

Abstract
Riboswitches are structured RNA motifs that recognize metabolites to alter the conformations of downstream sequences, leading to gene regulation. To investigate this molecular framework, we determined crystal structures of a preQ1-I riboswitch in effector-free and bound states at 2.00 Å and 2.65 Å-resolution. Both pseudoknots exhibited the elusive L2 loop, which displayed distinct conformations. Conversely, the Shine-Dalgarno sequence (SDS) in the S2 helix of each structure remained unbroken. The expectation that the effector-free state should expose the SDS prompted us to conduct solution experiments to delineate environmental changes to specific nucleobases in response to preQ1. We then used nudged elastic band computational methods to derive conformational-change pathways linking the crystallographically-determined effector-free and bound-state structures. Pathways featured: (i) unstacking and unpairing of L2 and S2 nucleobases without preQ1—exposing the SDS for translation and (ii) stacking and pairing L2 and S2 nucleobases with preQ1—sequestering the SDS. Our results reveal how preQ1 binding reorganizes L2 into a nucleobase-stacking spine that sequesters the SDS, linking effector recognition to biological function. The generality of stacking spines as conduits for effector-dependent, interdomain communication is discussed in light of their existence in adenine riboswitches, as well as the turnip yellow mosaic virus ribosome sensor.
INTRODUCTION
An emerging theme among nonprotein-coding (nc)RNAs that interact with cognate effectors is their propensity to use structural communication conduits that relay the effector-binding status in one domain to a distal region that modulates biological activity through conformational changes (1,2). Identifying and understanding these networks remains a central challenge in the ncRNA field (1–4). Riboswitches are ideal systems to explore ligand-induced ncRNA conformational changes due to their highly selective recognition of cognate effectors (5), a typically two-domain organization that features a conserved aptamer connected to a gene-regulatory expression platform (6,7), and their tractability for high-resolution structural analysis (8,9). Representative co-crystal structures have been determined for >30 distinct riboswitch classes bound to their cognate effectors (9), as well as to natural and synthetic compounds (10–14). Such knowledge has transformed our understanding of the molecular principles used by RNA to recognize small molecules (5,15–20) and, in some instances, has provided deeper insights into antibacterial design (12,21). However, only a handful of riboswitch structures are known in the effector-free state (3,4,14,22–27), and only a fraction of these reveal appreciable conformational differences compared to their effector-bound forms (3,4,22,24). Accordingly, structures that reveal conformational differences between effector-free and bound states are prized for their potential to reveal how effector recognition remodels the expression platform to reprogram biological activity (28,29).
The majority of known effector-free riboswitch structures correspond to transcriptional regulators. However, the Vibrio vulnificus (Vvu) add adenine riboswitch, the Gloeobacter violaceus guanidine-II riboswitch, the Thermobifida fusca guanidine-III riboswitch and the Thermoanaerobacter tengcongensis (Tte) preQ1-I riboswitch each regulate translation (Supplementary Table S1) (30–34). The Tte riboswitch structure is particularly informative because it harbors not only nucleotides required for preQ1 binding but also nucleobases of the Shine-Dalgarno sequence (SDS), thus allowing simultaneous visualization of aptamer and expression platform domains (3,30,35,36) (Figure 1A–C). To our knowledge, the Tte preQ1-I riboswitch is the only translational riboswitch crystallized in effector-free and bound states that includes the aptamer and expression platform (Supplementary Table S1). This organization has facilitated experimental and computational investigations of effector-mediated folding. Computational simulations suggested the formation of an in silico GpU platform between G11 and U12 that could facilitate unpairing of A32 and G33 from the anti-(a)SDS strand (37). Gō model simulations and experimental studies suggested that the Tte riboswitch adopts a mostly folded effector-binding pocket prior to preQ1 binding (22,31). Single-molecule experiments further revealed that both the preQ1-free and bound states underwent bursts of SDS accessibility to a 16S rRNA probe, wherein the timeframe of accessibility and burst frequency diminished with added preQ1 (38). This work provided insight into preQ1-dependent changes in riboswitch conformation and dynamics, supporting an induced-fit mode of effector binding. However, atomistic pathways used by riboswitches to sense and communicate effector binding to distal expression platforms remain an insufficiently studied area of the field.
Figure 1.
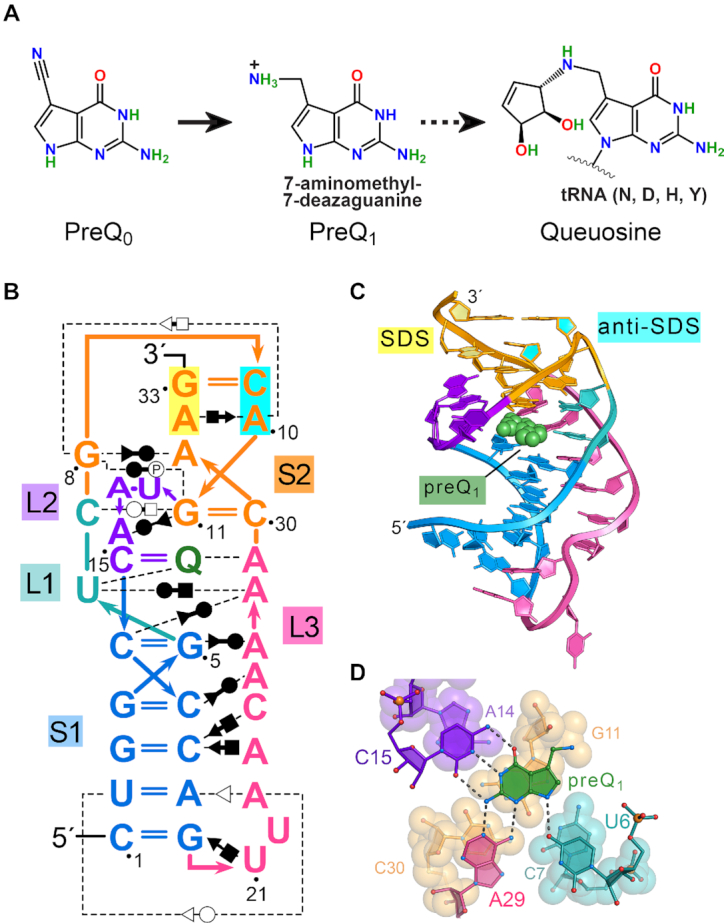
Queuosine (Q) synthesis, the Tte preQ1-I riboswitch fold and mode of effector binding. (A) The hypermodified base Q is synthesized by many bacteria starting from GTP and proceeds via pyrrolopyrimidine intermediates prequeuosine0 and prequeuosine1 (preQ1). PreQ1 is the last soluble metabolite prior to insertion into GUN anticodons of tRNAs decoding Asn, Asp, His or Tyr (broken arrow). In situ modifications are then added to the nucleotide to form Q (93). (B) Secondary structure of the wild-type preQ1-I riboswitch from Thermoanaerobacter tengcongensis (Tte) based on prior co-crystal structures (22,35). Color-codes correspond to specific pseudoknot stem (S) and loop (L) sequence as defined (94); preQ1 is shown as ‘Q’ (green). Non-canonical base pairing is indicated by the Leontis–Westhof symbols (95). Interactions with phosphate groups are denoted with the symbol P. The Shine-Dalgarno sequence (SDS) (expression platform) is highlighted in yellow; the opposing anti-(a)SDS is highlighted in cyan. (C) Ribbon diagram depicting the H-type pseudoknot fold based on a prior Tte co-crystal structure (PDB entry: 3q50) (22). Helix S2 comprises SDS-aSDS pairing (base and ribose rings filled yellow and cyan) that supports the gene-off conformation of the expression platform. PreQ1 is depicted as a CPK model (green). (D) Close-up view of the preQ1 binding pocket rotated −90° about the x-axis from panel C. Nucleotides that contribute to preQ1 specificity are depicted as ball-and-stick models; specificity base C15 of loop L2 forms Watson-Crick-like pairing to the effector face. Effector-recognition bases U6 and A29 contribute hydrogen bonds to the minor-groove (equivalent) edge of the effector. Here and elsewhere, putative hydrogen bonds are depicted as dashed lines. Nucleotides beneath preQ1 are depicted as CPK models.
To address this challenge, we determined two new crystal structures of the Tte preQ1-I riboswitch under low-salt conditions with bound Mn2+ ions. For the first time, loop L2 of the pseudoknot was ordered in electron density maps of both effector-free and bound states. The observation that the SDS remains base paired as part of helix S2—in all known crystal forms of translational preQ1-I riboswitches (22,35,36) — prompted us to conduct a series of solution experiments to probe preQ1-dependent conformational changes. These solution experiments included surface plasmon resonance (SPR), 2-aminopurine (2AP) fluorescence, SHAPE-seq (Selective 2′-Hydroxyl Acylation analyzed by Primer Extension) and DMS-seq using dimethylsulfate. We then conducted nudged elastic band (NEB) simulations using our crystal structures of the preQ1-free and preQ1-bound states as endpoints in an all-atom approach to model low-energy pathways describing the interconversion of the two conformations. The most plausible, experimentally-supported pathways exhibited dissolution of SDS-aSDS base pairs in S2 during the effector-free state — an attribute that differs markedly from the crystal structure. Importantly, the preQ1-occupied status of the binding pocket appears to be communicated by a ‘stacking spine’ of nucleobase interactions that is nucleated by preQ1 recognition in loop L2 (Figure 1D). We discuss the broader relevance of such spines, which are underscored by their presence in the Vvu add riboswitch, as well as the TLS-linker-UPD interaction found in the turnip yellow mosaic virus (TYMV) 3′-UTR.
MATERIALS AND METHODS
RNA purification
Wildtype Tte 33-mer RNA (Figure 1B) and other variants were synthesized by Dharmacon (Lafayette, CO, USA). Deprotection was performed as described by the manufacturer except that heating was for 1 h at 65 °C. Light sensitive precautions were taken to ensure the photostability of 2AP variants. RNA was purified by 15% denaturing PAGE and DEAE chromatography (39) with care to minimize UV exposure (40). After ethanol precipitation of pooled DEAE fractions, the RNA was dissolved in water and desalted on a PD-10 column (GE Healthcare). Sample quality was assessed by analytical PAGE stained with SYBR Gold (Thermo-Fisher) and visualized on a Gel Doc XR+ (BioRad). Yield was measured spectrophotometrically. Pure 2AP RNA variants were dissolved to a concentration of 5 μM in 20 mM Na-HEPES pH 7.0 and 0.1 mM EDTA and stored in 100 μl aliquots at −80 °C. All other RNA was stored as a lyophilized powder at −20 °C.
Structure determinations
The lyophilized riboswitch was dissolved in 60 μl folding buffer comprising 50 mM Na-HEPES pH 7.0, 0.10 M NaCl and 0.002 M MgCl2 (effector-free state) or 0.002 M MnCl2 (effector-bound state) and warmed to 65 °C for 3 min. The RNA was diluted to 1 ml with hot folding buffer and heated for an additional 3 min at 65 °C. The RNA was cooled to 24 °C. For the preQ1-bound state, 20 mM preQ1 (LeadGen Labs, LLC) in water was titrated into the riboswitch to achieve a 2:1 ligand–RNA molar ratio. RNA samples were concentrated to 0.4 mM by centrifugation using a PTFE Nanosep MF concentrator (Pall Corp., Inc). Crystals were grown from VDX plates (Hampton Research) by hanging-drop vapor-diffusion wherein 1 μl of RNA was combined with 1 μl of well-solution drawn from 1 ml. Effector-free crystals grew from solutions of 19% (w/v) PEG MME 2000, 0.15 M KCl, 0.05 M Na-cacodylate pH 7.0, 0.005 M MnCl2, 0.001 M MgCl2 and 0.002 M spermidine. Effector-bound crystals grew from 22% (w/v) PEG MME 2000, 0.15 M KCl, 0.05 M Na-cacodylate pH 7.0, 0.015 M MnCl2 and 0.002 M spermidine. Effector-free crystals grew in 3 weeks at 20 °C as hexagonal rods of size 0.3 mm × 0.075 mm × 0.075 mm. Effector-bound crystals grew in 1 week at 20 °C as hexagonal rods to a size of 0.12 mm × 0.05 mm × 0.05 mm. Crystals were cryo-protected by 3 min transfers into well solutions supplemented with 5% to 20% (v/v) glycerol. Single rods were captured in nylon loops using 16 mm copper pins (Hampton Research) with c* oriented parallel to the φ axis. Crystals were plunged into N2(l) for shipping to the Stanford Synchrotron Radiation Lightsource (SSRL). X-ray data collection is described in the Supplementary Methods.
Both bound-state and effector-free crystals were nearly isomorphous with the prior high-salt Tte preQ1-I riboswitch structure (Protein Data Bank entry 3q50). As such, each structure was determined using cycles of rigid body, positional, and temperature factor refinement in PHENIX (41). The prior test set was preserved for both new structures to maintain unbiased cross-validation as described (42). Both structures were rebuilt in COOT (43) and refined in PHENIX as necessary. Mn2+ ions were placed first into the higher-resolution effector-free structure to assist with coordination-sphere assignment. Distance and geometry restraints for Mn2+ were 2.07 ± 0.16 Å for Mn2+–O distance and 2.11 ± 0.13 Å for Mn2+–N distances with interaxial coordination angles approaching 90.0° (44,45). Placement of Mn2+ at sites I to VI was assisted by the anomalous signal, which ranged from 9.3 σ to 23.5 σ for the effector-free state and 5.6 σ to 16.0 σ for the preQ1-bound state. Waters were placed by manual building. Intensity and refinement statistics are provided in Table 1. Accessible surface area analysis and all-atom superpositions were performed in CCP4i using AreaiMol and LsqKab (46–48). Cartoons and structural models were drawn in PyMOL (Schrödinger LLC).
