tRNA chaperone mechanism by TruB which acts as an unfoldase inducing multiple folding/unfolding events in tRNA before slowly modifying it. Based on this proof-of-concept discovery, we hypothesize that other tRNA modification enzymes also perform a dual function in modifying and folding tRNA.
TrmA (former RumT) is an E. coli S-adenosylmethionine (SAM)-dependent methyltransferase that catalyzes the formation of the 5-methyluridine (m5U) modification at position 54 in the TΨC-loop in all E. coli tRNA species. Although this modification is not essential as the absence of trmA or its yeast homolog, trm2, does not impart a growth phenotype, m5U54 is a highly conserved modification. It is found across all three domains of life in almost all bacterial and eukaryotic tRNAs, much like Ψ55 (1,3,6), which suggests that there is an evolutionary significance for TrmA or m5U54, although precise roles of either at the cellular level are unclear. Given that TruB catalyzes a highly conserved modification adjacent to the TrmA target site and was recently established as a tRNA chaperone (4), we tested here the hypothesis that TrmA also functions as a tRNA chaperone and serves a dual function in modifying and folding tRNA. For modification of U54 to occur, TrmA requires opening of the T arm, which must disrupt tertiary interactions between the D arm and T arm, thus locally unfolding the tRNA (7).
Here, we first characterize tRNA binding by TrmA and identify two TrmA amino acid residues that are critical for tRNA binding by TrmA and for methylation of tRNA in vitro. With the goal of assessing the functional role of TrmA interacting with tRNA in vivo, we conducted bacterial co-culture competition assays to determine whether TrmA catalytic activity or tRNA binding and therefore folding are necessary for cellular fitness. In vivo, both TrmA catalytic activity and tRNA binding contribute to E. coli cellular fitness. Finally, we demonstrate that both wild-type and catalytically inactive TrmA promote tRNA folding in vitro, but this activity is lost for TrmA variants impaired in tRNA binding. In conclusion, we confirm the hypothesis that TrmA is a tRNA chaperone and provide mechanistic insight into tRNA binding by TrmA.
MATERIALS AND METHODS
Buffers and reagents
TAKEM4 buffer: 50 mM Tris–HCl, pH 7.5, 70 mM NH4Cl, 30 mM KCl, 1 mM EDTA and 4 mM MgCl2. [C5–3H]-UTP used in radioactive in vitro transcriptions was purchased from Moravek. Q5 DNA polymerase was purchased from New England Biolabs (NEB). Ecolite™ scintillation cocktail was purchased from MP Biomedicals. All other enzymes and chemicals were obtained from Fisher Scientific.
Protein expression and purification
QuikChange® site-directed mutagenesis (Stratagene) was used to mutate the E. coli trmA gene of the pCA24N(GFP minus)-TrmA plasmid (JW3937-AM) obtained from the National Bioresource E. coli Project ASKA (8) to encode the following single amino acid substitutions: R51A, F106A, F106E, H125A, H125E, Q190A, G220D, C324A and E358Q using overlapping primers (Supplementary Table S1). Double mutants R51A C324A, F106A C324A, F106E C324A, H125A C324A and H125E C324A were generated using the pCA24N(GFP minus)-TrmA C324A plasmid. Q5 DNA polymerase was used, and the PCR conditions are described in Supplementary Table S2.
All proteins were expressed and purified as previously described using Ni2+-affinity and size-exclusion chromatography (9). Final protein preparations were determined to be >90% pure by SDS-PAGE and RNA free as determined by A260. Circular dichroism spectra of each TrmA variant revealed no differences between the wild-type and mutant enzymes (Figure S3).
Circular dichroism spectroscopy
Enzymes were diluted to 1 μM in 50 mM sodium phosphate buffer (pH 7.5) and kept on ice prior to measurements. A Jasco J-815 CD spectrometer was initialized following a standard protocol according to Kelly et al. (10). Using a 1 mm cuvette, each sample was scanned eight times, with a 1 nm band width, from 190 nm to 320 nm. To ensure correct concentration, samples were analyzed by SDS-PAGE following CD analysis.
tRNA preparation
Tritium-labeled tRNAPhe was prepared as previously described (9). Briefly, the DNA encoding E. coli tRNAPhe gene was PCR amplified from the pCFO plasmid (Supplementary Table S4) and in vitro transcribed in the presence of 100 μM [5–3H]UTP. Tritium-labeled tRNAPhe was purified using a Nucleobond PC100 column.
Tritium release assay
Tritium-labeled tRNA was refolded in 1x TAKEM4 by heating at 65°C for 5 min followed by slow cooling to room temperature. The assay was conducted with 600 nM [3H]-tRNAPhe, 50 μM SAM and 5 μM or 20 nM enzyme for single-turnover and multiple-turnover, respectively. The single-turnover tritium release experiments were also conducted with TrmA wt and the binding variants F106E, and H125E with a higher concentration of enzyme, 20 μM, or 8.8 μM in the case of TrmA H125E. The amount of released tritium corresponding to m5U54 was determined through scintillation counting as previously described (9). All time courses were recorded in triplicate. Single-turnover experiments were analyzed by fitting with a single-exponential function to determine the apparent rate of catalysis (kapp):
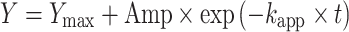 |
Nitrocellulose membrane filtration assay
Increasing concentrations (0–60 μM) of TrmA wt or TrmA variant was incubated in the presence of 10 nM [3H]-tRNA and 50 μM SAM in 1× TAKEM4 buffer at room temperature for 10 min. The reactions were filtered through a nitrocellulose membrane and the membrane was washed with 1 ml cold TAKEM4 buffer. The amount of [3H]-tRNA bound to TrmA was determined by scintillation counting. The nitrocellulose filter binding experiments to determine SAM binding were performed under similar conditions. Here, 50 nM [3H]-SAM was incubated with increasing enzyme concentrations with and without tRNA (5 μM) present. All binding assays were conducted three times. The dissociation constant (KD) was calculated by plotting the fraction of bound ligand against protein concentration and fitting the data to a hyperbolic function:
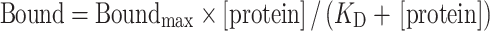 |
Co-culture competition assay
Plasmid pTrc99a was kindly provided by the Stansfield Laboratory (11). The DNA encoding trmA was cloned into the empty pTrc99a vector. Site-directed mutagenesis as described above was used to encode the single amino acid F106E, H125E, and C324A substitution using the pTrc99a-trmA vector. Empty pTrc99a was transformed into the parental E. coli Keio strain BW25113, whereas the plasmids bearing wild-type TrmA and variants were transformed into E. coli Keio ΔtrmA. The competition experiments were performed as described previously in triplicate (3,4,11). In brief, all E. coli co-cultures were cultured at 37°C in LB media containing carbenicillin (100 μg/ml). Strains were first grown individually overnight. Samples containing 1 OD600 of each cell strain were mixed to start the competition. The number of viable cells was determined by plating dilutions on LB plates with ampicillin (100 μg/ml) or kanamycin (50 μg/ml) in triplicate for each replicate.
