

Abstract
Diabetes has a significant and negative impact on wound healing, which involves complex interactions between multiple cell types. Keratinocytes play a crucial role in the healing process by rapidly covering dermal and mucosal wound surfaces to reestablish an epithelial barrier with the outside environment. Keratinocytes produce multiple factors to promote reepithelialization and produce factors that enhance connective tissue repair through the elaboration of mediators that stimulate angiogenesis and production of connective tissue matrix. Among the factors that keratinocytes produce to aid healing are transforming growth factor-β (TGF-β), vascular endothelial growth factor-A (VEGF-A), connective tissue growth factor (CTGF), and antioxidants. In a diabetic environment, this program is disrupted, and keratinocytes fail to produce growth factors and instead switch to a program that is detrimental to healing. Changes in keratinocyte behavior have been linked to high glucose and advanced glycation end products that alter the activities of the transcription factor, FOXO1. This review examines reepithelialization and factors produced by keratinocytes that upregulate connective tissue healing and angiogenesis and how they are altered by diabetes.
1. Reepithelialization
The epidermis covers the dermis, provides a barrier to pathogens, and regulates water release. Approximately 90% of cells in the epidermis are keratinocytes with the remainder consisting of leukocytes such as Langerhans cells, tactile epithelial (Merkel) cells, and melanocytes, which are separated from the underlying dermis by a basement membrane [1]. Barrier function is achieved by adherens junctions formed by cadherins, which are linked to actin filaments in the cytoplasm that link epidermal cells. As a barrier exposed to a complicated environment, the epidermis is primed to repair wounds when disrupted. Reepithelialization, which resurfaces the wound with new epithelium, is an important process in wound healing [2].
Reepithelialization of a skin wound occurs when basal keratinocytes migrate from the wound edge and dermal appendages (hair follicles, sweat glands, and sebaceous glands). Before migration occurs across an open wound, new granulation tissue must form. After initiating epithelial migration, keratinocytes proliferate, providing a sufficient number of cells for subsequent migration. Reepithelialization, thus, requires the formation of a provisional wound bed matrix and the migration and proliferation of keratinocytes.
Keratinocyte migration involves actin filament remodeling of the cytoskeleton and cell attachment and deattachment [2, 3]. For protrusion, actin filaments form lamellipodium that push forward; for traction, they assemble into antiparallel arrays with myosin II [4]. Keratinocyte migration also involves integrins [5] that consist of an alpha and a beta subunit that bind to specific extracellular matrix proteins with relatively high affinity. The integrins α6β4, α3β1, and αvβ6 participate in keratinocyte migration. Integrins α6β4 and α3β1 bind to laminin, while αvβ6 binds to fibronectin and tenascin [6, 7]. These specific integrin-extracellular matrix protein interactions are critical to reepithelialization since laminin, fibronectin, and tenacin are components of the provisional matrix in the wound bed. Studies examining mice with β6 gene deletion or mice treated with anti-αvβ6 antibody exhibit impaired reepithelialization, establishing a mechanistic relationship between their activity and keratinocyte migration in wound healing in vivo [8].
Matrix metalloproteinases (MMP) facilitate cell migration by enabling cell–matrix detachment, an essential aspect of movement [9]. The functional importance of MMPs in facilitating keratinocyte migration and wound healing has been shown by use of specific inhibitors and genetic deletion of MMP2 or MMP13. MMP inhibitors or MMP gene deletion delay keratinocyte migration and interfere with reepithelialization in vivo [10, 11].
Growth factors that stimulate keratinocyte migration include transforming growth factor-β (TGF-β), heparin-binding epidermal growth factor-like growth factor (HB-EGF), and fibroblast growth factor (FGF). Lineage-specific TGF-β1 deletion in keratinocytes impairs reepithelialization and wound healing [12]. Similarly, inhibition of TGF-β activity by antibody treatment results in diminished reepithelialization [13], while application of TGF-β can accelerate healing in diabetic animals where it is deficient [14, 15]. Mice with deletion of FGF2 have delays in wound healing linked to reduced keratinocyte migration and impaired lamellipodia formation [16]. A summary of growth factors produced by keratinocytes is provided in Table 1.
Table 1.
Examples of growth factors produced by keratinocytes that promote wound healing.
| Growth factor | Function | In vivo experiments and results | In vitro experiments and results |
|---|---|---|---|
| FGF2 | Reepithelialization and granulation tissue formation | Global deletion of FGF2 in mice cause delayed skin wound healing [16]. | FGF2 application stimulates human keratinocytes migration in vitro [88]. |
| TGF-β | Inflammation | Global deletion of TGF-β in mice cause delayed wound healing [12]; TGF-β application in rats at the wound site accelerates wound healing [14, 53]. | TGF-β application stimulates keratinocyte migration in skin explants by upregulating the expression of integrins [51, 52]. |
| Reepithelialization | |||
| Keratinocytes migration | |||
| Granulation tissue formation | |||
| VEGF-A | Angiogenesis | Keratinocyte-specific deletion of VEGF-A in mice reduces blood vessel formation at the wound site [74]. Topical VEGF in diabetic mice accelerate wound healing [74]. | VEGF-A stimulates endothelial cells in vitro to increase tube formation [74]. |
| HB-EGF | Reepithelialization | Keratinocyte-specific HB-EGF-deficient mice have delayed migration and wound closure [89]. | HB-EGF overexpression by epidermal keratinocytes increases motility [90]. |
2. Keratinocyte Migration, Proliferation, and Reepithelialization
Reepithelialization relies on the migration and proliferation of keratinocytes [17]. Migration of keratinocytes occurs within hours of wounding and precedes proliferation [18, 19]. Proliferation starts at day or so later and is needed to supply a sufficient number of keratinocytes to cover the wound surface [17]. Proliferating keratinocytes are located distal to the leading edge of the wound and are found in basal epidermal stem cells, hair follicle bulges, and sebaceous glands [18, 19]. The contribution of keratinocyte proliferation to reepithelialization has been shown through the use of inhibitors. Inhibition of proliferation by 5-fluorouracil (5-FU) or mitomycin-c impedes reepithelialization [19, 20]. In an alternative approach, genetic overexpression of cyclin-dependent kinase inhibitor 1B lowers the rate of epidermal migration, establishing the contribution of proliferation to reepithelialization [20]. Growth factors produced as a result of injury are released by a number of cell types to stimulate keratinocyte proliferation [21], and integrins on the keratinocyte surface facilitate accumulation of intracellular signaling mediators to enhance proliferation [17]. Even after epithelial closure, proliferation of keratinocytes continues [19, 22]. Growth factors that stimulate keratinocyte migration and proliferation are shown in Table 1.