Table 1.
Tte preQ1-I riboswitch data collection and refinement statistics
| Sample (PDB ID) | Effector free (6vuh) | PreQ 1 bound (6vui) |
|---|---|---|
| Data collectiona | ||
| Wavelength | 0.9537 | 1.700 |
| Resolut. range (Å) | 37.78–2.00 (2.05–2.00) | 37.62–2.68 (2.81–2.68) |
| Space group | P 63 2 2 | P 63 2 2 |
| a = b, c (Å) | 115.4, 57.7 | 112.3, 59.4 |
| α = β, γ (°) | 90.0, 120 | 90.0, 120 |
| Unique reflections | 15576 | 6544 |
| Multiplicity | 8.0 (7.6) | 7.5 (6.9) |
| Completeness (%) | 99.0 (92.2) | 99.8 (99.5) |
| Mean I/σ(I) | 13.2 (1.2) | 9.5 (1.5) |
| R merge (%)b | 5.7 (153.4) | 10.3 (127.6) |
| R p.i.m. (%)c | 2.2 (58.7) | 4.1 (51.4) |
| CC1/2d | 0.994 (0.626) | 0.985 (0.776) |
| Refinement | ||
| No. reflections (work/test) | 28736/2847 | 11709/1141 |
| R work/Rfree (%)e | 19.0/22.5 | 17.5/20.2 |
| No. of atoms | ||
| RNA | 702 | 702 |
| preQ1/Mn2+ | 6 | 13/6 |
| waters | 34 | 21 |
| R.M.S. deviations | ||
| Bonds (Å) | 0.007 | 0.003 |
| Angles (°) | 1.31 | 0.75 |
| Coord. error | 0.30 | 0.51 |
| Clashscoref | 0.94 | 0.92 |
| Molprobity scoref | 1.93 | 1.93 |
| Avg. B-factor (Å2) | ||
| RNA | 63.1 | 74.1 |
| preQ1 | N/A | 58.6 |
| Mn2+ | 79.7 | 87.7 |
| Waters | 61.2 | 63.7 |
aStatistics for the highest-resolution shell are shown in parentheses.
b R merge = ∑hkl∑i|Ii − |/∑hkl∑i |Ii|, where Ii is an intensity I for the ith measurement of a reflection with indices hkl and is the weighted mean of all measurements of I.
c
R
precision-indicating merging R-value = 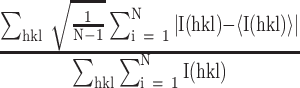 , where N is the redundancy of the data and
, where N is the redundancy of the data and 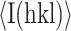 is the average intensity (90).
is the average intensity (90).
dCC1/2, Pearson correlation coefficient between intensities of random half-dataset (91).
e R work = Σhkl||Fobs(hkl)| –|Fcalc(hkl)||)/Σhkl|Fobs(hkl)| for the working set of reflections, Rfree is defined as Rwork for ∼10% of the reflections excluded from the refinement. All data from the available resolution ranges were used in the refinement.
fCalculated using the program Molprobity (92).
Surface plasmon resonance
CM5 chips conjugated to Neutravidin were prepared using reagents from GE Healthcare (22) on a BiaCore T200 (GE Healthcare, Inc.). Biotinylated Tte riboswitches and position 14 variants were deprotected by the manufacturer (Dharmacon, Inc.). Dry RNA was dissolved in 50 μl of 10 mM Na-cacodylate pH 7.0 and heated to 65 °C for 3 min; the RNA was added to 450 μl of 10 mM Na-cacodylate with 2 mM MgCl2 preheated to 65 °C, incubated for 3 min, and cooled in a heating block to 24 °C. RNA stocks ranging in concentration from 63 to 111 μM were diluted to 400 nM by a running buffer comprising 10 mM Na-HEPES pH 7.4, 0.10 M NaCl and 3 mM MgCl2. Each Tte riboswitch sample was immobilized within an individual flow cell to yield a surface density of ∼4000 response units (RU); the reference cell contained Neutravidin only. The kinetic experiments were run at a buffer flow rate of 100 μl min−1. PreQ1 (12.5 nM−400 nM) was injected for 180 s and allowed to dissociate for 600 s. The surface was regenerated using a 60 s injection of 3 M guanidine–HCl run at 30 μl min−1. Running buffer was passed over the chip for 160 s before beginning the next injection cycle. Experimental data were processed using the double-referencing method (49) and the buffer subtracted sensorgrams were fit to a 1:1 binding model to determine rate constants (kon and koff) and the equilibrium binding constant (KD). Table 2 shows average kinetic and equilibrium-binding parameters. SPR experiments on the A14(DAP) sample were performed in triplicate; all other samples were performed in quadruplicate.
Table 2.
Kinetic and equilibrium binding constants for Tte riboswitch A14-variants
| k on × 104 | S.E.a × 104 | k off × 10−4 | S.E. × 10−5 | K D × 10−9 | S.E. × 10−10 | t 1/2 | |
|---|---|---|---|---|---|---|---|
| M–1 s–1 | s–1 | M | min | ||||
| Wildtype | 11.60 | 1.13 | 2.12 | 1.6 | 1.84 | 0.8 | 54.5 |
| 2,6-Diaminopurine | 2.07 | 0.40 | 2.51 | 3.5 | 12.3 | 7.9 | 46.0 |
| 2-Aminopurine | 4.57 | 0.12 | 11.2 | 4.6 | 24.5 | 4.6 | 10.3 |
| Purine | 6.84 | 0.60 | 6.73 | 2.9 | 9.84 | 4.7 | 17.2 |
aStandard error estimated using S.E. = S.D./sqrt(N), where N is the number of experiments.
Isothermal titration calorimetry
Details of ITC are in the Supplemental Methods. Representative thermograms and curve fits are provided in Supplementary Figures S1A and S2. Average thermodynamic parameters are given in Supplementary Table S2.
Fluorescence emission
The 2AP variants were folded as follows, except where noted. An aliquot of 2AP RNA was thawed and heated to 65 °C for 3 min. The RNA was diluted to 1 ml with warm folding buffer (50 mM Na-HEPES pH 7.0, 100 mM NaCl and 2 mM MgCl2) to achieve a concentration of 500 nM, followed by incubation for an additional 3 min at 65 °C. The sample was cooled slowly to 24 °C. The A13(2AP) variant was folded by adding one part 500 nM A13(2AP)—preheated at 90 °C for 3 min followed by rapid cooling on ice for 2 min—to four parts chilled folding buffer. The sample was brought slowly to 24 °C. 20 mM PreQ1 dissolved in NANOpure™ UV/UF water was diluted serially to the desired concentrations with folding buffer.
Fluorescence emission of 2AP was measured for each variant as a function of preQ1 concentration as described (50,51). Briefly, each folded 100 nM riboswitch sample was added to a quartz cuvette (Starna Cells, Inc.). PreQ1 was pipetted into the RNA with manual mixing and allowed to equilibrate until three successive readings agreed to within 1%. Each point in a binding curve is the average of these readings. The total volume increase did not exceed 5%. Excitation and emission wavelengths were 320 nm and 390 nm attained with a Fluoromax-4 Spectrofluorometer (Horiba Scientific) using 10 trials per read, a 0.2% error rate and slit width of 5.0 nm. Argininamide was titrated into Tte variant A13(2AP) to assess non-specific binding (Supplementary Figure S1B). Titration of buffer into A32(2AP) labeled Tte riboswitch was also conducted (Supplementary Figure S1C). Both controls revealed no significant titration signal. Binding curves from three replicates were then used to generate a single average curve for each experiment.
Background fluorescence resulting from titration of preQ1 into buffer was subtracted from each emission measurement. To determine the apparent equilibrium dissociation constant, KD, corrected data were fit using Prism (GraphPad Software, Inc.) to a one-site binding model (Equation 1) with the following variables: Y is the observed emission at the ith titration; Rf is fluorescence intensity at zero preQ1 concentration; Rb is the fluorescence intensity at saturating preQ1; [preQ1] is the concentration of preQ1 for the ith titration; KD and R2 were derived from non-linear regression fitting using the Levenberg–Marquardt method. The broad range of counts per second (CPS) for replicate runs were normalized (Equation 2) such that the maximum CPS value of each replicate was 1.0 and the minimum was 0. The variables are: E, the normalized fluorescence emission at the ith titration; Z, the background corrected fluorescence emission at the ith titration (Y-average background at that point); A, the maximum emission for the replicate; B, the minimum emission for the replicate.
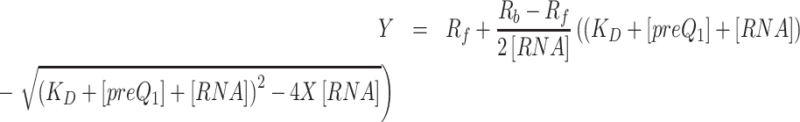 |
(1) |
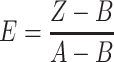 |
(2) |
Chemical modification of RNA
For chemical-modification experiments, the riboswitch was synthesized by T7 polymerase (39). The transcription product contained the 33-mer Tte riboswitch placed in a folding cassette. The 5′-end of the cassette possessed a short stable hairpin, whereas the 3′-end harbored a strong hairpin followed by a unique primer binding site as described (52). The cassette also provided a baseline level for SHAPE and DMS reactivity in duplex regions expected to be insensitive to preQ1. The 17-mer DNA primer and 90-mer DNA template were from IDT Inc. The transcript synthesized by T7 polymerase was: 5′-ggccuucgggccacCUGGGUCGCAGUAACCCCAGUUAACAAAACAAGucgauccgguucgccggauccaaaucgggcuucgguccgguuc-3′; the Tte riboswitch is capitalized and the primer binding site is italicized. The in vitro transcribed product was PAGE purified, desalted (39) and stored at −20 °C in 0.5× TE buffer.
For DMS alkylation, 20 pmol of pure RNA was dissolved in 24 μl of 0.5× TE buffer per modification condition. The RNA was heated to 95 °C for 2 min and placed on ice 2 min. A 12 μl volume of 3x RNA folding mix (333 mM HEPES pH 8.0, 333 mM NaCl and 20 mM MgCl2) was added; for the bound-state riboswitch, preQ1 was also added to a final concentration of 200 nM, yielding an RNA concentration of 0.01 nM. RNA was incubated at 24 °C for 20 min. A volume of 4 μl 1% (v/v) DMS (Sigma-Aldrich) diluted in neat ethanol was added to both free-state and bound-state samples to achieve a final concentration of 0.1% (v/v); control samples were treated with neat ethanol. Alkylation proceeded 20 min, followed by quenching with 200 μl of 30% (v/v) 2-mercaptoethanol, 200 mM NaCl, 0.25 mg ml−1 GlycoBlue (Thermo-Fisher Sci) and 2 mM EDTA pH 8.0 (53). Samples were ethanol-precipitated with 800 μl neat ethanol and stored at −20 °C. Precipitated RNA was harvested by centrifugation at 18 000 × g for 40 min at 4 °C. The air-dried pellet was dissolved in 20 μl of 0.5× TE buffer.
For SHAPE, the RNA was folded as described (above), except that the 24-μl-folded-RNA sample was split in half after addition of 12 μl of folding mix, yielding 18 μl of free-state or bound-state RNA per reaction condition. Acylation was performed with 2-methylnicotinic acid imidazolide (NAI) (Sigma-Aldrich) (54,55). A volume of 2 μl of 0.6 M NAI in DMSO was added to a concentration of 60 mM. Alternatively, 2 μl of DMSO was added to the control samples. The reactions proceeded 20 min at 24°C and were terminated by addition of a stop solution comprising 90 μl water, 5 μl 4 M NaCl, 1.5 μl GlycoBlue and 2 μl of 100 mM EDTA pH 8.0. The RNA was ethanol-precipitated by addition of 350 μl of neat ethanol. Pelleted RNA was recovered as described above. We conducted two or more replicates of specific chemical modification conditions, which were analyzed by PAGE. Subsequently, representative samples of each condition reported in the Supplementary Methods (i.e. gel lanes of Supplementary Figure S3) were subjected to next-generation sequencing.