To assess the cellular fitness of the E. coli co-cultures of Keio ΔtrmA pTrc99a-trmA F106E/H125E against Keio ΔtrmA pTrc99a-trmA C324A, 1 OD600 of the co-culture was collected after each 24-hour growth cycle and subjected to plasmid purification using the BioBasic EZ-10 spin column plasmid DNA miniprep kit. The trmA gene from purified plasmids was PCR amplified using TruTaq (Truin Science) using the primer set, TrmA pTrc99a sense and antisense. The PCR reactions contained 50 ng of plasmid as template, a final concentration of 3% DMSO and used 16 PCR cycles (see Supplementary Table S3 for PCR conditions). The PCR products were digested by HindIII for 3 h at 37°C and analyzed on a 2% agarose gel. Ratios of the band intensities of trmA C324A (top band) to trmA F106E or H125E (middle band) were determined via ImageJ. Ratios of Day 0 samples were set to 1 and ratios for subsequent samples in the time course were normalized accordingly. Each competition assay was conducted in triplicate.
Measuring tRNA folding by aminoacylation
Transfer RNA folding was measured using an adapted aminoacylation assay previous described (4,12). In vitro transcribed tRNAPhe was unfolded at 65°C and immediately added to 0°C reaction buffer containing 6 mM ATP, 1 mM DTT, 3 mM inorganic pyrophosphatase, 3 mM phosphoenolpyruvate, and 10 μg/ml pyruvate kinase. The tRNA (680 nM) was allowed to refold at 0°C for 0, 2, 5 or 20 min in the absence or presence of 200 nM TrmA. Following tRNA folding, [14C]Phe and Phe-tRNAPhe synthetase were added to the reaction mixture to final concentrations of 40 and 20 μM, respectively. Ten-microliter samples taken at different times from the aminoacylation reaction were spotted onto 5% (w/v) trichloroacetic acid (TCA) presoaked Whatman paper disks. Disks were allowed to dry, and then washed three times with 5% (w/v) TCA followed by 100% ethanol to remove free [14C]Phe. Following a drying step at 65°C, the amount of [14C]Phe-tRNAPhe on the disks was quantified via scintillation counting. The aminoacylation time courses were fit with a single-exponential function to determine the initial level of instantaneously aminoacylated tRNA (Y0)
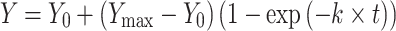 |
The initial level of folded and aminoacylated tRNA was then plotted against folding time. The resulting time courses were fit with the same exponential equation to obtain the rate of folding.
RESULTS
The objective of this study is to test the hypothesis that TrmA is a tRNA chaperone. To achieve this goal, it is necessary to distinguish between the catalytic activity of TrmA to methylate tRNA and its ability to bind tRNA which should result in disruption of tertiary interactions between the D and T arm, i.e. unfolding of tRNA. Therefore, we first characterized tRNA binding by TrmA to dissect the role of residues in the catalytic domain as well as the RNA binding domain for tRNA binding. Based on the crystal structure of TrmA bound to a T arm fragment (7), we focused on residues C324, E358, Q190 and G220 close to the active site to better understand the interplay of SAM and tRNA binding as well as binding of modified and unmodified tRNA to TrmA (Figure 1). In brief, C324 is essential for catalysis as its thiol group forms a covalent bond with carbon 6 of the uracil 54 in tRNA. Residue E358 is equally important since it acts as a general base to abstract a proton from carbon 5 of the methylated reaction intermediate to resolve the covalent bond to C324 (7,13,14). Glutamine 190 (Q190) forms a hydrogen bond with the uridine after it is flipped out of the T arm into the active site of TrmA (7). Although no electron density was observed for SAM in the TrmA crystal structure, modeling the position of the reaction product S-adenosyl-l-homocysteine (SAH) bound to RlmD (formerly RumA), a related E. coli rRNA methyltransferase, as well as comparing amino acid sequences to other methyltransferases predicts the SAM-binding surface in TrmA (7,15): in TrmA, the canonical glycine-rich region (GXGX), observed in other SAM-dependent methyltransferases, is located in a loop region between β11 and α6 and contains residues Gly220, Asn221, Gly222 and Asn223. Of these residues, we focused on G220 to analyze the interplay of SAM binding and tRNA binding. In addition to the catalytic domain, the tRNA is also interacting with the RNA-binding domain of TrmA. Alian et al. (7) crystallized TrmA bound to the T arm of tRNA and by docking the full-length tRNA onto the crystal structure, postulated that residues R51, F106 and H125 contact the D arm of tRNA (Figure 1). Arginine 51 (R51) may contribute to the destabilization of D arm and T arm interactions via side-chain clashes with G57 and G18. Phenylalanine 106 (F106) could have hydrophobic or stacking interactions with G18 of tRNA and the imidazole ring of H125 may pack against the G18 phosphate (7). Accordingly, these residues may be critical in disrupting the tertiary interactions between the D loop and the T loop and induce the local unfolding of tRNA without directly affecting the active site in the catalytic domain.
Figure 1.
