

Abstract
Liver, the major metabolic organ in the body, is known for its remarkable capacity to regenerate. Whereas partial hepatectomy (PHx) is a popular model for the study of liver regeneration, the liver also regenerates after acute injury, but less is known about the mechanisms that drive it.
Recent studies have shown that liver regeneration is critical for survival in acute liver failure (ALF), which is usually due to drug-induced liver injury (DILI). It is sometimes assumed that the signaling pathways involved are similar to those that regulate regeneration after PHx, but there are likely to be critical differences. A better understanding of regeneration mechanisms after DILI and hepatotoxicity in general could lead to development of new therapies for ALF patients and new biomarkers to predict patient outcome. Here, we summarize what is known about the mechanisms of liver regeneration and repair after hepatotoxicity. We also review the literature in the emerging field of liver regeneration biomarkers.
Keywords: Drug-induced liver injury, liver repair, acute liver failure, ischemic hepatitis, acetaminophen, carbon tetrachloride, alcohol, hepatocyte proliferation, growth factors, cytokines
Introduction
The liver is uniquely susceptible to toxic injury. Unlike other organs, it receives most of its blood supply (approximately 80%) from veins. Blood from the spleen, pancreas, stomach, and large intestine drains into the splenic vein which then merges with the superior mesenteric vein from the small intestine to form the hepatic portal vein. This unique arrangement ensures that everything absorbed from the stomach and intestines passes through the liver before reaching systemic circulation. The liver acts a chemical scrubber for the portal blood, catalyzing the conjugation of xenobiotics with highly water-soluble side groups to facilitate their excretion. This anatomy is so important for our health that it is completely conserved across vertebrates, from cyclostomata to placental mammals. Even cephalochordate lancelets possess a portal/liver- like system called the midgut diverticulum (Subbotin, 2017). However, in order to form those soluble conjugates, some chemicals must first pass through an intermediate step in which they form highly reactive metabolites. Those metabolites may bind to proteins or nucleic acids in the hepatocytes causing significant injury. The portal system also means that endotoxins produced by ingested bacteria reach the liver before any other organ, and the liver therefore bears the brunt of those toxins. Finally, the mixing of arterial and venous blood means that the liver is already hypoxic compared to other organs, and any further reduction in oxygen supply (e.g. ischemia) followed by sudden re-oxygenation can be catastrophic.
Fortunately, the liver also has phenomenal capacity for repair and regeneration after injury. This is most sticking demonstrated in partial hepatectomy (PHx) where as much as 70– 80% of the liver is removed and the remaining tissue will enlarge until the original functional mass is restored (Higgins and Anderson, 1931; Fausto et al., 2006; Michalopoulos, 2017, Sadler et al., 2007).
It is important to understand the mechanisms of liver repair and regeneration after hepatotoxicity. Most studies of liver regeneration have relied upon PHx model in rodents introduced by Anderson and Higgins (1931), but the concept of liver regeneration was actually introduced by clinicians who observed that patients with massive acute necrosis and atrophy of the liver can recover normal function over time (Milne, 1909). Importantly, there is strong evidence that regeneration is important for survival of acute liver failure (ALF), which is usually due to hepatotoxicity. For example, immunostaining for proliferating cell nuclear antigen (PCNA) is greater in liver tissue from ALF survivors than non-survivors (Kayano et al., 1992). While it has been said that the mechanisms of regeneration after acute necrosis are probably similar to PHx (Michalopoulos, 2013; 2014), there are clear differences in anatomical nature of the injury and in subsequent inflammation suggesting that differences in signaling are likely. A better understanding of the mechanisms involved may lead to new treatments for ALF.
The purpose of this chapter is to review the clinically relevant causes of acute liver injury (ALI) and ALF, and to summarize the mechanisms of regeneration after hepatotoxicity. At the end of this chapter, the reader should be able to 1) list the most common and severe causes of ALI, 2) describe major differences between PHx and hepatotoxicity, 3) discuss the mechanisms of liver repair and regeneration after hepatotoxicity, and 4) discuss biomarkers of liver regeneration and ALF prognosis.
Etiology and prognosis of acute liver injury
Ischemic hepatitis is the most common cause of severe ALI in the United States and other developed countries, defined as peak ALT ≥1,000 U/L. It accounts for approximately 50% of all cases (Galvin et al., 2015; Breu et al., 2018). Drug-induced liver injury (DILI) is the second most common cause, at approximately 15% (Galvin et al., 2015; Breu et al., 2018). Other common causes of severe ALI include viral hepatitis (10–15%), hepatobiliary diseases (e.g. gallstones) (5– 10%), and autoimmune hepatitis (1–5%) (Galvin et al., 2015; Breu et al., 2018).
Among those etiologies, DILI has the worst prognosis. Although ischemic hepatitis accounts for most cases of ALI, DILI is by far the most common single cause of ALF, responsible for nearly 60% of all cases (Lee, 2008). Furthermore, mortality in DILI-induced ALF is high, ranging from 25–75% depending on the causative drug (Ostapowicz et al., 2002; Larson et al., 2005; Lee, 2008; Reddy et al., 2016). While mortality in ischemic hepatitis is also high (approximately 50%) (Tapper et al., 2015), death is generally thought to be due to the underlying etiology (e.g. cardiovascular disease) and liver injury is often viewed as a secondary issue or merely a complication of the disease.
Acetaminophen (APAP) overdose is far and away the most common cause of DILI and DILI-induced ALF (Lee, 2008; Galvin et al., 2015; Breu et al., 2018). Approximately 50% of DILI cases in the US are attributed to APAP (Galvin et al., 2015), as well as about 80% of DILI- induced ALF (Lee, 2008). In fact, 80% may be an underestimate. Follow-up studies have revealed that some cases of ALF in which the cause cannot be determined using routine diagnostic tools are actually due to APAP overdose (Ganger et al., 2018). Although prognosis for APAP-induced ALF patients is generally better than for patients with ALF due to other etiologies, the sheer volume of cases means that APAP overdose is also the most common cause of ALF-related death (Ostapowicz et al., 2002). As a result, most studies of liver regeneration after DILI have relied on the mouse model of APAP overdose or samples from APAP overdose patients, though other models have also been used to provide general insight into the regeneration process after hepatotoxicity, or before APAP was recognized as the most common cause of ALF.
Major differences between hepatectomy and hepatotoxicity
The most obvious differences between PHx and xenobiotic hepatotoxicity are due to anatomy. While PHx involves the surgical removal of a large portion of the liver, xenobiotic hepatotoxicity causes necrosis that develops in pockets within the tissue. As a result, cell proliferation proceeds differently in the two cases. The process of cell division, which is at the center of liver regeneration, occurs in the remnant lobes after partial hepatectomy. In chemical- induced injury, hepatocytes surrounding the areas of necrosis enter the cell cycle and divide (Leevy et al., 1959; Apte et al., 2017). Hepatocyte proliferation after PHx is pan-lobular, beginning around the periportal areas and spreading outward in a wave and mainly in total absence of liver injury. The removal of a large piece of the liver in PHx also results in major hemodynamic changes. Loss of normal liver architecture due to necrosis likely also affects blood flow, but probably to a lesser extent. Finally, xenobiotic hepatotoxicity is usually accompanied by extensive sterile inflammation, which is not a consequence of PHx (Michalopoulos, 2013; 2014). Given these major, fundamental differences, it is not at all surprising that regeneration signaling differs at least somewhat between PHx and toxic injury.
