

Abstract
Palladium-catalyzed allylic C–H oxidation has been widely studied, but most precedents use acetic acid as the coupling partner. Here, we report a method compatible with diverse carboxylic acid partners. Use of a Pd0 pre-catalyst under aerobic reaction conditions leads to oxidation of Pd0 by O2 in the presence of the desired carboxylic acid to generate a PdII dicarboxylate that promotes acyloxylation of the allylic C–H bond. Good-to-excellent yields are obtained with ~1:1 ratio of the alkene and carboxylic acid reagents. Optimized reaction conditions employ 4,5-diazafluoren-9-one (DAF) as a ligand, in combination with a quinone/Fe(phthalocyanine) cocatalyst system to support aerobic catalytic turnover.
Keywords: palladium, aerobic, acyloxylation, homogeneous catalysis, C-H activation
Graphical Abstract
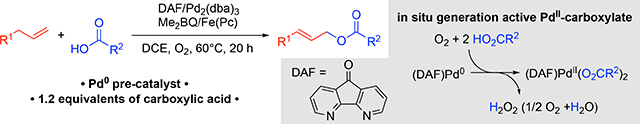
Use of a palladium(0) precatalyst provides the basis for in situ generation of an active palladium(II)-dicarboxylate catalyst under aerobic conditions in the presence of 4,5-diazafluoren-9-one (DAF). This tactic has been applied to allylic acyloxylation, enabling the formation of allylic esters with diverse nucleophilic carboxylate coupling partners from terminal linear alkenes and cyclic alkenes.
Oxidative cross-coupling reactions have been an area of extensive development in recent years. 1 Palladium-catalyzed allylic oxidation reactions are a prominent example of this reaction class.[2–8] Whereas many examples of allylic acetoxylation have been demonstrated, relatively few methods have been developed for related acyloxylation reactions that employ carboxylic acids other than acetic acid. Allylic acetoxylation was first reported in the early 1960s using stoichiometric palladium(II) salts in acetic acid with terminal olefins and cyclic alkenes.[2] Subsequent reports of palladium-catalyzed allylic acetoxylation employed stoichiometric oxidants such as benzoquinone,[2b,3] stoichiometric copper(II),[4] peroxides,[5] hypervalent iodine,[6] and, in certain cases, molecular oxygen.[7] These precedents are generally limited to acetoxylation because of the prevalent use of Pd(OAc)2 as the catalyst and acetic acid as the solvent or co-solvent (Figure 1a).
Figure 1.

(a) General method for allylic acetoxylation using Pd(OAc)2 as the catalyst and AcOH as the solvent or co-solvent; (b) Prior approaches to allylic acyloxylation using either Pd(OAc)2 or Pd(OBz)2 as the catalyst and the carboxylate in high quantities or incorporated through the oxidant; (c) Two approaches for the use of low stoichiometry carboxylic acid using palladium sources with non-coordinating anions.
Efforts to expand the scope of palladium-catalyzed allylic oxidation to incorporate other carboxylate groups have encountered several limitations. Pd(OAc)2 is one of the most common commercially available PdII sources and, therefore, is the most prevalent Pd source for allylic oxidation,[9,10] among other reactions.[11] Szabó successfully demonstrated two strategies for acyloxylation catalyzed by Pd(OAc)2: (a) using 4 equivalents of carboxylic anhydride,[9b] or (b) incorporating the carboxylate source into their oxidant by utilizing 3–3.5 equivalents of a hypervalent iodine oxidant with the corresponding lithium carboxylate salt.[6a,10] Hartwig used stoichiometric tert-butylbenzoyl peroxide and PdII benzoate to form allyl benzoates, reducing carboxylate loading to 2 equivalents.[ 12] The use of (co)solvent levels of carboxylic acid or the use of specialized carboxylate sources underlies the general limitation of these acyloxylation methods to the production of propionic, isobutyric, pivalic, or benzoic acid-derived allyl carboxylates (Figure 1b). An important strategy to address this limitation was introduced by White, who used the commercially available cationic [Pd(CH3CN)4](BF4)2 catalyst in combination with a tertiary amine base to enable use of diverse carboxylic acids as coupling partners (Figure 1c).[13] Recent efforts to implement this protocol in our lab, however, were complicated by the long reaction times (up to 72 h), high catalyst loading, and complicated workup to remove the stoichiometric quinone oxidant. Here, we introduce a new approach that addresses these challenges and enable the use of O2 as the terminal oxidant in the reactions.[14]