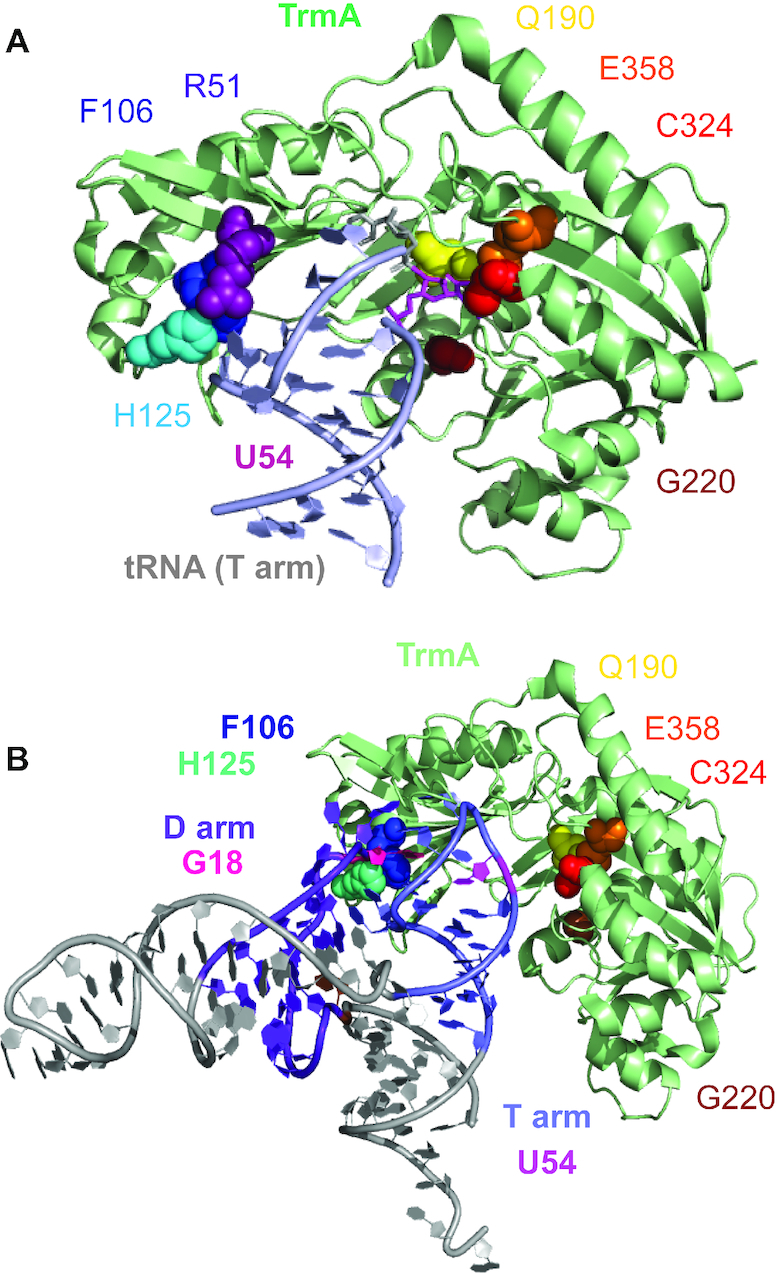
Structural representation of TrmA and its interaction with tRNA. (A) Structure of TrmA bound to T arm of tRNA (PDB ID: 3BT7). The target uridine 54 (magenta) is flipped into the active site comprising residues C324 (red) and E358 (orange) which are directly participating in catalysis of methylation. Residues G220 (brown) and Q190 (yellow) also contribute to catalysis by binding SAM (G220) and hydrogen-bonding with the flipped-out U54 (Q190). The tRNA elbow region has been predicted to interact with R51 (purple), F106 (blue) and H125 (cyan) (7). (B) Structural Model for the disruption of D arm and T arm tertiary interactions in tRNA by the F106-H125 wedge in the RNA-binding domain of TrmA. To model the initial interaction of TrmA with full-length tRNA, the T arm of the full-length tRNA structure (PDB ID: 4TRA) was aligned with the T arm bound to TrmA (PDB ID: 3BT7, PyMol). For clarity, the T arm bound to TrmA is not displayed. This structural model shows the steric clash between F106 (blue) and H125 (cyan) in the RNA-binding domain with the interface of the D arm (purple) and T arm (light blue) of the tRNA, in particular with nucleotide G18 (D arm, pink) which forms a tertiary interaction with Ψ55 in the T arm. Thus, F106 and H125 act like a wedge disrupting tertiary interactions within the elbow region in tRNA in order to allow the target U54 in the T arm to flip into the active site (Q190, E358, C324, G220). This tRNA unfolding is the mechanistic basis for the tRNA chaperone activity of TrmA.
The role of residues in the active site of TrmA for tRNA binding
We first used nitrocellulose filter binding to determine the affinity of wild-type TrmA for tRNA for subsequent comparison to TrmA variants containing substitutions in either the catalytic domain or the RNA binding domain. In the absence of SAM, the wild-type enzyme binds its substrate tRNAPhe extremely tight with a KD in the low nanomolar rage (Table 1), which reflects a combination of the binding affinity for both covalent and noncovalent complexes of substrate tRNA and TrmA as both complexes have been shown to be in equilibrium (13). In the presence of both tRNA and SAM which allows for methyl group transfer thus forming m5U-containing product tRNA, the binding affinity decreased ∼30-fold to a KD of 0.66 μM indicating that TrmA binds weaker to its product than its substrate (Table 1, Figure 2A).
Table 1.
Binding of TrmA wild-type and variants to tRNAPhe in the presence and absence of SAM. The dissociation constants (KD) and standard deviations were determined in triplicate by nitrocellulose filter binding (Figure 2, Supplementary Figure S1).
| TrmA variant | K D, μM | |
|---|---|---|
| [3H]-tRNA | [3H]-tRNA + SAM | |
| Wild-type (wt) | 0.02 ± 0.01a | 0.66 ± 0.11 |
| C324A | 1.08 ± 0.16 | 0.03 ± 0.02a |
| E358Q | 0.07 ± 0.04a | 0.011 ± 0.007 |
| Q190A | 0.28 ± 0.10 | 1.5 ± 0.2 |
| G220D | 0.22 ± 0.05 | 0.06 ± 0.03 |
| R51A | 0.14 ± 0.07 | 1.15 ± 0.34 |
| R51A C324A | 2.8 ± 0.7 | 0.16 ± 0.06 |
| F106A | 1.0 ± 0.4 | 2 ± 1 |
| F106A C324A | 18 ± 7 | 6 ± 2 |
| H125A | 4 ± 3 | 6 ± 2 |
| H125A C324A | 47 ± 27 | 3 ± 1 |
| F106E | 1.6 ± 0.9 | 3.2 ± 1.0 |
| H125E | 1.4 ± 0.4 | 2.1 ± 0.8 |
aNote that KDs in the very low nanomolar range have not been determined with high accuracy as very few data points are found in this region of the binding curve.
Figure 2.
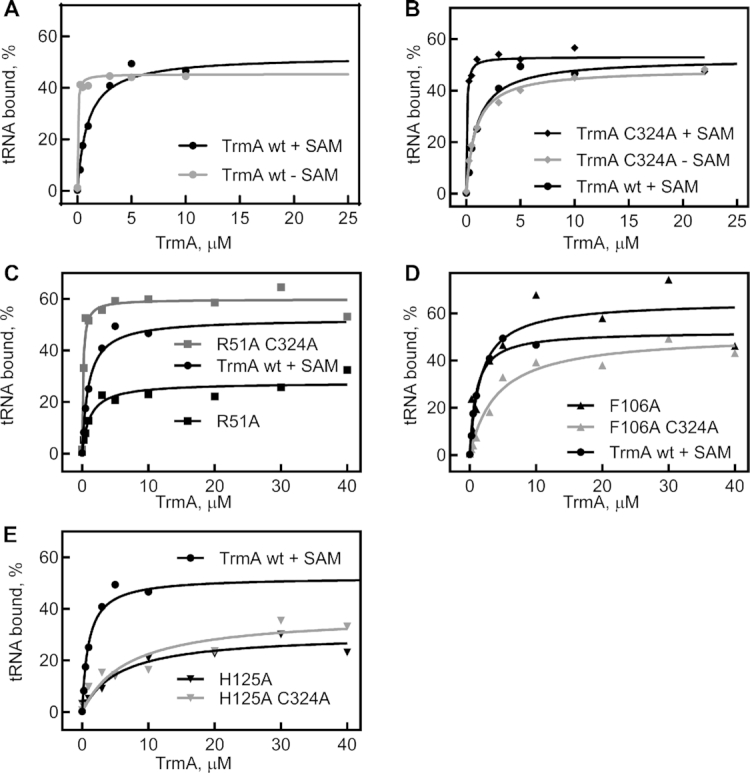
tRNA binding by TrmA wild-type and variants. To determine the affinity of TrmA for tRNA in the presence or absence of 50 μM SAM, 10 nM of radioactively labeled tRNAPhe was incubated with increasing concentrations of TrmA wt or variants. The percentage of bound RNA was determined by nitrocellulose filtration, and the data were fitted to a hyperbolic equation to determine the dissociation constant (KD). A representative binding curve is shown for each data set, and dissociation constants measured in at least three independent experiments are summarized in Table 1. (A) Binding of tRNA to TrmA wt in the presence (black) or absence (gray) of SAM. (B) tRNA binding by the catalytically inactive TrmA C324A variant in the presence (black) and absence (gray) of SAM. The experiments in C to E show tRNA binding in the presence of 50 μM SAM for the following TrmA variants: (C) TrmA R51A (black) and TrmA R51A C324A (gray). (D) TrmA F106A (black) and TrmA F106A C324A (gray). (E) TrmA H125A (black) and TrmA H125A C324A (gray). For comparison, the same binding curve for TrmA wt in the presence of SAM is shown in all panels.