Samples for next-generation sequencing were prepared in groups of four. These groups included RNA analyzed in the presence and absence of preQ1, as well as matched controls prepared in the presence and absence of DMS or NAI. Each sample was subjected to reverse transcription using Superscript III (Thermo-Fisher Sci) based on the primer 5′-GAACCGGACCGAAGCCCG-3′. DNA library preparation and analysis was conducted using the SHAPE-Seq v2.1 workflow (56). Sequencing adapters were ligated to the cDNA using CircLigase (Lucigen). Separate PCR reactions using Phusion DNA Polymerase (NEB) were mixed for each sample and amplified 15 cycles with an annealing temperature of 65 °C with 15 s extension. Sample pairs (+ preQ1 and –preQ1) contained identical Illumina indexes for sequencing; however, the (+) and (–) samples were distinguished using a unique selection primer for each as follows: CTTTCCCTACACGACGCTCTTCCGATCTRRRY (+) and CTTTCCCTACACGACGCTCTTCCG ATCTYYYR (–). Following PCR, samples were purified using Agencourt Ampure XP beads (Beckman Coulter). The concentration and quality were assessed using the Qubit fluorometer (ThermoFisher) and Bioanalyzer (Agilent). Samples were pooled equimolar and were sequenced on an Illumina MiSeq system with a paired-end-run recipe of 2 × 35 bp. Following sequencing, Spats-v1.9.30 (56,57) was used to demultiplex the positive and negative signals and calculate SHAPE reactivities based on the target sequence: TGGCCTTCGGGCCACCTGGGTCGCAGTAACCCCAGTTAACAAAACAAGTCGATCCGGTTCGCCGGATCCAAATCGGGCTTCGGTCCGGTTC. Of the reads, >60% (∼600,000) mapped to the target. Reactivity spectra were generated using Spats. Individual reactivities were plotted as a function of nucleotide position (Supplementary Figure S4). Differential reactivities, defined as [Reactivity]Effector-Bound − [Reactivity]Efffector-Free (54,58), were plotted as a function of nucleotide position and mapped onto specific crystal structures using Pymol (Schrödinger LLC). We emphasize that all chemical modification reactions were conducted under the same buffer conditions but mapping of the reactivity values as ρ or differential ρ was based upon the identification of structures (i.e. low-salt preQ1-free, low-salt preQ1-bound or high-salt preQ1-bound) that best explained the respective experimental results.
Computational analysis
To identify minimum energy pathways for Tte riboswitch interconversion between the effector-free and preQ1-bound states, we used the nudged elastic band (NEB) method (59,60) as implemented in Amber 18 (61) using explicit solvent (62). Fixed endpoints were derived from crystal structures of the effector-free state (this study) and a prior high-salt, preQ1-bound state (PDB entry 3q50) (22). Three Mn2+ cations coordinated to N7 of G5 (site III), G8 (site II) and G33 (site Ia) in the effector-free crystal structure were retained as Mg2+ cations in both structures (Figure 2A); similar Mn2+ coordination was observed in the new bound-state crystal structure herein (Figure 2B). C7 was replaced with an N3(H+) cytidine to stabilize the C7•G11-C30 base triple, as observed in other RNAs (63), prior preQ1-I riboswitch MD simulations (37), as well as MD simulations of the effector-free Tte riboswitch performed here. Each structure was solvated in a truncated octahedron of OPC water (64) with 12 Å padding from the RNA. A total of 25 K+ ions were added to neutralize the system, and then 10 K+ and 10 Cl− ions were added to model KCl at a bulk concentration of 150 mM (65). To make the NEB endpoints chemically identical, 431 water molecules were removed from the effector-free system, leaving each riboswitch with 7039 waters total.
Figure 2.
Ribbon diagrams of the preQ1-free and preQ1-bound states of the Tte preQ1-I riboswitch and select Mn2+ binding sites. (A) Ribbon diagram depicting the H-type pseudoknot fold of the preQ1-I riboswitch based on the preQ1-free Tte crystal structure of this investigation. Six Mn2+ binding sites (maroon spheres) are labeled. Structural elements are colored and labeled as in Figure 1. (B) Ribbon diagram of the preQ1-bound co-crystal structure from this investigation; preQ1 is depicted as a CPK model (green); the site Ib ion is observed only in the preQ1-bound state. (C) Ball-and-stick and ribbon diagram depicting the site II Mn2+ ion, which binds similarly in the preQ1-free and bound-state structures. Water molecules are denoted by wat. (D) Diagram depicting the site III Mn2+ ion, which is compatible with the preQ1-free conformation. (E) Diagram depicting the site IV Mn2+ ion, which is compatible with both free and bound-state conformations.
Each solvated structure was minimized using 2000 steps each of steepest descent and conjugate gradient minimization with 1 kcal mol−1 Å−2 Cartesian restraints on non-hydrogen atoms in the RNA. Then, this minimization was repeated without Cartesian restraints. The systems were equilibrated in three steps: (i) heating from 0 K to 298.15 K over 100 ps at constant volume; (ii) running dynamics at 298.15 K for 200 ps at 1 atm pressure (controlled by a Monte Carlo barostat) and (iii) running dynamics at 298.15 K for 100 ps at constant volume. All three equilibration steps used a Langevin integrator with a collision frequency of 5 ps−1, 1 kcal mol−1 Å−2 Cartesian restraints on all RNA atoms and a time step of 2 fs. The final frame of the final equilibration step was subjected to 4000 steps each of steepest descent and conjugate gradient minimization with no Cartesian restraints to produce the coordinates of the NEB endpoints.
All simulations used the Amber ff99+bsc0+ChiOL3 RNA force field (66–68), the Li-Merz OPC-HFE parameters for monovalent ions (69), and the 12–6–4 parameters for Mg2+ ions (70). Since these Mg2+ parameters were tuned for TIP4P-Ew water, the pairwise Lennard–Jones parameters for the interaction between Mg2+ and the oxygen of OPC water were modified to copy the pairwise Lennard–Jones parameters for the interaction between Mg2+ and the oxygen of TIP4P-Ew water in that model. Parameters for N3(H+) cytidine were taken from the residue name ‘CP’ from all_prot_nucleic10.lib in Amber 18. Electrostatics were computed using the Particle Mesh Ewald method (71,72) with a 10 Å direct space cutoff. The SHAKE algorithm (73) was used to constrain the bond lengths of covalent bonds to hydrogen.
NEB pathways were found using simulated annealing (74). Solvent atoms were retained for the constant-volume NEB calculation, but NEB spring forces were applied only to RNA heavy atoms. The calculation used 24 images, with 12 images started from the coordinates of each of the fixed endpoints. First, the images were heated from 0 K to 298.15 K over 100 ps using an NEB spring constant of 10 kcal mol−1 Å−2 and a time step of 2 fs. Subsequent NEB steps used a spring constant of 25 kcal mol−1 Å−2 and a time step of 1 fs. Next, the images were equilibrated at 298.15 K for 200 ps, heated from 298.15 K to 498.15 K over 100 ps, equilibrated at 498.15 K for 200 ps, cooled from 498.15 K to 298.15 K over 100 ps, and then equilibrated at 298.15 K for 200 ps. Finally, quenched dynamics were run for 200 ps at 0 K with a time step of 2 fs. The total simulation time for the NEB protocol was 1100 ps. The final frame of the quenched dynamics simulation was taken as the coordinates of the image in the minimal energy path.
A second NEB calculation was conducted in which the aSDS-SDS base pairings of helix S2 were preserved. We applied upper-boundary harmonic restraints with an energy constant of 20 kcal mol−1 Å−2 on the heavy atoms involved in 6 hydrogen bonds observed in both the preQ1-bound and effector-free crystal structures; the restraints included: G33N1–C9N3, G33N2–C9O2, C9N4–G33O6, A32N6–A10N3, A32N6–A10O2′ and A10C2–A32N7. Two improper torsion restraints for each base pair were used to enforce coplanarity: C9N1–C9C4–G33N3–G33N7, C9C6–C9C2–G33N1–G33N9, A10N9–A10N1–A32N1–A32N9 and A10N7–A10N3–A32N7–A32N3. The restraints were flat around the values observed in the NEB endpoints with a width of 15° for the C9–G33 pair and 30° for the A10•A32 pair; outside of the flat region, the restraint was harmonic with an energy constant of 5 kcal mol−1 Å−2.
Analysis of NEB trajectories, including principal component analysis and visualization of principal components on endpoint structures, was performed using LOOS (75).
RESULTS
Quality control and folds of effector-free and preQ1-bound Tte riboswitch structures
Structures of riboswitches in effector-free states are valued for their potential to reveal conformational changes induced by ligand binding (28) (Supplementary Table S1). We previously determined the crystal structure of the effector-free Tte translational riboswitch at 2.85 Å-resolution (22). The results revealed that A14 of loop L2 enters the binding pocket, where it overlaps the site of effector recognition. However, electron-density maps were unclear for the remainder of loop L2. The positions of U12 and C15 were uncertain and A13 was excluded from the model due to conformational disorder. Parallel observations were made for loop L2 of the B. subtilis transcriptional preQ1-I riboswitch, as well as more recent Tte preQ1-I riboswitch structures captured in the effector-free and preQ1-bound states (36,76,77). Accordingly, we sought to elucidate changes in loop L2 by screening for new crystallization conditions. We found that solutions of poly(ethylene) glycol and Mn2+ could replace high levels of Li2SO4 and Mg2+ used previously (22). These ‘low salt’ Tte riboswitch crystals were prepared in the preQ1-bound and effector-free states, and diffracted X-rays to 2.65 Å and 2.00 Å-resolution (Table 1).
The new preQ1-bound and effector-free Tte riboswitch crystal structures are of high quality, as shown by their Rwork/Rfree values of 17.5%/20.2% and 19.0%/22.5% with acceptable geometries, clash scores, and MolProbity metrics (Table 1). The observation that crystals of the effector-free state diffract better than the preQ1-bound state defies prior trends in which higher disorder was associated with ligand-free crystal forms (24). Electron-density for each structure revealed no chain breaks, allowing construction of complete models with unprecedented L2 loop details (Supplementary Figure S5A and B). Both structures exhibited the H-type pseudoknot fold (Figure 2A and B) (16) characterized by A-form helical stem S1 and pseudoknotted upper stem S2, wherein canonical pairs G33–C9 and G11–C30 flank intervening non-canonical pairs A10•A32 and G8•A31 (Figure 1B). Each structure also exhibited multiple A-amino kissing interactions and an inclined A-minor interaction between the A-rich tail of L3 and the minor groove of S1, which support preQ1 readout by A29 (Supplementary Figure S6 and Figure 1D). A pairwise all-atom superposition between the new structures produced a root-mean-square deviation (rmsd) deviation of 1.1 Å; the maximum deviation was 8.3 Å, which occurred at preQ1-specificity base C15 of the L2 loop (described below). Both bound and effector-free structures revealed A13 bulged away from the core fold (maximum rmsd values of 13.8 Å and 8.6 Å), in contrast to a prior high-salt, preQ1-bound structure wherein A13 stacks inside the L2 loop (22) (Figures 1C, 2A and B).
PreQ1 binding influences Mn2+ coordination near the effector-binding pocket
Our choice of Mn2+ for crystallization was empirical since this ion produced superior diffraction compared to mother liquors containing Mg2+ alone. At present, we do not know why Mg2+ could not be completely supplanted by Mn2+ in the preQ1-bound state (see Materials and Methods). Prior work showed that Mg2+ can bind to N7 of purine—albeit infrequently—whereas Mn2+ readily binds N7 and has a higher affinity for this site than Mg2+ (45). Indeed, our ITC experiments revealed that preQ1 binds the wildtype Tte riboswitch with 3-fold higher affinity in 4 mM Mn2+ compared to 4 mM Mg2+ (ΔΔG = −0.8 kcal mol−1); the average KD values were 2.5 ± 1.0 and 8.1 ± 0.9 nM (Supplementary Table S2). A total of six Mn2+ binding sites were observed in the effector-free and preQ1-bound, low-salt Tte crystal structures, designated sites I to VI (Figure 2A, B and Supplementary Figure S5C–E), wherein five of six sites showed inner sphere N7 coordination. Overall, the improved binding affinity of Mn2+ at N7 of guanine and integration of these ions into crystal contacts appeared to contribute favorably to X-ray diffraction.
The high-salt Li2SO4 conditions used in our prior Tte riboswitch structure hindered Mg2+ binding to specific sites and interfered with riboswitch recognition of the methylamine moiety of preQ1 (22). The absence of observable Mg2+ ions in our earlier work makes it worthwhile to highlight new details of Mn2+ coordination here. In both structures, sites Ia and V mediate crystal contacts, and site VI participates in a sharp U-bend between S1 and L3 (Supplementary Figure S7). The site Ib Mn2+ is found only in the preQ1-bound state and likely neutralizes charge by coordinating between the non-bridging pro-Sp oxygen of A32 and the pro-Rp oxygen of G33; no other ligands can be seen at site Ib and the occupancy is likely weak based on its elevated temperature factor. In contrast, Mn2+ sites II, III and IV are particularly well defined and interesting because each is located in the major groove flanking the preQ1 pocket. The site II cation is present in both the preQ1-bound and effector-free states. The Mn2+ ion coordinates N7 of G8 by an inner sphere contact (Figure 2C). The remaining coordination sphere comprises five waters that mediate Mn2+ contacts to the non-bridging phosphate oxygens of C7, G8 and A10. The ion stabilizes a turn that transitions from the pseudoknot L1 loop to helix S2. A10 is part of the aSDS sequence that hydrogen bonds to A32 of the SDS via sugar-edge to major-groove interactions (Figure 2C). In contrast, the Mn2+ ion at site III was observed only in the effector-free state. The cation coordinates directly to N7 of G5, which forms the floor of the binding pocket (Figure 2D). Five waters complete the coordination sphere. One water mediates contacts between a non-bridging oxygen of A14—the base that occupies the preQ1 site in the effector-free state—and the O6 keto of G5. Another water contacts N7 of A14, while a third contacts a non-bridging oxygen of G5. Altogether the site III ion knits together L2 and S1 in the effector-free state. Notably, coordination of Mn2+ at site III appears to overlap the preQ1 binding site due to coordination of the ion at N7 of G5. In the preQ1-bound state, the latter imine likely hydrogen bonds to the methylamine of preQ1, which has a positive charge (Figure 2E). Although this interaction is not clear in 2mFo – DFc electron density maps, it is better defined in noise-filtered, feature-enhanced maps (Supplementary Figure S5B). An alternative model in which the methylamine hydrogen bonds to the 2′-OH of G11 was not supported by the electron density and mutation of this nucleotide to 2′-deoxy G11 had no appreciable effect on preQ1 affinity (22). Guanine is conserved at position 5 of the consensus sequence in type I and II preQ1-I riboswitches (7), supporting the N7 interactions posited here in terms of preQ1 recognition and divalent ion coordination in the effector-free state. Such detail on divalent ion binding was not observed previously due to the 4.0 M concentration of Li+ used in crystallization (22).
