

Abstract
Objective
Increased breathing rate, apnea and respiratory failure is associated with sudden unexpected death in epilepsy (SUDEP). We recently demonstrated the progressive nature of epilepsy and mortality in Kcna1−/− mice a model of temporal lobe epilepsy and SUDEP. Here, we tested the hypothesis that respiratory dysfunction progresses with age in Kcna1−/− mice, thus increasing risk of respiratory failure and sudden death (SD).
Methods
Respiratory parameters were determined in conscious mice at baseline and following increasing doses of methacholine (MCh) using non-invasive airway mechanics (NAM) systems. Kcna1+/+, Kcna1+/− and Kcna1−/− littermates were assessed during three age ranges when up to ~30%, ~55% and ~90%of Kcna1−/− mice have succumbed to SUDEP: postnatal day (P) 32-36, P40-46 and P48-56, respectively. Saturated arterial O2 (SaO2) was determined with pulse oximetry. Lung and brain tissues were isolated and Kcna1 gene and protein expression were evaluated by RT-qPCR and Western blot techniques. Airway smooth muscle responsiveness was assessed in isolated trachea exposed to MCh.
Results
Kcna1−/− mice experienced an increase in basal respiratory drive, chronic oxygen desaturation, frequent apnea-hypopnea (A-H), an atypical breathing sequence of A-H-tachypnea-A-H, increased tidal volume and hyperventilation induced by MCh. The MCh-provoked hyperventilation was dramatically attenuated with age. Interestingly, only Kcna1−/− mice developed seizures following exposure to MCh. Seizures were provoked by lower concentrations of MCh as Kcna1−/− mice approached sudden death (SD). MCh-induced seizures experienced by a subset of younger Kcna1−/− mice triggered death. Respiratory parameters of these younger Kcna1−/− mice resembled older near-SD Kcna1−/− mice. Kcna1 gene and protein were not expressed in Kcna1+/+ and Kcna1+/− lungs and MCh-mediated airway smooth muscle contractions exhibited similar EC50’s in isolated Kcna1+/+ and Kcna1−/− trachea.
Significance
The Kcna1−/− model of SUDEP exhibits progressive respiratory dysfunction which suggests a potential increased susceptibility for respiratory failure during severe seizures that may result in sudden death.
Keywords: Kv1.1 knockout, mortality, survival, hypopnea, seizures, apnea
Introduction
The lifetime incidence of epilepsy is 1:26 people.1 Approximately one third of people with epilepsy have seizures that are refractory to current anti-seizure drugs and the estimated risk for Sudden Unexpected Death in Epilepsy (SUDEP) in the refractory population is dramatically increased from 1:1000 to ~1:150 people each year.2,3 The pathological mechanisms mediating SUDEP events are not understood, however, risk factors include uncontrolled seizures, generalized tonic-clonic (GTC) seizures, age and cardiac arrhythmias.2,4–7
The mechanisms underlying SUDEP are elusive primarily because it is rarely clinically witnessed. Of these witnessed cases, the majority report a partial or GTC seizure,4–7 which is associated with dysfunctional respiratory response reflexes8, lowered blood oxygen saturation followed by a period of increased breathing rate, cardiac arrhythmia/bradycardia, apnea, respiratory arrest and asystole.4–7 Although it is clear that respiratory arrest contributes to the terminal SUDEP event, it is unknown whether a progressive respiratory dysfunction develops in susceptible individuals that may contribute to the pathology that culminates in death.
The Kcna1 gene encodes for the Kv1.1 alpha subunit of the delayed rectifier potassium channel, which participates in the repolarization of the neuronal membrane potential, action potential properties, neurotransmitter release, synaptic integration and network oscillatory behavior.9,10 Kcna1-null (Kcna1−/−) mice develop spontaneous recurrent seizures including myoclonic seizures, clonus and generalized tonic-clonic seizures around the third week of their life.9,11,12 We have recently demonstrated that seizures progressively become more frequent and severe until the mice die at the early age of postnatal day (P) 42 ± 1.312, although the background strain of the mouse influences the severity of seizures and mortality.11 A genetic variant in Kcna1 has been identified in a clinical SUDEP case.13 Kcna1−/− mice phenocopy human SUDEP and experience a GTC seizure, cardiac arrhythmias and asystole prior to death.14 While chemically-provoked seizures elicited minimal cardiorespiratory effects in wild type mice, similarly provoked seizures in Kcna1−/− mice often result in peri-ictal apneas, decreased blood oxygen saturation and sudden spreading depolarization in the dorsal medulla followed by death.15 These studies indicate that the respiratory responses of Kcna1−/− mice ultimately fail following the final GTC. Here, we tested the hypothesis that Kcna1−/− mice exhibit progressive respiratory dysfunction which is associated with age-dependent susceptibility to sudden death (SD).
Material and Methods
Animals
Kcna1-null mice on a C3HeB/FeJ congenic background were bred and housed in the Animal Resource Facilities at Creighton University School of Medicine in a temperature (25°C) and humidity (50-60%) controlled, pathogen-free environment on a 12-hr light/dark cycle. Food and water were provided ad libitum. Male and female mice were used. No gender differences were detected in this study. Genotype was determined (Transnetyx Inc., Cordova, TN, U.S.A.). Kcna1−/− mice seize and thus experiments are not blinded to the experimenter. Procedures were in accordance with National Institutes of Health guidelines, the EU Directive 2010/63/EU and were approved by the Institutional Animal Care and Use Committee at Creighton University School of Medicine.
Respiratory Measurements
Non-invasive airway mechanics techniques were used to measure respiratory parameters in non-anesthetized mice (NAM; Buxco Finepointe/DSI Inc, Minneapolis, MN, USA). Kcna1+/+, Kcna1+/− and Kcna1−/− littermates were assessed at three age bins: P32-36, P40-46 and P48-56 when up to ~30%, ~55% and ~90% of Kcna1−/− have succumbed to SUDEP.12 Mice were allowed to acclimate in the NAM chamber for 10-15 min, exposed to nebulized phosphate buffered saline (PBS), then challenged with increasing doses of MCh (6, 12, 24, 48, 100 and 200 mg ml−1, 45μl for 30 sec). Each challenge was followed immediately by a 3-min recording period (Buxco FinePointe acquisition software V2.3.1.9). The independent measurement of both nasal and thoracic air flow allows the NAM to measure several more respiratory parameters than whole body plethysmography. Accordingly, peak expiratory flow (PEF), peak inspiratory flow (PIF), time of inspiration (Ti), time of expiration (Te), frequency of breathing (f), minute ventilation (VE), tidal volume (VT), respiratory drive (VT/Ti) and specific airway resistance (sRaw) were determined. To account for varying body weights, f, PIF, PEF, VE and VT were normalized to individual animals’ weights.
Seizure Monitoring
The NAM chamber physically restricted the ability to conduct electroencephalography (EEG) recordings, therefore only convulsive seizures were quantified. Seizures were video-monitored during recording sessions and continued for 20-30 min after removal from the NAM. Seizures were scored using a modified Racine scale (0-normal; 1-immobility; 2-head bobbing; 3-forelimb/hindlimb clonus and tail extension; 4- generalized clonus with forelimb tonic extension; 5-severe tonic-clonic seizures with hindlimb tonic extension) as we have previously described.11,12,16,17
Apnea/Hypopnea Analysis
Respiratory cycles were determined for each animal. The duration of two respiratory cycles was calculated for each individual animal at baseline using the equation: (frequency of breaths per min/60 sec) * 2. An apnea event was defined as the decrease of airflow ≥ 90% for a period ≥ two complete respiratory cycles. Hypopnea was defined as the reduction of airflow ≥ 30% for a period ≥ two complete respiratory cycles (i.e. shallow breathing).