Regeneration after drug-induced liver injury
Carbon tetrachloride hepatotoxicity.
Although carbon tetrachloride (CCl4) is not a drug, it has been used as a model for DILI, and hepatotoxicity in general, for decades (McGill and Jaeschke, 2019). The mechanisms of injury and repair after carbon tetrachloride (CCl4) hepatotoxicity have been studied since the 1950s (Leevy et al., 1959). The biotransformation of CCl4 produces toxic products able to cause severe necrosis in the liver. Cytochrome P450s cleave CCl4 at the carbon- halogen bond, creating the trichloromethyl radical (CCl3•) which can react with lipids, proteins, and DNA (Weber et al., 2003). The resulting lipid peroxidation and protein-lipid cross linking leads to cell dysfunction and cell membrane destruction (Weber et al., 2003).
Several studies indicate that cytokines, presumably secreted in response to liver injury, help to drive regeneration after CCl4 hepatotoxicity. In the PHx model, serum tumor necrosis factor-alpha (TNFα) increases rapidly (Akerman et al., 1992; Trautwein et al., 1996), and inhibition or disruption of TNFα signaling blunts regeneration (Akerman et al., 1992; Yamada et al., 1997). Interleukin-6 (IL-6) expression also increases early after PHx, though with a slight delay compared to TNFα (Trautwein et al., 1996). According the the ‘priming and progression’ theory of liver regeneration, it is thought that the earlier TNFα signaling stimulates IL-6 production through NFκB-regulated transcription, and IL-6 is important for liver regeneration in part through activation of signal transducer and activator of transcription (Stat3). Importantly, Stat3 stimulation and cell proliferation are impaired in IL-6-deficient mice post-PHx (Cressman et al., 1996), while injecting the same mice with IL-6 restores those effects (Cressman et al., 1996). Tumor necrosis factor (TNF) receptor 1 (TNFR1)-deficient mice have reduced Stat3 signaling and significantly delayed regeneration after CCl4 treatment compared to WT animals (Yamada and Fausto, 1998). Some data also indicate that hepatocyte proliferation is reduced after CCl4 in liver-specific Stat3 KO mice (Moh et al., 2007). Although it is difficult to directly study the role of IL-6 in regeneration after CCl4 toxicity due to early protective effects of IL-6 KO in that model, there is some evidence that IL-6 deficiency delays regeneration in a model of chronic CCl4 toxicity (Río et al., 2008). Altogether, it appears that TNFα and IL-6 signaling are also important for recovery after CCl4-induced liver injury.
There is also evidence for role of growth factors in regeneration after CCl4 treatment, as in PHx. For example, direct manipulation of hepatocyte growth factor (HGF) and its receptor c-Met have revealed that HGF is important. Treatment with an antibody against HGF dramatically reduces DNA synthesis after CCl4-induced injury (Burr et al., 1998). Interestingly, that effect was not observed in c-Met KO mice, but reduction in necrotic areas during recovery did appear to be delayed in those animals (Huh et al., 2004). Furthermore, mice with Cre adenovirus-driven hepatocyte-specific HGF KO displayed reduced DNA synthesis after CCl4 treatment, similar to the HGF antibody-treated mice (Phaneuf et al., 2004). Indirect evidence for the importance of HGF in CCl4 hepatotoxicity is also available. For example, plasmin is important for maturation and release of HGF from the extracellular matrix (ECM), and plasmin-deficient mice have reduced c-Met signaling after CCl4 treatment, and DNA synthesis is reduced in plasmin-deficient hepatocytes cultured with HGF (Shanmukhappa et al., 2009). Overall, it seems clear that HGF is important for regeneration after CCl4 hepatotoxicity. On the other hand, liver regeneration after CCl4 treatment is similar between epidermal growth factor receptor (EGFR) KO and WT mice (Scheving et al., 2015). That indicates that, unlike the PHx model, EGFR is not very important for regeneration in CCl4 hepatotoxicity, though it should be noted that combined KO of EGFR and c-Met reduced regeneration at one early time point more than c-Met KO alone (Scheving et al., 2015).
With regard to metabolism, Rao and Mehendale (1989) reported that fructose 1,6- diphosphate treatment can increase regeneration in CCl4 hepatotoxicity. On the basis of those results and results from similar studies, the authors concluded that fructose aids regeneration by acting as an energy source. Consistent with that, a recent study demonstrated that the energy sensor AMP-activated protein kinase (AMPK) is activated during CCl4 hepatotoxicity, and treatment with an AMPK inhibitor reduces liver regeneration (Huang et al., 2018). On the other hand, glucose loading and diabetes are both known to reduce recovery in rodents with CCl4 toxicity (Chanda and Mehendale, 1995).
One of the most interesting results from studies of liver regeneration after hepatotoxicity caused by CCl4 and other industrial chemicals was the discovery of the phenomenon sometimes referred to as the “threshold effect.” Mehendale and coworkers observed that administration of a low dose of CCl4 protects against lethality of a later large dose by stimulating repair mechanisms and promoting hepatocyte proliferation (Kodavanti et al., 1989a; 1989b; Thakore and Mehendale, 1991). The initial insult need not be from the same hepatotoxicant. For example, administration of a low dose of thioacetamide protects against APAP hepatotoxicity by increasing hepatocyte proliferation (Chanda et al., 1995), likely through Wnt/β-catenin signaling (Dadhania et al., 2017). Numerous other stressors are now known to have the same effect (Apte et al., 2017). They also discovered that pre-treatment with chlordecone abolishes the early phase of hepatocyte proliferation after CCl4 treatment and increases mortality (Mehendale, 1989). These observations, combined with other discoveries such as the possible role of Ca2+-dependent proteases in the spread of necrosis and inhibition of those proteases by endogenous calpastatin (Limaye et al., 2003; 2006), led to the idea that the outcome of hepatotoxicity is determined by a balance between mechanisms of injury and mechanisms of repair. That idea is supported by the discovery that hepatocyte proliferation increases with increasing severity of hepatotoxicity until a threshold is reached (Mangipudy et al., 1995; Rao et al., 1997; Sathanandam et al., 2004). Beyond the threshold, proliferation is impaired and mortality increases (Mangipudy et al., 1995; Rao et al., 1997; Sathanandam et al., 2004). This observation is also the basis of the incremental dose model of APAP hepatotoxicity mentioned in the next section.
Overall, some mechanisms of regeneration appear to be common between PHx and CCl4 toxicity, but there may also be major differences. In particular, EGFR signaling may not be as important in the CCl4 model. Many other mechanisms have yet to be explored in CCl4 toxicity, and more differences may be revealed over time.
Acetaminophen hepatotoxicity.
The mechanisms of acetaminophen (APAP) hepatotoxicity have been thoroughly investigated, though large gaps in our knowledge remain. The toxicity starts with formation of a reactive metabolite that binds to proteins (McGill and Jaeschke, 2013; Kennon-McGill and McGill, 2018). Binding to mitochondrial proteins in particular causes mitochondrial dysfunction and oxidative stress (McGill et al., 2012a; Xie et al., 2015; McGill and Jaeschke, 2013). The initial oxidative stress is thought to activate the c-Jun N-terminal kinases 1/2 (JNK) (Gunawan et al., 2006; Du et al., 2015), which then translocate into mitochondria and exacerbate the mitochondrial damage (Hanawa et al., 2008; Ramachandran et al., 2011a) leading to loss of mitochondrial membrane potential and decreased respiration (Meyers et al., 1988; Kon et al., 2004; Reid et al., 2005; Ramachandran et al., 2011b). The mitochondria then swell and rupture (Placke et al., 1987), releasing endonucleases that translocate to the nucleus and cleave nuclear DNA (Bajt et al., 2006). The result is oncotic necrosis of hepatocytes (Gujral et al., 2002). Importantly, the mechanisms of injury appear to be the same in mice and humans (McGill et al., 2011; 2012b; 2014; Xie et al., 2014).