3. The Impact of Diabetes on Reepithelialization
Diabetes is a metabolic disease characterized by hyperglycemia and has two major forms, type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM). T1DM results from an absolute insulin secretion deficiency, while T2DM is caused by insulin resistance and inadequate compensatory insulin secretion [23]. Diabetic wounds have a microenvironment with elevated levels of glucose, advanced glycation end products (AGEs), reactive oxygen species (ROS), and inflammatory cytokines. High glucose levels reduce keratinocyte migration and proliferation in vivo and in scratch wound assays in vitro [24]. Advanced glycation end products (AGEs) impair keratinocyte proliferation and migration with effects that are similar to those induced by high glucose [25]. Interestingly, the reduced migration is linked to high production of MMP9 and reduced expression of tissue inhibitor of matrix metalloproteinase, TIMP. Oxidative stress is a disturbance in prooxidant and antioxidant balance and is characterized by increased levels of reactive oxygen species (ROS) [26]. Diabetes increases ROS levels that are harmful to wound healing and inhibit keratinocyte migration and proliferation [27]. Furthermore, increased ROS levels stimulate apoptosis, which may be an additional factor that impairs the healing response in diabetes [28]. High levels of ROS induce the production of inflammatory cytokines such as TNFα. When TNF is inhibited in diabetic wounds, reepithelialization is significantly enhanced [29]. In addition, diabetes causes prolonged chemokine expression that leads to difficulty in downregulating inflammation, contributing to poor healing outcomes [30]. In diabetic animals, high levels of TNF and reduced levels of TGF-beta are linked to poor healing outcomes that are also characterized by a high percentage of M1 macrophages relative to M2 macrophages at later time points [31]. When TNF is inhibited, there is restoration of M2 macrophage levels and improved healing [31]. Thus, the failure to switch from an M1 to M2 phenotype may interfere with resolution of inflammation and delayed diabetic wound healing.
Diabetic wounds that heal slowly are more susceptible to the formation of a biofilm on the wound surface and the formation of chronic, nonhealing wounds [32]. All dermal and mucosal wounds must cope with the presence of bacteria. Slowly healing wounds are susceptible to biofilm formation at the wound site [33, 34]. Wound infection may impair healing through prolonged inflammation that interferes with the transition from the inflammatory and proliferative phases to the maturation phase of healing [35, 36]. In addition, high levels of ROS contribute to reduced microbial diversity that increases the likelihood that a biofilm-forming specie will dominate the wound, promote biofilm formation, and prevent wound closure [37]. Thus, reversing the redox imbalance at wound sites may one day be an adjunct to limit the formation of chronic wounds [38].
In human dermal wounds, the dominant bacteria are Staphylococci (25%), Corynebacterium (20%), Clostridiales (18%), and Pseudomonas (12%) [39]. Staphylococcus aureus is particularly linked to delayed reepithelialization and closure [40]. High throughput sequencing studies indicate that strain-level variations of Staphylococcus aureus as well as Corynebacterium striatum and Alcaligenes faecalis correspond with poor wound healing outcomes in diabetic humans [41]. Moreover, due to antibiotic resistance, antibiotics are less effective than debridement in reducing wound biofilm and improving healing in diabetic patients [41]. Bacteria can influence the events of healing by inducing inflammation that retards reepithelialization as described above. Bacteria can also have direct effects on keratinocytes by stimulating apoptosis, reducing migration, and decreasing proliferation [42]. Thus, the presence of bacteria disrupts the careful interplay between keratinocytes and immune cells that is necessary for initial inflammation, subsequent resolution of inflammation, and successful transition to later stages in healing. An effective host response is particularly important in oral wounds as deletion of IL-1 has a deleterious effect on oral mucosal healing that is rescued by antibiotic treatment [43]. In contrast, IL-1 deletion has only a small effect on dermal wounds.
4. FOXO1, Diabetes, and Reepithelialization
Transcription factors organize cellular activity to orchestrate a coordinated response to wound healing. The forkhead box O1 (FOXO1) transcription factors stimulate a diverse array of cellular activities including differentiation, apoptosis, DNA repair, response to oxidative stress, inflammation, and the expression of growth factors [44, 45]. There are four different isoforms of FOXO, FOXO1, FOXO3, FOXO4, and FOXO6. For wound healing, this review will focus on FOXO1 specifically, since it is the best studied. Activation of FOXO1 results in its translocation to the nucleus and regulation of gene transcription. FOXO1 expression and activation are significantly increased in keratinocytes by wounding [44, 46, 47]. Keratinocyte-specific FOXO1 deletion results in delayed dermal and mucosal healing [15, 47–49] since FOXO1 activity in keratinocytes is needed for normal keratinocyte migration, reepithelialization and keratinocyte-stimulated connective tissue formation, and angiogenesis in the wound bed [48, 50].