Oximetry
Blood oxygen saturation was measured noninvasively using MouseOx Plus infrared pulse oximetry (Starr Life Sciences Corp, Oakmont, PA, USA). The hair around the neck was removed and a CollarClip Sensor (Starr Life Sciences Corp) was fitted in Kcna1+/+ and Kcna1−/− mice (P40-42). Eight parameters including oxygen saturation (SaO2), pulse rate and respiratory rate were acquired at 5 Hz and mice were recorded for 1 hr. Only measurements that were error-free for all parameters were included in the analyses. Error-free measurements proved more difficult to obtain for Kcna1−/− mice resulting in a lower, but reasonable, number of usable data points compared to Kcna1+/+ mice (1411 ± 600 vs. 2939 ± 214, n = 4, p = 0.079, unpaired t-test). The total number of measurements for individual Kcna1−/− mice did not correlate with the SaO2 (R2 = 4×10−6, p = 0.998, Pearson correlation coefficient) or the percent of measurements considered hypoxic (< 90% SaO2) (R2 = 0.037, p = 0.81) indicating that the difference in error-free measurements between Kcna1+/+ and Kcna1−/− mice did not skew the results. State of vigilance was video recorded and scored as active or rest (immobile and eyes closed) by a trained technician.
Assessment of airway smooth muscle reactivity
Kcna1+/+ and Kcna1−/− mice (P39-41) were sacrificed, tracheas were removed and placed in Krebs’ solution (composition in mM: NaCl-120; KCl-5.5; CaCl2-2.5; NaH2PO4-1.4; MgCl2-1.2; NaHCO3-25; dextrose-11.1; CaNa2-EDTA-0.027) equilibrated with 95% O2-5% CO2 at pH 7.4. Tracheas were cut into 2-mm ring segments, mounted via 2 stainless steel pins through the lumen, and placed in water-jacketed glass muscle chambers containing Krebs’ solution maintained at 37°C and gassed with 95% O2-5% CO2, pH 7.4. One pin was attached to a Grass FT.03 force transducer (Quincy, MA) for measurement of isometric contraction. After 1-hr equilibration at 300 mg resting tension, ring segments were challenged with a maximal contractile concentration (60 mM) of KCl, washed for 15 min. This was repeated until the response was within 10% of the previous contraction. After 30 min of washing, cumulative concentration-response curves for MCh-induced contraction were obtained. Airway reactivity was determined as the contractile sensitivity to MCh, which was quantified as the half-maximal effective concentration (EC50) for MCh-induced contraction as previously described.
Reverse transcriptase qPCR
Tissue was homogenized in Trizol, RNA was extracted via phenol chloroform, treated with DNase, suspended in 30μL of nuclease free H2O and quantified (NanoDrop 2000 spectrophotometer, Thermo Scientific, USA). cDNA synthesis (4μg) was completed using Improm II reverse transcriptase kit (Promega, Madison, WI) and a C1000™ Thermo Cycler (25°C-5 min, 42°C-60 min and 72°C-15 min, Bio-Rad Labs, Hercules, CA). Quantitative PCR was performed for Kcna1 and housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (100 ng), 10μl SYBR green PCR master mix (Agilent Technologies, Santa Clara, CA) and 1μl of forward and 1μl of reverse primers (10 pmol/μl) (Integrated DNA Technologies, Coralville, IA), 7.9μl nuclease free water, mixed together and using a AriaMx Real Time PCR instrument (Agilent Technologies, Santa Clara, CA). Primers14 were Kcna1 forward, 5’-GCATCGACAACACCACAGTC-3’; Kcna1 reverse, 5’-CGGCGGCTGAGGTCACTGTCAGAGGCTAAGT-3’; Gapdh forward, 5’-TGAAGGTCGGTGTGAACGGATTT GGC-3’; Gapdh reverse, 5’-ATGTAGGCCATGAGGTCCACCAC-3’ (IDT’s Primer Quest software2). Cycling conditions: 95°C-10min, 40cycles of 95°C-30sec, 72°C-30sec. Each real-time PCR was performed using three biological samples in technical triplicates and melt curve analyses were completed to ensure the specificity of amplification. Calculations of the Kcna1 gene expression relative to Gapdh were based on the differences in threshold cycles using the 2– ΔΔCt method.18
Immunofluorescent western blot
Mouse brain and lungs were isolated and flash frozen in liquid nitrogen. Tissue was homogenized in buffer (in mM: 4-HEPES pH-7.4, 320-sucrose, 2-EGTA, 10-Na pyrophosphate, 1-Na orthovanadate, 10-NaF, 0.1-PMSF, phosphatase inhibitor cocktail). Following centrifugation, protein (15μg) (in Laemmli loading buffer and β-mercaptoethanol) was separated by electrophoresis (12% Mini-PROTEAN TGX gels; Biorad, Hercules, CA), transferred to Immobilon-FL PVDF membranes (EMD Millipore, Billerica, MA, U.S.A.) and blocked with Odyssey blocking buffer (OBB; Li-Cor Biosciences, Lincoln, NE, U.S.A.)/PBS at a 1:1 ratio. Membranes were incubated overnight with primary antibodies rabbit anti-Kv1.1 (1:1000; ab32433, Abcam) or mouse anti-beta actin (1:8000; 926-42212 LiCor Biosciences) at 4°C. Following PBS-T washes, membranes were incubated in secondary antibodies for one hour: goat anti-rabbit (1:5,000; 926-32221, Li-Cor Biosciences) or goat anti-mouse (1:20,000-1:40,000; 926-32210, Li-Cor Biosciences). Samples were run in duplicates, images were captured on an Odyssey FC (Licor, Lincoln, NE). Kv1.1 protein signal was normalized to β-actin values.
Reagents and Statistical Analysis
Unless otherwise specified, all reagents were purchased from Sigma-Aldrich. Data are presented as the mean ± S.E. Statistical significance was determined with Prism6 software (Graphpad Software Inc., La Jolla, CA, U.S.A.) using either an unpaired t-test, one-way ANOVA or two-way ANOVA for genotype and age with an appropriate post hoc test. EC50 values for MCh were calculated from concentration-response curves by nonlinear regression analysis.
Results
Kcna1−/− mice have increased respiratory drive and blood oxygen desaturation with increased probability of SD
We determined respiratory endpoints during three age ranges: P32-36, P40-46 and P48-56 when up to ~30%, ~55% and ~90%, respectively, of Kcna1−/− mice have succumbed to SD12. For simplicity, we refer to these groups as SD30, SD55 and SD90. Most baseline breathing parameters were similar. The most notable change occurred in the SD90 Kcna1−/− mice cohort which had the highest probability of imminent SD at the time of the respiratory experiments12. VT was significantly increased in SD90Kcna1−/− mice when compared to age-matched Kcna1+/+ controls (p < 0.01; Kcna1+/− resembled Kcna1+/+; Figure 1A). Also as the probability of SD rose, Kcna1−/− respiratory drive (VT/Ti) and peak inspiratory flow was significantly increased (p < 0.01 and p < 0.05, respectively; Figure 1B and C).
Figure 1. Basal respiration and chemoresponsive instability in Kcna1−/− mice.