As noted, the methylamine group of preQ1 interacts with N7 of G5, blocking coordination of the site III Mn2+. Due to conformational differences between the bound and effector-free states, the Mn2+ at site III is missing key ligands when A14 moves out of the binding pocket. Instead, Mn2+ binding is favored at nearby site IV in the preQ1-bound state, wherein inner-sphere coordination shifts to N7 of G4 (Figure 2E). Like the other Mn2+ sites, five waters complete the coordination sphere, mediating contacts to N7 of G3 as well as O6 and non-bridging oxygens of G4. Such coordination is typical of divalent ion coordination at tandem guanine sites in RNA duplexes (44). Overall, the new models of the preQ1-bound and effector-free states provide a solid foundation for experiments designed to explore the conformational changes associated with preQ1 recognition and communication of the effector-bound status of the binding pocket to the distal expression platform.
Loop L2 adopts three conformations influenced by preQ1 binding
The largest difference between the effector-free and preQ1-bound structure was observed at L2 (Figure 3A and B). A pairwise, all-atom superposition yielded an average rmsd of 1.1 Å but the rmsd for nucleotides U12 through C15 was 5.1 Å. The backbone in the effector-free state adopts an S-shaped curve that extrudes U12, A13, and specificity base C15; A14 enters the aptamer pocket concomitantly and overlaps the site of preQ1 binding. The Watson–Crick face of A14 mimics the minor-groove edge of preQ1, as observed under high-salt conditions (22). In contrast, U6 of loop L1 and A29 of loop L3 remained stationary in the pocket, where the O4 keto group of U6 hydrogen bonds to N6 of A14 and the N6 of A29 hydrogen bonds to N1 of A14 (Figure 3A). The nucleobase interactions with A14 are reminiscent of preQ1 readout in both the low- and high-salt preQ1-bound states (Figure 3B,C). Although preQ1 recognition in the low-salt, bound-state structure is similar to that of the prior high-salt structure (all-atom rmsd of 0.65 Å), the backbone conformations differ at L2 (average rmsd of 4.2 Å). Specifically, A13 bulges away from the core in the low-salt structure, whereas A13 stacks between A14 and A32 in the prior high-salt model (Figure 3B and C).
Figure 3.
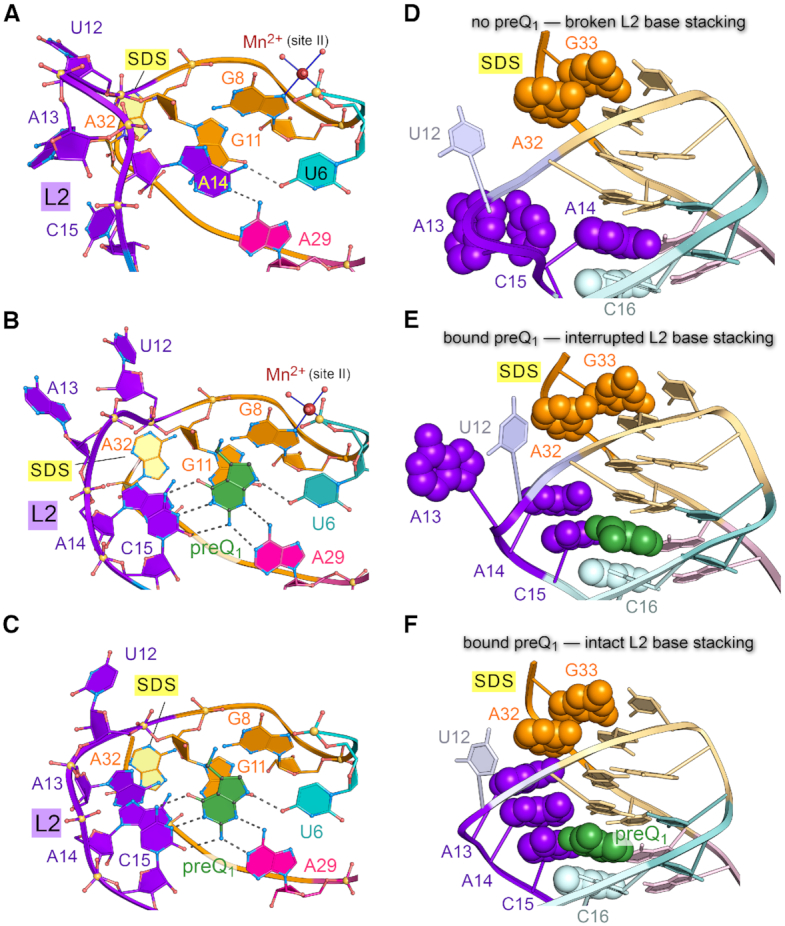
Effector-induced stacking conformations in the L2 loop of the Tte preQ1-I riboswitch depend on preQ1 binding. (A) Close-up of loop L2 and the preQ1-binding pocket for the effector-free riboswitch of this investigation. A14 forms hydrogen bonds to bases U6 and A29. (B) Close-up of L2 in the preQ1-bound, low-salt crystal form of this investigation. A14 stacks atop specificity base C15, which recognizes preQ1. (C) Close-up of L2 in the preQ1-bound, high-salt crystal form described previously (22). (D) Close-up view of loop L2 in the effector-free preQ1-I riboswitch structure of this investigation; the view is rotated ∼90° about the x-axis relative to panel A. Base stacking is emphasized by CPK models for A32 and G33 of the SDS, A13, A14 and C15 of L2, and C16 of stem S1. In the effector-free state shown, the nucleobase stacking interaction is disrupted. (E) Close-up view of loop L2 and the mode of preQ1-binding by the low-salt Tte riboswitch structure of this investigation. In the preQ1-bound state shown, nucleobase stacking within loop L2 is interrupted by bulged A13. (F) Close-up view of loop L2 and the mode of preQ1-binding by the high-salt Tte riboswitch structure (22). In this preQ1-bound state, L2 exhibits continuous base stacking that links preQ1 binding by C15 to the SDS at A32.
The distinct L2 conformations show key differences in core base stacking that have implications for communicating the occupancy status of the effector-binding pocket to the distal expression platform. The intervening long-range interaction network can be visualized when the L2 bases are depicted as CPK models (Figure 3D-F). In the effector-free state, A13 stacked upon specificity base C15 but the bases were oriented orthogonally to the helical axis (Figure 3D). A14 simultaneously occupied the preQ1 pocket while A32 and G33 of the SDS remained stacked and base paired to A10 and C9 to form helix S2 of the pseudoknot.
In the preQ1-bound state under low-salt, a different L2 loop conformation was observed. C15 moved inside the pocket for preQ1 recognition, partially stacking atop C16 in the pocket floor (Figure 3E); A14 simultaneously exited the pocket and stacked upon C15. Unexpectedly, A13 protruded from the pocket and apparently bulged into solution. Inspection of local crystal contacts revealed that A13 actually stacked between A32′ and A14′ within the L2′ loop of a neighboring molecule (Supplementary Figure S8A). This interaction formed a non-canonical, intermolecular A13′•A31 pair comparable to the intramolecular A13•A31 pair observed in the prior high-salt structure (22) (Supplementary Figure S8B and C). In contrast, A13 did not engage in an analogous interaction in the effector-free state (Supplementary Figure S8D). These observations highlight the preference of loop L2 to adopt a continuous nucleobase stack when preQ1 occupies the effector-binding pocket.
The various conformations observed in the crystal structures underscore the flexibility of the L2 loop. However, the full extent of preQ1-dependent stabilization of L2 was realized fully in the context of the original high-salt, bound-state structure wherein continuous intramolecular base stacking was observed (Figure 3F). Namely, π-stacking occurred amongst A13, A14 and C15. This stack is flanked by C16 of helix S1 on one side and A32 of helix S2 on the other side. A32 and G33 represent the first two bases of the SDS that must base pair with the 3′-end of the 16S rRNA for translation initiation. An important implication of our observations is that continuous stacking by the L2 nucleobases buttresses the SDS in response to preQ1, supporting a gene-off state. In addition, S2 bases are likely to be more dynamic than implied by the effector-free crystal structure, wherein the SDS is buried. For this reason, we conducted solution-based experiments to test this hypothesis.
Variations at position 14 of loop L2 influence preQ1 binding and dissociation kinetics
As a first step toward characterizing the flexibility of L2 loop nucleotides in solution, we first tested the observation that A14 occupies the effector-binding pocket, ostensibly competing with preQ1 (Figure 3A,D). We tested this hypothesis by altering the number of hydrogen bonds formed between position 14 and aptamer-core bases, which is predicted to alter the kinetics of preQ1 association (kon) and dissociation (koff), as measured by SPR. The wildtype preQ1-free state reveals two hydrogen bonds between A14 and preQ1-recognition bases U6 and A29 (Figure 4A), whereas the preQ1-bound state shows that A14 makes two hydrogen bonds to the G11 minor-groove edge. Wildtype preQ1 binding yielded a kon of (11.60 ± 1.13) × 104 M−1 s−1 and koff of (2.12 ± 0.16) × 10−4 s−1 (Table 2). These rate constants and the KD of 1.84 ± 0.08 nM agree well with prior SPR measurements on the wildtype Tte riboswitch, which yielded a kon of (7.77 ± 0.04) × 104 M−1 s−1, a koff of (1.53 ± 0.01) × 10−4 s−1, and KD of 2.05 ± 0.29 nM (22). Accordingly, we substituted the adenine analogue 2,6-diaminopurine (DAP) at position 14 with the prediction that it would deter preQ1 binding due to its ability to form an additional hydrogen bond to A29 in the effector-free state (Figure 4B). Indeed, A14(DAP) slowed kon by ∼6-fold compared to wildtype (Table 2). In contrast, koff remained nearly unchanged—an ∼1.2-fold effect—as reflected by the comparable wildtype and A14(DAP) half-lives of dissociation, which were 55 min and 46 min. Similarities in koff support the structure-based analysis that suggests both A14 and A14(DAP) share common Watson-Crick features in the context of L2 that allow each base to engage in nearly identical hydrogen-bonding patterns with the minor-groove edge of G11 in the preQ1-bound state (Figure 4B).
Figure 4.

Kinetic and affinity analysis of preQ1 binding by the wildtype Tte riboswitch and position 14 variants. (A) Representative sensorgrams from surface plasmon resonance (SPR) showing preQ1 association and dissociation to the wildtype riboswitch. Hydrogen-bonding diagrams for A14 are depicted for the effector-free state (binding pocket view) and preQ1-bound state (L2 loop view) based on the crystal structures of this investigation (Figure 2A and B). PreQ1 concentrations are shown in the key and colored lines represent background-subtracted data; black lines indicate the global fit to a 1:1 binding model. The chi2 (RU2) for the fit was 1.8. The kon and koff rates and the apparent KD values are in Table 2. (B) Representative sensorgrams for preQ1 interaction with the A14(DAP) (2,6-diaminopurine) variant. The chi2 (RU2) for the fit was 0.81. Putative hydrogen-bonding interactions for A14(DAP) are depicted for the effector-free state (binding pocket view) and preQ1-bound state (L2 loop view) based on the wildtype crystal structures of this investigation. (C) Representative sensorgrams showing preQ1 interactions with the A14(2AP) (2-aminopurine) variant. The chi2 (RU2) for the fit was 1.5. Putative hydrogen-bonding interactions for A14(2AP) are depicted. (D) Representative sensorgrams showing preQ1 interactions with the A14(Pur) (purine) variant. The chi2 (RU2) for the fit was 0.85. Putative hydrogen-bonding interactions for A14(Pur) are depicted.
We then sought to alter the preQ1 binding kinetics when position 14 makes fewer hydrogen bonds to preQ1 recognition bases. The adenine analogue 2-aminopurine (2AP) was substituted at position 14 (A14(2AP)), where it is predicted to form two hydrogen bonds to A29 but no contacts to U6 in the preQ1-free state (Figure 4C). This substitution resulted in a faster kon than A14(DAP) by 2.2-fold but a slower kon than wildtype by 2.5-fold (Table 2). The results suggest that multiple hydrogen bonds between the nucleobase at position 14 and A29 reduce the preQ1 association rate, possibly by stabilization of position 14 in the binding pocket. Conversely, A14(2AP) was predicted to make only one hydrogen bond to G11 in the preQ1-bound state—rather than two as observed for wildtype. This weaker interaction provides a plausible explanation for the faster koff of A14(2AP), which is ∼5-fold faster than wildtype with a t1/2 of 10.3 min (Table 2). Unexpectedly, substitution of purine for adenine at position 14 (A14(Pur)) showed trends similar to A14(2AP) in terms of slowing preQ1 association (Table 2). A14(Pur) is predicted to make only one hydrogen bond to A29, although an additional favorable interaction could occur in the pocket to slow effector binding. A14(Pur) is also predicted to form a single hydrogen bond to G11 in the preQ1-bound state (Figure 4D). Like 2AP, A14(Pur) elicits a 3.2-fold faster preQ1 dissociation rate compared to wildtype with a t1/2 of 17.2 min (Table 2). Overall these experiments support the supposition that A14 enters the preQ1-binding pocket but must exit for efficient preQ1 association. Moreover, the pattern of hydrogen bonds made by A14 to G11 in the preQ1-bound state suggests that A14 can stack inside the aptamer core (Figure 3F) and influences effector dissociation.