FOXO1 regulates several genes that participate in wound repair. TGF-β1 is an important FOXO1 gene target. FOXO1 deletion results in significantly reduced TGF-β1 expression in keratinocytes in vivo [15, 50]. This regulation is significant since TGF-β1 plays a primary role in reepithelialization. TGF-β treatment enhances reepithelialization in porcine cutaneous wounds in vivo and stimulates human keratinocyte closure of scratch wounds in vitro and keratinocyte migration in transwell assays [51, 52]. TGF-β application can also accelerate incisional wound closure [14, 53]. TGF-β stimulates keratinocyte migration in part, by inducing integrin expression and by recruiting macrophages and fibroblasts to wound areas. TGF-β1 rescues impaired reepithelialization caused by FOXO1 deletion, demonstrating that TGF-β1 is critical for optimal reepithelialization in vivo. FOXO1 induces TGF-β1 by binding to the TGF-β1 promotor to upregulate its transcriptional activity [15, 50]. FOXO1 also regulates the expression of integrins-β6 and -α3 needed for keratinocyte migration [8]. Deletion of FOXO1 in vitro reduces expression of these integrins, which causes decreased migration that is rescued by integrin overexpression [15]. FOXO1 also protects keratinocytes from oxidative stress by activating antioxidant defense and DNA repair enzymes. Lineage-specific FOXO1 deletion in keratinocytes in vivo increases oxidative damage and in vitro enhances ROS levels, increases oxidative damage, enhances apoptosis, and interferes with keratinocyte migration [15]. The significance of antioxidant expression in keratinocyte migration is shown by rescue of migration in FOXO1deleted keratinocytes by application of exogenous antioxidants [15]. In contrast to results above, global haploinsufficiency of (FOXO1+/− mice) has been reported to accelerate healing [54]. This may indicate that the FOXO1 expression plays different roles in the healing process in different cell types (global vs. lineage-specific deletion) [15, 54].
Diabetes impairs reepithelialization by inhibiting keratinocyte migration in both skin and mucosal wounds [47, 50]. There are several mechanisms through which diabetes exerts this effect. As discussed above, diabetes is associated with high levels of glucose, greater formation of advanced glycation end products (AGEs), and enhanced production of inflammatory mediators such as TNF, all of which can inhibit keratinocyte migration [47]. High glucose and AGEs cause molecular changes that diminish the capacity FOXO1 to stimulate TGF-β1 expression (Figure 1) [47]. Diabetes also enhances the production of factors that interfere with wound healing when they are produced at high levels (Figure 1). In type 2 diabetic foot ulcers, there is an excessive MMP expression and activity, with increased levels of MMP1, -2, -8, and -9 and reduced levels of TIMP-2 [55]. High levels of MMPs degrade extracellular matrix and limit the ability of keratinocytes to migrate over the wound bed [9]. Interestingly, the negative effect of high glucose on keratinocyte migration is improved by addition of an MMP9 inhibitor or by reducing FOXO1 activity [56]. In type 1 diabetic animals, FOXO1 ablation in vivo reduces high levels of the MMP9 expression [56]. In vitro experiments demonstrate that high glucose increases FOXO1 binding to the MMP9 promoter and enhances MMP9 luciferase reporter activity through a FOXO1-dependent mechanism [56]. It is also noteworthy that the absolute level of MMP activity is important, as the absence of MMPs is also problematic. Thus, like many factors, MMP levels need to be tightly regulated at an optimal level for healing to progress since both their absence or overexpression can be damaging to wound healing [57].
Figure 1.
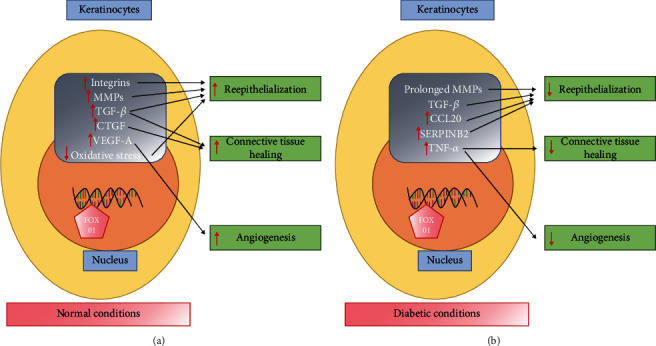
(a) Under normal conditions, FOXO1 promotes reepithelialization through upregulating the expression of integrins, MMPs, TGF-β, and antioxidants. FOXO1 promotes connective tissue healing through induction of the TGF-β and CTGF expression and stimulates angiogenesis via upregulating VEGF-A. (b) In diabetic conditions, FOXO1 exhibits reduced binding to the TGF-β1 promoter to diminish the TGF-β1 expression. However, its interaction with a number of factors that inhibit healing when expressed at high levels is increased including MMP-9, CCL20, IL-36γ, and SERPINB2 which hamper reepithelialization. FOXO1 is induced by high levels of glucose, advanced glycation end products, and TNF that are elevated in diabetic wounds.
FOXO1 in diabetic conditions can enhance the expression of factors that inhibit reepithelialization. In particular, high glucose, AGEs, or inflammation increase FOXO1 binding to the promoters of proinflammatory factors such as CCL20 and IL-36γ [50]. High levels of CCL20 and IL-36γ interfere with keratinocyte migration and are significantly elevated in keratinocytes in type 1 diabetic mucosal wounds compared to normoglycemic wounds [50]. Inhibition of both CCL20 and IL-36γ significantly improve keratinocyte migration when keratinocytes are tested in high glucose or AGE-supplemented media. Like MMP9, the normal expression of IL-36γ is likely to be important for reepithelialization as someIL-36γ is needed for adequate keratinocyte proliferation in healing wounds [58, 59]. Thus, a minimum level of IL-36γ may be required for normal wound healing, while too much IL-36γ may be detrimental and contribute to proinflammatory processes as in psoriatic plaques [60] and in response to bacterial infection [61]. Although IL-36γ expression has been shown to increase in keratinocytes in diabetic mucosal wounds in vivo and by high glucose and AGES in vitro [50], another study showed that the expression of IL-36γ was not elevated in full thickness diabetic wounds from skin biopsies [59]. The reason for the apparent discrepancy is unknown, but it could reflect differences between mucosal and skin wounds.