The earliest studies of regeneration after APAP overdose in mice revealed that pro- regenerative cytokines play an important role. Although several earlier attempts were made to study the mechanisms of liver regeneration after APAP overdose in rats, we now know that rats are a poor model for APAP hepatotoxicity compared to mice (McGill et al., 2012a). Those initial mouse studies revealed that IL-6 expression increases early as 4 h after APAP overdose (James et al., 2003), and Ju et al. (2002) demonstrated that depletion of Kupffer cells with liposomal clodronate reduces expression of TNFα, IL-6, and other cytokines and exacerbates the toxicity at both early and late time points. Shortly thereafter, it was discovered that both IL-6 KO and TNFR KO mice have similar injury compared to wild-type mice at early time points after APAP overdose but delayed recovery later on (James et al., 2003; 2005). Furthermore, pretreatment of the IL-6 KO mice with exogenous IL-6 restored hepatocyte proliferation at a late time point (James et al., 2003). Interestingly, it appears that in the absence of IL-6, other cytokines can become upregulated to compensate (James et al., 2003; Masubuchi et al., 2003).
Surprisingly, the role of other signaling pathways in regeneration after APAP hepatotoxicity has only recently been explored. It was demonstrated that β-catenin is activated as early as 1 h after APAP overdose in mice and remains active until at least 6 h (Apte et al., 2009). Moreover, the increase in β-catenin activation is preceded by increased phosphorylation (and therefore inactivation) of GSK3β (Apte et al., 2009). Importantly, β-catenin-deficient mice that were induced to express Cyp2e1 had reduced expression of regeneration markers at 24 h after APAP despite similar injury (Apte et al., 2009), and staining for nuclear β-catenin was found to correlate with proliferating cell nuclear antigen (PCNA) expression in APAP-induced ALF patients (Apte et al., 2009). Consistent with those data, Bhushan et al. (2014) later used an incremental dosing model to demonstrate that nuclear localization of β-catenin and GSK3β phosphorylation are reduced at a dose of APAP that inhibits liver regeneration, while overexpression of stable β-catenin improves it. Finally, treatment with a highly specific GSK3β inhibitor enhanced liver regeneration after APAP overdose (Bhushan et al., 2017a). Thus, Wnt/β- catenin signaling is clearly important.
Less is known about the importance of growth factors after APAP hepatotoxicity, but some data are available. Similar to PHx, EGFR is activated within 1–6 h after APAP overdose, and treatment with an EGFR inhibitor at a late time point impaired recovery and survival (Bhushan et al., 2017b). Also similar to the PHx model, vascular endothelial growth factor (VEGF) increases later after APAP overdose than other growth factors, but inhibition of VEGF signaling also blunts regeneration (Donahower et al., 2006; 2010). Despite those similarities, there do appear to be differences in growth factor signaling between PHx and APAP overdose. Although the role of HGF in regeneration after acetaminophen toxicity has not been directly investigated, there is evidence that it may not be as important as in the PHx model. Deficiency for plasminogen activator inhibitor 1 (PAI-1), which inhibits the plasminogen activators responsible for plasmin activation and therefore HGF liberation from ECM, unexpectedly increased injury and reduced markers of liver regeneration at multiple time points after APAP treatment (Bajt et al., 2008; Sullivan et al., 2012). In contrast, inhibition of PAI-1 enhances regeneration after PHx (Watanabe et al., 2007). Although another group reported that PAI-1 KO mice also have reduced regeneration after PHx, no quantitation of regeneration markers was provided to support that claim (Beier et al., 2016). In addition, similar to EGFR, the incremental dose model does not support a role for HGF signaling (Bhushan et al., 2014). Finally, serum HGF values are actually 2–4 fold higher in non-survivors of APAP-induced liver injury than in survivors (Hughes et al., 1994), and it is known that over-activation of the HGF receptor c-met can trigger cell death (Zhao, 2007). Therefore, it appears that HGF is either not involved in regeneration in APAP hepatotoxicity or at least that it is not a complete mitogen in the APAP model, though further testing is required for verification.
There is some evidence for metabolic signaling in recovery from APAP-induced liver injury. Bile acids are elevated in both mice and humans after APAP overdose (Woolbright et al., 2012; Bhushan et al., 2013), and cholic acid dietary supplementation greatly accelerates regeneration while cholestyramine reduces it (Bhushan et al., 2013). More recently, it has been suggested that phosphatidic acid (PA) and possibly lysophosphatidic acid (LPA) are involved in liver regeneration in the APAP model (Lutkewitte et al., 2018). PA is dramatically elevated in both liver tissue and plasma after APAP overdose in mice and humans due to deactivation of lipin 1 and 2, and inhibition of PA and LPA synthesis reduces expression of regeneration markers at late time points (Lutkewitte et al., 2018), possibly through an effect on Wnt/β-catenin. Finally, it has been suggested that β-oxidation is important as an energy source for hepatocyte proliferation after APAP overdose similar to PHx (Bhattacharyya et al., 2013), but that hypothesis has not yet been tested.
Overall, we now know that cytokines, Wnt/β-catenin and EGFR are important for liver regeneration in APAP hepatotoxicity, and emerging data also suggest a role for bile acids and lipid second messengers like PA and LPA. Other factors have also been suggested to play a role, such as β-oxidation and sterile inflammation, but more research is needed in those areas. However, it appears that HGF may not be as important in APAP hepatotoxicity as in the PHx model.
Other acute hepatotoxicants.
It is clear that regeneration determines outcome after treatment with numerous other hepatotoxicants, including thioacetaminde, diallyl sulfide, allyl alcohol, bromobenzene and others (Mehendale, 2005). It is also clear that nutrition, age and other factors have a major influence on liver regeneration in these models (Mehendale, 2005). Unfortunately, it is not yet known if growth factors, cytokines, or metabolic intermediates are important in the mechanisms that lead to hepatic recovery in those models. Furthermore, increases in growth factors like HGF, TGFα and VEGF have been reported in several studies of galactosamine hepatotoxicity (Okajima et al., 1990; Webber et al., 1993), but no mechanistic studies have been done to determine whether or not they play a role in regeneration in that model. Additional research with these models could help us to understand the importance particular regeneration signaling pathways in other forms of acute xenobiotic-induced liver injury. Finally, it could be interesting to study hepatocyte proliferation in a model of idiosyncratic DILI (IDILI), including the recently developed Utrecht-Pohl IDILI model involving breakage of immune tolerance (McGill and Jaeschke, 2019).
Alcoholic hepatitis.