We next assessed the contribution of the catalytic residues, C324 and E358, to TrmA binding affinity for tRNA. The variant TrmA C324A is unable to form a covalent complex with tRNA, and consequently displays a 50-fold reduction in binding affinity for tRNA compared to the wild-type enzyme in absence of SAM (Figure 2B, Table 1). However, upon addition of SAM, the affinity of TrmA C324A to tRNA was dramatically increased to the low nanomolar range although no covalent bond could be formed. This result demonstrates that SAM binding strongly improves tRNA binding to TrmA, and the opposite is true as well: the affinity for SAM is significantly higher in the presence compared to the absence of tRNA (Supplementary Table S5, Figure S1). Hence, the two substrates of TrmA, SAM and tRNA, bind in a positive allosteric mode to this enzyme.
The TrmA E358Q variant cannot undergo proton abstraction after methyltransfer and therefore retains the covalent enzyme-tRNA bond. Measuring the affinity for tRNA revealed very tight binding of tRNA to TrmA E358Q in the both presence and absence of SAM with a KD of 0.011 μM and 0.07 μM, respectively (Table 1, Supplementary Figure S1). Thus, covalent complex formation is the major contributor to tRNA affinity masking the positive allosteric effect of SAM binding.
Alanine substitution of residue Q190, that hydrogen bonds to the target uracil, results in a 14-fold reduction in affinity for tRNA compared to TrmA wt in the absence of SAM (Tables 1, 2 and Supplementary Table S1, Figure S1). The tRNA binding defect of the TrmA Q190A variant is partially rescued by binding of SAM leading to tRNA methylation as only a two-fold increase in KD is observed relative to TrmA wt (1.5 μM versus 0.066 μM).
Table 2.
Single-turnover rates of methylation by TrmA wild-type and variants. Rates were determined by fitting duplicates of tritium release time courses with a single exponential function (Figure 3)
| TrmA variant | rate, min−1 |
|---|---|
| Wild-type (wt)a | 5.4 ± 0.6 |
| C324A | Not active |
| E358Q | Not active |
| Q190A | 1.33 ± 0.09 |
| G220D | 0.11 ± 0.01 |
| R51A | 1.28 ± 0.13 |
| R51A C324A | Not active |
| F106A | 0.23 ± 0.01 |
| F106A C324A | Not active |
| F106E | Not active |
| H125A | 0.30 ± 0.02 |
| H125A C324A | Not active |
| H125E | Not active |
aRate determined by rapid quench-flow analysis (Figure 3A).
Lastly, substituting G220 with aspartic acid completely impairs SAM binding as expected (Supplementary Table S5, Figure S1) confirming the role of this residue. Notably, this substitution also lowers the affinity to tRNA in the absence of SAM by 10-fold compared to TrmA wt, possibly due to the negative charge introduced in the vicinity of tRNA (Table 1). In the presence of SAM, the TrmA G220Q variant will not bind SAM and will not modify tRNA such that a relatively high affinity for unmodified tRNA is observed (0.06 μM) rather than the lower affinity of TrmA wt for modified tRNA measured under these conditions.
Residues F106 and H125 are critical for tRNA binding
To identify important contacts of TrmA with tRNA outside the catalytic domain, we analyzed the effects of substitutions of R51, F016 and H125 on tRNA binding. Knowing such critical interactions will be helpful to distinguish the role of tRNA binding and methyltransfer for a potential tRNA chaperone function of TrmA. Initially, the three residues were substituted with alanine. However, for these single-residue substitutions within TrmA, covalent bond formation between C324 and tRNA can still occur masking the contributions of non-covalent interactions between R51, F106 and H125 with tRNA. Therefore, we also create the double-residue substitutions with C324A to identify the role of these residues independent of covalent bond formation.
Residue R51 seems to play only a minor role in tRNA binding. The variant TrmA R51A exhibited only slightly decreased binding affinities to tRNA compared to the wild-type enzyme in both the presence and absence of SAM, with a KD of 0.14 μM (7-fold increase compared to TrmA wt) and 1.15 μM (twofold increase), respectively (Figure 2C, Table 1). Similar effects were observed for the TrmA R51A C324A double mutant which cannot form a covalent bond to tRNA; compared to TrmA C324A, the affinity for unmodified tRNA of the TrmA R51A C324A variant was increased 2- and 5-fold in the absence and presence of SAM, respectively. In conclusion, residue R51 in the RNA binding domain of TrmA contributes only mildly to tRNA binding as substitutions result in 2- to 7-fold changes in affinity.
In contrast to residue 51, we observe very strong defects in tRNA binding for all TrmA variants harboring a substitutions of residue F106 or H125. The single-substitution TrmA F106A and H125A variants exhibit a more than 50-fold decrease in binding affinities for tRNA in the absence of SAM compared to TrmA wt with dissociation constants of 1.0 and 4 μM, respectively (Figure 2D, E, Table 1). In the presence of SAM TrmA F106A and H125A can modify tRNA (vide infra), which is reflected in the further increased dissociation constants relative to the KD values measured in the absence of SAM (Table 1). The strong effect of the single-substitution variants of TrmA on tRNA binding is remarkable, as these proteins are still able to form a covalent bond between C324 and tRNA. Therefore, we next determined the affinity of TrmA double-substitution variants lacking C324 for tRNA binding. Notably, the double-substitution variants TrmA F106A C324A and H125A C324A display extremely low affinities of 18 and 47 μM in absence of SAM due to the combined loss of the covalent bond to tRNA and the interactions with the RNA binding domain (Figure 2D and E, Table 1). Compared to the single-subsitution TrmA C324A, the double-substitution TrmA variants thus display an 18- and 47-fold reduction in tRNA affinity in the absence of SAM. Finally, to create TrmA variants that are essentially unable to bind tRNA without affecting the active site, we generated the single-substitution TrmA F106E and H125E where the substitution with a negatively charged glutamate residue should repel the tRNA. Interestingly, no further decrease in activity of TrmA F016E and TrmA H125E was observed compared to variants TrmA F106A and H125A (Table 1). In conclusion, our data indicate that residues F106 and H125, but not R51, are highly important for tRNA binding.