PreQ1 binding influences the stacking of nucleotides in the L2 loop and S2 helix
We next tested the hypothesis that bases within loop L2 and adjacent to helix S2 change their base stacking in response to preQ1. We replaced individual adenines at positions 10, 13, 14 and 32 with the fluorescent isomer of adenine, 2AP, and monitored changes in emission as a function of preQ1 concentration (Figure 5). The fluorescence of 2AP depends on its environment such that solvation leads to greater fluorescence emission, whereas quenching occurs in less hydrated, base-stacked environments (78). We first analyzed A14(2AP) because it provided an independent means to validate SPR data (above) while providing a benchmark for other 2AP variants.
Figure 5.

Fluorescence emission analysis of Tte preQ1-I riboswitch bases within loop L2 and stem S2. (A) Diagram illustrating the location of bases A10, A13, A14 and A32 (ball-and-stick models) in the context of the effector-free structure (ribbon diagram) of this investigation. (B) Diagram illustrating the location of bases A10, A13, A14 and A32 in the context of the preQ1-bound structure (PDB entry 3q50) (22). (C) Titration of preQ1 into a 2-aminopurine-labeled (A14(2AP)) Tte riboswitch. Fluorescence emission was monitored at 390 nm. Here and elsewhere filled circles represent the experimental data resulting from three independent measurements including standard errors of the mean. Smooth trend curves show the fit of a one-site-binding model, used to calculate the apparent KD and standard deviation for preQ1 binding. The goodness-of-fit between the model and data is indicated by R2. (D) Titration of preQ1 into the A13(2AP) Tte riboswitch. (E) Titration of preQ1 into the A10(2AP) Tte riboswitch. (F) Titration of preQ1 into the A32(2AP) Tte riboswitch.
The results revealed that A14(2AP) loses fluorescence emission with added preQ1 (Figure 5C), suggesting that position 14 is less stacked and more solvated in the effector-free state. The preQ1-dependent change in position 14 solvent accessibility and stacking agrees to some extent with the observed environmental differences in our structures (Figure 5A and B). Although A14 exhibits a lower solvent accessible surface area in the effector-free state (41 Å2) compared to the effector-bound states prepared under low-salt and high-salt conditions (195 and 123 Å2), the diminution in A14(2AP) fluorescence suggests that the nucleobase makes dynamic excursions into and out of the binding pocket in the absence of preQ1. A similar change in A14(2AP) fluorescence emission was observed independently using stopped-flow methods (79). To ascertain that the A14(2AP) modification does not impair Tte riboswitch folding, we calculated an apparent KD from the A14(2AP) fluorescence emission curve, which yielded a value of 34.2 ± 1.6 nM (Figure 5C). This value falls within 1.8-fold of the KD of 19.3 ± 0.8 nM measured independently for A14(2AP) by ITC (Supplementary Table S2), and within 1.4-fold of the KD of 24.5 ± 4.6 nM measured by SPR (Table 2). Experiments for A13(2AP) revealed comparable fluorescence decreases with increasing preQ1 (Figure 5D). The apparent KD of 25.7 ± 2.4 nM from the emission spectrum agreed well with the value of 21.6 ± 1.1 nM attained by ITC (Supplementary Table S2). These observations support an A13 conformation wherein adenine stacks between A14 and A32 when preQ1 binds, as observed in the high-salt co-crystal structure (Figure 5B).
We next probed changes in the nucleobase environment within the aSDS and SDS of pseudoknot stem S2. Although our crystal structures suggest that these positions form identical hydrogen bonds in the effector-free and bound states, we hypothesized that these nucleobases are flexible in solution in the absence of preQ1. Individual 2AP substitutions at positions 10 and 32 of the aSDS and SDS each yielded fluorescence emission decreases with added preQ1 (Figure 5E and F). The results imply effector-dependent conformational changes to the nucleobase environments such that they experience increased solvent shielding and greater base stacking with added effector. The apparent KD values from fitting the A10(2AP) and A32(2AP) fluorescence decay curves (i.e. 13.9 ± 0.7 and 12.3 ± 1.4 nM) were in good agreement with those from ITC (i.e. 12.6 ± 2.0 and 10.5 ± 2.4 nM) (Supplementary Table S2).
The site-specific fluorescence-emission analysis here supports preQ1-mediated conformational changes in bases 13 and 14 of the L2 loop. These observations are mostly consistent with base stacking observed in the Tte riboswitch crystal structure (Figure 5B and Supplementary Figure S8). Namely, the high-salt preQ1-bound state features nucleobase stacking that starts with the C15-preQ1 pair, and proceeds through A14 and A13 (Figure 5B). In contrast, the effector-free structure, and prior biophysical analysis, implied that helix S2 retains its fold in the absence of preQ1—perhaps undertaking only modest induced-fit adjustments upon sensing the effector (22,31,38). By comparison, the solution experiments here demonstrated that loop L2 bases, as well as A10 and A32 of the aSDS and SDS, are dynamic with measurable unstacking in the absence of preQ1 (Figure 5E and F). This observation is corroborated in part by an independent NMR analysis of the G11–C30 pair in the binding pocket ceiling that revealed at least two S2 conformations in the absence of preQ1 (79).
PreQ1 reduces global riboswitch flexibility and increases base pairing in helix S2
The possibility that A10, A13, A14 and A32 spend more time exposed to solvent due to greater overall dynamics of the fold in the preQ1-free state prompted us to conduct a global analysis of preQ1-dependent flexibility changes. We first used SHAPE-seq to reveal how effector recognition alters 2′-hydroxyl acylation, where increased reactivity was interpreted as an indicator of greater nucleotide flexibility (80,81). SHAPE-seq analysis of the effector-free Tte riboswitch revealed several flexible regions: A13 and C15 of loop L2; U21, U22 and A23 at the S1-L3 transition; and C9 and A10 of the aSDS as well as A32 and G33 of the SDS (Supplementary Figure S4A). The reactivities (ρ values) (56) at these positions were markedly higher than those of stems comprising canonical Watson–Crick base pairs (e.g. S1 of the Tte riboswitch or hairpins S0 and S3 of the SHAPE cassette that flanks the riboswitch) (Supplementary Figure S4A). Mapping the acylation reactivity of the preQ1-free riboswitch onto the preQ1-free crystal structure revealed numerous flexible nucleotides outside stem S1, most notably in loop L2 segments adjacent to the binding pocket and helix S2 harboring the SDS (Figure 6A).
Figure 6.

Structural mapping of preQ1-dependent 2′-hydroxyl group acylation and base methylation for the Tte riboswitch. (A) SHAPE-seq reactivity (ρ) values mapped onto the preQ1-free crystal structure (Figure 2A). (B) SHAPE-seq reactivity (ρ) values mapped onto the preQ1-bound structure (Figure 2B). Here and elsewhere, preQ1 is shown as a green ball-and-stick model but no reactivity values were measured (C) Differential SHAPE reactivities mapped onto the preQ1-bound high-salt structure (PDB entry: 3q50) (22). The differential reactivity key is: light blue means little or no flexibility change between preQ1-free and bound states; red equals the greatest increase in flexibility with added preQ1; dark blue equals the largest decrease in flexibility with added preQ1. (D) Plot of differential SHAPE reactivity (Δρ) versus sequence position. The Tte riboswitch sequence encompasses nucleotides 1–33. Nucleotides on the 5′- and 3′-ends were derived from a folding cassette (52). The Δρ values were mapped to the structure in panel C. Here and elsewhere canonical helical regions in the cassette and riboswitch (i.e. S0, S1 and S3) were used to calculate standard deviations of Δρ; these are drawn as orange, dashed lines to indicate signal-to-noise thresholds. The colors of the bars match the colors of the corresponding base in panel C. Bars corresponding to bases in the folding cassette (see Materials and Methods) are colored gray. (E) DMS-seq ρ values mapped onto the preQ1-free crystal structure (Figure 2A). (F) DMS-seq ρ values mapped onto the preQ1-bound structure (Figure 2B). (G) DMS Δρ mapped onto the preQ1-bound high-salt structure (PDB entry: 3q50) (22). The differential reactivity key is: green means little change in Watson-Crick face accessibility between preQ1-free and bound states; red equals the greatest increase in accessibility with added preQ1; dark blue equals the largest decrease in accessibility with added preQ1. (H) Plot of DMS Δρ versus sequence position. The Δρ values were mapped to the structure in panel G. N.d. means not determined. Individual reactivity values are provided in Supplementary Figure S4.
Mapping the SHAPE reactivity of the preQ1-bound sample onto the preQ1-bound crystal structure showed that most nucleotides have appreciably lower reactivities than the effector-free state (Figure 6A, B and Supplementary Figure S4A, B). Regions of reduced reactivity included: the L1 loop harboring ligand-recognition base U6; the SDS–aSDS pairs within stem S2; and the A-rich tail of loop L3. The sharp turn joining S1 and L3 remained reactive—as observed for the effector-free state — suggesting the nucleotides are predisposed to acylation (Supplementary Figure S6C). The most reactive position was A13 in loop L2, which produced a higher acylation level than the preQ1-free state (Supplementary Figure S4A, B). The effector-free model reveals that the 2′-OH of A13 hydrogen bonds to a non-bridging phosphate oxygen of C15 (Supplementary Figure S9A), potentially reducing acylation. This interaction does not occur in the effector-bound structures because C15 enters the pocket to recognize preQ1, presumably leaving the A13 2′-OH exposed. Specifically, in the preQ1-bound, high-salt conformation the A13 2′-OH hydrogen bonds to O4′ of A14 (Supplementary Figure S9B), whereas the preQ1-bound, low-salt structure reveals no contacts to the 2′-OH of A13, supporting the high-degree of acylation (Supplementary Figure S9C).
To pinpoint changes in Tte riboswitch flexibility resulting from preQ1 binding, we performed a differential reactivity (Δρ) analysis. Here values corresponding to ρbound − ρfree were mapped onto the high-salt preQ1-bound structure and plotted as a function of nucleotide position (Figure 6C and D). In accord with the role of the Tte riboswitch in translational regulation (22,35,38), nucleotides within aSDS-SDS pairs (i.e. C9–G33 and A10•A32) became less flexible with added preQ1. A14 and C15 also decreased subtly in flexibility, consistent with participation of L2 nucleobase in stacking and effector recognition (Figures 1D and 3B, C, E, F). In general, the A-rich tail from A23 to A28 showed decreased flexibility as well, consistent with its stabilizing minor-groove interactions with S1 (35,76,77) and preQ1 readout by position A29 (Supplementary Figure S6).
We next conducted a DMS-seq analysis to investigate the dependence of riboswitch base pairing on preQ1 binding. In particular, reduced DMS methylation of adenine N1 and cytosine N3 was seen as an indicator of Watson–Crick pairing (56). Our results revealed susceptibility to methylation in the absence of effector in the following regions: C9 and A10 of the aSDS; A13 of loop L2; the A-rich tail of loop L3; and the SDS base A32 (Figure 6E and Supplementary Figure S4C). In the presence of preQ1, most bases showed markedly reduced methylation. However, A10 of the helix S2 (i.e. the aSDS) and A13 of loop L2 (Figure 6F and Supplementary Figure S4D) remained reactive, consistent with the absence of Watson–Crick pairing by these bases in both effector-free and bound-state crystal structures (Supplementary Figures S7A, S8C, D and S9). As expected, helical regions with Watson-Crick pairing, such as S1, were well protected from DMS methylation, regardless of added preQ1.
To identify changes in base-pairing that resulted from preQ1 binding, we performed a differential DMS-reactivity (Δρ) analysis (Figure 6G,H). The results reveal that C9, as well as C30, A31 and A32 became less susceptible to methylation with added preQ1, consistent with greater helical character in S2—in support of a gene-off conformation that buries the SDS. In general, the loop L3 nucleobases became less susceptible to methylation as well. This observation suggests that the A-amino kissing interactions in the minor groove of S1 play an integral role in conformational stability of the riboswitch, wherein the Watson–Crick faces of the L3 loop bases are sequestered upon preQ1 binding (Supplementary Figure S6); several L3 adenines that show direct hydrogen bonding between N1 and the S1 minor groove (i.e. A23, A26, A27 and A28) exhibited reduced methylation (Figure 6G and H). Similarly, A13 of loop L2 also showed decreased methylation, consistent with hydrogen bonding of its N1 imine to the 2′-OH of A31, while contributing to the continuous loop-L2-nucleobase stack that buttresses A32 and G33 of the SDS (Supplementary Figure S9B).