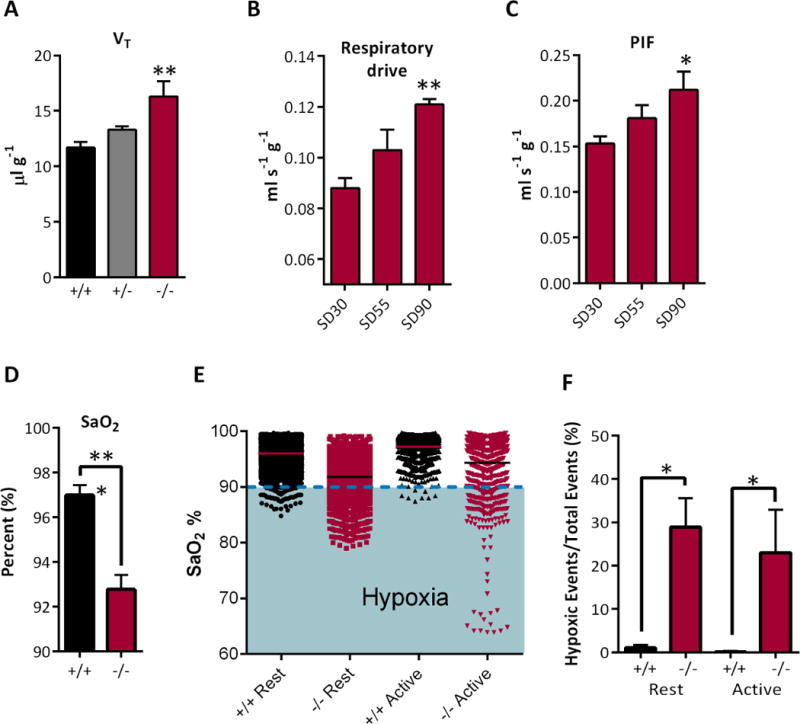
A) Tidal volume (VT) is increased in SD90 Kcna1−/− mice (F (2,14) = 6.9). (B) As Kcna1−/− mice age respiratory drive (VT/Ti: F (2,14) = 8.9) and (C) PIF (F (2,14) = 3.5) are elevated. Significance was determined by two way ANOVA with Dunnett’s multiple comparisons test, n = 5-7. (D) Pulse oximetry indicates that arterial oxygen saturation (SaO2) is lower in SD55 Kcna1−/− mice compared to age matched Kcna1+/+ mice (data expressed as mean per animal ± SEM, n = 4 mice). (E) Plotting all SaO2 measurements indicates oxygen desaturation during different behavioral states by ~4% and illustrates frequent excursions into hypoxia when SaO2 < 90%. (F) Quantification of the percentage of measurements with SaO2 < 90%, unpaired Mann-Whitney test; n = 8, *p < 0.05, **p < 0.01, *** p < 0.001.
An increase in respiratory drive can be triggered by blood gas instability. To assess stability we measured SaO2 by oximetry over one hour in freely moving mice. SaO2 was reduced by ~4% in SD55 Kcna1−/− mice when compared to age-matched Kcna1+/+ mice (92.8 ± 0.7% vs. 97 ± 0.5%, n = 4, p < 0.001) (Figure 1D). SaO2 was reduced by a similar degree during both rest and activity. Plotting all of the raw data revealed that a large number of Kcna1−/− measurements were hypoxic (i.e. SaO2 drops below 90%) (Figure 1E). The percent of hypoxic measurements was determined for each subject. Kcna1+/+ mice rarely experienced hypoxia (with only 0.6 ± 0.3% of measurements having SaO2 < 90%), whereas 26 ± 0.7% of the measurements from Kcna1−/− mice were considered hypoxic (n = 4, Figure 1F). These data suggest that Kcna1−/− mice experience frequent hypoxia.
Apnea and hypopnea is severe in Kcna1−/− mice
Hypoxia can be caused by apnea or hypopnea (referred to as A-H). A-H frequently occur during/after generalized tonic-clonic seizures and during the final events leading up to SUDEP in humans and in the Kcna1−/− model.6,15 We hypothesized that Kcna1−/− mice are more prone to A-H events at ages with increased probability SD. Raw traces of eupnea, apnea and hypopnea are depicted in Figure 2A-C, respectively. In the SD30 group, there were no differences in the number of apnea events among genotypes. A trend emerged in the SD55 group (p = 0.09; Kcna1+/+ vs. Kcna1−/−). SD90 Kcna1−/− mice experienced significantly more apnea events when compared to age-matched Kcna1+/+ and Kcna1+/− littermates (Figure 2D). Interestingly, hypopnea was highest in the SD30 Kcna1−/− cohort and significantly diminished with age (Figure 2E). A-H rarely occurred in Kcna1+/+ and Kcna1+/− mice. Kcna1−/− mice spent 8-18% of time monitored in A-H, compared with only 1-4% for Kcna1+/+ and Kcna1+/− mice (Figure 2F). It is important to note that these measurements occurred in the absence of seizures. These data suggest a critical period exists and the potential conversion of hypopnea events to apnea may indicate a dysfunction in circuitry and systems responsible for preventing apnea onset.
Figure 2. Kcna1−/− mice experience more apnea and hypopnea events.
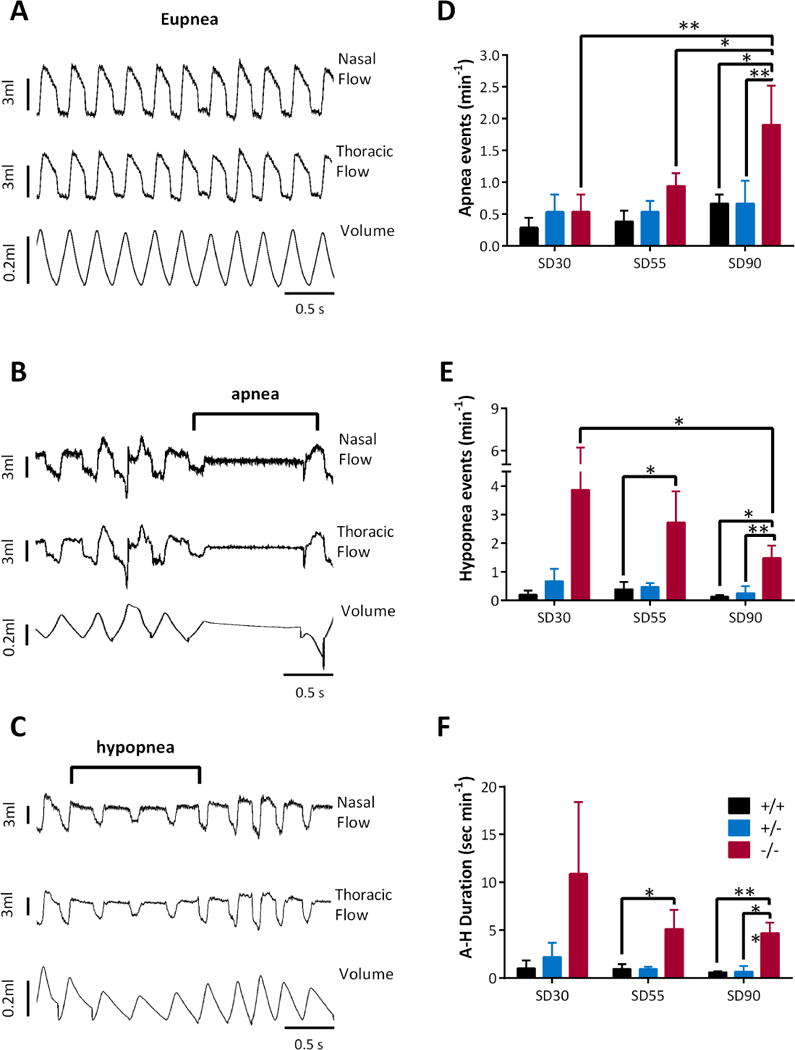
Example traces of (A) normal breathing (eupnea), (B), apnea and (C) hypopnea. Kcna1−/− mice display (D) more apnea events and (E) fewer hypopnea events as they age. (F) Kcna1−/− mice spend significantly more time in apnea-hypopnea (A-H). Significance was determined by a two-way ANOVA for age and genotype followed by Holm-Sidak’s multiple comparisons test, n = 4-7, * p < 0.05, ** p < 0.01.