Loss of tRNA binding correlates with reduced methylation activity
To assess the effect of substitutions in the catalytic and the RNA-binding domain on the methylation activity of TrmA, we conducted tritium release assays where a tritium at position C5 of the target uracil is released upon addition of the methyl group (Figure 3 and Supplementary Figure S2) (9,16). By measuring methylation activity under single-turnover conditions with enzyme excess, we first determined directly the rate of methylation (Table 2). For TrmA wild-type, these experiments were conducted after rapid mixing in a quench-flow apparatus to obtain accurate rates for this fast reaction (Figure 3A). With increasing concentrations of TrmA from 2.5 to 10 μM, we always observe a rate of about 5.4 min−1 suggesting that the reaction is not limited by TrmA binding to tRNA under these conditions such that the observed rate reflects the rate constant for methylation. As expected, the catalytically inactive variants, TrmA C324A and E358Q, showed no methylation of tRNA (Figure 3B and C) whereas the substitution G220D resulted in a drastic reduction of methylation (Figure 3B, Table 2). Notably, the variant TrmA Q190A only reduced the methylation rate 4-fold indicating that the hydrogen bond to the target uracil is not critical for catalysis (Figure 3B).
Figure 3.
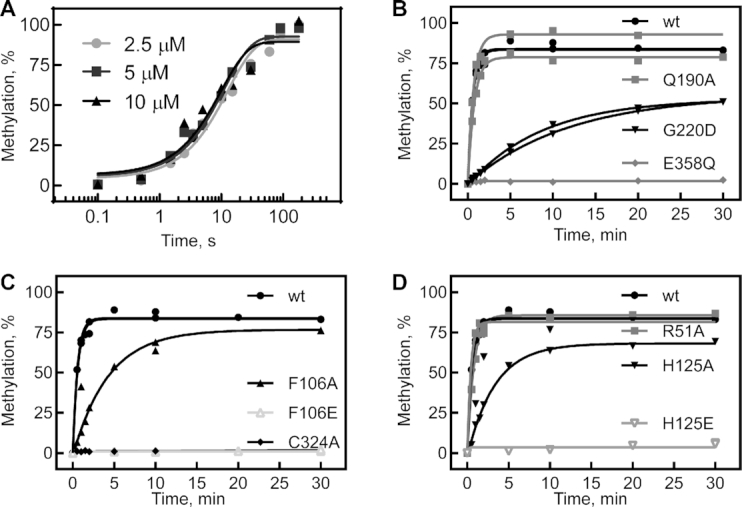
5-methyluridine formation by TrmA wt and variants under single-turnover conditions. (A) To quantitatively determine the single-turnover rate for methylation by TrmA wt, time courses were measured in the quench-flow instrument by rapidly mixing tritium-labeled tRNA (final concentration 1 μM) with 2.5 μM (light gray circles), 5 μM (dark gray squares), and 10 μM (black triangles) TrmA wild-type (preincubated with 50 μM cold SAM) before being quenched with HCl. The resulting data was fitted with a single exponential function to determine the apparent rate of methylation which is independent of the TrmA concentration and therefore not rate-limited by binding of TrmA to tRNA. (B) Single-turnover conditions (5 μM TrmA and 50 μM SAM versus 600 nM tRNA) were used for determining methylation activity rates for TrmA wt (black circles) and variants with substitutions in the active site: Q190A (gray squares), G220D (black triangles), and E358Q (gray diamonds). Rates of methylation for the TrmA variants are determined by fitting to a single-exponential function and are summarized in Table 2. (C) The same single-turnover conditions were applied to compare methylation by TrmA wt (black circles) with the catalytically inactive variant C324A (diamonds) and TrmA F106A (black triangles) and F106E (open gray triangles). (D) The same time course for methylation by TrmA wt is compared to the reactions with R51A (gray squares), H125A (black inverted triangle) and H125E (open inverted gray triangle). All time courses were conducted in duplicate with both data sets shown.
When analyzing the TrmA variants with substitutions in the RNA-binding domain, we observed a modification activity similar to wild-type for variant TrmA R51A with only a 4-fold reduction in activity; this effect is consistent with the negligible loss of tRNA binding by this variant (Figure 3D, Table 2). Conversely, substituting alanine for F106 and H125 resulted in an 18- to 23-fold reduction in the rate of methylation, and substituting F106 and H125 with glutamate completely abolished modification activity (Figure 3C and D, Table 2). Altogether, our data clearly demonstrate that of the three predicted tRNA binding residues, two residues, namely F106 and H125, are critical for tRNA binding by TrmA. As tRNA binding is obviously a prerequisite for tRNA modification, the disruption of tRNA binding by substitutions of F106 and H125 results in a loss of catalysis. In addition, even when tRNA is bound to the TrmA F106E and H125E variants, the tRNA might bind in an incorrect orientation due to the electrostatic repulsion thus hindering tRNA methylation which is supported by the observation of very little enzyme activity even at high protein concentrations (Supplementary Figure S2).
Catalytic activity and tRNA binding by TrmA are critical for bacterial fitness
Based on our observations for the tRNA pseudouridine synthase TruB (4), we hypothesized that impaired tRNA binding by TrmA should prevent tRNA folding by this putative tRNA chaperone and in turn affect cellular fitness. Therefore, we conducted co-culture competition assays in order to determine the roles of TrmA catalytic activity and tRNA binding by TrmA in vivo. Therein, we compared the fitness of E. coli wt to E. coli ΔtrmA strains expressing the wild-type or variants of TrmA. All E. coli strains in this experiment express the pTrc99a plasmid (empty or encoding TrmA) such that the metabolic burden of maintaining the plasmid is the same for all strains. As expected, the E. coli knockout strain ΔtrmA bearing the empty plasmid, pTrc99a, is eventually outcompeted by wild-type E. coli over the time course of 12 days, suggesting that loss of TrmA imparts a cellular fitness disadvantage (Figure 4A). As a control, we show that introducing pTrc99a carrying the wild-type TrmA enzyme alleviates the growth phenotype. Interestingly, E. coli ΔtrmA strains expressing either TrmA F106E, H125E or C324A were rapidly outcompeted by E. coli wild-type (Figure 4A). Because all three variants strongly reduced tRNA methylation in the tritium release assay (Figure 3), we conclude that TrmA-catalyzed modification of tRNA is critical for cellular fitness in vivo. Therefore, this competition assay cannot directly reveal the contribution of tRNA binding for cellular fitness as it is masked by the importance of tRNA methylation.