Pd(OAc)2 is a readily available catalyst precursor, but its use in acyloxylation reactions with other carboxylic acids requires an excess of carboxylic acid coupling partner to promote formation of the desired Pd-dicarboxylate species and minimize allylic acetate side-product formation (Figure 2a).[9a] An alternative approach would be to generate the desired PdII-dicarboxylate species from a non-carboxylate-containing precursor, as reflected by the approach of White and co-workers in Figure 1c.[13b] We reported that Pd2(dba)3 (dba = dibenzylideneacetone) can form a PdII-peroxo under O2 in the presence of a bidentate nitrogen-containing ligand,[15a] and carboxylic acids react with PdII-peroxo species to afford the corresponding PdII-carboxylate species (Figure 2b).[15] Yu and co-workers demonstrated that Pd2(dba)3 is an effective catalyst for aerobic C–H oxidative imidoylation,[16] and subsequent mechanistic investigations revealed that a key intermediate in the reaction is a PdII-peroxo, which reacts with an acidic N–H bond to afford the key PdII-amidate species.[17] These precedents suggested to us that Pd2(dba)3 could be used for the in situ formation of a PdII-dicarboxylate catalyst in the presence of O2 and thereby provide the basis for aerobic acyloxylation.
Figure 2.

(a) Formation of a PdII-dicarboxylate via reaction of Pd(OAc)2 with other carboxylic acids, RCO2H. (b) Formation of the a PdII-dicarboxylate from a Pd0 source, O2 and two equivalents of carboxylic acid.
Molecular oxygen has been shown to be a competent oxidant for allylic acetoxylation.[7] Kaneda and co-workers used O2 as the sole oxidant at elevated pressure,[7b] and we later reported a method compatible with 1 atm O2 promoted by using 4,5-diazafluoren-9-one (DAF) as an ancillary ligand.[7c,18] Even earlier, however, Bäckvall demonstrated a co-catalytic approach to achieve aerobic allylic acetoxylation of cyclohexene by using Pd(OAc)2 in combination with hydrobenzoquinone and iron(II) phthalocyanine [Fe(Pc)] as co-catalysts.[7a] In these systems, benzoquinone is proposed to oxidize the Pd0 to PdII resulting in the formation of hydroquinone, and Fe(Pc) catalyzes the reoxidation of hydroquinone by O2. Following this initial report, numerous multicomponent cocatalyst systems have been developed for aerobic acyloxylation.[19]
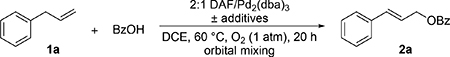
Building on our previously reported aerobic oxidation method, which used DAF as an ancillary ligand,[7c] we tested a series of conditions for the reaction of allylbenzene (1a) with benzoic acid using a Pd2(dba)3/DAF-based catalyst system (Table 1; see Tables S1–S3 in the Supporting Information for additional screening conditions). Initial tests showed significant improvement in the product yield upon addition of the cocatalysts 2,6-dimethylbenzoquinone (Me2BQ) and Fe(Pc) to the reaction mixture (entries 1–3). Separate studies showed that inclusion of a Brønsted base does not improve the product yield (see Table S1 in the Supporting Information). Optimization of the cocatalyst quantities led to the formation of cinnamyl acetate 2a in >90% yield (e.g., entries 3–6), and a high yield could be obtained with only 1.2 equiv of BzOH relative to allylbenzene (Table 1, entry 7). The ability to use lower amounts of the carboxylic acid coupling partner is especially appealing when the carboxylic acid is expensive or available in limited quantities. Negligible product yield was observed in the absence of DAF as an ancillary ligand (entry 8).
Table 1.