Respiratory responses surrounding A-H events are dysregulated in Kcna1−/− mice
We examined the respiration parameters prior to and following each A-H event. We were able to detect bronchoconstriction prior to approximately half of all A-H events, as indicated by an increase in sRaw values (Kcna1+/+ 21 ± 4%; Kcna1+/− 42 ± 10%; Kcna1−/− 33 ± 3% higher than baseline sRaw). Irrespective of whether bronchoconstriction occurred, we classified three distinct respiratory patterns surrounding all A-H events (Types 1-3). The incidence of each Type was similar across ages (data not shown) and differed among genotypes. As depicted in Figure 3A, Type 1 A-H events were preceded by tachypnea (rapid breathing and increased VT), and were then followed by eupnea (normal breathing). Type 2 A-H events were preceded and followed by eupnea (Figure 3B) and Type 3 events were preceded by eupnea then followed by tachypnea (Figure 3C). A majority of Kcna1+/+ and Kcna1+/− A-H events were Type 1 (29/33 Kcna1+/+ and 32/35 Kcna1+/− events; Figure 3D).
Figure 3. All genotypes display three types of Apnea-Hypopnea events.
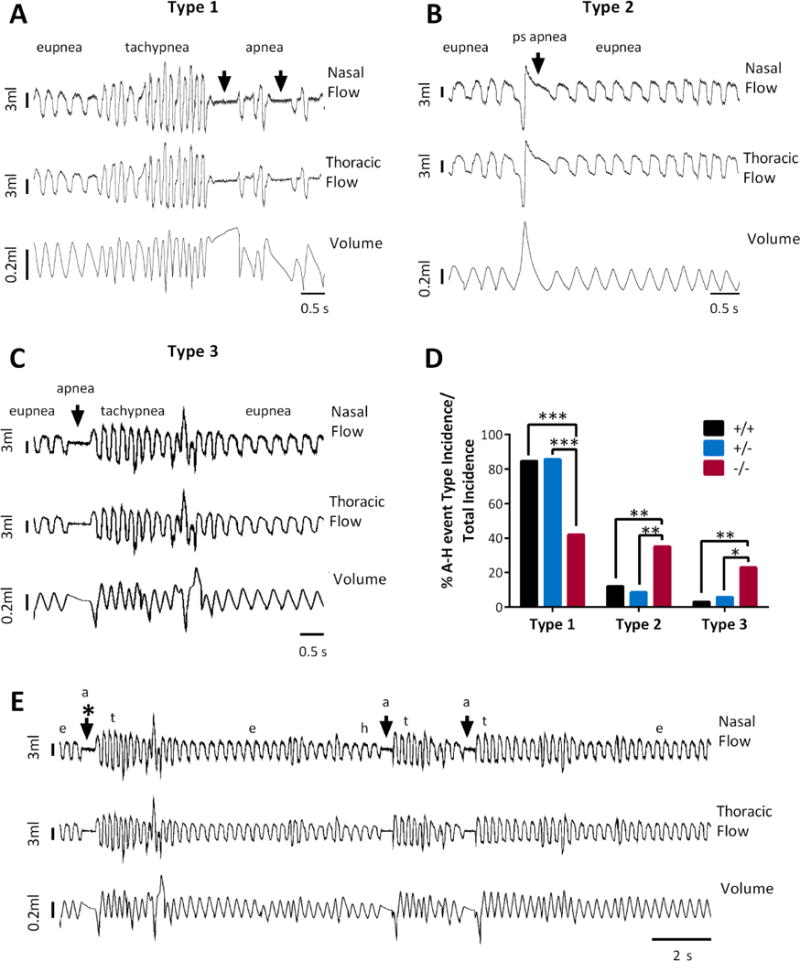
(A) An example of a Type 1 event which begins with eupnea, then tachypnea, then apnea or hypopnea. (B) Type 2 events are preceded and followed by eupnea. Shown is an example of a post sigh (ps) apnea. (C) Type 3 events are preceded by eupnea and followed by tachypnea. (D) In comparison to Kcna1+/+ and Kcna1+/− mice, Kcna1−/− mice had decreased Type 1 events and increased Type 2 and 3 events. Significance determined by Fisher’s exact test, * p < 0.05, ** p < 0.01, *** p < 0.001. (E) Example trace of sequential A-H events. Eupnea (e), apnea (a), tachypnea (t). Arrows indicate apnea events. Asterisk indicates the Type 3 event shown in C on a shorter time scale.
In contrast, Knca1−/− mice had two differences in their A-H phenotypes. Knca1−/− mice had significantly fewer Type 1 A-H events (42%) and a greater number of Types 2 and 3 A-H events (35% and 23% of events, respectively; Figure 3D). Interestingly, in Kcna1−/− mice it was not uncommon to observe an A-H event that was either preceded or followed by tachypnea, to then be followed by another A-H event, in a repeating pattern (Figure 3E). Collectively, these data suggest that Kcna1−/− mice are more prone to A-H events and the typical respiratory responses surrounding apnea events may be dysregulated or these responses are compensatory in Kcna1−/− mice.
MCh paradoxically provokes hyperventilation and seizures in Kcna1−/− mice
Respiratory endpoints were assessed following exposure to increasing doses of nebulized MCh, a muscarinic cholinergic receptor agonist that induces bronchoconstriction. To compare responses, we normalized each response to its corresponding PBS baseline. Kcna1+/+ and Kcna1+/− mice responded similarly with hypoventilation across all ages: indicated by reduced breathing rates, tidal volume, minute ventilation, peak inspiratory and expiratory flow and respiratory drive; while inspiration time, expiration time and specific airway resistance were increased. Because there were no differences across ages, data were combined across age for both Kcna1+/+ and Kcna1+/− mice for each endpoint (depicted in grey in Figure 4).
Figure 4. Paradoxical hyperventilation provoked by MCh attenuates with age in Kcna1−/− mice.