Figure 4.
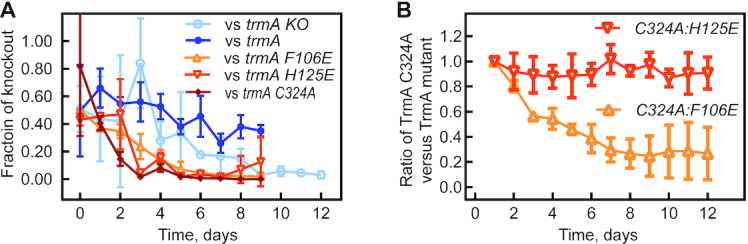
Contribution of tRNA binding and catalytic activity by TruB to bacterial fitness. (A) Bacterial co-culture competition between E. coli wt and E. coli ΔtrmA (open circles), the ΔtrmA strain expressing TrmA wt protein (closed circles), TrmA F106E (open gray triangles), TrmA H125E (open inverted gray triangles) or TrmA C324A (diamonds). (B) Restriction enzyme and PCR-based bacterial co-culture competition assay between E. coli ΔtrmA expressing the catalytic inactive TrmA C324A variant and E. coli ΔtrmA expressing the tRNA binding variants TrmA F106E or H125E.
Thus, to resolve the significance of tRNA binding and therefore tRNA folding and chaperone activity relative to the methylation activity, we conducted direct competition assays between E. coli ΔtrmA strains expressing two different TrmA variants, one being impaired in catalysis (TrmA C324A) and the other being impaired in tRNA binding (TrmA F106E or H125E). To differentiate between the two strains, which both harbor a kanamycin resistance cassette, we PCR amplified the trmA gene from the isolated plasmids and digested the PCR products with HindIII to distinguish trmA C324A (no HindIII site) from trmA H125E and F106E (one HindIII site) (Figure 4B and Supplementary Figure S4). By comparing the band intensities of trmA F106E or H125E with respect to trmA C324A, we can estimate the approximate ratio of the strain bearing the tRNA binding variant to the strain bearing the catalytically inactive variant. Interestingly, the TrmA F106E variant outcompetes the TrmA C324A variant (Figure 4B). This suggests that the fitness defect upon losing methylation by the TrmA C324A variant is larger than the fitness defect of impaired tRNA binding by TrmA F106E which may result from residual methylation activity of TrmA F106E. In contrast, after 12 days of co-culture, the catalytically inactive TrmA C324A variant is present in a comparable ratio to the TrmA H125E (Figure 4B). Thus, neither strain can out-compete the other suggesting that the loss of tRNA binding by TrmA H125E and the accompanying decrease in methylation is affecting cellular fitness to the same extent as abolishing methylation activity alone (TrmA C324A). Due to the high importance of methylation activity by TrmA for cellular fitness, it is therefore impossible to distinguish the relative in vivo contribution of tRNA binding and possibly tRNA folding by TrmA for E. coli.
TrmA folds tRNA in vitro independent of its modification activity
To directly test our hypothesis that TrmA is a tRNA chaperone and facilitates tRNA folding, we used an in vitro tRNA folding assay (4,12). Therein, correctly folded tRNA is rapidly aminoacylated whereas unfolded or misfolded tRNA is only slowly aminoacylated allowing us to quantify the folded tRNA population. Briefly, tRNAPhe was unfolded at 65°C and slowly folded at 0°C in the presence or absence of wild-type enzyme or TrmA variant. At selected folding time points, tRNA folding was measured by determining the fraction of instantaneously aminoacylated tRNA upon addition of an excess of aminoacyl-tRNA synthetase (12). As previously observed (4), tRNA folding seems to be biphasic with one tRNA population folding rapidly before our first data point (∼30 s), whereas another tRNA population folds slower (Figure 5A). Compared to tRNA folding in the absence of any protein, the fraction of rapidly folding tRNA increases almost two-fold in the presence of TrmA wild-type indicating that TrmA facilitates rapid tRNA folding (Figure 5A). To assess the importance of methylation for tRNA folding, we repeated the assay in the presence of the inactive TrmA C324A variant which displayed similar tRNA folding abilities as TrmA wild-type (Figure 5A). Finally, we examined tRNA folding in presence of TrmA F106E and H125E which are significantly compromised in tRNA binding. Whereas TrmA H125E shows a strongly reduced ability to fold tRNA compared to TrmA wild-type, TrmA F106E cannot assist in tRNA folding and the tRNA folding time course resembles the reaction in absence of any protein (Figure 5B). Overall, our experiments demonstrate that TrmA can fold tRNA in vitro independent of its modification activity and that tRNA folding by TrmA is dependent on tRNA binding.
Figure 5.
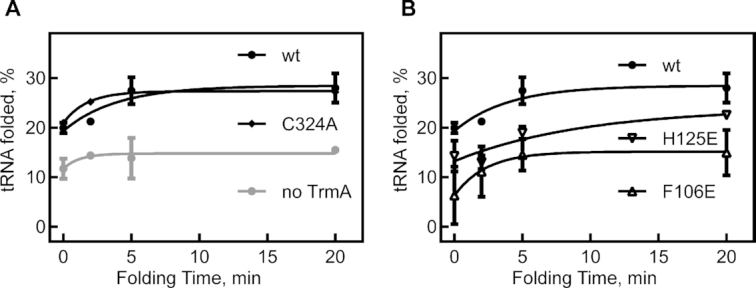
tRNA folding by TrmA in vitro observed by aminoacylation efficiency. (A) Unfolded tRNAPhe was allowed to refold on ice in the presence or absence of TrmA for different folding times. The folded tRNA was then quantified as the percentage of tRNA that was instantaneously aminoacylated upon addition of an excess of [14C]phenylalanine and phenylalanine-tRNA-synthetase. Both catalytically active TrmA wt and the inactive variant TrmA C324A are promoting tRNA folding. (B) The tRNA folding assay was conducted with the TrmA variants H125E and F106E which are impaired in binding tRNA. As expected, the tRNA folding activity is reduced (H125E) or abolished (F106E).
DISCUSSION
In summary, we have here characterized the properties of the tRNA methyltransferase TrmA with respect to tRNA binding and catalysis identifying critical structure-function relationships and tested the hypothesis that TrmA is a tRNA chaperone similar to the pseudouridine synthase TruB. We identify an important contact between two conserved residues, F106 and H125, in the RNA-binding domain of TrmA and the elbow region of tRNA. Single-amino acid substitutions of these residues severely impair tRNA binding and in turn tRNA methylation. Through E. coli co-culture competition assays, we demonstrate that the methylation activity of TrmA is critical for cellular fitness. Finally, we prove the hypothesis that TrmA is a tRNA chaperone by showing that TrmA significantly improves tRNA folding in vitro. The tRNA chaperone activity of TrmA is independent of its catalytic activity, but it depends on tRNA binding.