Very little effort has been made to study the role of liver regeneration or hepatocyte proliferation in alcohol toxicity, but some data are available. The proportion of hepatocytes in S-phase increases in rats on the Lieber-DeCarli diet, peaks at approximately 10% after 2 weeks, and then declines back to normal levels while evidence of injury persists (Apte et al., 2004). Those data indicate that continued ethanol exposure inhibits liver regeneration and recovery from alcohol-associated liver disease, so early progression of the disease may be due in part to failure of regeneration. Consistent with that, it is well known that heavy ethanol consumption can inhibit regeneration after both PHx and acute hepatotoxicity (Wands et al., 1979; Duguay et al., 1982; Chen et al., 1998; Diehl, 1999; Juskeviciute et al., 2016). Furthermore, regeneration and Stat3 signaling appear to be suppressed in patients with alcoholic cirrhosis compared to other cirrhosis etiologies (Horiguchi et al., 2007), which suggests a role for IL-6 in regeneration after ethanol-induced hepatotoxicity. Interestingly, experiments with zebrafish suggest that VEGF has a detrimental role in alcohol-associated liver disease through activation of stellate cells (Zhang et al., 2016). Finally, recent data demonstrate that augmenter of liver regeneration (ALR) has a role in reducing injury and progression of alcoholic liver disease (Kumar et al., 2016).
Biomarkers of liver regeneration
Biomarkers of liver regeneration may be useful for prediction of survival in ALF, particularly drug-induced ALF. As mentioned, mortality in drug-induced ALF is high. For most ALF patients, a liver transplant is the only life-saving treatment. While N-acetylcysteine (NAC) can be used to treat APAP overdose, NAC loses efficacy beyond 24 h (Rumack et al., 1981). Furthermore, the decision to perform a liver transplant must be made quickly, as the median time from onset of ALF to death is only 2–5 days (Reddy et al., 2016). Unfortunately, the current clinical laboratory markers of liver injury do not predict outcome in acute liver injury (Antoine et al., 2012; McGill et al., 2014; McGill, 2016). Better biomarkers could help clinicians making rapid treatment decisions.
Some studies of liver regeneration biomarkers in ALF have already been performed. It has been known for many years that recovery of hepatic function, reflected by markers like prothrombin time (or INR) and bilirubin, are predictive of outcome. In fact, the commonly used Model for End Stage Liver Disease (MELD) score as well as the King’s College Criteria for poor prognosis in ALF include both (O’Grady et al., 1989; Malinchoc et al., 2000). However, although those markers display good operating characteristics, recovery of liver function typically does not occur until after the peak of injury. Given the rapid progression of ALF, changes are usually too late to be very useful. Thus, novel biomarkers are needed. Schmidt and Dalhoff (2005) reported that circulating α-fetoprotein (AFP), a marker of hepatocyte proliferation, is dramatically elevated in survivors of APAP-induced ALF compared to non- survivors (Schmidt and Dalhoff, 2005). These data were later reproduced in patients with ALF due to other etiologies (Schiødt et al., 2006). Unfortunately, AFP values do not increase until 1–3 days after the peak of liver injury, similar to prothrombin time and bilirubin. Another study found that α-NH-butyric acid is elevated in serum after PHx in mice, and that it is higher in child and adolescent survivors of ALF (Rudnick et al., 2009). AFP and α-NH-butyric acid have the advantage of already being available in some clinical laboratories. AFP is often measured as a biomarker of hepatocellular carcinoma, while α-NH-butyric acid can be measured in some hospitals that have biochemical genetics laboratories with the capability to screen for and monitor metabolic diseases.
Several biomarkers of regeneration that are not yet widely available may be more useful for prediction of patient outcome than current tests. Data from small studies indicate that serum leukocyte cell-derived chemotaxin 2 (Lect2), which is elevated in mice undergoing regeneration after PHx (Sato et al., 2004b), are far lower in non-survivors of ALF (Sato et al., 2004a). More impressively, Church et al. (2018) recently reported that osteopontin could predict poor outcome in a large cohort of DILI patients. Finally, our lab recently discovered that the lipid phosphatidic acid (PA) is important for liver regeneration after APAP hepatotoxicity, and that it is elevated in serum from APAP overdose patients with liver injury (Lutkewitte et al., 2018). However, it is not yet known if circulating PA values correlate with outcome.
Recently, novel computational methods have been used to aid interpretation of liver injury biomarkers. So far, these methods have primarily been used to understand changes in biomarkers retrospectively, after they have been observed in human subjects (Longo et al., 2017; Woodhead et al., 2017), but it may be possible to predict such changes prospectively. Furthermore, consideration of regeneration has been incorporated into such models to estimate the amount of time needed to restore the hepatocytes lost due to injury (Longo et al., 2017; Church and Watkins, 2018). In the future, it may be possible to use related techniques to predict the extent and kinetics of liver regeneration, and therefore patient outcome, using patient data in real-time from monitoring of serum injury biomarkers.
Overall, the future of liver regeneration biomarkers looks bright, but a better understanding of basic regeneration mechanisms after acute injury such as DILI is needed. Also, larger and more detailed studies of existing regeneration biomarkers are needed to fully evaluate their clinical utility.
Conclusion
Liver regeneration after PHx has been thoroughly investigated, but far fewer studies have addressed the mechanisms that drive liver regeneration after DILI. While there is considerable overlap, there are also clear differences such as the relative importance of HGF and EGFR in PHx, APAP, and CCl4 hepatotoxicity. Mechanistically, additional research is needed on the roles of cell signaling and metabolism in regeneration after toxic injury, as well as on the role of regeneration in alcohol-associated liver disease. Clinically, biomarkers of liver regeneration show promise for prediction of outcome in ALF, but more studies are needed to validate them.
Figure 1. Major features of regeneration or repair between partial hepatectomy and hepatotoxicity models.