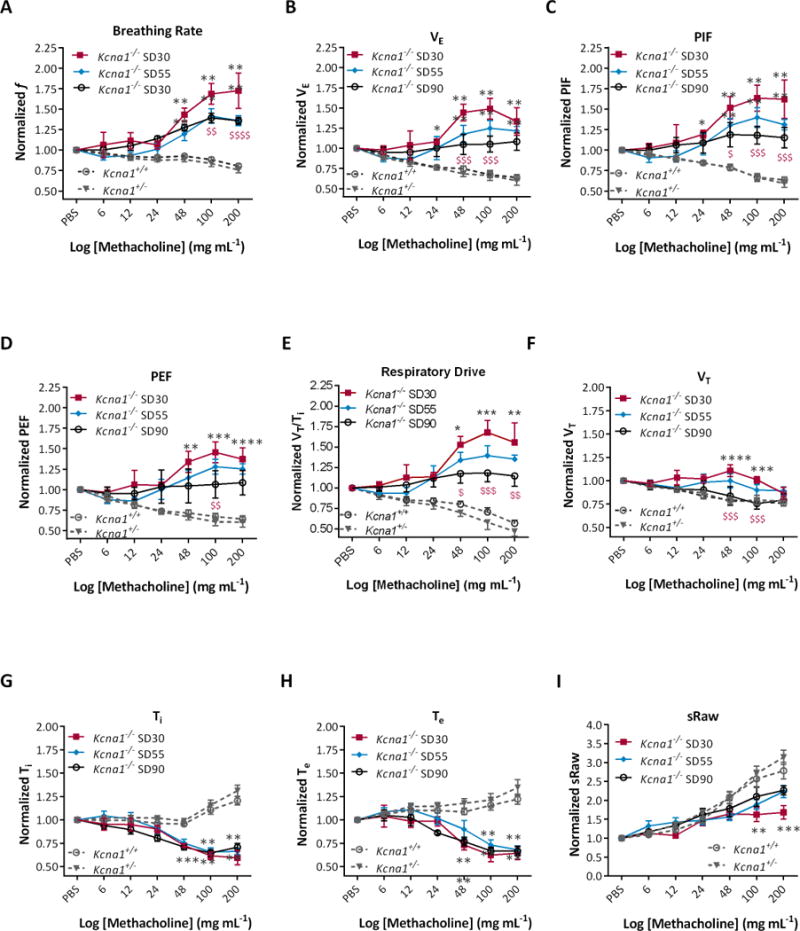
In Kcna1−/− mice, exposure to increasing concentrations of nebulized increased (A) respiration rate (f: F (2,203) = 105.5, p < 0.0001), (B) minute ventilation (VE: F (2,203) = 64.3, p < 0.0001), (C) peak inspiratory flow (PIF: F (2,203) = 73.9, p < 0.0001), (D) peak expiratory flow (PEF F (2,203) = 41.2, p < 0.0001) and (E) respiratory drive (VT/Ti: F (2,203) = 31.9, p < 0.0001); and decreased (F) tidal volume (VT: F (2,203) = 22.5, p < 0.0001), (G) inspiratory time (Ti: F (2,203) = 54.8, p < 0.0001) and (H) expiratory time (Te: F (2,203) = 41.3, p < 0.0001), with (I) little effect on specific airway resistance (sRaw: F (2,203) = 5.7, p < 0.005). Significance was determined by two-way ANOVA with repeated measures followed by Tukey’s multiple comparisons test; n = 5-7, grey * p < 0.05, ** p < 0.01, *** p < 0.001 indicates P32-36 and P48-56 Kcna1−/− cohorts differ from Kcna1+/+. Red $ p < 0.05, $$ p < 0.01, $$$ p < 0.001, $$$$ p < 0.0001 indicates P48-56 differs from P32-36 Kcan1−/− mice.
In contrast, MCh provoked hyperventilation in Kcna1−/− mice: indicated by increased breathing rate, minute ventilation, peak inspiratory and expiratory flow and respiratory drive, along with a decrease in inspiration time and expiration time (Figure 4A-I). Interestingly, the overall response was attenuated as Kcna1−/− mice aged. MCh provoked seizures in only Kcna1−/− mice in a concentration-dependent manner. All Kcna1−/− mice experienced seizures by 200 mg ml−1 MCh (Figure 5A).
Figure 5. Seizures are triggered by lower MCh concentrations in SD90 Kcna1−/− mice.
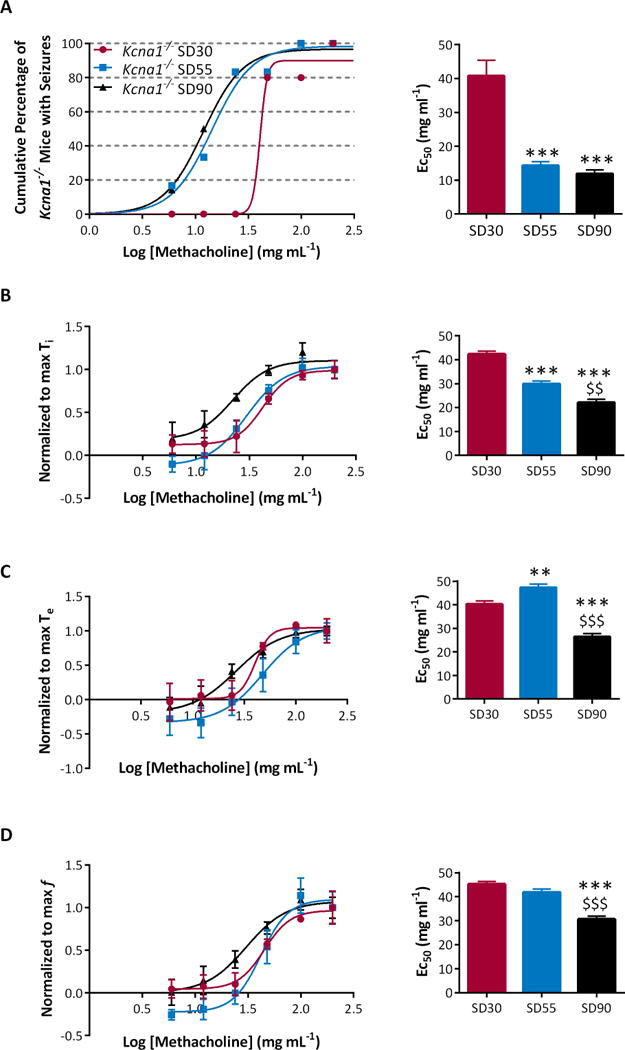
(A) MCh induced seizures at lower concentrations in older Kcna1−/− mice. (B) MCh concentration response curves exhibited a leftward shift in SD90 Kcna1−/− mice for inspiratory time (Ti), (C) expiratory time (Te), and (D) and respiration rate (f). This resulted in significantly reduced MCh EC50’s (right). Significance was determined by a one-way ANOVA followed by Tukey’s multiple comparisons test, ** p < 0.01, *** p < 0.001 v. P32-36 and $$ p < 0.01, $$$ p < 0.001 v. P42-46, n = 5-6.
Kcna1−/− mice become more sensitive to MCh-induced tachypnea and seizures with age
Kcna1−/− mice developed greater sensitivity to MCh as they aged. The EC50 for seizure induction in the SD30 group was 40.8 mg ml−1. This significantly decreased to 11.9 mg ml−1, representing a 71% overall reduction with age (p < 0.0001; Figure 5A). In the SD30 cohort, the EC50s for inspiration time, expiration time and breathing rate were ~40-45 mg ml−1 and decreased 35-50% as mice approached SD (p < 0.0001, p < 0.0001, p < 0.0001; Figure 5B-D).
MCh-induced seizures resulted in the death of one SD30 and two SD55 Kcna1−/− mice. These three mice died at P36, P45 and P46 after experiencing severe seizures. This presented the opportunity to determine whether these susceptible mice had distinguishing respiratory characteristics, the hypothesis being that they would resemble the SD90 cohort. One baseline parameter that distinguished SD90 Kcna1−/− mice from age-matched Kcna1+/+ and Kcna1+/− mice was tidal volume. The tidal volume of the Kcna1−/− mice that died young was 15.0 ± 1.3 μl g−1, resembling the SD90 cohort (16.3 ± 1.4 μl g−1). In addition, the sensitivity to MCh between the two cohorts were similar. The EC50s for expiration time was 24.9 ± 1.3 and for breathing rate was 34.7 ± 1.3 mg ml−1, similar to the SD90 group (26.4 ± 1.3 and for f was 30.6 ± 1.3 mg ml−1). Collectively, these data indicate that Kcna1−/− mice at higher risk of SUDEP have greater respiratory sensitivity to MCh.