Our experiments provide insight into the functional role of active site residues of TrmA and reveal cooperative binding of the tRNA and SAM substrates to TrmA. By analyzing tRNA and SAM binding using the catalytically inactive variants TrmA C324A, which cannot form a covalent bond to tRNA, we are not only confirming the importance of covalent bond formation for tight tRNA binding as expected, but we also uncover that SAM binding has a positive allosteric effect on tRNA binding and vice versa. Accordingly, even in the absence of a covalent bond, tRNA binds tightly to TrmA with nanomolar affinity (KD = 30 nM) when SAM is present. These findings suggest that binding of either SAM or tRNA induce conformational changes in the active site of TrmA that facilitate binding of the second substrate.
Further analysis of active site residues uncovers that Q190 forming hydrogen bonds with the target U54 is not essential for methylation activity as only a modest four-fold decrease in methylation activity was observed upon substitution Q190 with alanine. This Q190 residue is more important for binding tRNA as a 10-fold change in the dissociation constant compared to wild-type was observed. These results agree with an earlier in vivo study that recorded a reduction, but not elimination of m5U formation in E. coli for TrmA Q190A and that could not observe a TrmA Q190A tRNA complex in vivo (14). In contrast, the homologous residue Q265 in the rRNA methyltransferase RumA (now RlmD) is highly important for its methylation activity (15). Moreover, we directly confirm that residue G220 in TrmA is critical for SAM binding as predicted and thus also for tRNA methylation in agreement with the in vivo characterization of TrmA G220D (14). The variant G220D also displays a 10-fold reduction in tRNA binding in the absence of SAM; however, tRNA binding is restored in the presence of high SAM concentrations. Within E. coli, the SAM concentration is between 1 and 13 μM and thus insufficient to compensate for the G220D mutation explaining the previous observation that no TrmA G220D-tRNA complex is detected in E. coli (14).
In addition to characterizing the structure-function relationship for catalytic residues, we have also identified the role of critical residues in the RNA-binding domain of TrmA. The crystal structure of TrmA in complex with the T arm of tRNA predicts interactions of the RNA-binding domain of TrmA with the elbow region of tRNA, most likely the D arm (7). Specifically, the author proposed that R51 could intercalate between G18 and G19 after disruption of the tertiary base-pairs between G18 and Ψ55 as well as G19 and C56 in the D loop and the T loop. Similarly, F106 was predicted to form hydrophobic or stacking interactions with G18, and H125 could interact with the G18 phosphate (7). Our biochemical analysis demonstrates that R51 is not critical for tRNA binding and therefore is unlikely to intercalate between G18 and G19 of the D arm. However, we confirm that both F106 and H125 are highly important for the interaction of TrmA with tRNA and likely interact with G18 in the D loop similarly as predicted. Thereby, residues F106 and H125 are ideally positioned to disrupt the tertiary interaction between G18 and Ψ55 which is necessary for the T arm to interact with TrmA in the conformation observed in the crystal structure and for U54 to insert into the active site (7). It is thus reasonable to assume that upon binding of tRNA to TrmA, the contacts of tRNA with F106 and H125 in the RNA-binding domain are required to locally unfold the elbow region of tRNA in order to allow the subsequent accommodation of the T arm and the target site U54 in the active site of TrmA.
Here, we confirm the hypothesis that TrmA is a tRNA chaperone like the pseudouridine synthase TruB. Independent of its catalytic activity, the interaction of TrmA with tRNA facilitates correct and rapid folding of tRNA as evident in instantaneous aminoacylation in the in vitro folding assay (Figure 5). Based on the structure of TrmA in complex with the T arm and the vicinity of U54 targeted by TrmA and U55 modified by TruB, both enzymes likely follow a similar mechanism in promoting tRNA folding (4,7). As shown for TruB, upon initial binding of tRNA to the modifying enzyme, the tRNA undergoes an induced-fit rearrangement to insert the target based into the active site (4). According to the structures for both TruB and TrmA, this conformational change in the tRNA must include disruption of tertiary interactions between the D loop and the T loop in the elbow region of tRNA. We aligned the structure of the T arm of full-length tRNA with the T arm bound to TrmA to model the initial interaction of TrmA with tRNA (Figure 1B). This model clearly illustrates the steric clash of F106 and H125 with G18 at the interface of D arm and T arm. Therefore, we propose that F106 and H125 act as a wedge to disrupt tertiary interactions in the tRNA elbow region thereby causing the tRNA to unfold (Figure 1B). Importantly, both tRNA modifying enzymes, TruB and TrmA, are likely to also unfold incorrect tRNA structures. This unfolding of tertiary structure within the tRNA by TrmA or TruB, provides the tRNA with a second chance at correct folding. Accordingly, TrmA and TruB would act like protein chaperones called unfoldases that repeatedly bind, unfold and release their substrates (17). For TruB, rapid-kinetic fluorescent stopped-flow experiments using 2-aminopurine labeled tRNA indeed uncovered a repeated unfolding/refolding cycle of tRNA while bound to TruB prior to slow pseudouridine formation (4). However, no fluorescent signal is observed upon interaction of TrmA with tRNA containing a 2-aminopurine at position 57 (unpublished data); this is likely because the base 57 is not in direct contact with TrmA in contrast to TruB. Therefore, there is no readily available reporter to directly observe tRNA unfolding by TrmA. However, it is interesting that some tRNA folding activity remained for TrmA H125E whereas TrmA F106E had lost this activity entirely. This observation agrees with the suggestion that H125 interacts with the phosphate of G18 whereas F106 may directly contact the base of G18 (Figure 1B); accordingly, F106 may be more important for disrupting tertiary interactions between the D loop and the T loop than H125.
It is interesting to consider the relevance of the tRNA chaperone activity of TrmA within the cell. As shown by the competition assays (Figure 4), the catalytic activity of TrmA is essential for bacterial fitness rendering it difficult to independently assess the importance of tRNA binding and folding in E. coli. Based on our findings for TruB (4), it is likely that both functions of TrmA, the methylation and the folding of tRNA, contribute to cellular fitness. Notably, previous studies in Saccharomyces cerevisiae support the suggestion that Trm2, the homolog of E. coli TrmA, acts as a tRNA chaperone in vivo (18). Therein, the authors discovered that catalytically inactive Trm2 protein can stabilize mutant forms of tRNASer. Interestingly, yeast strains with the mutant tRNASer were also dependent on stabilization by the pseudouridine synthase Pus4, the homolog of the E. coli enzyme TruB which we showed to be a tRNA chaperone as well (4). A recent study found that in bovine, the mitochondrial Trmt2B protein is conserved, yet inactive as the catalytic cysteine is mutated suggesting that the presence of Trmt2B alone, which can likely still function as a tRNA chaperone, is important and conserved in contrast to Trmt2B’s catalytic activity (19). Thus, it is likely that the tRNA m5U54 methyltransferase is a conserved tRNA chaperone in different organisms which stabilizes tRNAs and enhances cellular fitness. While the enzyme is non-essential and dispensable under optimal growth conditions, it was likely conserved throughout evolution because TrmA/Trm2 can fold and stabilize tRNAs under non-optimal conditions which organisms regularly encounter in their natural environment.