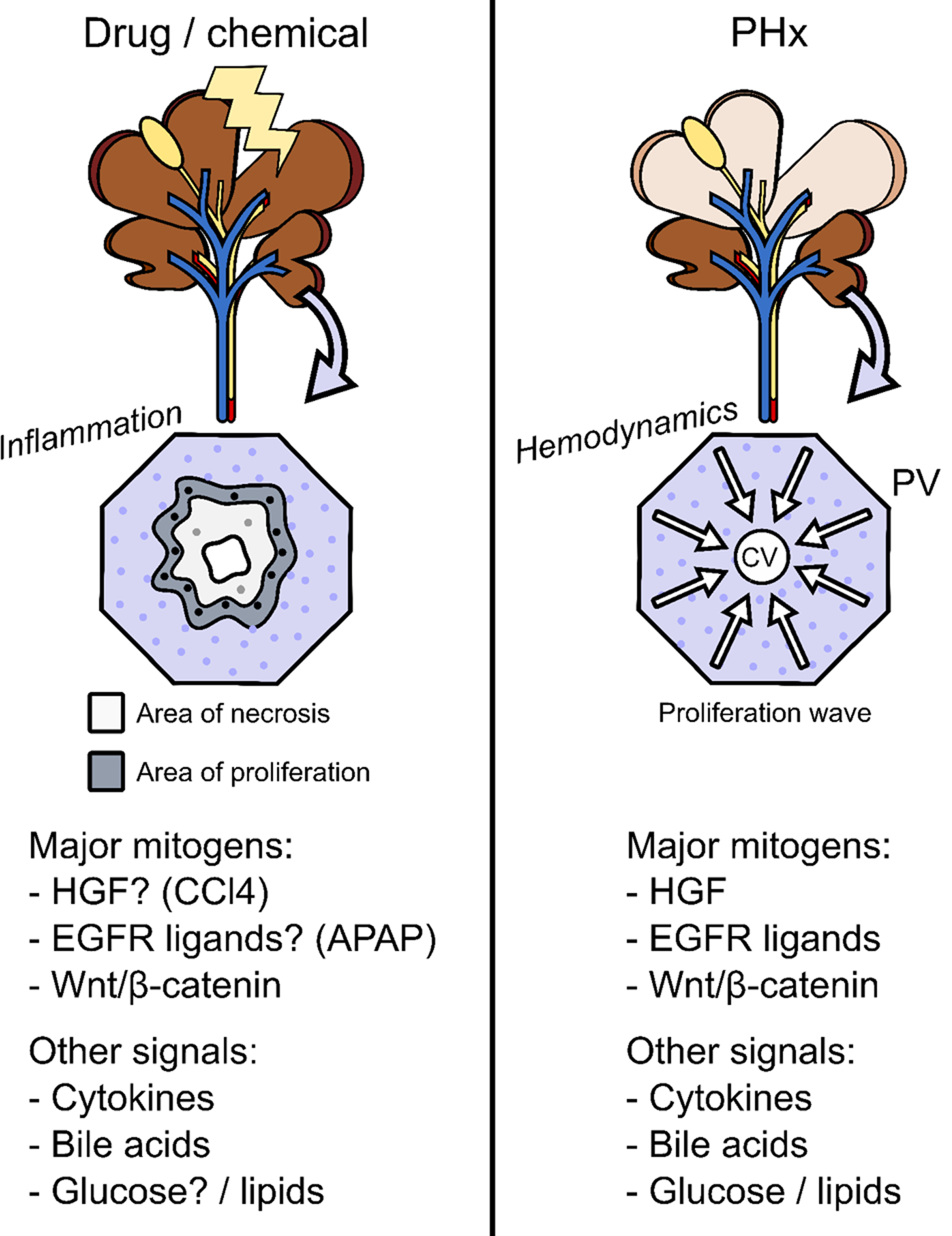
In xenobiotic hepatotoxicity, areas of necrosis gradually develop and hepatocytes surrounding those areas begin to proliferate. The necrosis typically results in extensive sterile inflammation. The roles of different mitogens and other signaling mediators appears to be dependent upon the specific hepatotoxicant in question and even on the dose of the hepatotoxicant. In the partial hepatectomy model (PHx), approximately 70% of the total liver mass is surgically removed. Removal of liver tissue is relatively fast, and results in large changes in blood flow and blood pressure in the liver. The most important mitogens in PHx are hepatocyte growth factor (HGF) and other c-Met ligands, epidermal growth factor receptor (EGFR) ligands, and β-catenin. Other signals, especially cytokines like tumor necrosis factor, also have important roles.
Acknowledgements
This work was supported in part by a Pinnacle Research Award from the American Association for the Study of Liver Diseases (AASLD) Foundation (MRM), start-up funds from the University of Arkansas for Medical Sciences (MRM), and by National Institutes of Health grants R01 DK098414 (UA), P20 GM103549 (UA), P30 GM118247 (UA), and T32 GM106999 (MMC).
ABBREVIATIONS
- APAP
Acetaminophen
- ALF
Acute Liver Failure
- ALI
Acute liver injury
- AFP
Alpha-fetoprotein
- DILI
Drug-induced liver injury
- CCl4
Carbon tetrachloride
- GSK
Glycogen synthase kinase
- HGF
Hepatocyte growth factor
- IL-6
Interleukin-6
- EGFR
Endothelial growth factor receptor
- PHx
Partial hepatectomy
- PA
Phosphatidic acid
- PAI-1
Plasminogen activator inhibitor 1
- PCNA
Proliferating cell nuclear antigen
- TNF
Tumor necrosis factor
- VEGF
Vascular endothelial growth factor
References
- Akerman P, Cote P, Yang SQ, McClain C, Nelson S, Bagby GJ, Diehl AM (1992) Antibodies to tumor necrosis factor-alpha inhibit liver regeneration after partial hepatectomy. Am J Physiol. 263, G579–85. [DOI] [PubMed] [Google Scholar]
- Anand SS, Mehendale HM (2004) Liver regeneration: a critical toxicodynamic response in predictive toxicology. Environ Toxicol Pharmacol. 18, 149–160. [DOI] [PubMed] [Google Scholar]
- Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, Craig DG, Simpson KJ, Jaeschke H, Park BK (2012) Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol. 56, 1070–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Apte U, Bhushan B, Dadhania V (2017) Hepatic defenses against toxicity: tissue regeneration and repair In Roth RA (Ed.), Comprehensive Toxicology. Amsterdam: Elsevier. [Google Scholar]
- Apte U, McCree R, Ramaiah SK (2004) Hepatocyte proliferation is the possible mechanism for the transient decrease in liver injury during steatosis stage of alcoholic liver disease. Toxicol Pathol. 32, 567–76. [DOI] [PubMed] [Google Scholar]
- Apte U, Singh S, Zeng G, Cieply B, Virji MA, Wu T, Monga SP (2009) Beta-catenin activation promotes liver regeneration after acetaminophen-induced injury. Am J Pathol. 175, 1056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajt ML, Cover C, Lemasters JJ, Jaeschke H (2006) Nuclear translocation of endonuclease G and apoptosis-induced factor during acetaminophen-induced liver cell injury. Toxicol Sci. 94, 217–25. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Yan HM, Farhood A, Jaeschke H (2008) Plasminogen activator inhibitor-1 limits liver injury and facilitates regeneration after acetaminophen overdose. Toxicol Sci. 104, 419– 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier JI, Guo L, Ritzenthaler JD, Joshi-Barve S, Roman J, Arteel GE (2016) Fibrin- mediated integrin signaling plays a critical role in hepatic regeneration after partial hepatectomy in mice. Ann Hepatol. 15, 762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Pence L, Beger R, Chaudhuri S, McCullough S, Yan K, Simpson P, Hennings L, Hinson J, James L (2013) Acylcarnitine profiles in acetaminophen toxicity in the mouse: comparison to toxicity, metabolism and hepatocyte regeneration. Metabolites. 3, 606–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan B, Borude P, Edwards G, Walesky C, Cleveland J, Li F, Ma X, Apte U (2013) Role of bile acids in liver injury and regeneration following acetaminophen overdose. Am J Pathol. 183, 1518–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan B, Chavan H, Borude P, Xie Y, Du K, McGill MR, Lebofsky M, Jaeschke H, Krishnamurthy P, Apte U (2017b) Dual role of epidermal growth factor receptor in liver injury and regeneration after acetaminophen overdose in mice. Toxicol Sci. 155, 363–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan B, Poudel S, Manley MW Jr., Roy N, Apte U (2017a) Inhibition of glycogen synthase kinase 3 accelerated liver regeneration after acetaminophen-induced hepatotoxicity in mice. Am J Pathol. 187, 543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan B, Walesky C, Manley M, Gallagher T, Borude P, Edwards G, Monga SP, Apte U (2014) Pro-regenerative signaling after acetaminophen-induced acute liver injury in mice identified using a novel incremental dose model. Am J Pathol. 184, 3013–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breu AC, Patwardhan VR, Nayor J, Ringwala JN, Devore ZG, Ganatra RB, Hathorn KE, Horton L, Iriana S, Tapper EB (2018) A Multicenter Study Into Causes of Severe Acute Liver Injury. Clin Gastroenterol Hepatol. doi: 10.1016/j.cgh.2018.08.016. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Burr AW, Toole K, Chapman C, Hines JE, Burt AD (1998) Anti-hepatocyte growth factor antibody inhibits hepatocyte proliferation during liver regeneration. J Pathol. 185:298– 302. [DOI] [PubMed] [Google Scholar]
- Chanda S, Mangipudy RS, Warbritton A, Bucci TJ, Mehendale HM (1995) Stimulated hepatic tissue repair underlies heteroprotection by thioacetamide against acetaminophen- induced lethality. Hepatology. 21, 477–486. [PubMed] [Google Scholar]
- Chanda S, Mehendale HM (1995) Nutritional impact on the final outcome of liver injury inflicted by model hepatotoxicants: effect of glucose loading. FASEB J. 9, 240–5. [PubMed] [Google Scholar]
- Chen J, Ishac EJ, Dent P, Kunos P, Gao B (1998) Effects of ethanol on mitogen-activated protein kinase and stress-activated protein kinase cascades in normal and regenerating liver. Biochem J. 334, 669–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church RJ, Watkins PB (2018) In silico modeling to optimize interpretation of liver safety biomarkers in clinical trials. Exp Biol Med (Maywood). 243, 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church RJ, Kullak-Ublick GA, Aubrecht J, Bonkovsky HL, Chalasani N, Fontana RJ, Goepfert JC, Hackman F, King NMP, Kirby S, Kirby P, Marcinak J, Ormarsdottir S, Schomaker SJ, Schuppe-Koistinen I, Wolenski F, Arber N, Merz M, Sauer JM, Andrade RJ, van Bömmel F, Poynard T, Watkins PB (2018) Candidate biomarkers for diagnosis and prognosis of drug-induced liver injury: an international collaborative effort. Hepatology. doi: 10.1002/hep.29802. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R (1996) Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 274, 1379–83. [DOI] [PubMed] [Google Scholar]