In summary, diabeteshas several negative effects on keratinocytes that are caused by a change in FOXO1 activity modulated by high glucose or AGEs. Thus, FOXO1 exerts different effects under normal compared to diabetic conditions. On a molecular level, high glucose and AGEs increase FOXO1 binding to the promoter regions of some genes, while reducing FOXO1 interaction with the promoter regions of other genes. For example, high glucose or AGEs diminish FOXO1 binding to the promoter region of TGF-β1 and reduce TGF-β1 expression. However, high glucose or AGEs increase FOXO1 interaction with the promoters of MMP9, CCL20, and IL-36γ to increase their expression to high levels, which interferes with keratinocyte migration [50, 62]. Studies in progress suggest that high glucose and AGEs modify FOXO1-DNA interactions through epigenetic mechanisms that may involve changes in DNA methylation as well as histones.
5. Keratinocytes, Connective Tissue Healing, Diabetes, and FOXO1
The crosstalk between epidermal keratinocytes and dermal fibroblasts is needed for normal connective tissue healing [63]. Keratinocytes stimulate fibroblasts and myofibroblasts through production of growth factors such as TGF-β and CTGF (Table 1) [49]. Fibroblasts play a critical role in connective tissue healing by producing and remodeling extracellular matrix [64]. Myofibroblasts, a subset of fibroblasts, are responsible for connective-tissue compaction and wound contraction [65]. In vitro experiments show that TGF-β1 production and activation are upregulated in keratinocytes to stimulate fibroblasts in cocultures [66]. Interestingly, keratinocyte-specific deletion of FOXO1 reduces keratinocyte-produced TGF-β1 and impairs connective tissue healing, demonstrating the importance of keratinocytes in the wound healing process [15]. Thus, without FOXO1 produced by keratinocytes, there is diminished numbers of fibroblasts and myofibroblasts caused by reduced proliferation of these cells [49]. Mechanistically, this is tied to FOXO1-induced TGF-β1 as defective connective tissue matrix formation in mice with keratinocyte-specific FOXO1 deletion is rescued by addition of exogenous TGF-β1 [15]. However, addition of TGF-β1 has little effect on connective tissue healing of wounds in control mice, showing that under normal conditions, production of TGF-β1 is sufficient and that the addition of more TGF-β1 is not helpful.
TGF-β can stimulate connective tissue formation by induction of connective tissue growth factor (CTGF), also known as cellular communication network factor-2 (CCN-2) [49, 68]. Application of CTGF in vivo stimulates fibroblast proliferation and deposition of collagen [69]. Furthermore, CTGF inhibition reduces the quantity and quality of granulation tissue [69]. In addition to TGF-β1, keratinocytes secrete TGF-β2. In vivo experiments show that TGF-β2 induces recruitment of fibroblasts to the wound site and increases collagen deposition and scar formation in vivo [70]. Thus, TGF-β produced by keratinocytes can induce CTGF in keratinocytes and in cells in the connective tissue of healing wounds [49, 71]. FOXO1 ablation reduces keratinocyte-produced TGF-β1, which leads to a reduction in the CTGF expression [49] and less vigorous production of connective tissue matrix in diabetic wounds [49]. In contrast, the excessive TGF-β1 expression may be a key factor in wound fibrosis [67].
6. Keratinocytes, Angiogenesis, and FOXO1
Angiogenesis is essential in wound healing and is triggered by hypoxia that results from vascular disruption in wounded tissue [72]. Vascular endothelial growth factor-A (VEGF-A) is the primary isoform of VEGF in wounds and stimulates endothelial cells to proliferate and form vessels. Keratinocytes promote angiogenesis in mucosal and skin wounds through secretion of growth factors [48, 73]. Studies in human wounds and animal models demonstrate that VEGF is produced by keratinocytes during the healing process. Mice with lineage-specific VEGF-A deletion in keratinocytes have delayed wound closure and reduced angiogenesis [74]. In addition to VEGF-A, keratinocytes produce the proangiogenic factor, mitogen-regulated protein 3 (MRP3) [73], and TGF-β1, which are needed for normal angiogenesis [75].
Hypoxia-inducible factor-1 (HIF-1) is a transcription factor that stimulates the VEGF-A expression [72]. FOXO1 also participates in VEGF-A transcriptional regulation and stimulates wound angiogenesis by stimulating the VEGF-A expression [48]. FOXO1 deletion reduces VEGF-A protein levels in wounded mucosal epithelium in vivo [48], which results in reduced vascular density and a diminished number of proliferating endothelial cells in vivo at wound sites [48]. Interestingly, FOXO1 in chondrocytes also controls VEGF-A expression and angiogenesis in long bone fracture healing [76]. FOXO1 directly interacts with the VEGF-A promoter thereby inducing VEGF-A promoter activity and VEGF-A expression [48]. A large animal model further supports the role of FOXO1 in promoting angiogenesis as shown by reduced neovascularization when a FOXO1-specific inhibitor is applied [48]. Global deletion of FOXO1 causes embryonic lethality due to vascular failure [77], and FOXO1-deficient endothelial cells show impaired angiogenesis in vivo [78].
Diabetes negatively affects wound healing by interfering with angiogenesis [72]. Diabetic human and animal wounds with reduced angiogenesis have diminished vascularity and capillary density. Defective VEGF production has been reported in keratinocytes from diabetic db/db mice compared with keratinocytes from normal mice [79]. Reduced M2 macrophage levels in diabetic wounds may also contribute to impaired angiogenesis [72]. In addition to having reduced expression of proangiogenic factors, diabetic wounds also have increased antiangiogenic factors produced by keratinocytes [80, 81]. Thrombospondin-1(TSP-1) is an angiogenesis inhibitor [82] and produced in greater amounts by keratinocytes in high-glucose environment [83]. Thus, it is possible that an increase in TSP-1 acts as a brake in the formation of new blood vessels in diabetic wounds. FOXO1 may contribute to diabetes-impaired angiogenesis [84]. FOXO1 enhances apoptosis of microvascular endothelial cells in diabetic animals in vivo and induces apoptosis in microvascular cell endothelial cells and pericytes exposed to high glucose in vitro [85]. The increased apoptosis may negatively affect angiogenesis. In high glucose media, FOXO1 disrupts human microvascular endothelial cell formation of vascular tubes [86]. Local injection of adenovirus-expressing FOXO1 in type 2 diabetic mice with skin wounds reduces vascular density [87]. Thus, in a high glucose, diabetic environment FOXO1 may affect endothelial cells to disrupt angiogenesis and impair wound healing.