Computational modeling reveals a simple pathway for a free- to bound-state transition
We next considered how the preQ1-free state—wherein A14 overlaps the preQ1 binding site—could interconvert to the effector bound-state. Although unbiased MD has captured microsecond timescales of preQ1 riboswitch conformational changes (37,82), it is insufficient to observe large-scale motions, such as pathways connecting ligand-free and bound states (37). Accordingly, our ability to resolve the first intact structures of the effector-free and preQ1-bound states prompted us to use NEB (59)—a method that delineates low-energy pathways joining two molecular conformations. The fixed endpoints for each NEB pathway were assigned to be the preQ1-free coordinates of this investigation (Figure 2A) and the high-salt, preQ1-bound coordinates (Figure 1C) (22) with the effector removed. Since NEB is not deterministic, we generated four independent pathways in the absence of restraints on the pathways (Figure 7A–C and Supplementary Movies S1–S4). The potential energy of the atoms subject to NEB spring forces by image (Supplementary Figure S10A) suggests that the independent pathways pass over potential energy barriers at different rates. Therefore, a principal component analysis (PCA) of all unrestrained NEB trajectories grouped together was used to identify common features of progress coordinates that allow the effector-free riboswitch to adopt a conformation that accommodates preQ1 in the binding pocket (Figure 7D-F and Supplementary Figure S10). The first two principal components (PCs) explain 61% and 23% of the variance across pooled images, indicating that these two PCs are sufficient to visualize the NEB trajectories in a reduced 2-dimensional projection (Supplementary Figure S10B). We interpret PC1 as a progress coordinate that describes the atomistic motions that must occur for the effector-free riboswitch to adopt a conformation that can accommodate preQ1 in the binding pocket observed in the bound-state crystal structure. PC2 describes an orthogonal set of atomistic motions that perturbs the endpoint structures in a way that permits the productive motions associated with PC1 to be favorable.
Figure 7.

Atomistic motions associated with interconversion of the Tte preQ1-I riboswitch from the preQ1-free to the preQ1-bound state. (A–C) Images from a representative nudged elastic band (NEB) calculation (blue path in panels D–F) without restraints. The complete trajectory is provided as Supplementary Movie S1. Images shown are: (A) image 1, corresponding to the preQ1-free endpoint, (B) image 9, corresponding to a low-energy intermediate, and (C) image 24, corresponding to the preQ1-bound endpoint. RNA residues are colored as described in Figure 1. Dashed, gray lines represent distances between nucleobases that form either stacking interactions in loop L2 or base pairs in stem S2. (D–F) Nucleobase distances (between the centroids of nucleobase atoms C2, C4, and C6) are plotted as a function of a progress coordinate — the first principal component (PC) from the unrestrained NEB pathway — to facilitate comparison between trajectories. Vertical lines indicate the value of this progress coordinate for the images depicted in panels A–C: cyan for image 1 (A), yellow for image 9 (B), and purple for image 24 (C). Nucleobase distances plotted are (D) C9-G33, representing the breaking of the aSDS-SDS base pairs, (E) A13-A14, representing the formation of the L2 nucleobase stacking interactions, and (F) A13-A32, representing docking of the L2 stacking interactions into stem S2. (G–H) Visualization of the first (G) and second (H) PCs, describing 61% and 23% of the variance, as a projection onto the preQ1-bound endpoint. Red lines represent the direction of the PC eigenvector for every atom. In (H), the magnitude of the red lines is scaled by the relative variances between the second and first PCs (0.38).
We next sought to identify which atomistic motions were associated with these two PCs. Projecting PC1 onto the structure of the preQ1-free endpoint revealed that this coordinate involves motion of A14 out of the preQ1-binding pocket, reorganization of residues in loop L2, and contraction of the riboswitch along the helical axis from S1 to S2 (Figure 7G). A similar projection of PC2 onto the same structure revealed that this coordinate involves motion of residues in S2—including nucleotides within the SDS and aSDS—in a direction orthogonal to the helical axis (Figure 7H). By visualizing low-energy minima along a representative pathway (Figure 7A, Supplementary Movie S1 and Supplementary Figure S11A-C) and plots of these atomistic motions as a function of the progress coordinate PC1 (Figure 7D–F and Supplementary Figure S11D–G), a coherent description of the sequence of events along this pathway emerges.
In contrast to the effector-free crystal structure, in which helix S2 is paired, all four NEB trajectories revealed early breaking of the two aSDS-SDS base pairs C9–G33, A10•A32, and G8•A31 during the preQ1-free to preQ1-bound transition (Figure 7B, D and Supplementary Figure S11D, E). These base pairs, present in both endpoint structures, remained broken until late in the pathway. Following disruption of these pairs from the effector-free state, A14 leaves the preQ1 binding pocket and stacks between L2 bases A13 and C15 (Figure 7B,E and Supplementary Figure S11B, F). Next, an interaction between A13 of loop L2 and A32 of the SDS completes long-range nucleobase stacking observed in the preQ1-removed endpoint (Figure 7C and F). Finally, the disrupted base pairs C9–G33, A10•A32 and G8•A31 return to their paired positions (Figure 7D and Supplementary Figure S11D, E). Although the four NEB pathways disagree about the extent to which the residues in helix S2 deviate from the base-paired conformations observed in the endpoint structures and about the details of nucleotide motions in some steps, the broader sequence of events is common to all trajectories.
We next generated plausible NEB pathways in which helix S2 base pairs cannot open (Supplementary Movies S5-S8 and Supplementary Figure S12), as seen in the effector-free crystal structure (Figure 2A). Restraints were applied to enforce interbase hydrogen bonds and coplanarity of aSDS–SDS base pairs C9–G33 and A10•A32. Projecting these restrained NEB trajectories onto the first two PCs from the unrestrained NEB simulations revealed that the restrained trajectories adopt the same parabolic shape as the unrestrained trajectories (Supplementary Figure S10C and D); however, the motion of the restrained trajectories along PC2 was restricted compared to the unrestrained trajectories (Supplementary Figure S10B and D). Accordingly, the restrained base pairs as well as the G8•A31 pair in S2 do not exhibit the SDS–aSDS fraying seen in the unrestrained pathway (Supplementary Figure S12B, D, H, J, K). Continuous base stacking involving C15, A14, A13, A32 and G33 proceeds in a manner similar to the unrestrained trajectories. However, greater variability exists between the restrained pathways in terms of the sequence of interactions formed (Supplementary Figure S12E, F, L,M).
Importantly, the restrained NEB trajectories show inconsistencies with our experimental observations. SHAPE-seq analysis demonstrated decreased flexibility at C9 and A10 of the aSDS as well as A32 and G33 of the SDS in the presence of preQ1 (Figure 6C and D). DMS-seq revealed reduced methylation at C9, A31 and A32 when preQ1 is bound (Figure 6G and H). A10 and A32 also changed from less stacked to more stacked environments with added preQ1 (Figure 5E and F). As such, the unrestrained NEB trajectories showed better agreement with experimental observations. PreQ1 addition also reduced S2 flexibility and increased base-pairing according to SHAPE-seq and DMS-seq (Figure 6). On the other hand, DMS-seq and 2AP fluorescence analysis at positions 13 and 14 support both the unrestrained and restrained NEB pathways wherein these bases unstacked early in the trajectory but coalesce as part of a continuous L2 loop nucleobase stack as they move toward the preQ1-bound state. Hence, although it is feasible to undertake interconversion pathways in which helix S2 remains base paired, these pathways are not as well aligned with solution experiments as the unrestrained pathways. Notably, neither restrained nor unrestrained NEB pathways indicated formation of the G11pU12 platform that was previously posited to unpair A32 and G33 from the aSDS strand (37).
DISCUSSION
The principles of molecular recognition used by riboswitches to bind their cognate effectors are well understood (9). However, a knowledge gap exists in our understanding of how ligand binding leads to conformational changes that reprogram associated expression platforms that control gene expression (2). To address this challenge, we undertook local and global analyses of preQ1-dependent changes in nucleotide stacking, flexibility and base pairing. Although previous biophysical and computational studies suggested that the Tte preQ1-I riboswitch is mostly pre-folded without its effector (22,31,37), our results provide greater atomistic understanding of how the riboswitch reorganizes a partially unfolded state to bind preQ1 and convey this occupancy status to the distal expression platform. Accordingly, helix S1 at the base of the pseudoknot maintains canonical base pairing—as demonstrated by its low flexibility and lack of methylation in the presence or absence of preQ1 (Figure 6 and Supplementary Figure S4). In contrast, SDS-aSDS pairing in helix S2 and loop L3 showed significant decreases in flexibility and methylation upon preQ1 binding. To attain a coherent picture of the associated effector-dependent conformational changes, we leveraged the new, effector-free crystal structure of the Tte preQ1-I riboswitch and a prior preQ1-bound structure as endpoints for NEB calculations (Supplementary Movies S1–S4). An obvious feature that emerged early in NEB pathways was a constellation of stacked bases in loop L2 comprising A13, A14 and C15 (Figure 7B,E, Supplementary Figure S11B,F and Supplementary Movies S1–S4). In general, the results of the unrestrained NEB pathways showed agreement between independent trajectories, and between computational trajectories and solution experiments, providing confidence that the proposed pathways are reasonable approximations of riboswitch motion — albeit in the absence of induced-fit adjustments due to the ligand. Importantly, single-molecule experiments performed on the Tte preQ1-I riboswitch demonstrated that transitions between open and closed SDS conformations occur multiple times in the presence or absence of preQ1 binding (38).
Despite the success of NEB, two caveats potentially limit the interpretation of our computationally predicted interconversion pathways. First, NEB identifies pathways connecting two states that pass through intermediate conformations with low potential energy. Yet, NEB is blind to the free energy of these intermediate conformations. Although the unrestrained and restrained NEB trajectories exhibit similar potential energy barriers (Supplementary Figure S10A and C), additional simulations would be needed to understand the barriers in free energy in order to discriminate between the feasibility of the respective pathways without reference to solution experiments. Second, NEB requires chemically identical endpoints. To meet this requirement, preQ1 was removed from the endpoint derived from the preQ1-bound structure. The absence of the ligand likely causes our simulations to underestimate the favorability of conformations in which direct interaction with preQ1 is expected.
The collective NEB and solution experiments imply that the riboswitch adopts a third conformation—distinct from the effector-free and preQ1-bound crystal structures reported here—wherein SDS–aSDS base pairs in helix S2 are broken. In the absence of the effector, the riboswitch likely samples an equilibrium between two conformations in which the SDS is either accessible or sequestered in base pairs with the aSDS, the latter of which is captured in the effector-free crystal structure. PreQ1 binding then shifts this equilibrium toward a conformation containing SDS-aSDS base pairs, captured in the preQ1-bound crystal structure. This binding model is consistent with the decrease in 2AP fluorescence (Figure 5) and changes in SHAPE reactivity (Figure 6C and D) observed upon titration of preQ1 and the exploration of conformations in which the SDS is accessible in the unrestrained NEB pathways (Figure 7B and D). Altogether, these findings allow us to propose that loop L2 acts as a molecular communication conduit that connects preQ1 sensing by C15 with SDS-aSDS base pairing in helix S2, located >10 Å from the binding pocket. Based on strong experimental evidence, the S2 helix is most likely unpaired in the absence of effector, as indicated in the early stages of unrestrained NEB pathways.
Importantly, the local L2 loop nucleobase stack observed in the preQ1-bound conformation forms part of a global nucleobase ‘stacking spine’ that spans the length of the riboswitch structure (Figure 8A, inset). This network is expected to impart global fold stability, as underscored by a recent study demonstrating preQ1-dependent stabilization of helix S2 in the presence of helicase (83). A key feature of the sensing network is the position of specificity base C15 inside the L2 loop where it base pairs with preQ1, thereby completing a coaxial stack between helices S1 and S2 (Figure 8A). Same-strand stacking provides a structural buttress for the SDS, which sits directly atop loop L2. In contrast, L2 unstacks in the absence of effector (Supplementary Figure S13A,B), breaking the nucleobase stacking-spine network and exposing the SDS for translation initiation. In short, the stacking spine serves as molecular network that links effector binding to the distal SDS while imparting global stability to the riboswitch fold.
Figure 8.

Long-range nucleobase interaction spines in riboswitches and the TYMV 3′-UTR. (A) Stacking spine of the preQ1-bound Tte riboswitch (PDB entry: 3q50) (22). The spine exhibits continuous nucleobase stacking (hash marks) supported by preQ1 binding at C15 in loop L2; the resulting stacking spine communicates effector binding in the sensing region (aptamer) to the distal SDS (expression platform) to support a gene-off functional state; U12 was omitted for clarity. Inset, preQ1 favors a continuous stacking spine (space-filling model) that spans the length of the riboswitch. (B) Stacking spine of the Vvu add riboswitch in complex with adenine (PDB entry 5swe) (3). Adenine mediates nucleobase stacking contacts between switch-helix P1 and J1–2 nucleobases (hash marks). Inset, a continuous stacking spine spans the length of the riboswitch and includes adenine, which stabilizes the fold to achieve gene-regulation. (C) Stacking spine of the TYMV TLS-linker-UPD domain interaction (PDB entry: 6mj0) (1). In the absence of ribosomes, the UPD sensing domain remains intact and favors continuous base stacking that extends from the UPD, through the linker and into the TLS (hash marks). The U8•U24 pair (U8 in green) is analogous to effector binding by a riboswitch, albeit several other UPD bases influence stability of this domain (12). Inset, a continuous stacking spine spans the length of the structure. In each example, the designation ‘spine’ is justified due to the global stability imparted by the motif to the entire fold; in this manner, the spine provides a tangible communication conduit that links a sensing region with a functional domain (1).
A search for analogous nucleobase spines led to the Vvu add riboswitch structure, which also exhibits a same-strand-stacking network that spans the length of the structure (Figure 8B, inset). This network depends upon adenine recognition to complete co-axial helical stacking of the three-way helical junction—the archetypal fold of purine riboswitches (84). Serial crystallography and spectroscopic analysis revealed that the effector-free conformation of the multi-helix junction differs substantially from the effector-bound state (3). Upon binding, adenine stabilizes switch-helix P1 via cross-strand and same-strand stacking (Figure 8B). Linker J1–2 forms a network of nucleobase interactions akin to loop L2 of the Tte preQ1-I riboswitch (Figure 8A,B). This network is interrupted in the absence of adenine as revealed by the effector-free conformation (Supplementary Figure S13C, D). Unlike the Tte preQ1 riboswitch, which integrates the SDS into its pseudoknot fold, adenine binding by the Vvu riboswitch affects the underlying P1 switch helix, which stabilizes the distal SDS.