- Dadhania VP, Bhushan B, Apte U, Mehendale HM (2017) Wnt/β-Catenin Signaling Drives Thioacetamide-Mediated Heteroprotection Against Acetaminophen-Induced Lethal Liver Injury. Dose Response. 15, 1559325817690287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl AM (1999) Effect of ethanol on tumor necrosis factor signaling during liver regeneration. Clin Biochem. 32, 571–8. [DOI] [PubMed] [Google Scholar]
- Du K, Xie Y, McGill MR, Jaeschke H (2015) Pathophysiological significance of c-jun N- terminal kinase in acetaminophen hepatotoxicity. Expert Opin Drug Metab Toxicol. 11, 1769– 1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguay L, Coutu D, Hetu C, Joly JG (1982) Inhibition of liver regeneration by chronic alcohol administration. Gut. 23, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahower BC, McCullough SS, Hennings L, Simpson PM, Stowe CD, Saad AG, Kurten RC, Hinson JA, James LP (2010) Human recombinant vascular endothelial growth factor reduces necrosis and enhances hepatocyte regeneration in a mouse model of acetaminophen toxicity. J Pharmacol Exp Ther. 334, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahower B, McCullough SS, Kurten R, Lamps LW, Simpson P, Hinson JA, James LP (2006) Vascular endothelial growth factor and hepatocyte regeneration in acetaminophen toxicity. Am J Physiol Gastrointest Liver Physiol. 291, G102–9. [DOI] [PubMed] [Google Scholar]
- Fausto N (2000) Liver regeneration. J Hepatol. 32, 19–31. [DOI] [PubMed] [Google Scholar]
- Galvin Z, McDonough A, Ryan J, Stewart S (2015) Blood alanine aminotransferase levels >1,000 IU/l - causes and outcomes. Clin Med (Lond). 15, 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganger DR, Rule J, Rakela J, Bass N, Reuben A, Stravitz RT, Sussman N, Larson AM, James L, Chiu C, Lee WM; Acute Liver Failure Study Group. (2018) Acute liver failure of indeterminate etiology: a comprehensive systematic approach by an expert committee to establish causality. Am J Gastroenterol. 113, 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H (2002) Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci. 67, 322–8. [DOI] [PubMed] [Google Scholar]
- Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N (2006) c-Jun N- terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 131, 165–78. [DOI] [PubMed] [Google Scholar]
- Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N (2008) Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 283, 13565–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins GM, Anderson RM (1931) Experimental pathology of the liver. Arch Pathol. 12, 186–202. [Google Scholar]
- Horiguchi N, Ishac EJ, Gao B (2007) Liver regeneration is suppressed in alcoholic cirrhosis: correlation with decreased STAT3 activation. Alcohol. 41, 271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang D, Lin L, Jiang R, Dai J, Tang L, Yang Y, Ge P, Wang B, Zhang L (2018) Potential roles of AMP-activated protein kinase in liver regeneration in mice with acute liver injury. Mol Med Rep. 17, 5390–95. [DOI] [PubMed] [Google Scholar]
- Hughes RD, Zhang L, Tsubouchi H, Daikuhara Y, Williams R (1994) Hepatocyte growth factor and biliprotein levels and outcome in fulminant hepatic failure. J Hepatol. 20, 106–11. [DOI] [PubMed] [Google Scholar]
- Huh CG, Factor VM, Sánchez A, Uchida K, Conner EA, Thorgeirsson SS (2004) Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci USA. 101, 4477–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James LP, Kurten RC, Lamps LW, McCullough S, Hinson JA (2005) Tumour necrosis factor receptor 1 and hepatocyte regeneration in acetaminophen toxicity: a kinetic study of proliferating cell nuclear antigen and cytokine expression. Basic Clin Pharmacol Toxicol. 97, 8–14. [DOI] [PubMed] [Google Scholar]
- James LP, Lamps LW, McCullough S, Hinson JA (2003) Interleukin 6 and hepatocyte regeneration in acetaminophen toxicity in the mouse. Biochem Biophys Res Commun. 309, 857–63. [DOI] [PubMed] [Google Scholar]
- Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, Pohl LR (2002) Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. 15, 1504–13. [DOI] [PubMed] [Google Scholar]
- Juskeviciute E, Dippold RP, Antony AN, Swarup A, Vadigepalli R, Hoek JB (2016) Inhibition of miR-21 rescues liver regeneration after partial hepatectomy in ethanol-fed rats. Am J Physiol Gastrointest Liver Physiol. 311, G794–G806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayano K, Yasunaga M, Kubota M, Takenaka K, Mori K, Yamashita A, Kubo Y, Sakaida I, Okita K, Sanuki K (1992) Detection of proliferating hepatocytes by immunohistochemical staining for proliferating cell nuclear antigen (PCNA) in patients with acute hepatic failure. Liver. 12, 132–6. [DOI] [PubMed] [Google Scholar]
- Kennon-McGill S, McGill MR (2018) Extrahepatic toxicity of acetaminophen: critical evaluation of the evidence and proposed mechanisms. J Clin Transl Res. 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodavanti PR, Joshi UM, Young RA, Bell AN, Mehendale HM (1989a) Role of hepatocellular regeneration in chlordecone potentiated hepatotoxicity of carbon tetrachloride. Arch Toxicol. 63, 367–75. [DOI] [PubMed] [Google Scholar]
- Kodavanti PR, Joshi UM, Young RA, Meydrech EF, Mehendale HM (1989) Protection of hepatotoxic and lethal effects of CCl4 by partial hepatectomy. Toxicol Pathol. 17, 494–505. [DOI] [PubMed] [Google Scholar]
- Kon K, Kim JS, Jaeschke H, Lemasters JJ (2004) Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 40, 1170–9. [DOI] [PubMed] [Google Scholar]
- Kumar S, Wang J, Rani R, Gandhi CR (2016) Hepatic deficiency of augmenter of liver regeneration exacerbates alcohol-induced liver injury and promotes fibrosis in mice. PLoS One. 11, e0147864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiødt FV, Ostapowicz G, Shakil AO, Lee WM; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 42, 1364–1372. [DOI] [PubMed] [Google Scholar]
- Lee WM (2008) Etiologies of acute liver failure. Semin Liver Dis. 28, 142–52. [DOI] [PubMed] [Google Scholar]
- Leevy CM, Hollister RM, Schmid R, MacDonald RA, Davidson CS (1959) Liver regeneration in experimental carbon tetrachloride intoxication. Proc Soc Exp Biol Med. 102, 672–5. [DOI] [PubMed] [Google Scholar]