Notably, two tRNA modifying enzymes, TrmA and TruB, have now been revealed to have dual functions and to also assist tRNA in adopting its correct three-dimensional structure (4). As discussed, in both cases structural and biochemical evidence suggests that the tRNA modifying enzymes are disrupting tertiary interactions in the tRNA elbow region thereby providing tRNA with a second chance at correct folding. Therefore, it seems likely that other RNA modifying enzymes and other RNA binding proteins may also act as RNA chaperones explaining the wide conservation of RNA modifying enzymes even in relatively simple organisms. It is now recognized that all RNAs both non-coding and coding have structural features which are supporting their function (20), and there is a plethora of RNA binding proteins that induce a conformational change in their bound RNA (21). Our study demonstrates that RNA binding and modification are functionally linked to RNA folding, and it seems therefore only plausible that several other RNA binding proteins will have similar dual functionality acting as RNA chaperones like TrmA and TruB.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the National BioResource Project (NIG, Japan) for the TrmA expression plasmids and the ΔtrmA deletion strain, and the Stansfield lab for plasmid pTrc99a.
Contributor Information
Laura Carole Keffer-Wilkes, University of Lethbridge, Alberta RNA Research and Training Institute (ARRTI), Department of Chemistry and Biochemistry, Lethbridge, AB T1K 3M4, Canada.
Emily F Soon, University of Lethbridge, Alberta RNA Research and Training Institute (ARRTI), Department of Chemistry and Biochemistry, Lethbridge, AB T1K 3M4, Canada.
Ute Kothe, University of Lethbridge, Alberta RNA Research and Training Institute (ARRTI), Department of Chemistry and Biochemistry, Lethbridge, AB T1K 3M4, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Natural Sciences and Engineering Research Council of Canada [Discovery Grant RGPIN-2014-05954]; Alberta Innovates [Strategic Research Chair 2015]; University of Lethbridge [School of Graduate Studies Fellowship to L.K.-W.]. Funding for open access charge: NSERC Discovery Grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Sprinzl M., Horn C., Brown M., Ioudovitch A., Steinberg S.. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998; 26:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Phizicky E.M., Alfonzo J.D.. Do all modifications benefit all tRNAs. FEBS Lett. 2010; 584:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gutgsell N., Englund N., Niu L., Kaya Y., Lane B.G., Ofengand J.. Deletion of the Escherichia coli pseudouridine synthase gene truB blocks formation of pseudouridine 55 in tRNA in vivo, does not affect exponential growth, but confers a strong selective disadvantage in competition with wild-type cells. RNA. 2000; 6:1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Keffer-Wilkes L.C., Veerareddygari G.R., Kothe U.. RNA modification enzyme TruB is a tRNA chaperone. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:14306–14311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishitani R., Yokoyama S., Nureki O.. Structure, dynamics, and function of RNA modification enzymes. Curr. Opin. Struct. Biol. 2008; 18:330–339. [DOI] [PubMed] [Google Scholar]
- 6. Nurse K., Wrzesinski J., Bakin A., Lane B.G., Ofengand J.. Purification, cloning, and properties of the tRNA psi 55 synthase from Escherichia coli. RNA. 1995; 1:102–112. [PMC free article] [PubMed] [Google Scholar]
- 7. Alian A., Lee T.T., Griner S.L., Stroud R.M., Finer-Moore J.. Structure of a TrmA–RNA complex: A consensus RNA fold contributes to substrate selectivity and catalysis in m5U methyltransferases. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:6876–6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kitagawa M., Ara T., Arifuzzaman M., Ioka-Nakamichi T., Inamoto E., Toyonaga H., Mori H.. Complete set of ORF clones of Escherichia coli ASKA library (A Complete Set of E. coli K-12 ORF Archive): Unique resources for biological research. DNA Res. 2005; 12:291–299. [DOI] [PubMed] [Google Scholar]
- 9. Wright J.R., Keffer-Wilkes L.C., Dobing S.R., Kothe U.. Pre-steady-state kinetic analysis of the three Escherichia coli pseudouridine synthases TruB, TruA, and RluA reveals uniformly slow catalysis. RNA. 2011; 17:2074–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kelly S.M., Jess T.J., Price N.C.. How to study proteins by circular dichroism. Biochim. Biophys. Acta. 2005; 1751:119–139. [DOI] [PubMed] [Google Scholar]
- 11. Kinghorn S.M., O’Byrne C.P., Booth I.R., Stansfield I.. Physiological analysis of the role of truB in Escherichia coli: a role for tRNA modification in extreme temperature resistance. Microbiology. 2002; 148:3511–3520. [DOI] [PubMed] [Google Scholar]
- 12. Bhaskaran H., Rodriguez-Hernandez A., Perona J.J.. Kinetics of tRNA folding monitored by aminoacylation. RNA. 2012; 18:569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kealey J.T., Gu X., Santi D.V.. Enzymatic mechanism of tRNA (m5U54)methyltransferase. Biochimie. 1994; 76:1133–1142. [DOI] [PubMed] [Google Scholar]
- 14. Urbonavicius J., Jäger G., Björk G.R.. Amino acid residues of the Escherichia coli tRNA(m5U54)methyltransferase (TrmA) critical for stability, covalent binding of tRNA and enzymatic activity. Nucleic Acids Res. 2007; 35:3297–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee T.T., Agarwalla S., Stroud R.M.. A unique RNA Fold in the RumA-RNA-cofactor ternary complex contributes to substrate selectivity and enzymatic function. Cell. 2005; 120:599–611. [DOI] [PubMed] [Google Scholar]
- 16. Santi D.V., Hardy L.W.. Catalytic mechanism and inhibition of tRNA (uracil-5-)methyltransferase: evidence for covalent catalysis. Biochemistry. 1987; 26:8599–8606. [DOI] [PubMed] [Google Scholar]
- 17. Mattoo R.U., Goloubinoff P.. Molecular chaperones are nanomachines that catalytically unfold misfolded and alternatively folded proteins. Cell. Mol. Life Sci. 2014; 71:3311–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johansson M.J., Bystrom A.S.. Dual function of the tRNA(m5U54)methyltransferase in tRNA maturation. RNA. 2002; 8:324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Powell C.A., Minczuk M.. TRMT2B is responsible for both tRNA and rRNA m(5)U-methylation in human mitochondria. RNA Biol. 2020; 17:451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Incarnato D., Oliviero S.. The RNA epistructurome: uncovering RNA function by studying structure and post-transcriptional modifications. Trends Biotechnol. 2017; 35:318–333. [DOI] [PubMed] [Google Scholar]
- 21. Hentze M.W., Castello A., Schwarzl T., Preiss T.. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018; 19:327–341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.