A well-characterized nucleobase-stacking spine has been described previously for the TLS-linker-UPD ncRNA of TYMV (85). The UPD folds as a pseudoknot that acts as a structure-based sensor of ribosomes. The folded UPD enhances translation via a long-range interaction network that stabilizes the upstream TLS domain (1). A short linker comprising unpaired nucleotides acts as an interdomain communication conduit (Figure 8C). The intact UPD favors formation of a stacked spine that runs from the UPD, through the linker, and into the TLS variable domain and D-loop (Figure 8C, inset). Analogous spines span the lengths of the Tte preQ1-I and Vvu add riboswitches, as noted (Figure 8A and B, insets).
Each riboswitch aptamer is functionally analogous to the UPD of TYMV, which senses the ribosome rather than a small-molecule effector. Accordingly, the interdomain linker joining the TLS and UPD is analogous to L2 of the Tte preQ1-I riboswitch and J1–2 of the Vvu add riboswitch, which change conformation in response to effector binding (Supplementary Figure S13). In each case, the folding status of a key linker or loop impacts the stability of a distal element that controls function—i.e., the SDS of the Tte preQ1-I riboswitch, the P1 switch helix of the Vvu add riboswitch, or the TLS of the TYMV 3′-UTR. Although stacking spines are not universally conserved communication conduits in ncRNAs, additional investigations are needed to identify and understand the role of such networks in gene-regulation.
Even though we established that the Tte preQ1-I riboswitch undergoes specific effector-dependent structural changes, the overall fold is still considered largely pre-folded relative to other family members. Indeed, the B. subtilis and Fusobacterium nucleatum (Fnu) preQ1-I riboswitches were significantly unfolded without preQ1 (22,31,37,86). The stability of the Tte preQ1-I riboswitch is achieved to some degree by overlap of nucleobase A14 with the preQ1 binding site, giving rise to a closed state (Supplementary Figure S14A). Our SPR experiments revealed that base variations at position 14 influenced preQ1 association and dissociation rates (Figure 4), supporting the hypothesis that A14 makes excursions into and out of the binding pocket. NEB trajectories further revealed that A14 exits the pocket via simple pathways, giving rise to open conformations that can accommodate preQ1. Prior single-molecule analysis indicated that the SDS of the Tte preQ1-I riboswitch continuously toggles between accessible and inaccessible states in the absence or presence of preQ1 (38). Accordingly, the ability of the riboswitch pocket to toggle between open and closed conformations provides a plausible mechanism to prevent structural collapse in the absence of effector. Similar ‘placeholder’ nucleobases that overlap effector-binding pockets have been observed for A46 of the Tte SAM-I riboswitch and U48 of the Vvu adenine riboswitch (3,4) (Supplementary Figure S14B,C). Like the Tte preQ1-I riboswitch, these riboswitches significantly alter their backbone trajectories to achieve binding-pocket closure.
Conversely, analysis of known effector-free transcriptional riboswitches (Supplementary Table S1) revealed no comparable placeholder nucleobases. The Fnu FMN riboswitch uses same-strand stacking and nucleobase pairing between J3–4 and J4–5 to maintain an open pocket receptive to effector binding (24). Unlike loop L2 of the Tte preQ1-I riboswitch, J3–4 of the Fnu riboswitch appears to serve as flexible gate that becomes less stacked to accommodate FMN. Other aptamers including the lysine, glycine, PRPP and SAM-IV riboswitches (Supplementary Table S1) showed no appreciable differences between their effector-free and bound-state conformations, suggesting they are completely pre-folded. Overall, this comparison illustrates that placeholder nucleobases, flexible gates and high levels of pre-folding provide a continuum of riboswitch strategies to facilitate effector binding.
CONCLUSION
The Tte preQ1-I riboswitch is a metabolite-sensing gene regulator that controls translation. Our experimental results support a gene-regulatory pathway in which loop L2 and stem S2 of the pseudoknot exhibit reduced base pairing and less same-strand stacking in the absence of preQ1. Complementary NEB calculations provide new atomistic details about the conformations sampled during interconversion of the effector-free and preQ1-bound states. The collective results pinpoint the location and types of chemical interactions that likely change during the proposed induced-fit recognition of preQ1 (31). Effector recognition by specificity base C15 reorganizes loop L2, leading to a bridge of continuously stacked bases that buttress the SDS. The importance of L2 is further underscored by synthetic ligands that target preQ1-I riboswitches in a manner that forces L2 bases into an unstacked effector-free-like conformation (36). Such compounds are not as effective in functional assays compared to preQ1, most likely because they interrupt the long-range stacking spine that links effector binding to the expression platform. Similar uncoupling was reported for specific mutants of the Lactobacillus rhamnosus preQ1-II riboswitch, which bound tightly to preQ1 but exhibited poor gene regulation in live cells (2). The broader utility of stacking spines for interdomain communication is exemplified by their presence in the Vvu add riboswitch and the TLS-linker-UPD ribosome sensor within the TYMV 3′-UTR. Nonetheless, such stacking spines are not universally conserved in riboswitches. The class III preQ1 riboswitch positions its expression platform within a pseudoknot oriented orthogonally to its aptamer, precluding a continuous stacking-spine interaction (87). The thiamine pyrophosphate riboswitch uses two parallel helical domains that sandwich the effector within the molecular interface (88,89). Accordingly, riboswitches likely use many different conduits to link effector binding with biological function. This remains an insufficiently studied but fundamentally important aspect of the switching mechanism.
DATA AVAILABILITY
Crystallographic coordinates and structure factor amplitudes have been deposited into the Protein Data Bank as entry 6vuh (effector free) and entry 6vui (preQ1 bound). Sequence reads for SHAPE-seq and DMS-seq were deposited into the Sequence Read Archive hosted by NCBI/NIH with accession codes: SRX7726568, SRX7726569, SRX7726570, SRX7726571, SRX7726572 and SRX7726573.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Wedekind lab for technical assistance. We thank Dr. Andrew Torelli for identifying the low-salt Mn2+ crystallization conditions used in this investigation. We thank Rachel Bonn-Breach for writing the Python code to integrate ρ values into PDB coordinates. We thank Dr Alan Grossfield for useful discussions about the computational analysis of NEB pathways. Computer time was provided by the University of Rochester Center for Integrated Research Computing.
Notes
Present address: Debapratim Dutta, Department of Biophysics, Johns Hopkins University, Baltimore, MD 21218, USA.
Contributor Information
Griffin M Schroeder, Department of Biochemistry & Biophysics, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA; Center for RNA Biology, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA.
Debapratim Dutta, Department of Biochemistry & Biophysics, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA; Center for RNA Biology, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA.
Chapin E Cavender, Department of Biochemistry & Biophysics, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA; Center for RNA Biology, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA.
Jermaine L Jenkins, Department of Biochemistry & Biophysics, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA; Center for RNA Biology, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA.
Elizabeth M Pritchett, Genomics Research Center, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA.
Cameron D Baker, Genomics Research Center, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA.
John M Ashton, Genomics Research Center, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA.
David H Mathews, Department of Biochemistry & Biophysics, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA; Center for RNA Biology, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA.
Joseph E Wedekind, Department of Biochemistry & Biophysics, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA; Center for RNA Biology, University of Rochester School of Medicine & Dentistry, Rochester, NY 14642, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM132185 to D.H.M., GM063162 to J.E.W]; C.E.C. and G.M.S. from National Institutes of Health [T32 AI049815 to J.E.W., T32 GM118283 to K.L.B.]; Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Basic Energy Sciences [DE-AC02-76SF00515]; The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences [P41 GM103393]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hartwick E.W., Costantino D.A., MacFadden A., Nix J.C., Tian S., Das R., Kieft J.S.. Ribosome-induced RNA conformational changes in a viral 3′-UTR sense and regulate translation levels. Nat. Commun. 2018; 9:5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dutta D., Wedekind J.E.. Nucleobase mutants of a bacterial preQ1-II riboswitch that uncouple metabolite sensing from gene regulation. J. Biol. Chem. 2020; 295:2555–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stagno J.R., Liu Y., Bhandari Y.R., Conrad C.E., Panja S., Swain M., Fan L., Nelson G., Li C., Wendel D.R. et al.. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature. 2017; 541:242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stoddard C.D., Montange R.K., Hennelly S.P., Rambo R.P., Sanbonmatsu K.Y., Batey R.T.. Free state conformational sampling of the SAM-I riboswitch aptamer domain. Structure. 2010; 18:787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chavali S.S., Bonn-Breach R., Wedekind J.E.. Face-time with TAR: Portraits of an HIV-1 RNA with diverse modes of effector recognition relevant for drug discovery. J. Biol. Chem. 2019; 294:9326–9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Serganov A., Nudler E.. A decade of riboswitches. Cell. 2013; 152:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roth A., Breaker R.R.. The structural and functional diversity of metabolite-binding riboswitches. Annu. Rev. Biochem. 2009; 78:305–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garst A.D., Edwards A.L., Batey R.T.. Riboswitches: structures and mechanisms. Cold Spring Harb. Perspect. Biol. 2011; 3:a003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McCown P.J., Corbino K.A., Stav S., Sherlock M.E., Breaker R.R.. Riboswitch diversity and distribution. RNA. 2017; 23:995–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Serganov A., Huang L., Patel D.J.. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature. 2009; 458:233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Howe J.A., Wang H., Fischmann T.O., Balibar C.J., Xiao L., Galgoci A.M., Malinverni J.C., Mayhood T., Villafania A., Nahvi A. et al.. Selective small-molecule inhibition of an RNA structural element. Nature. 2015; 526:672. [DOI] [PubMed] [Google Scholar]
- 12. Vicens Q., Mondragon E., Reyes F.E., Coish P., Aristoff P., Berman J., Kaur H., Kells K.W., Wickens P., Wilson J. et al.. Structure-activity relationship of Flavin analogues that target the Flavin mononucleotide riboswitch. ACS Chem. Biol. 2018; 13:2908–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thore S., Frick C., Ban N.. Structural basis of thiamine pyrophosphate analogues binding to the eukaryotic riboswitch. J. Am. Chem. Soc. 2008; 130:8116–8117. [DOI] [PubMed] [Google Scholar]
- 14. Serganov A., Huang L., Patel D.J.. Structural insights into amino acid binding and gene control by a lysine riboswitch. Nature. 2008; 455:1263–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Edwards A.L., Reyes F.E., Héroux A., Batey R.T.. Structural basis for recognition of S-adenosylhomocysteine by riboswitches. RNA. 2010; 16:2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peselis A., Serganov A.. Structure and function of pseudoknots involved in gene expression control: Structure and function of pseudoknots. Wiley Interdiscip. Rev. RNA. 2014; 5:803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Belashov I.A., Dutta D., Salim M., Wedekind J.E.. Tails of three knotty switches: How PreQ1 riboswitch structures control protein translation. eLS. 2014; John Wiley & Sons, Ltd; doi.org/10.1002/9780470015902.a0021031. [Google Scholar]