Optimization of Pd0-catalyzed aerobic allylic acyloxylation.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | %[DAF/Pd][a] | 1a [M] |
BzOH [eq] |
%Me2BQ | %Fe(Pc) | % Yield[b] |
| 1 | 2.5 | 1.08 | 3 | ---- | ---- | 24 |
| 2 | 2.5 | 1.08 | 3 | 5 | ---- | 56 |
| 3 | 2.5 | 1.08 | 3 | 5 | 2.5 | 72 |
| 4 | 2.5 | 1.08 | 3 | 5 | 0.5 | 78 |
| 5 | 2.5 | 1.08 | 3 | 1.25 | 0.5 | 51 |
| 6 | 5 | 1.08 | 2 | 10 | 0.5 | 98 |
| 7 | 5 | 1.08 | 1.2 | 10 | 0.5 | 93 |
| 8[c] | 5 | 1.08 | 1.2 | 10 | 0.5 | 4 |
[DAF/Pd] = 2:1 DAF:Pd2(dba)3 Reported mol% DAF and mol% total Pd
NMR yield relative to 1,3,5-trimethoxybenzene
No DAF.
The reaction time-profile for the benzoyloxylation of allylbenzene was compared with three different catalyst sources under similar aerobic conditions: Pd2(dba)3, Pd(OAc)2 and Pd(OBz)2. The data in Figure 3 show that the Pd0 source (Pd2(dba)3) exhibits activity very similar to the PdII sources, and it avoids the formation of the undesired allylic acetate byproduct, which forms in 7% yield when Pd(OAc)2 as the precatalyst. The similar activity exhibited by Pd2(dba)3 and Pd(OBz)2 is noteworthy because it indicates that it is not necessary to independently synthesize a PdII-dicarboxylate precatalyst for oxidative coupling reactions with carboxylic acids that lack commercially available Pd(O2CR)2 species. The active PdII catalyst may be generated in situ from Pd0, O2, and two equivalents of RCO2H.
Figure 3.

Product formation over time when catalyzed by different palladium sources. [Pd2(dba)3] = 27 mM, [Pd(OBz)2] = [Pd(OAc)2] = 54 mM, [Me2BQ] = 108 mM, [Fe(Pc)] = 5.4 mM, [allylbenzene] = 1.08 M, DCE (6 mL), O2 (1 atm), at 60 °C for 20 h. The lines reflect smooth fits to the data to guide the eye.
Having demonstrated the viability of Pd2(dba)3 as an effective precatalyst, this approach was tested with a series of other carboxylic acids, using only slight excess relative to the alkene (1.2 equiv). Moderate-to-excellent yields of the corresponding E-allylic carboxylates were obtained from these reactions (Figure 4). Effective substrates included a number of substituted benzoic acid derivatives and cinnamic acid (2a-2d). Good yields were also obtained with aliphatic carboxylic acid derivatives (2e-h), although only moderate yield was observed with octanoic acid (2i). 2-Thiophenecarboxylic acid also an effective coupling partner, affording 2j. The reaction was also performed successfully on a 1 g scale, generating the cinnamate derivative 2b in 85% yield.
Figure 4.

Carboxylic acid scope. NMR yield relative to 1,3,5-trimethoxybenzene standard (isolated yield in parentheses). Average of two reactions. [a] Isolated yield on 1 g scale: 85%.
This method also provides good to excellent yields for a number of other terminal alkenes (Figure 5). Both electron rich and electron poor allylbenzene derivatives gave excellent yields (2a, 3a-d). Aliphatic substrates produced good yields (3e) including those bearing the acid sensitive Boc group (3f). Substrates with homoallylic carbonyl groups gave mixed results. An excellent yield was achieved with the homoallylic ester (3g), but the homoallylic amide resulted in a poor yield (3h), possibly reflecting complications from the N-H group. Product 3a was obtained in 80% yield when the reaction was scaled up to 1 g of 4-allyltoluene. For all compounds in Figures 4 and 5, the E isomer was the major product observed (≥10:1 E:Z; see Supporting Information for details).
Figure 5.

Substrate scope. NMR % yield relative to 1,3,5-trimethoxybenzene (isolated % yield in parentheses). Average of two reactions. [a] Isolated yield on 1 g scale: 80% [b] Yields obtained using t-butylbenzoquinone due to co-elution with 2,6-dimethylbenzoquinone during column chromatography purification.