- Limaye PB, Apte UM, Shankar K, Bucci TJ, Warbritton A, Mehendale HM (2003) Calpain released from dying hepatocytes mediates progression of acute liver injury induced by model hepatotoxicants. Toxicol Appl Pharmacol. 191, 211–226. [DOI] [PubMed] [Google Scholar]
- Limaye PB, Bhave VS, Palkar PS, Apte UM, Sawant SP, Yu S, Latendresse JR, Reddy JK, Mehendale HM (2006) Upregulation of calpastatin in regenerating and developing rat liver: role in resistance against hepatotoxicity. Hepatology. 44, 379–388. [DOI] [PubMed] [Google Scholar]
- Longo DM, Generaux GT, Howell BA, Siler SQ, Antoine DJ, Button D, Caggiano A, Eisen A, Iaci J, Stanulis R, Parry T, Mosedale M, Watkins PB. (2017) Refining Liver Safety Risk Assessment: Application of Mechanistic Modeling and Serum Biomarkers to Cimaglermin Alfa (GGF2) Clinical Trials. Clin Pharmacol Ther. 102, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutkewitte AJ, Schweitzer GG, Kennon-McGill S, Clemens MM, James LP, Jaeschke H, Finck BN, McGill MR (2018) Lipin deactivation after acetaminophen overdose causes phosphatidic acid accumulation in liver and plasma in mice and humans and enhances liver regeneration. Food Chem Toxicol. 115, 273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinchoc M, Kamath PS, Gordon FD, Peine CJ, Rank J, ter Borg PC (2000) A model to predict poor survival in patients undergoing transjugular intrahepatic portosystemic shunts. Hepatology. 31, 864–71. [DOI] [PubMed] [Google Scholar]
- Mangipudy RS, Chanda S, Mehendale HM (1995) Tissue repair response as a function of dose in thioacetamide hepatotoxicity. Environ Health Perspect. 103, 260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masubuchi Y, Bourdi M, Reilly TP, Graf ML, George JW, Pohl LR (2003) Role of interleukin-6 in hepatic heat shock protein expression and protection against acetaminophen- induced liver disease. Biochem Biophys Res Commun. 304, 207–12. [DOI] [PubMed] [Google Scholar]
- Mehendale HM (1989) Mechanism of the lethal interaction of chlordecone and CCl4 at non- toxic doses. Toxicol Lett. 49, 215–241. [DOI] [PubMed] [Google Scholar]
- Milne LS (1909) The histology of liver tissue regeneration. J Pathol Bacteriol. 13, 127–158. [Google Scholar]
- McGill MR (2016) The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 15, 817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Jaeschke H (2013) Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 30, 2174–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Jaeschke H (2019) Animal models of drug-induced liver injury. Biochim Biophys Acta Mol Basis Dis. doi: 10.1016/j.bbadis.2018.08.037 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H (2012b) The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 122, 1574–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Staggs VS, Sharpe MR, Lee WM, Jaeschke H; Acute Liver Failure Study Group. (2014) Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology. 60, 1336–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H (2012a) Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol. 264, 387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H (2011) HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology. 53, 974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehendale HM (2005) Tissue repair: an important determinant of final outcome of toxicant- induced injury. Toxicol Pathol. 33, 41–51. [DOI] [PubMed] [Google Scholar]
- Meyers LL, Beierschmitt WP, Khairallah EA, Cohen SD (1988) Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol Appl Pharmacol. 93, 378–87. [DOI] [PubMed] [Google Scholar]
- Michalopoulos GK (2013) Principles of liver regeneration and growth homeostasis. Compr Physiol. 3, 485–513. [DOI] [PubMed] [Google Scholar]
- Michalopoulos GK (2014) Advances in liver regeneration. Expert Rev Gastroenterol Hepatol. 8, 897–907. [DOI] [PubMed] [Google Scholar]
- Michalopoulos GK (2017) Hepatostat: liver regeneration and normal liver tissue maintenance. Hepatology. 65, 1384–92. [DOI] [PubMed] [Google Scholar]
- Moh A, Iwamoto Y, Chai GX, Zhang SS, Kano A, Yang DD, Zhang W, Wang J, Jacoby JJ, Gao B, Flavell RA, Flu XY (2007) Role of STAT3 in liver regeneration: survival, DNA synthesis, inflammatory reaction and liver mass recovery. Lab Invest. 87, 1018–28. [DOI] [PubMed] [Google Scholar]
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R (1989) Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 97, 439–45. [DOI] [PubMed] [Google Scholar]
- Okajima A, Miyazawa K, Kitamura N (1990) Primary structure of rat hepatocyte growth factor and induction of its mRNA during liver regeneration following hepatic injury. Eur J Biochem. 193, 375–81. [DOI] [PubMed] [Google Scholar]
- Ostapowicz G, Fontana RJ, Schiødt FV, Larson A, Davern TJ, Han SH, McCashland TM, Shakil AO, Hay JE, Hynan L, Crippin JS, Blei AT, Samuel G, Reisch J, Lee WM; U.S. Acute Liver Failure Study Group. (2002) Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 137, 947–954. [DOI] [PubMed] [Google Scholar]
- Phaneuf D, Moscioni AD, LeClair C, Raper SE, Wilson JM (2004) Generation of a mouse expressing a conditional knockout of the hepatocyte growth factor gene: demonstration of impaired liver regeneration. DNA Cell Biol. 23, 592–603. [DOI] [PubMed] [Google Scholar]
- Placke ME, Ginsberg GL, Wyand DS, Cohen SD (1987) Ultrastructural changes during acute acetaminophen-induced hepatotoxicity in the mouse: a time and dose study. Toxicol Pathol. 15, 431–8. [DOI] [PubMed] [Google Scholar]
- Ramachandran A, Lebofsky M, Baines CP, Lemasters JJ, Jaeschke H (2011b) Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic Res. 45, 156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Lebofsky M, Weinman SA, Jaeschke H (2011a) The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 251, 226–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SB, Mangipudy RS, Mehendale HM (1997) Tissue injury and repair as parallel and opposing responses to CCl4 hepatotoxicity: a novel dose-response. Toxicology. 118, 181–193. [DOI] [PubMed] [Google Scholar]
- Rao SB, Mehendale HM (1989) Protective role of fructose 1,6-bisphosphate during CCl4 hepatotoxicity in rats. Biochem J. 262, 721–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SB, Mehendale HM (1991) Effect of colchicine on hepatobiliary function in CCl4 treated rats. Biochem Pharmacol. 42, 2323–32. [DOI] [PubMed] [Google Scholar]
- Reddy KR, Ellerbe C, Schilsky M, Stravitz RT, Fontana RJ, Durkalsi V, Lee WM; Acute Liver Failure Study Group. Determinants of outcome among patients with acute liver failure listed for liver transplantation in the United States. Liver Transpl. 2016;22:505–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AB, Kurten RC, McCullough SS, Brock RW, Hinson JA (2005) Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Pharmacol Exp Ther. 312, 509–16. [DOI] [PubMed] [Google Scholar]