- 18. Liberman J.A., Wedekind J.E.. Base ionization and ligand binding: how small ribozymes and riboswitches gain a foothold in a protein world. Curr. Opin. Struct. Biol. 2011; 21:327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Serganov A., Patel D.J.. Amino acid recognition and gene regulation by riboswitches. Biochim. Biophys. Acta. 2009; 1789:592–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wedekind J.E., Dutta D., Belashov I.A., Jenkins J.L.. Metalloriboswitches: RNA-based inorganic ion sensors that regulate genes. J. Biol. Chem. 2017; 292:9441–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Warner K.D., Hajdin C.E., Weeks K.M.. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018; 17:547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jenkins J.L., Krucinska J., McCarty R.M., Bandarian V., Wedekind J.E.. Comparison of a PreQ1 riboswitch aptamer in metabolite-bound and free states with implications for gene regulation. J. Biol. Chem. 2011; 286:24626–24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ren A., Xue Y., Peselis A., Serganov A., Al-Hashimi H.M., Patel D.J.. structural and dynamic basis for low-affinity, high-selectivity binding of L-glutamine by the glutamine riboswitch. Cell Rep. 2015; 13:1800–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vicens Q., Mondragon E., Batey R.T.. Molecular sensing by the aptamer domain of the FMN riboswitch: a general model for ligand binding by conformational selection. Nucleic Acids Res. 2011; 39:8586–8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peselis A., Serganov A.. ykkC riboswitches employ an add-on helix to adjust specificity for polyanionic ligands. Nat. Chem. Biol. 2018; 14:887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang L., Serganov A., Patel D.J.. Structural insights into ligand recognition by a sensing domain of the cooperative glycine riboswitch. Mol. Cell. 2010; 40:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garst A.D., Héroux A., Rambo R.P., Batey R.T.. Crystal structure of the lysine riboswitch regulatory mRNA element. J. Biol. Chem. 2008; 283:22347–22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liberman J.A., Wedekind J.E.. Riboswitch structure in the ligand-free state. Wiley Interdiscip. Rev. RNA. 2012; 3:369–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones C.P., Ferre-D’Amare A.R.. Long-range interactions in riboswitch control of gene expression. Annu. Rev. Biophys. 2017; 46:455–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lemay J.F., Desnoyers G., Blouin S., Heppell B., Bastet L., St-Pierre P., Masse E., Lafontaine D.A.. Comparative study between transcriptionally- and translationally-acting adenine riboswitches reveals key differences in riboswitch regulatory mechanisms. PLoS Genet. 2011; 7:e1001278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suddala K.C., Rinaldi A.J., Feng J., Mustoe A.M., Eichhorn C.D., Liberman J.A., Wedekind J.E., Al-Hashimi H.M., Brooks C.L., Walter N.G.. Single transcriptional and translational preQ1 riboswitches adopt similar pre-folded ensembles that follow distinct folding pathways into the same ligand-bound structure. Nucleic Acids Res. 2013; 41:10462–10475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang K., Li S., Kappel K., Pintilie G., Su Z., Mou T.C., Schmid M.F., Das R., Chiu W.. Cryo-EM structure of a 40 kDa SAM-IV riboswitch RNA at 3.7 Å resolution. Nat. Commun. 2019; 10:5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang L., Wang J., Lilley D.M.J.. The structure of the guanidine-II riboswitch. Cell Chem. Biol. 2017; 24:695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang L., Wang J., Wilson T.J., Lilley D.M.J.. Structure of the guanidine III riboswitch. Cell Chem. Biol. 2017; 24:1407–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spitale R.C., Torelli A.T., Krucinska J., Bandarian V., Wedekind J.E.. The structural basis for recognition of the PreQ0 metabolite by an unusually small riboswitch aptamer domain. J. Biol. Chem. 2009; 284:11012–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Connelly C.M., Numata T., Boer R.E., Moon M.H., Sinniah R.S., Barchi J.J., Ferré-D’Amaré A.R., Schneekloth J.S.. Synthetic ligands for PreQ1 riboswitches provide structural and mechanistic insights into targeting RNA tertiary structure. Nat. Commun. 2019; 10:1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Banáš P., Sklenovský P., Wedekind J.E., Šponer J., Otyepka M.. Molecular mechanism of preQ1 riboswitch action: a molecular dynamics study. J. Phys. Chem. B. 2012; 116:12721–12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rinaldi A.J., Lund P.E., Blanco M.R., Walter N.G.. The Shine-Dalgarno sequence of riboswitch-regulated single mRNAs shows ligand-dependent accessibility bursts. Nat Comm. 2016; 7:8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lippa G.M., Liberman J.A., Jenkins J.L., Krucinska J., Salim M., Wedekind J.E.. Crystallographic analysis of small ribozymes and riboswitches. Methods Mol. Biol. 2012; 848:159–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kladwang W., Hum J., Das R.. Ultraviolet shadowing of RNA can cause significant chemical damage in seconds. Sci. Rep. 2012; 2:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kleywegt G.J., Brunger A.T.. Checking your imagination: applications of the free R value. Structure. 1996; 4:897–904. [DOI] [PubMed] [Google Scholar]
- 43. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wedekind J.E. Sigel A., Sigel R., Sigel H.. Structural aspects of metal ion binding in small natural and artificial RNA enzymes. Metal Ions in Life Sciences: Structural and Catalytic Roles of Metals in RNA. 2011; 9:Wiley-Blackwell; 299–345. [PubMed] [Google Scholar]
- 45. Leonarski F., D’Ascenzo L., Auffinger P.. Mg2+ ions: do they bind to nucleobase nitrogens. Nucleic Acids Res. 2017; 45:987–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al.. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Cryst. A. 1976; 32:922–923. [Google Scholar]
- 48. Lee B., Richards F.M.. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971; 55:379–400. [DOI] [PubMed] [Google Scholar]
- 49. Myszka D.G. Improving biosensor analysis. J. Mol. Recognit. 1999; 12:279–284. [DOI] [PubMed] [Google Scholar]
- 50. Belashov I.A., Crawford D.W., Cavender C.E., Dai P., Beardslee P.C., Mathews D.H., Pentelute B.L., McNaughton B.R., Wedekind J.E.. Structure of HIV TAR in complex with a lab-evolved RRM provides insight into duplex RNA recognition and synthesis of a constrained peptide that impairs transcription. NucleicAcids Res. 2018; 46:6401–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bradrick T.D., Marino J.P.. Ligand-induced changes in 2-aminopurine fluorescence as a probe for small molecule binding to HIV-1 TAR RNA. RNA. 2004; 10:1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilkinson K.A., Merino E.J., Weeks K.M.. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006; 1:1610–1616. [DOI] [PubMed] [Google Scholar]
- 53. Tijerina P., Mohr S., Russell R.. DMS footprinting of structured RNAs and RNA–protein complexes. Nat. Protoc. 2007; 2:2608–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wilkinson K.A., Merino E.J., Weeks K.M.. RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNA(Asp) transcripts. J. Am. Chem. Soc. 2005; 127:4659–4667. [DOI] [PubMed] [Google Scholar]
- 55. Spitale R.C., Crisalli P., Flynn R.A., Torre E.A., Kool E.T., Chang H.Y.. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013; 9:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Watters K.E., Yu A.M., Strobel E.J., Settle A.H., Lucks J.B.. Characterizing RNA structures in vitro and in vivo with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Methods. 2016; 103:34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aviran S., Trapnell C., Lucks J.B., Mortimer S.A., Luo S., Schroth G.P., Doudna J.A., Arkin A.P., Pachter L.. Modeling and automation of sequencing-based characterization of RNA structure. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:11069–11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dutta D., Belashov I.A., Wedekind J.E.. Coupling green fluorescent protein expression with chemical modification to probe functionally relevant riboswitch conformations in live bacteria. Biochemistry. 2018; 57:4620–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jonsson H., Mills G., Jacobsen K.W.. Berne B.J., Ciccoti G., Coker D.F.. Nudged elastic band method for finding minimum energy paths of transitions. Classical and Quantum Dynamics in Condensed Phase Simulations. 1998; Singapore: World Scientific. [Google Scholar]
- 60. Mills G., Jónsson H.. Quantum and thermal effects in H2 dissociative adsorption: evaluation of free energy barriers in multidimensional quantum systems. Phys. Rev. Lett. 1994; 72:1124. [DOI] [PubMed] [Google Scholar]
- 61. Case D.A., Ben-Shalom I.Y., Brozell S.R., Cerutti D.S., Cheatham I.T.E., Cruzeiro V.W.D., Darden T.A., Duke R.E., Ghoreishi D., Gilson M.K. et al.. AMBER 2018. 2018; San Francisco: University of California. [Google Scholar]
- 62. Bergonzo C., Campbell A.J., Walker R.C., Simmerling C.. A partial nudged elastic band implementation for use with large or explicitly solvated systems. Int. J. Quant. Chem. 2009; 109:3781–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nixon P.L., Rangan A., Kim Y.-G., Rich A., Hoffman D.W., Hennig M., Giedroc D.P.. Solution structure of a luteoviral P1–P2 frameshifting mRNA pseudoknot. J Molec Biol. 2002; 322:621–633. [DOI] [PubMed] [Google Scholar]
- 64. Izadi S., Anandakrishnan R., Onufriev A.V.. Building water models: a different approach. J. Phys. Chem. Lett. 2014; 5:3863–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schmit J.D., Kariyawasam N.L., Needham V., Smith P.E.. SLTCAP: A simple method for calculating the number of ions needed for MD simulation. J. Chem. Theory Comput. 2018; 14:1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang J., Cieplak P., Kollman P.A.. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules. J. Computat. Chem. 2000; 21:1049–1074. [Google Scholar]
- 67. Perez A., Marchan I., Svozil D., Sponer J., Cheatham T.E. 3rd, Laughton C.A., Orozco M.. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007; 92:3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zgarbová M., Otyepka M., Sponer J., Mládek A., Banáš P., Cheatham T.E. 3rd, Jurečka P.. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 2011; 7:2886–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li P., Song L.F., Merz K.M. Jr. Systematic parameterization of monovalent ions employing the nonbonded model. J. Chem. Theory Comput. 2015; 11:1645–1657. [DOI] [PubMed] [Google Scholar]
- 70. Panteva M.T., Giambasu G.M., York D.M.. Force field for Mg2+, Mn2+, Zn2+, and Cd2+ ions that have balanced interactions with nucleic acids. J. Phys. Chem. B. 2015; 119:15460–15470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Darden T., York D., Pedersen L.. Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993; 98:10089–10092. [Google Scholar]
- 72. Salomon-Ferrer R., Götz A.W., Poole D., Le Grand S., Walker R.C.. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013; 9:3878–3888. [DOI] [PubMed] [Google Scholar]
- 73. Ryckaert J.P., Ciccotti G., Berendsen H.J.C.. Numerical-integration of cartesian equations of motion of a system with constraints - molecular-dynamics of N-Alkanes. J. Comput. Phys. 1977; 23:327–341. [Google Scholar]
- 74. Mathews D.H., Case D.A.. Nudged elastic band calculation of minimal energy paths for the conformational change of a GG non-canonical pair. J. Mol. Biol. 2006; 357:1683–1693. [DOI] [PubMed] [Google Scholar]
- 75. Romo T.D., Leioatts N., Grossfield A.. Lightweight object oriented structure analysis: tools for building tools to analyze molecular dynamics simulations. J. Comput. Chem. 2014; 35:2305–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Klein D.J., Edwards T.E., Ferré-D’Amaré A.R.. Cocrystal structure of a class I preQ1 riboswitch reveals a pseudoknot recognizing an essential hypermodified nucleobase. Nat. Struct. Mol. Biol. 2009; 16:343–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kang M., Peterson R., Feigon J.. Structural Insights into riboswitch control of the biosynthesis of queuosine, a modified nucleotide found in the anticodon of tRNA. Mol. Cell. 2009; 33:784–790. [DOI] [PubMed] [Google Scholar]
- 78. Jones A.C., Neely R.K.. 2-Aminopurine as a fluorescent probe of DNA conformation and the DNA-enzyme interface. Q. Rev. Biophys. 2015; 48:244–279. [DOI] [PubMed] [Google Scholar]
- 79. Frener M., Micura R.. Conformational rearrangements of individual nucleotides during RNA-ligand binding are rate-differentiated. J. Am. Chem. Soc. 2016; 138:3627–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gherghe C.M., Shajani Z., Wilkinson K.A., Varani G., Weeks K.M.. Strong correlation between SHAPE chemistry and the generalized NMR order parameter (S2) in RNA. J. Am. Chem. Soc. 2008; 130:12244–12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. McGinnis J.L., Dunkle J.A., Cate J.H., Weeks K.M.. The mechanisms of RNA SHAPE chemistry. J. Am. Chem. Soc. 2012; 134:6617–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Aytenfisu A.H., Liberman J.A., Wedekind J.E., Mathews D.H.. Molecular mechanism for preQ1-II riboswitch function revealed by molecular dynamics. RNA. 2015; 21:1898–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lund P.E., Chatterjee S., Daher M., Walter N.G.. Protein unties the pseudoknot: S1-mediated unfolding of RNA higher order structure. Nucleic Acids Res. 2020; 48:2107–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Porter E.B., Marcano-Velázquez J.G., Batey R.T.. The purine riboswitch as a model system for exploring RNA biology and chemistry. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2014; 1839:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Matsuda D., Dreher T.W.. The tRNA-like structure of Turnip yellow mosaic virus RNA is a 3′-translational enhancer. Virology. 2004; 321:36–46. [DOI] [PubMed] [Google Scholar]
- 86. Rieder U., Kreutz C., Micura R.. Folding of a transcriptionally acting preQ1 riboswitch. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:10804–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Liberman J.A., Suddala K.C., Aytenfisu A., Chan D., Belashov I.A., Salim M., Mathews D.H., Spitale R.C., Walter N.G., Wedekind J.E.. Structural analysis of a class III preQ1 riboswitch reveals an aptamer distant from a ribosome-binding site regulated by fast dynamics. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E3485–E3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Edwards T.E., Ferre-D’Amare A.R.. Crystal structures of the thi-box riboswitch bound to thiamine pyrophosphate analogs reveal adaptive RNA-small molecule recognition. Structure. 2006; 14:1459–1468. [DOI] [PubMed] [Google Scholar]
- 89. Thore S., Leibundgut M., Ban N.. Structure of the eukaryotic thiamine pyrophosphate riboswitch with its regulatory ligand. Science. 2006; 312:1208–1211. [DOI] [PubMed] [Google Scholar]
- 90. Weiss M.S. Global indicators of X-ray data quality. J. Appl. Crystallogr. 2001; 34:130–135. [Google Scholar]
- 91. Karplus P.A., Diederichs K.. Linking crystallographic model and data quality. Science. 2012; 336:1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. McCarty R.M., Bandarian V.. Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin. Chem. Biol. 2008; 15:790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Peselis A., Serganov A.. Structure and function of pseudoknots involved in gene expression control. Wiley Interdiscip. Rev. RNA. 2014; 5:803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Leontis N.B., Westhof E.. Geometric nomenclature and classification of RNA base pairs. RNA. 2001; 7:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic coordinates and structure factor amplitudes have been deposited into the Protein Data Bank as entry 6vuh (effector free) and entry 6vui (preQ1 bound). Sequence reads for SHAPE-seq and DMS-seq were deposited into the Sequence Read Archive hosted by NCBI/NIH with accession codes: SRX7726568, SRX7726569, SRX7726570, SRX7726571, SRX7726572 and SRX7726573.