Kv1.1 is not expressed in lung tissue and does not participate in airway smooth muscle reactivity
Kcna1 expression in lung tissue was investigated to determine the potential contribution of Kv1.1 to the intrinsic function of this organ. We used brain tissue as a positive control. RT-qPCR and western blots revealed that Kcna1 mRNA and protein were highly expressed in both Kcna1+/+ and Kcna1+/− brain (Figure 6A, B). The loss of one allele resulted in a ~70% reduction of mRNA and ~60% reduction of protein in Kcna1+/− brain. Kcna1 mRNA or protein were not detectable in Kcna1+/+ or Kcna1+/− lung tissue. There was no Kcna1 mRNA or protein in Kcna1−/− brain or lung.
Figure 6. Kcna1 mRNA and protein is not present in lung, and does not participate in airway smooth muscle reactivity to MCh.
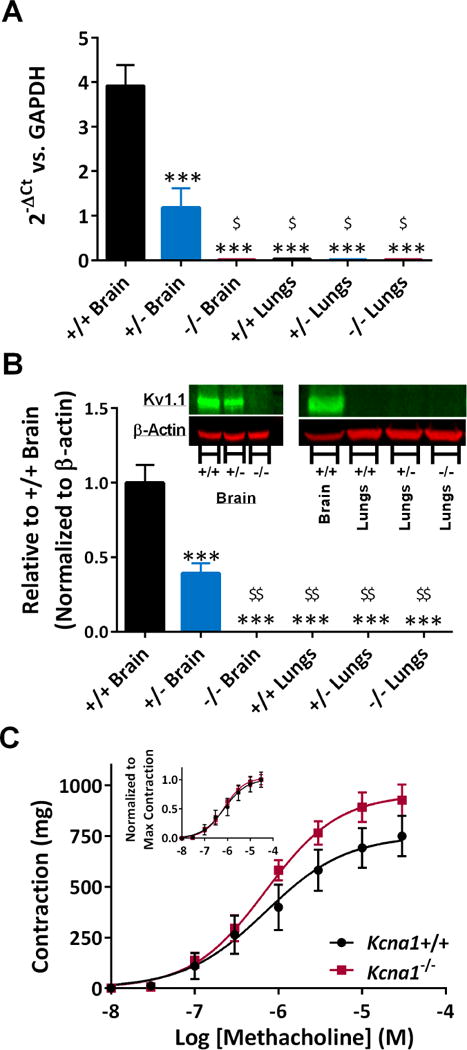
Kcna1 (A) mRNA and (B) protein were expressed to ~30-40% of wild-type levels in Kcna1+/− brain tissue and absent in Kcna1−/− mice brains. In contrast, Kcna1 mRNA and protein were not detected in lung tissue from all three genotypes. Significance was determined by a two-way ANOVA for tissue and genotype followed by Tukey’s multiple comparisons test, *** p < 0.001 v. Kcna1+/+ and $ p < 0.05, $$ p < 0.01 v. Kcna1+/−, n = 3 per genotype. (C) Concentration-response curves demonstrating the maximal contractile response and the sensitivity to MCh of isolated trachea did not differ between Kcna1+/+ and Kcna1−/− mice (n = 4). Inset, concentration response curves normalized to the maximum contraction for each genotype. Significance was determined by a two-way ANOVA repeated measures for MCh concentration and genotype followed by Tukey’s multiple comparisons test.
In vitro experiments with isolated trachea from Kcna1+/+ and Kcna1−/− mice were conducted to determine whether airway smooth muscle responsiveness to increasing concentrations of MCh differed between genotypes. Mean concentration-response curves for MCh-stimulated contraction of isolated tracheas were constructed and EC50 values calculated (Figure 6C). Kcna1+/+ and Kcna1−/− trachea had similar EC50s (0.85 ± 0.4 μM v.0.65 ± 0.2 μM, respectively; n = 4 each genotype), consistent with muscarinic-cholinergic receptor activation. These data indicate that the absence of Kv1.1 does not affect airway smooth muscle reactivity to MCh.
Discussion
This is the first study demonstrating that changes in respiratory function are progressive with age in Kcna1−/− mice which may promote respiratory failure prior to SD. (1) Kcna1−/− mice experience chronic blood gas instability as evidenced by frequent seizures, chronic oxygen desaturation, frequent A-H, the unusual breathing sequence of repeating patterns of tachypnea-A-H, increased tidal volume and the paradoxical MCh-induced hyperventilation. Blood gas instability triggers a compensatory increase in respiratory drive. Thus, the increase in basal respiratory drive as Kcna1−/− mice aged may indeed be compensatory. (2) The MCh challenge unveiled further respiratory abnormalities in Kcna1−/− mice. Following MCh, the hyperventilation response of Kcna1−/− mice was dramatically attenuated with age. Interestingly, only Kcna1−/− mice developed seizures following exposure to MCh and only Kcna1−/− mice became more sensitive to MCh as they approached sudden death. Indeed, MCh induced sudden death in a subset of SD30 and SD55 Kcna1−/− mice that exhibited respiratory parameters resembling SD90 Kcna1−/− mice.
Kcna1−/− mice exhibit chemoresponsive instability
Limited data are available concerning the role of Kv1.1 in respiratory peripheral organs including the trachea and lungs. We did not detect Kcna1 mRNA or Kv1.1 protein in lung tissue and there were no differences in contractility of Kcna1+/+ Kcna1−/− trachea. Our data further support previous findings. Despite Kcna1 mRNA expression in trachea-bronchial airway smooth muscle cells, it does not contribute to contractile function.19,20 Kv1.1 alpha subunit protein is located in neural tissue and its presence and function is established in the vagal and phrenic nerves.14,21 Hyperexcitable phrenic nerves of Kcna1−/− mice may contribute to increased respiration rate, an endpoint that was similar across age. We focused on endpoints that changed with age as these may implicate additional factors and have relevance to SUDEP susceptibility.
Our data is consistent with the notion that Kcna1−/− mice exhibit chemoresponsive instability. Kcna1−/− mice experience chronic oxygen desaturation, frequent seizures and A-H, both of which have been shown to be associated with elevated CO2 (hypercapnia) and hypoxia.4,8,22–24 In the seminal report by Ryvlin et al. the terminal GTC seizure was followed by a brief period of tachypnea, then bouts of apnea and finally complete respiratory arrest9. A similar breathing sequence of increased breathing rate then apnea was detected in Kcna1−/− mice. Tachypnea is increased respiration rate (reflected by elevated breathing rate, and shortened inspiration and expiration time). Hyperventilation is an increase in respiration rate and depth (reflected by increased tidal volume and peak inspiratory and expiratory flow). Tachypnea or hyperventilation is typically followed by arousal, not apnea. Hyperventilation-induced apnea is rare and is the result of chemoresponsive instability. Hyperventilation-induced apnea has been reported to occur in people with central apnea or respiratory failure.25–28 This suggests that chemoresponsive instability may occur in patients prior to SUDEP and if it occurs early, hyperventilation-induced apnea may provide a novel marker of SUDEP risk.
A previous study has reported that Kcna1−/− mice have normal (and slightly elevated) response to hypoxia which is thought to be due to the absence of Kv1.1 in the peripheral carotid bodies.29 Loss of Kv1.1 did not change the hypercapnia ventilator response, 29 which is thought to be centrally mediated. These data suggest a central mechanism may be responsible for chemoresponsive stability in SD90 Kcna1−/− mice.