Preliminary results suggest that this method could be effective with cyclic substrates. A number of early studies demonstrated allylic acetoxylation of cyclohexene and other simple cyclic alkenes;[2b,5c,7a,20] however, studies with substituted cyclic alkenes are rare.[4a,10,21] A noteworthy exception is a recent study by Szabó,[10] who reported allylic trifluoroacetoxylation of 5- and 6-membered rings using catalytic Pd(OAc)2 with PhI(O2CCF3)2 as the oxidant. The reaction affords high trans diastereoselectivity with mono-substituted substrates. A simple Pd(OAc)2/Me2BQ cocatalyst system (conditions A, Figure 6) enables effective aerobic allylic acetoxylation of the cyclopentene derivative 4, generating 5-OAc in good yield (75%) with exclusive formation of the trans diastereomer. Use of the Pd2(dba)3/DAF catalyst system described above (conditions B, Figure 6) with acetic acid as the coupling partner afforded 5-OAc in good yield (69%), with selectivity of 1:1.3 cis:trans. Use of benzoic acid as the coupling partner again affords the product in good yield (5-OBz, 81%), but with a small preference for the cis diastereomer (1.6:1). To our knowledge, this result is the first example of cis-selectivity in Pd-catalyzed allylic acyloxylation. Preliminary efforts to improve the cis diastereoselectivity further were not successful, but these results provide an important starting point for future studies to probe the origin of diastereoselectivity in these reactions. These results suggest an ancillary ligand may be used to modulate reaction selectivity, for example, by changing the mechanism of nucleophilic attack by the carboxylic acid nucleophile on the allyl-PdII intermediate (i.e., inner sphere versus outer sphere nucleophilic attack). Another valuable preliminary result was obtained with the Diels-Alder-derived cyclohexene 6, which undergoes allylic oxidation under conditions B to afford the cis benzoate derivative 7 in 73% yield. This result contrasts the recently reported dehydrogenation of 6 to afford 8 under aerobic conditions with a different Pd catalyst system.[22] Together these results highlight prospects for ligand/catalyst-modulation of reaction selectivity in the oxidation of cyclic alkenes.
Figure 6.

Pd2(dba)3-catalyzed aerobic allylic acyloxylation of cyclic substrates. NMR yields relative to 1,3,5-trimethoxybenzene. Average of two reactions.
The mechanism in Scheme 1 provides a general mechanism for the allylic C–H acyloxylation of terminal alkenes catalyzed by DAF/Pd0 (Scheme 1). The on-cycle PdII(O2CR)2 species is proposed to arise from the reaction of Pd2(dba)3 with O2 in the presence of DAF (cf. Figure 2).15 This step could also be promoted by the quinone. The DAF/PdII(O2CR)2 catalyst is then expected to promote allylic C–H oxidation via a sequence of well established reaction steps.7,8 Catalyst reoxidation is proposed to occur according to a multistep proton-coupled electron-transfer sequence in which the 2,6 -dimethylbenzoquinone (denoted “BQ”) oxidizes the Pd0, the Feox(Pc) species oxidizes the 2,6-dimethylhydroquinone (H2Q), and O2 oxidizes the resulting Fered(Pc).7a
Scheme 1.

Proposed mechanism for DAF/Pd0-catalyzed aerobic allylic C–H acyloxylation of linear terminal alkenes.
In summary, we have demonstrated that commercially available Pd2(dba)3 is an effective Pd0 precatalyst that enables effective coupling of allylic C–H bonds and various carboxylic acids in approximately 1:1 ratio. In situ reaction of the Pd0 precatalyst with O2 (or Me2BQ), followed by reaction of the resulting Pd/O2 (or Pd/Me2BQ) adduct is proposed to provide the basis for generation of the active PdII-dicarboxylate catalyst that participates in the allylic C–H activation and coupling the alkene substrate. This method represents the first example of Pd-catalyzed allylic acyloxylation effective with O2 as the terminal oxidant and carboxylates other than acetate. Preliminary results with cyclic alkenes set the stage for further methodological and mechanistic studies to develop catalyst-controlled methods for stereoselective aerobic oxidation of allylic C–H bonds.