- Río A, Gassull MA, Aldeguer X, Ojanguren I, Cabré E, Fernández E (2008) Reduced liver injury in the interleukin-6 knockout mice by chronic carbon tetrachloride administration. Eur J Clin Invest. 38, 306–16. [DOI] [PubMed] [Google Scholar]
- Rudnick DA, Dietzen DJ, Turmelle YP, Shepherd R, Zhang S, Belle SH, Squires R; Pediatric Acute Liver Failure Study Group. (2009) Serum alpha-NH-butyric acid may predict spontaneous survival in pediatric acute liver failure. Pediatr Transplant. 13, 223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumack BH, Peterson RC, Koch GG, Amara IA (1981) Acetaminophen overdose. 662 cases with evaluation of oral acetylcysteine treatment. Arch Intern Med. 141, 380–5. [DOI] [PubMed] [Google Scholar]
- Sadler KC, Krahn KN, Gaur NA, Ukomadu C (2007) Liver growth in the embryo and during liver regeneration in zebrafish requires the cell cycle regulator, uhrf1. Proc Natl Acad Sci USA. 104, 1570–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Watanabe H, Kameyama H, Kobayashi T, Yamamoto S, Takeishi T, Hirano K, Oya H, Nakatsuka H, Watanabe T, Kokai H, Yamagoe S, Suzuki K, Oya K, Kojima K, Hatakeyama K (2004a) Serum LECT2 level as a prognostic indicator in acute liver failure. Transplant Proc. 36, 2359–61. [DOI] [PubMed] [Google Scholar]
- Sato Y, Watanabe H, Kameyama H, Kobayashi T, Yamamoto S, Takeishi T, Hirano K, Oya H, Nakatsuka H, Watanabe T, Kokai H, Yamagoe S, Suzuki K, Oya K, Kojima K, Hatakeyama K (2004b) Changes in serum LECT 2 levels during the early period of liver regeneration after adult living related donor liver transplantation. Transplant Proc. 36, 2357– 8. [DOI] [PubMed] [Google Scholar]
- Shanmukhappa K, Matte U, Degen JL, Bezerra JA (2009) Plasmin-mediated proteolysis is required for hepatocyte growth factor activation during liver repair. J Biol Chem. 284, 12917– 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheving LA, Zhang X, Stevenson MC, Threadgill DW, Russell WE (2015) Loss of hepatocyte EGFR has no effect alone but exacerbates carbon tetrachloride-induced liver injury and impairs regeneration in hepatocyte Met-deficient mice. Am J Physiol Gastrointest Liver Physiol. 308, G364–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiødt FV, Ostapowicz G, Murray N, Satyanarana R, Zaman A, Munoz S, Lee WM (2006) Alpha-fetoprotein and prognosis in acute liver failure. Liver Transpl. 12, 1776–81. [DOI] [PubMed] [Google Scholar]
- Schmidt LE, Dalhoff K (2005) Alpha-fetoprotein is a predictor of outcome in acetaminophen- induced liver injury. Hepatology. 41, 26–31. [DOI] [PubMed] [Google Scholar]
- Subbotin VM. Arguments on the origin of the vertebrate liver and the Amphioxus hepatic diverticulum: a hypothesis on evolutionary novelties. Pisma v Vavilovskii Zhurnal. 2017. http://www.bionet.nsc.ru/vogis/download/hypothesis/appx1.pdf Accessed on 09/12/2018. [Google Scholar]
- Sullivan BP, Kassel KM, Jone A, Flick MJ, Luyendyk JP (2012) Fibrin(ogen)- independent role of plasminogen activators in acetaminophen-induced liver injury. Am J Pathol. 180, 2321–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper EB, Sengupta N, Bonder A (2015) The Incidence and Outcomes of Ischemic Hepatitis: A Systematic Review with Meta-analysis. Am J Med. 128, 1314–1321. [DOI] [PubMed] [Google Scholar]
- Thakore KN, Mehendale HM (1991) Role of hepatocellular regeneration in CCl4 autoprotection. Toxicol Pathol. 19, 47–58. [DOI] [PubMed] [Google Scholar]
- Trautwein C, Rakermann T, Niehof M, Rose-John S, Manns MP (1996) Acute-phase response factor, increased binding, and target gene transcription during liver regeneration. Gastroenterology. 110, 1854–62. [DOI] [PubMed] [Google Scholar]
- Wands JR, Carter EA, Bucher NL, Isselbacher KJ (1979) Inhibition of hepatic regeneration in rats by acute and chronic ethanol intoxication. Gastroenterology. 77, 528–31. [PubMed] [Google Scholar]
- Watanabe K, Togo S, Takahashi T, Matsuyama R, Yamamoto H, Shimizu T, Makino H, Matsuo K, Morioka D, Kubota T, Nagashima Y, Shimada H (2007) PAI-1 plays an important role in liver failure after excessive hepatectomy in the rat. J Surg Res. 143, 13–99. [DOI] [PubMed] [Google Scholar]
- Webber EM, FitzGerald MJ, Brown PI, Bartlett MH, Fausto N (1993) Transforming growth factor-alpha expression during liver regeneration after partial hepatectomy and toxic injury, and potential interactions between transforming growth factor-alpha and hepatocyte growth factor. Hepatology. 18, 1422–31. [PubMed] [Google Scholar]
- Weber LW, Boll M, Stampfl A (2003) Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 33, 105–136. [DOI] [PubMed] [Google Scholar]
- Woodhead JL, Brock WJ, Roth SE, Shoaf SE, Brouwer KL, Church R, Grammatopoulos TN, Stiles L, Siler SQ, Howell BA, Mosedale M, Watkins PB, Shoda LK. (2017) Application of a Mechanistic Model to Evaluate Putative Mechanisms of Tolvaptan Drug-Induced Liver Injury and Identify Patient Susceptibility Factors. Toxicol Sci. 155, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolbright BL, Ramachandran A, McGill MR, Yan HM, Bajt ML, Sharpe MR, Lemasters JJ, Jaeschke H (2012) Lysosomal instability and cathepsin B release during acetaminophen hepatotoxicity. Basic Clin Pharmacol Toxicol. 111, 417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Du K, Dorko K, Kumer SC, Schmitt TM, Ding WX, Jaeschke H (2015) Mitochondrial protein adducts formation and mitochondrial dysfunction during N- acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol Appl Pharmacol. 289, 213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H (2014) Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol. 279, 266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Fausto N (1998) Deficient liver regeneration after carbon tetrachloride injury in mice lacking type 1 but not type 2 tumor necrosis factor receptor. Am J Pathol. 152, 1577–89. [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Kirillova I, Peschon JJ, Fausto N (1997) Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA. 94, 1441–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Ellis JL, Yin C (2016) Inhibition of vascular endothelial growth factor signaling facilitates liver repair from acute ethanol-induced injury in zebrafish. Dis Model Mech. 9, 1383–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Difrancesca D, Wang X, Zarnegar R, Michalopoulos GK, Yin XM (2007) Promotion of Fas-mediated apoptosis in Type II cells by high doses of hepatocyte growth factor bypasses the mitochondrial requirement. J Cell Physiol. 213, 556–63. [DOI] [PMC free article] [PubMed] [Google Scholar]