7. Summary
Wound healing is a complex process with many cells and factors involved. Keratinocytes play crucial roles in this process by reepithelialization of open wounds and by the production of factors that influence connective tissue healing and angiogenesis (Table 1). FOXO1 is essential in upregulating the wound healing activity in keratinocytes by stimulating expression of factors that promote healing including TGF-β1, integrins, and antioxidants. In diabetic wounds, FOXO1 activity changes to take on a negative role. This is due to the influence of conditions present in the diabetic wound such as high levels of glucose, increased AGEs, and increased TNF levels. These factors decrease the interaction of FOXO1 with the TGF-β1 promoter and have a detrimental effect on both reepithelialization and formation of new connective tissue. Furthermore, diabetic conditions increase FOXO1 interactions with the promoters of other genes to enhance their expression. This has a negative effect since high levels of MMP9, CCL20, and IL-36γ impede keratinocyte migration and interfere with reepithelialization.
Acknowledgments
This work was supported by a grant from the NIDCR (DTG) (R01DE019108 and R01DE017732).
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
Dana Graves conceived the manuscript. Yulan Wang and Dana Graves wrote the manuscript, and Dana Graves edited it.
References
- 1.Fuchs E. Epithelial skin Biology. Current Topics in Developmental Biology. 2016;116:357–374. doi: 10.1016/bs.ctdb.2015.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woodley D. T. In The molecular and cellular biology of wound repair. Springer; 1988. Reepithelialization; pp. 339–354. [Google Scholar]
- 3.Henry G., Li W., Garner W., Woodley D. T. Migration of human keratinocytes in plasma and serum and wound re-epithelialisation. Lancet. 2003;361(9357):574–576. doi: 10.1016/S0140-6736(03)12510-X. [DOI] [PubMed] [Google Scholar]
- 4.Small J. V., Resch G. P. The comings and goings of actin: coupling protrusion and retraction in cell motility. Current Opinion in Cell Biology. 2005;17(5):517–523. doi: 10.1016/j.ceb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Martins-Green M. The yin and yang of integrin function in re-epithelialization during wound healing. Advances in Wound Care. 2013;2(3):75–80. doi: 10.1089/wound.2011.0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sehgal B. U., DeBiase P. J., Matzno S., et al. Integrin β4 regulates migratory behavior of keratinocytes by determining laminin-332 organization. Journal of Biological Chemistry. 2006;281(46):35487–35498. doi: 10.1074/jbc.M606317200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds L. E., Conti F. J., Silva R., et al. α3β1 integrin-controlled smad7 regulates reepithelialization during wound healing in mice. Journal of Clinical Investigation. 2008;118(3):965–974. doi: 10.1172/jci33538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang X., Wu J., Spong S., Sheppard D. The integrin alphavbeta6 is critical for keratinocyte migration on both its known ligand, fibronectin, and on vitronectin. Journal of Cell Science. 1998;111, Part 15:2189–2195. doi: 10.1242/jcs.111.15.2189. [DOI] [PubMed] [Google Scholar]
- 9.Rohani M. G., Parks W. C. Matrix remodeling by MMPs during wound repair. Matrix Biology. 2015;44-46:113–121. doi: 10.1016/j.matbio.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Hattori N., Mochizuki S., Kishi K., et al. MMP-13 plays a role in keratinocyte migration, angiogenesis, and contraction in mouse skin wound healing. American Journal of Pathology. 2009;175(2):533–546. doi: 10.2353/ajpath.2009.081080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jansen P. L., Rosch R., Jansen M., et al. Regulation of MMP-2 gene transcription in dermal wounds. Journal of Investigative Dermatology. 2007;127(7):1762–1767. doi: 10.1038/sj.jid.5700765. [DOI] [PubMed] [Google Scholar]
- 12.Crowe M. J., Doetschman T., Greenhalgh D. G. Delayed wound healing in immunodeficient TGF-β1 knockout mice. Journal of Investigative Dermatology. 2000;115(1):3–11. doi: 10.1046/j.1523-1747.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 13.Lu L., Saulis A. S., Liu W. R., et al. The temporal effects of anti-TGF-β1, 2, and 3 monoclonal antibody on wound healing and hypertrophic scar formation. Journal of the American College of Surgeons. 2005;201(3):391–397. doi: 10.1016/j.jamcollsurg.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 14.Puolakkainen P. A., Reed M. J., Gombotz W. R., Twardzik D. R., Abrass I. B., Helene Sage E. Acceleration of wound healing in aged rats by topical application of transforming growth factor-beta1. Wound Repair and Regeneration. 1995;3(3):330–339. doi: 10.1046/j.1524-475x.1995.t01-1-30314.x. [DOI] [PubMed] [Google Scholar]
- 15.Ponugoti B., Xu F., Zhang C., Tian C., Pacios S., Graves D. T. FOXO1 promotes wound healing through the up-regulation of TGF-β1 and prevention of oxidative stress. Journal of Cell Biology. 2013;203(2):327–343. doi: 10.1083/jcb.201305074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ortega S., Ittmann M., Tsang S. H., Ehrlich M., Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(10):5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pastar I., Stojadinovic O., Yin N. C., et al. Epithelialization in wound healing: a comprehensive review. Advances in Wound Care. 2014;3(7):445–464. doi: 10.1089/wound.2013.0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tumbar T., Guasch G., Greco V., et al. Defining the epithelial stem cell niche in skin. Science. 2004;303(5656):359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aragona M., Dekoninck S., Rulands S., et al. Defining stem cell dynamics and migration during wound healing in mouse skin epidermis. Nature Communications. 