Supplementary Material
Acknowledgements
This work was funded by the NSF (CHE-1665120). NMR instrumentation was supported by NSF CHE-1048642 and a generous gift from Paul J. and Margaret M. Bender. GC instrumentation was supported by NIH 1S10OD020022-1. C. V. K. was supported in part by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1256259. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Experimental Section
Full experimental details and product characterization data are given in the supporting information.
References
- [1].For representative reviews on oxidative heterofunctionalization of C–H bonds, emphasizing those compatible with O2 as the oxidant, see:Liu C, Zhang H, Shi W, Lei A, Chem. Rev 2011, 111, 1780–1824;Shi Z, Zhang C, Tang C, Jiao N, Chem. Soc. Rev 2012, 41, 3381–3430;Wang D, Weinstein AB, White PB, Stahl SS, Chem. Rev 2018, 118, 2636–2679.
- [2].a) Vagaftik MN, Moiseev II, Syrkin YK, Izv. Akad. Nauk SSR 1962, 5, 930–931; [Google Scholar]; b) Anderson CB, Winstein S, J. Org. Chem 1963, 28, 605–606. [Google Scholar]
- [3].For leading references of Pd-catalyzed allylic acetoxylation using stoichiometric benzoquinone see:Akermark B, Hansson S, Rein T, Vagberg J, Heumann A, Backvall J-E, J. Organomet. Chem 1989, 369, 433–444;Chen MS, White MC, J. Am. Chem. Soc 2004, 126, 1346–1347;Chen MS, Prabagaran N, Labenz NA, White MC, J. Am. Chem. Soc 2005, 127, 6970–6971;Fraunhoffer KJ, Prabagaran N, Sirois LE, White MC, J. Am. Chem. Soc 2006, 128, 9032–9033;Covell DJ, White MC, Angew. Chem. Int. Ed 2008, 47, 6448–6451;Angew. Chem. Int. Ed. Engl 2008, 120, 6548–6551;Stang EM, White MC, Nat. Chem 2009, 1, 547–551;Lin B-L, Labinger JA, Bercaw JE, Can. J. Chem 2009, 87, 264–271;Kondo H, Yu F, Yamaguchi J, Liu G, Itami K, Org. Lett 2014, 16, 4212–4215;Malik HA, Taylor BLH, Kerrigan JR, Grob JE, Houk KN, Du Bois J, Hamann LG, Patterson AW, Chem. Sci 2014, 5, 2352–2361.
- [4].a) Heumann A, Reglier M, Waegell B, Angew. Chem 1982, 94, 397; [Google Scholar]; Angew. Chem. Int. Ed. Engl 1982, 21, 366–367; [Google Scholar]; b) Firdoussi LE, Baqqa A, Allaoud S, Allal BA, Karim A, Castanet Y, Mortreux A, J. Mol. Catal. A: Chem 1998, 135, 11–22. [Google Scholar]
- [5].a) Uemura S, Fukuzawa S, Toshimitsu A, Okano M, Tet. Lett 1982, 23, 87–90; [Google Scholar]; b) Akermark B, Larsson EM, Oslob JD, J. Org. Chem 1994, 59, 5729–5733; [Google Scholar]; c) Jia C, Muller P, Mimoun H, J. Mol. Catal. A: Chem 1995, 101, 127–136. [Google Scholar]
- [6].a) Pilarski LT, Selander N, Bose D, Szabo KJ, Org. Lett 2009, 11, 5518–5521; [DOI] [PubMed] [Google Scholar]; b) Check CT, Henderson WH, Wray BC, Vanden Eynden MJ, Stambuli JP, J. Am. Chem. Soc 2011, 133, 18503–18505. [DOI] [PubMed] [Google Scholar]
- [7].a) Backvall J-E, Hopkins RB, Grennberg H, Mader MM, Awasthi AK, J. Am. Chem. Soc 1990, 112, 5160–5166. [Google Scholar]; b) Mitsudome T, Umetani T, Nosaka N, Mori K, Mizugaki T, Ebitani K, Kaneda K, Angew. Chem 2006, 118, 495–499; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. Engl 2006, 45, 481–485;16323234 [Google Scholar]; c) Campbell AN, White PB, Guzei IA, Stahl SS, J. Am. Chem. Soc. 2010, 132, 15116–15119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liron F, Oble J, Lorion MM, Poli G, Eur. J. Org. Chem 2014, 2014, 5863–5883. [Google Scholar]