Respiratory drive is an endogenous tool that is increased to promote blood gas stability. For example, a buildup of CO2 in the lungs will elevate blood CO2 levels to signal peripheral and central chemoreceptors to increase respiration rate which in turn stabilizes blood gasses. Indeed, as Kcna1−/− mice approach SD, basal respiratory drive and peak inspiratory flow are uniquely increased. Respiratory drive reflects the volume of air which enters and exits the lungs in one cycle normalized to inspiration time. Increased peak inspiratory flow indicates deeper inspiration. Respiratory drive and inspiration can be influenced by central input to the medulla where pre-inspiratory, inspiratory and expiratory neurons control the timing and air flow of inspiration and expiration. Orexin neurons in the lateral hypothalamus project to medullary pre-inspiratory and inspiratory neurons and are upstream of intercostal muscle-, phrenic nerve- and diaphragm-ventilator responses, influencing volume, inspiration and overall respiratory drive.30–33
Chemoresponsive orexin neurons increase firing in response to hypercapnia and hypoxia to increase breathing rate.34–37 We have previously found that orexin protein levels are elevated in the lateral hypothalamus of Kcna1−/− mice.40 Therefore, increased orexin neuronal activation may be a central driver of increased respiratory responses of Kcna1−/− mice. Indeed, orexin crosses the blood brain barrier and is elevated in the plasma of patients with epilepsy and in patients with hypercapnic respiratory failure.38,39 Elevation of orexin may be a compensatory mechanism to increase respiratory drive in people with respiratory failure. Elevated orexin levels in people with epilepsy and in Kcna1−/− mice may also be reflective of an endogenous compensatory mechanism to increase respiratory drive.40 Ongoing studies are exploring the potential role of orexin in the respiratory dysfunction of Kcna1−/− mice.
MCh challenge unveils abnormal responses in Kcna1−/− mice
MCh is routinely used to diagnose asthma in humans and animals and does not cause seizures in normal humans or animals. However, the MCh-challenge is contraindicated for people with epilepsy suggesting an abnormal, potentially dangerous response. MCh is a charged quarternary amine which prevents it from crossing the blood brain barrier (BBB). Seizures can make the BBB permeable and we have found Kcna1−/− BBB to be leaky40; therefore, it is possible for MCh to enter the Kcna1−/− brain. Administration of a mechanistically similar drug, pilocarpine is used to induce seizures in animal models. Though, it is unlikely that the relatively small amount of MCh inhaled reaches concentrations in the CNS necessary to elicit a seizure. A more probable cause for MCh-induced seizures is the effect of hyperventilation on blood gases. Hyperventilation can lower seizure threshold and cause seizures in people with epilepsy. In fact, it is commonly used as a diagnostic tool.
MCh increases bronchoconstriction and airway resistance.18 Kcna1+/+, Kcna1+/− and Kcna1−/− mice exhibited similar, non-asthmatic airway resistance responses to MCh consistent with our in vitro trachea data and previous reports indicating that the absence of Kv1.1 does not affect airway smooth muscle responses to MCh.19,20 Kcna1+/+ and Kcna1+/− mice responded to increasing MCh with expected respiratory depression. In stark contrast, MCh triggered hyperventilation and increased respiratory drive in Kcna1−/− mice. Furthermore, the MCh EC50 decreased with age, indicating increased sensitivity to MCh as Kcna1−/− mice approached SD. Hyperventilation is triggered by blood gas instability. Thus, the hyperventilation further supports the notion of a compensatory response to endogenous blood gas instability. Interestingly, this potentially compensatory response to MCh is significantly attenuated in SD90 Kcna1−/− mice. This loss of or reduced ability to trigger deep breathing (hyperventilation), may indicate the compensatory mechanism becomes less reliable as Kcna1−/− mice approach sudden death.
Conclusion
One SUDEP case study with long-term monitoring reported a change in heart rate variability preceding the final seizure and sudden death when compared to within subject data seven months prior.7 Although it was not clear how early the change occurred, it suggests a temporal window to detect within subject changes may occur in people at high risk for SUDEP. Data reported herein indicate a temporal window of opportunity may exist to identify chemoresponsive instabilities to possibly identify/develop/implement treatments to postpone or prevent SUDEP, however, additional pre-clinical and clinical studies are required.
Key Bullet Points.
Kcna1−/− mice experience chronic blood gas instability, which triggered a compensatory increase in respiratory drive as mice aged.
The MCh challenge unveiled respiratory abnormalities in Kcna1−/− mice.
Only Kcna1−/− mice developed seizures following exposure to MCh and only Kcna1−/− mice became more sensitive to MCh as they approached sudden death.
The progression of respiratory dysfunction with age may promote respiratory failure when challenged by a severe seizure, thereby increasing susceptibility of Kcna1−/− mice to sudden death.
Data reported herein indicate a temporal window of opportunity may exist to identify chemoresponsive instabilities to possibly identify/develop/implement preventative treatments to postpone or prevent SUDEP
Acknowledgments
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. We wish to thank Stephanie Matthews for her technical support. This work was supported by National Institutes of Health (NIH) NS072179 (KAS), NIH NS085389 (TAS), and the Citizens United for Research in Epilepsy Foundation (KAS) and (TAS). This work was also supported by the National Center for Research Resources grant G20RR024001. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. Please note, KAS has published under the names K Dorenbos, KA Fenoglio, KA Fenoglio-Simeone, and KA Simeone.
Footnotes
The authors have no conflicts of interest.