2017;8(1) doi: 10.1038/ncomms14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park S., Gonzalez D. G., Guirao B., et al. Tissue-scale coordination of cellular behaviour promotes epidermal wound repair in live mice. Nature Cell Biology. 2017;19(3):155–163. doi: 10.1038/ncb3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Safferling K., Sütterlin T., Westphal K., et al. Wound healing revised: a novel reepithelialization mechanism revealed by in vitro and in silico models. Journal of Cell Biology. 2013;203(4):691–709. doi: 10.1083/jcb.201212020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rousselle P., Braye F., Dayan G. Re-epithelialization of adult skin wounds: cellular mechanisms and therapeutic strategies. Advanced Drug Delivery Reviews. 2019;146:344–365. doi: 10.1016/j.addr.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 23.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;36(Supplement_1):S67–S74. doi: 10.2337/dc13-s067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lan C.-C. E., Liu I. H., Fang A. H., Wen C. H., Wu C. S. Hyperglycaemic conditions decrease cultured keratinocyte mobility: implications for impaired wound healing in patients with diabetes. British Journal of Dermatology. 2008;159(5):1103–1115. doi: 10.1111/j.1365-2133.2008.08789.x. [DOI] [PubMed] [Google Scholar]
- 25.Zhu P., Yang C., Chen L.-H., Ren M., Lao G.-j., Yan L. Impairment of human keratinocyte mobility and proliferation by advanced glycation end products-modified BSA. Archives of Dermatological Research. 2011;303(5):339–350. doi: 10.1007/s00403-010-1102-z. [DOI] [PubMed] [Google Scholar]
- 26.Dissemond J., Goos M., Wagner S. N. Die Bedeutung von oxidativem stress in der Genese und Therapie chronischer Wunden. Hautarzt. 2002;53(11):718–723. doi: 10.1007/s00105-001-0325-5. [DOI] [PubMed] [Google Scholar]
- 27.OʼTOOLE E. D. E. L. A., GOEL M. I. M. I., WOODLEY D. A. V. I. D. T. Hydrogen peroxide inhibits human keratinocyte migration. Dermatologic Surgery. 1996;22(6):525–529. doi: 10.1111/j.1524-4725.1996.tb00368.x. [DOI] [PubMed] [Google Scholar]
- 28.Al-Mashat H. A., Kandru S., Liu R., Behl Y., Desta T., Graves D. T. Diabetes enhances mRNA levels of proapoptotic genes and caspase activity, which contribute to impaired healing. Diabetes. 2006;55(2):487–495. doi: 10.2337/diabetes.55.02.06.db05-1201. [DOI] [PubMed] [Google Scholar]
- 29.Siqueira M. F., Li J., Chehab L., et al. Impaired wound healing in mouse models of diabetes is mediated by TNF-α dysregulation and associated with enhanced activation of forkhead box o1 (FOXO1) Diabetologia. 2010;53(2):378–388. doi: 10.1007/s00125-009-1529-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naguib G., Al-Mashat H., Desta T., Graves D. T. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. Journal of Investigative Dermatology. 2004;123(1):87–92. doi: 10.1111/j.0022-202X.2004.22711.x. [DOI] [PubMed] [Google Scholar]
- 31.Al-Mulla F., Leibovich S. J., Francis I. M., Bitar M. S. Impaired TGF-β signaling and a defect in resolution of inflammation contribute to delayed wound healing in a female rat model of type 2 diabetes. Molecular BioSystems. 2011;7(11):3006–3020. doi: 10.1039/c0mb00317d. [DOI] [PubMed] [Google Scholar]
- 32.Percival S. L., McCarty S. M., Lipsky B. Biofilms and wounds: an overview of the evidence. Advances in Wound Care. 2015;4(7):373–381. doi: 10.1089/wound.2014.0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhall S., Do D., Garcia M., et al. A Novel Model of Chronic Wounds: Importance of Redox Imbalance and Biofilm-Forming Bacteria for Establishment of Chronicity. PLoS ONE. 2014;9(10):p. e109848. doi: 10.1371/journal.pone.0109848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robson M. C. Wound infection. A failure of wound healing caused by an imbalance of bacteria. Surgical Clinics of North America. 1997;77(3):637–650. doi: 10.1016/S0039-6109(05)70572-7. [DOI] [PubMed] [Google Scholar]
- 35.Guo S., Dipietro L. A. Factors affecting wound healing. Journal of Dental Research. 2010;89(3):219–229. doi: 10.1177/0022034509359125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edwards R., Harding K. G. Bacteria and wound healing. Current Opinion in Infectious Diseases. 2004;17(2):91–96. doi: 10.1097/00001432-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 37.Kim J. H., Ruegger P. R., Lebig E. G., et al. High levels of oxidative stress create a microenvironment that significantly decreases the diversity of the microbiota in diabetic chronic wounds and promotes biofilm formation. Frontiers in Cellular and Infection Microbiology. 2020;10 doi: 10.3389/fcimb.2020.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dhall S., Do D. C., Garcia M., et al. Generating and reversing chronic wounds in diabetic mice by manipulating wound redox parameters. Journal of Diabetes Research. 2014;2014:18. doi: 10.1155/2014/562625.562625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank D. N., Wysocki A., Specht-Glick D. D., et al. Microbial diversity in chronic open wounds. Wound Repair and Regeneration. 2009;17(2):163–172. doi: 10.1111/j.1524-475x.2009.00472.x. [DOI] [PubMed] [Google Scholar]
- 40.Seth A. K., Geringer M. R., Galiano R. D., Leung K. P., Mustoe T. A., Hong S. J. Quantitative comparison and analysis of species-specific wound biofilm virulence using an in vivo, rabbit-ear model. Journal of the American College of Surgeons. 2012;215(3):388–399. doi: 10.1016/j.jamcollsurg.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 41.Kalan L. R., Meisel J. S., Loesche M. A., et al. Strain- and Species-Level Variation in the Microbiome of Diabetic Wounds Is Associated with Clinical Outcomes and Therapeutic Efficacy. Cell Host & Microbe. 2019;25(5):641–655.e5. doi: 10.1016/j.chom.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhattacharya R., Xu F., Dong G., et al. Effect of Bacteria on the Wound Healing Behavior of Oral Epithelial Cells. PLoS ONE. 2014;9(2):p. e89475. doi: 10.1371/journal.pone.0089475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graves D. T., Nooh N., Gillen T., et al. IL-1 plays a critical role in oral, but not dermal, wound healing. Journal of Immunology. 2001;167(9):5316–5320. doi: 10.4049/jimmunol.167.9.5316. [DOI] [PubMed] [Google Scholar]