- [9].a) Thiery E, Aouf C, Belloy J, Harakat D, Le Bras J, Muzart J, J. Org.Chem 2010, 75, 1771–1774; [DOI] [PubMed] [Google Scholar]; b) Pilarski LT, Janson PG, Szabo KJ, J. Org. Chem 2011, 76, 1503–1506. [DOI] [PubMed] [Google Scholar]
- [10].Alam R, Pilarski LT, Pershagen E, Szabo KJ, J. Am. Chem. Soc 2012, 134, 8778–8781. [DOI] [PubMed] [Google Scholar]
- [11].Grennberg H, Foot JS, Banwell MG, Roman DS, Palladium(II) Acetate. In Encyclopedia of Reagents for Organic Synthesis. 2015, doi: 10.1002/047084289X.rp001.pub3 [DOI] [Google Scholar]
- [12].Litman ZC, Sharma A, Hartwig JF, ACS Catal 2017, 7, 1998–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Covell DJ, Vermeulen NA, Labenz NA, White MC, Angew. Chem 2006, 118, 8397–8400; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. Engl 2006, 45, 8217–8220; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vermeulen NA, Delcamp JH, White MC, J. Am. Chem. Soc. 2010, 132, 11323–11328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].For discussion of safety issues associated with scalable aerobic oxidations, see:Osterberg PM, Niemeier JK, Welch CJ, Hawkins JM, Martinelli JR, Johnson TE, Root TW, Stahl SS, Org. Process Res. Dev 2015, 19, 1537–1543.
- [15].a) Stahl SS, Thorman JL, Nelson RC, Kozee MA, J. Am. Chem. Soc 2001, 123, 7188–7189; [DOI] [PubMed] [Google Scholar]; b) Konnick MM, Guzei IA, Stahl SS, J. Am. Chem. Soc 2004, 126, 10212–10213; [DOI] [PubMed] [Google Scholar]; c) Konnick MM, Gandhi BA, Guzei IA, Stahl SS, Angew. Chem. Int. Ed 2006, 45, 2904– 2907. [DOI] [PubMed] [Google Scholar]
- [16].Liu Y-J, Xu H, Kong W-J, Shang M, Dai H-X, Yu J-Q. Nature 2014, 515, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Dang Y, Deng X, Guo J, Song C, Hu W, Wang Z-X, J. Am. Chem. Soc 2016, 138, 2712–2723. [DOI] [PubMed] [Google Scholar]; b) Tereniak SJ, Stahl SS. J. Am. Chem. Soc 2017, 139, 14533–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) White PB, Jaworski JN, Fry CG, Dolinar BS, Guzei IA, Stahl SS, J. Am. Chem. Soc 2016, 138, 4869–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jaworski JN, McCann SD, Guzei IA, Stahl SS, Angew. Chem. Int. Ed 2017, 56, 3605– 3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].See, for example:Larsson EM, Akermark B, Tet. Lett 1993, 34, 2523–2526Goncalves JA, Gusevskaya EV, Appl. Catal., A 2004, 258, 93–98Henderson WH, Check CT, Proust N, Stambuli JP, Org. Lett 2010, 12, 824–827.
- [20].a) Heumann A, Akermark B, Angew. Chem 1984, 96, 443; [Google Scholar]; Angew. Chem. Int. Ed. Engl 1984, 23, 453–454 [Google Scholar]; b) Grennberg H, Bergstad K, Backvall J-E, J. Mol. Catal. A: Chem 1996, 113, 355–358 [Google Scholar]; c) Grennberg H, Backvall J-E, Chem. Eur. J 1998, 4, 1083–1089. [Google Scholar]
- [21].a) McMurry JE, Kočovsky P, Tet. Lett 1984, 25, 4187–4190; [Google Scholar]; b) Hansson S, Heumann A, Rein T, Akermark B, J. Org. Chem 1990, 55, 975–984. [Google Scholar]
- [22].Iosub AV, Stahl SS, J. Am. Chem. Soc 2015, 137, 3454–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.