References
- 1.Hesdorffer DC, Logroscino G, Benn EK, et al. Estimating risk for developing epilepsy: a population-based study in Rochester, Minnesota. Neurology. 2011;76:23–27. doi: 10.1212/WNL.0b013e318204a36a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Surges R, Thijs RD, Tan HL, et al. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol. 2009;5:492–504. doi: 10.1038/nrneurol.2009.118. [DOI] [PubMed] [Google Scholar]
- 3.Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia. 2014;55:1479–1485. doi: 10.1111/epi.12666. [DOI] [PubMed] [Google Scholar]
- 4.Nashef L, Walker F, Allen P, et al. Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry. 1996;60:297–300. doi: 10.1136/jnnp.60.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walczak TS, Leppik IE, D’Amelio M, et al. Incidence and risk factors in sudden unexpected death in epilepsy: a prospective cohort study. Neurology. 2001;56:519–25. doi: 10.1212/wnl.56.4.519. [DOI] [PubMed] [Google Scholar]
- 6.Ryvlin P, Montavont A, Kahane P. Sudden unexpected death in epilepsy: from mechanisms to prevention. Curr Opin Neurol. 2006;19:194–199. doi: 10.1097/01.wco.0000218238.90711.f4. [DOI] [PubMed] [Google Scholar]
- 7.Jeppesen J, Fuglsang-Frederiksen A, Brugada R, et al. Heart rate variability analysis indicates preictal parasympathetic overdrive preceding seizure-induced cardiac dysrhythmias leading to sudden unexpected death in a patient with epilepsy. Epilepsia. 2014;55:e67–71. doi: 10.1111/epi.12614. [DOI] [PubMed] [Google Scholar]
- 8.Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. 2008;131:3239–3245. doi: 10.1093/brain/awn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smart SL, Lopantsev V, Zhang CL, et al. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- 10.Simeone TA, Simeone KA, Samson KK, et al. Loss of the Kv1.1 potassium channel promotes pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices. Neurobiol Dis. 2013;54:68–81. doi: 10.1016/j.nbd.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wenzel HJ, Vacher H, Clark E, et al. Structural consequences of Kcna1 gene deletion and transfer in the mouse hippocampus. Epilepsia. 2007;48:2023–2046. doi: 10.1111/j.1528-1167.2007.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simeone KA, Matthews SA, Rho JM, et al. Ketogenic Diet increases longevity in a model of Sudden Unexpected Death in Epilepsy, the Kv1.1KO mice. Epilepsia. 2016;57:178–82. doi: 10.1111/epi.13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagnall RD, Crompton DE, Semsarian C. Genetic Basis of Sudden Unexpected Death in Epilepsy. Front Neurol. 2017;20:8–348. doi: 10.3389/fneur.2017.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glasscock E, Yoo JW, Chen TT, et al. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aiba I, Noebels JL. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med. 2015;7:282ra46. doi: 10.1126/scitranslmed.aaa4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simeone KA, Matthews SA, Samson KK, et al. Targeting deficiencies in mitochondrial respiratory complex I and functional uncoupling exerts anti-seizure effects in a genetic model of temporal lobe epilepsy and in a model of acute temporal lobe seizures. Exp Neurol. 2014;251:84–90. doi: 10.1016/j.expneurol.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simeone TA, Matthews SA, Samson KK, et al. Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp Neurol. 2017;287:54–64. doi: 10.1016/j.expneurol.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Donohue HA, Abel PW, Bockman CS. Pharmacological properties of serotonin receptor subtypes mediating contraction of bovine inferior alveolar arteries. Arch Oral Biol. 2004;49:223–32. doi: 10.1016/j.archoralbio.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Adda S, Fleischmann BK, Freedman BD, et al. Expression and function of voltage-dependent potassium channel genes in human airway smooth muscle. J Biol Chem. 1996;271:13239–13243. doi: 10.1074/jbc.271.22.13239. [DOI] [PubMed] [Google Scholar]
- 20.Park WS, Firth AL, Han J, et al. Patho-, physiological roles of voltage-dependent K+ channels in pulmonary arterial smooth muscle cells. J Smooth Muscle Res. 2010;46:89–105. doi: 10.1540/jsmr.46.89. [DOI] [PubMed] [Google Scholar]
- 21.Zhou L, Messing A, Chiu SY. Determinants of excitability at transition zones in Kv1.1-deficient myelinated nerves. J Neurosci. 1999;19:5768–5781. doi: 10.1523/JNEUROSCI.19-14-05768.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seyal M, Bateman LM, Albertson TE, et al. Respiratory changes with seizures in localization-related epilepsy: analysis of periictal hypercapnia and airflow patterns. Epilepsia. 2010;51:1359–1364. doi: 10.1111/j.1528-1167.2009.02518.x. [DOI] [PubMed] [Google Scholar]
- 23.Blum AS, Ives JR, Goldberger AL, et al. Oxygen desaturations triggered by partial seizures: implications for cardiopulmonary instability in epilepsy. Epilepsia. 2000;41:536–541. doi: 10.1111/j.1528-1157.2000.tb00206.x. [DOI] [PubMed] [Google Scholar]
- 24.Hewertson J, Boyd SG, Samuels MP, et al. Hypoxaemia and cardiorespiratory changes during epileptic seizures in young children. Dev Med Child Neurol. 1996;38:511–522. doi: 10.1111/j.1469-8749.1996.tb12112.x. [DOI] [PubMed] [Google Scholar]
- 25.Xie A, Wong B, Phillipson EA, et al. Interaction of hyperventilation and arousal in the pathogenesis of idiopathic central sleep apnea. Am J Respir Crit Care Med. 1994;150:489–495. doi: 10.1164/ajrccm.150.2.8049835. [DOI] [PubMed] [Google Scholar]
- 26.Xie A, Rankin F, Rutherford R, et al. Effects of inhaled CO2 and added dead space on idiopathic central sleep apnea. J Appl Physiol. 1997;82:918–926. doi: 10.1152/jappl.1997.82.3.918. [DOI] [PubMed] [Google Scholar]
- 27.Munemoto T, Masuda A, Nagai N, et al. Prolonged post-hyperventilation apnea in two young adults with hyperventilation syndrome. Biopsychosoc Med. 2013;7:9. doi: 10.1186/1751-0759-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinsky MR. Functional hemodynamic monitoring. Crit Care Clin. 2015;31:89–111. doi: 10.1016/j.ccc.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kline DD, Buniel MC, Glazebrook P, et al. Kv1.1 deletion augments the afferent hypoxic chemosensory pathway and respiration. J Neurosci. 2005;25:3389–3399. doi: 10.1523/JNEUROSCI.4556-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugita T, Sakuraba S, Kaku Y, et al. Orexin induces excitation of respiratory neuronal network in isolated brainstem spinal cord of neonatal rat. Respir Physiol Neurobiol. 2014;200:105–109. doi: 10.1016/j.resp.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Badami VM, Rice CD, Lois JH, et al. Distribution of hypothalamic neurons with orexin (hypocretin) or melanin concentrating hormone (MCH) immunoreactivity and multisynaptic connections with diaphragm motoneurons. Brain Res. 2010;1323:119–126. doi: 10.1016/j.brainres.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dias MB, Li A, Nattie EE. Antagonism of orexin receptor-1 in the retrotrapezoid nucleus inhibits the ventilatory response to hypercapnia predominantly in wakefulness. J Physiol. 2009;587:2059–2067. doi: 10.1113/jphysiol.2008.168260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stettner GM, Kubin L. Antagonism of orexin receptors in the posterior hypothalamus reduces hypoglossal and cardiorespiratory excitation from the perifornical hypothalamus. J Appl Physiol. 2013;114:119–130. doi: 10.1152/japplphysiol.00965.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazarenko RM, Stornetta RL, Bayliss DA, et al. Orexin A activates retrotrapezoid neurons in mice. Respir Physiol Neurobiol. 2011;175:283–287. doi: 10.1016/j.resp.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burdakov D, Karnani MM, Gonzalez A. Lateral hypothalamus as a sensor-regulator in respiratory and metabolic control. Physiol Behav. 2013;121:117–124. doi: 10.1016/j.physbeh.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams RH, Jensen LT, Verkhratsky A, et al. Control of hypothalamic orexin neurons by acid and CO2. Proc Natl Acad Sci U S A. 2007;104:10685–10690. doi: 10.1073/pnas.0702676104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamaguchi K, Futatsuki T, Ushikai J, et al. Intermittent but not sustained hypoxia activates orexin-containing neurons in mice. Respir Physiol Neurobiol. 2015;206:11–14. doi: 10.1016/j.resp.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 38.Zhu LY, Summah H, Jiang HN, et al. Plasma orexin-a levels in COPD patients with hypercapnic respiratory failure. Mediators Inflamm. 2011;754847 doi: 10.1155/2011/754847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaciński M, Budziszewska B, Lasoń W, Zając A, et al. Level of S100B protein, neuron specific enolase, orexin A, adiponectin and insulin-like growth factor in serum of pediatric patients suffering from sleep disorders with or without epilepsy. Pharmacol Rep. 2012;64:1427–1433. doi: 10.1016/s1734-1140(12)70940-4. [DOI] [PubMed] [Google Scholar]
- 40.Roundtree HM, Simeone TA, Johnson C, et al. Orexin Receptor Antagonism Improves Sleep and Reduces Seizures in Kcna1-null Mice. Sleep. 2016;39:357–368. doi: 10.5665/sleep.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]