- 44.Hameedaldeen A., Liu J., Batres A., Graves G., Graves D. FOXO1, TGF-β Regulation and Wound Healing. International Journal of Molecular Sciences. 2014;15(9):16257–16269. doi: 10.3390/ijms150916257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rajendran N. K., Dhilip Kumar S. S., Houreld N. N., Abrahamse H. Understanding the perspectives of forkhead transcription factors in delayed wound healing. Journal of Cell Communication and Signaling. 2019;13(2):151–162. doi: 10.1007/s12079-018-0484-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Markus Roupé K., Alberius P., Schmidtchen A., Sørensen O. E. Gene expression demonstrates increased resilience toward harmful inflammatory stimuli in the proliferating epidermis of human skin wounds. Experimental Dermatology. 2010;19(8):e329–e332. doi: 10.1111/j.1600-0625.2009.01038.x. [DOI] [PubMed] [Google Scholar]
- 47.Zhang C., Ponugoti B., Tian C., et al. FOXO1 differentially regulates both normal and diabetic wound healing. Journal of Cell Biology. 2015;209(2):289–303. doi: 10.1083/jcb.201409032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeon H. H., Yu Q., Lu Y., et al. FOXO1 regulates VEGFA expression and promotes angiogenesis in healing wounds. Journal of Pathology. 2018;245(3):258–264. doi: 10.1002/path.5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang C., Lim J., Liu J., et al. FOXO1 expression in keratinocytes promotes connective tissue healing. Scientific Reports. 2017;7(1) doi: 10.1038/srep42834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu F., Othman B., Lim J., et al. FOXO1 inhibits diabetic mucosal wound healing but enhances healing of normoglycemic wounds. Diabetes. 2014;64(1):243–256. doi: 10.2337/db14-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gailit J., Clark R. A. F., Welch M. P. TGF-β1 Stimulates Expression of Keratinocyte Integrins During Re-Epithelialization of Cutaneous Wounds. Journal of Investigative Dermatology. 1994;103(2):221–227. doi: 10.1111/1523-1747.ep12393176. [DOI] [PubMed] [Google Scholar]
- 52.Hebda P. A. Stimulatory effects of transforming growth factor-beta and epidermal growth factor on epidermal cell outgrowth from porcine skin explant cultures. Journal of Investigative Dermatology. 1988;91(5):440–445. doi: 10.1111/1523-1747.ep12476480. [DOI] [PubMed] [Google Scholar]
- 53.Mustoe T., Pierce G., Thomason A., Gramates P., Sporn M., Deuel T. Accelerated healing of incisional wounds in rats induced by transforming growth factor-beta. Science. 1987;237(4820):1333–1336. doi: 10.1126/science.2442813. [DOI] [PubMed] [Google Scholar]
- 54.Mori R., Tanaka K., de Kerckhove M., et al. Reduced foxo1 expression accelerates skin wound healing and attenuates scarring. American Journal of Pathology. 2014;184(9):2465–2479. doi: 10.1016/j.ajpath.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muller M., Trocme C., Lardy B., Morel F., Halimi S., Benhamou P. Y. Matrix metalloproteinases and diabetic foot ulcers: the ratio of MMP-1 to TIMP-1 is a predictor of wound healing. Diabetic Medicine. 2008;25(4):419–426. doi: 10.1111/j.1464-5491.2008.02414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang C., Lim J., Jeon H. H., et al. FOXO1 deletion in keratinocytes improves diabetic wound healing through MMP9 regulation. Scientific Reports. 2017;7(1):p. 10565. doi: 10.1038/s41598-017-10999-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bullard K. M., Lund L., Mudgett J. S., et al. Impaired wound contraction in stromelysin-1-deficient mice. Annals of Surgery. 1999;230(2):260–265. doi: 10.1097/00000658-199908000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang Z., Liu Y., Li C., et al. IL-36γ induced by the tlr3-slug-vdr axis promotes wound healing via reg3a. Journal of Investigative Dermatology. 2017;137(12):2620–2629. doi: 10.1016/j.jid.2017.07.820. [DOI] [PubMed] [Google Scholar]
- 59.Wu Y., Quan Y., Liu Y., et al. Hyperglycaemia inhibits REG3A expression to exacerbate TLR3-mediated skin inflammation in diabetes. Nature Communications. 2016;7(1) doi: 10.1038/ncomms13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ainscough J. S., Macleod T., McGonagle D., et al. Cathepsin S is the major activator of the psoriasis-associated proinflammatory cytokine IL-36γ. Proceedings of the National Academy of Sciences of the United States of America; 2017; pp. E2748–E2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huynh J., Scholz G. M., Aw J., et al. IRF6 regulates the expression of IL-36γ by human oral epithelial cells in response to Porphyromonas gingivalis. Journal of Immunology. 2016;196(5):2230–2238. doi: 10.4049/jimmunol.1501263. [DOI] [PubMed] [Google Scholar]
- 62.Graves D. T. In Pressure injury, diabetes and negative pressure wound therapy. Springer; 2017. FOXO1 has a dual function to promote normal but inhibit diabetic wound healing; pp. 57–67. [Google Scholar]
- 63.Werner S., Krieg T., Smola H. Keratinocyte–fibroblast interactions in wound healing. Journal of Investigative Dermatology. 2007;127(5):998–1008. doi: 10.1038/sj.jid.5700786. [DOI] [PubMed] [Google Scholar]
- 64.Tracy L. E., Minasian R. A., Caterson E. J. Extracellular matrix and dermal fibroblast function in the healing wound. Advances in Wound Care. 2016;5(3):119–136. doi: 10.1089/wound.2014.0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li B., Wang J. H.-C. Fibroblasts and myofibroblasts in wound healing: force generation and measurement. Journal of Tissue Viability. 2011;20(4):108–120. doi: 10.1016/j.jtv.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shephard P., Martin G., Smola-Hess S., Brunner G., Krieg T., Smola H. Myofibroblast differentiation is induced in keratinocyte-fibroblast co-cultures and is antagonistically regulated by endogenous transforming growth factor-β and interleukin-1. American Journal of Pathology. 2004;164(6):2055–2066. doi: 10.1016/S0002-9440(10)63764-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Montastruc J. L., Cathala B., Richard J. P., et al. Drugs and traffic accidents. Evaluation of a survey realized in a hospital environment. Thérapie. 1988;43(4):313–315. [PubMed] [Google Scholar]
- 68.Leask A., Holmes A., Black C. M., Abraham D. J. Connective tissue growth factor gene regulation. Journal of Biological Chemistry. 2003;278(15):13008–13015. doi: 10.1074/jbc.M210366200. [DOI] [PubMed] [Google Scholar]
- 69.Alfaro M. P., Deskins D. L., Wallus M., et al. A physiological role for connective tissue growth factor in early wound healing. Laboratory Investigation. 2013;93(1):81–95. doi: 10.1038/labinvest.2012.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barrientos S., Stojadinovic O., Golinko M. S., Brem H., Tomic-Canic M. PERSPECTIVE ARTICLE: Growth factors and cytokines in wound healing. Wound Repair and Regeneration. 2008;16(5):585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 71.Vivar R., Humeres C., Muñoz C., et al. FOXO1 mediates TGF-beta1-dependent cardiac myofibroblast differentiation. Biochimica et Biophysica Acta. 2016;1863(1):128–138. doi: 10.1016/j.bbamcr.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 72.Okonkwo U., DiPietro L. Diabetes and wound angiogenesis. International Journal of Molecular Sciences. 2017;18(7):p. 1419. doi: 10.3390/ijms18071419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mitchell K., Szekeres C., Milano V., et al. Alpha3beta1 integrin in epidermis promotes wound angiogenesis and keratinocyte-to-endothelial-cell crosstalk through the induction of MRP3. Journal of Cell Science. 2009;122(11):1778–1787. doi: 10.1242/jcs.040956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson K. E., Wilgus T. A. Vascular endothelial growth factor and angiogenesis in the regulation of cutaneous wound repair. Advances in Wound Care. 2014;3(10):647–661. doi: 10.1089/wound.2013.0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dickson M. C., Martin J. S., Cousins F. M., Kulkarni A. B., Karlsson S., Akhurst R. J. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121(6):1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 76.Zhang C., Feinberg D., Alharbi M., et al. Chondrocytes promote vascularization in fracture healing through a foxo1-dependent mechanism. Journal of Bone and Mineral Research. 2018;34(3):547–556. doi: 10.1002/jbmr.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dharaneeswaran H., Abid M. R., Yuan L., et al. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circulation Research. 2014;115(2):238–251. doi: 10.1161/circresaha.115.303227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Furuyama T., Kitayama K., Shimoda Y., et al. Abnormal angiogenesis in FOXO1 (FKHR)-deficient mice. Journal of Biological Chemistry. 2004;279(33):34741–34749. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- 79.Frank S., Hübner G., Breier G., Longaker M. T., Greenhalgh D. G., Werner S. Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. Journal of Biological Chemistry. 1995;270(21):12607–12613. doi: 10.1074/jbc.270.21.12607. [DOI] [PubMed] [Google Scholar]
- 80.Hu S. C.-S., Lan C.-C. E. High-glucose environment disturbs the physiologic functions of keratinocytes: focusing on diabetic wound healing. Journal of Dermatological Science. 2016;84(2):121–127. doi: 10.1016/j.jdermsci.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 81.Rafehi H., El-Osta A., Karagiannis T. C. Genetic and epigenetic events in diabetic wound healing. International Wound Journal. 2011;8(1):12–21. doi: 10.1111/j.1742-481X.2010.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. Journal of Cellular and Molecular Medicine. 2002;6(1):1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lan C.-C. E., Huang S.-M., Wu C.-S., Wu C.-H., Chen G.-S. High-glucose environment increased thrombospondin-1 expression in keratinocytes via DNA hypomethylation. Translational Research. 2016;169:91–101.e3. doi: 10.1016/j.trsl.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 84.Tsuchiya K., Ogawa Y. Forkhead box class o family member proteins: the biology and pathophysiological roles in diabetes. Journal of Diabetes Investigation. 2017;8(6):726–734. doi: 10.1111/jdi.12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Behl Y., Krothapalli P., Desta T., Roy S., Graves D. T. FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes. 2009;58(4):917–925. doi: 10.2337/db08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lim J. C., Ko K. I., Mattos M., et al. TNFα contributes to diabetes impaired angiogenesis in fracture healing. Bone. 2017;99:26–38. doi: 10.1016/j.bone.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang X., Sun J., Chen G., et al. Resveratrol promotes diabetic wound healing via SIRT1-FOXO1-c-Myc signaling pathway-mediated angiogenesis. Frontiers in Pharmacology. 2019;10:p. 421. doi: 10.3389/fphar.2019.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sogabe Y., Abe M., Yokoyama Y., Ishikawa O. Basic fibroblast growth factor stimulates human keratinocyte motility by Rac activation. Wound Repair and Regeneration. 2006;14(4):457–462. doi: 10.1111/j.1743-6109.2006.00143.x. [DOI] [PubMed] [Google Scholar]
- 89.Shirakata Y. Heparin-binding EGF-like growth factor accelerates keratinocyte migration and skin wound healing. Journal of Cell Science. 2005;118(11):2363–2370. doi: 10.1242/jcs.02346. [DOI] [PubMed] [Google Scholar]
- 90.Poumay Y., De Rouvroit C. L. HB-EGF, the growth factor that accelerates keratinocyte migration, but slows proliferation. Journal of Investigative Dermatology. 2012;132(9):2129–2130. doi: 10.1038/jid.2012.225. [DOI] [PubMed] [Google Scholar]