


Abstract
Transcriptome-wide association studies (TWASs) integrate expression quantitative trait loci (eQTLs) studies with genome-wide association studies (GWASs) to prioritize candidate target genes for complex traits. Several statistical methods have been recently proposed to improve the performance of TWASs in gene prioritization by integrating the expression regulatory information imputed from multiple tissues, and made significant achievements in improving the ability to detect gene-trait associations. Unfortunately, most existing multi-tissue methods focus on prioritization of candidate genes, and cannot directly infer the specific functional effects of candidate genes across different tissues. Here, we propose a tissue-specific collaborative mixed model (TisCoMM) for TWASs, leveraging the co-regulation of genetic variations across different tissues explicitly via a unified probabilistic model. TisCoMM not only performs hypothesis testing to prioritize gene-trait associations, but also detects the tissue-specific role of candidate target genes in complex traits. To make full use of widely available GWASs summary statistics, we extend TisCoMM to use summary-level data, namely, TisCoMM-S2. Using extensive simulation studies, we show that type I error is controlled at the nominal level, the statistical power of identifying associated genes is greatly improved, and the false-positive rate (FPR) for non-causal tissues is well controlled at decent levels. We further illustrate the benefits of our methods in applications to summary-level GWASs data of 33 complex traits. Notably, apart from better identifying potential trait-associated genes, we can elucidate the tissue-specific role of candidate target genes. The follow-up pathway analysis from tissue-specific genes for asthma shows that the immune system plays an essential function for asthma development in both thyroid and lung tissues.
INTRODUCTION
Over the last decade, GWASs have achieved remarkable successes in identifying genetic susceptible variants for a variety of complex traits (1). However, the biological mechanisms to understand these discoveries remain largely elusive as majority of these discoveries are located in non-coding regions (2). Recent expression quantitative trait loci (eQTLs) studies indicate that the expression regulatory information may play a pivotal role in bridging both genetic variants and traits (3–5). Cellular traits in comprehensive eQTL studies can serve as reference data, providing investigators with an opportunity to examine the regulatory role of genetic variants on gene expression. For example, the Genotype-Tissue Expression (GTEx) Project (6) has provided DNA sequencing data from 948 individuals and collected gene-expression measurements of 54 tissues from these individuals in the recent V8 release.
Transcriptome-wide association studies (TWASs) have been widely used to integrate the expression regulatory information from these eQTL studies with GWASs to prioritize genome-wide trait-associated genes (7–9). A variety of TWAS methods have been proposed using different prediction models for expression imputation, including the parametric imputation models, e.g. PrediXcan (7), TWAS (8), CoMM (10) and CoMM-S2 (11), and the nonparametric imputation model, e.g. Tigar (12). These methods have been used for analyzing many complex traits with expression profiles from different tissues, successfully enhancing the discovery of genetic risk loci for complex traits (9,13).
To further improve the power of identifying potential target genes, two recent studies were proposed by leveraging the substantial shared eQTLs across different tissues, i.e., MultiXcan (14) and UTMOST (15). They use a step-wise procedure: they first conduct imputation for gene expressions across multiple tissues and then perform subsequent association analysis using a multivariate regression that pools information across different tissues. Compared to the single-tissue methods, these multi-tissue strategies enhance the imputation accuracy for gene expression and thus improve the power of identifying potential target genes.
Despite their successes, the existing multi-tissue methods have several limitations. First, MultiXcan and UTMOST cannot be used to identify the tissue-specific gene-trait associations. Many studies have shown that genes associated with complex traits are always regulated in a tissue-specific manner (9,16–18). For example, a recent study across 44 tissues confirmed this phenomenon in 18 complex traits (19), implying the persuasive role of tissue-specific regulatory effects in a wide range of complex traits. Using a single-tissue test, one can easily reach a false conclusion regarding which tissue that a gene affects traits through. Second, both MultiXcan and UTMOST rely on a step-wise inference framework, ignoring the uncertainty in the process of expression imputation and thus losing power, especially when cellular-heritability is small (10). Recently, CoMM (10) and its variant for summary-level data, CoMM-S2 (11), have been proposed to account for uncertainty in the process of expression imputation. These studies demonstrate that the statistical power can be largely improved in a unified probabilistic model. Third, MultiXcan and UTMOST do not make efficient use of the shared patterns of eQTLs across tissues, where MultiXcan uses principal component analysis (PCA) regularization on the predicted expression data, and UTMOST uses penalized regularization on coefficients for eQTL effects. A study of GTEx revealed these shared patterns (20), and later many efforts have been made to take advantage of them in the analysis for GTEx data. For example, Urbut et al. proposed statistical methods for estimating and testing eQTL effects explicitly incorporating this extensively tissue-shared patterns (21), shedding light on how to account for the tissue-shared eQTLs in statistical modeling successfully.
To overcome these limitations, we propose a tissue-specific collaborative mixed model (TisCoMM) for TWASs, providing a principled way to perform gene-trait joint and tissue-specific association tests across different tissues. Our method allows us not only to perform hypothesis testing to prioritize gene–trait association but also to uncover the tissue-specific role of candidate genes. By conditioning on the trait-relevant tissues, one could largely remove the spurious associations due to highly correlated gene expressions among multiple tissues. As a unified model, TisCoMM jointly conducts the ‘imputation’ and the association analysis, pooling expression regulatory information across multiple tissues explicitly. Furthermore, we extend TisCoMM to use summary statistics from a GWAS, namely, TisCoMM-S2. In simulations, we show that both TisCoMM and TisCoMM-S2 provide correctly controlled type I error and are more powerful than existing multi-tissue methods. More importantly, our methods can be used to test for the tissue-specific role of candidate genes. We illustrate the benefits of our methods using summary-level GWASs data in 33 complex traits. Results show that our findings have biologically meaningful implications. The follow-up pathway analysis from tissue-specific genes for asthma shows that the regulated immune system in both thyroid and lung tissues could have significant impact on asthma development.
MATERIALS AND METHODS
Methods for comparison
We conducted comprehensive simulations and real data analysis to gauge the performance of different methods by performing gene–trait joint and tissue-specific tests across different tissues.
To detect gene-trait association, we compared the performance of three methods in the main text: (i) our TisCoMM and TisCoMM-S2 implemented in the R package TisCoMM; (ii) MultiXcan and S-MultiXcan implemented in the MetaXcan package available at http://gene2pheno.org/; (iii) UTMOST available at https://github.com/Joker-Jerome/UTMOST/.
To detect the tissue-specific effect, we compared the performance of TisCoMM tissue-specific test with three single-tissue methods that include (i) CoMM available at https://github.com/gordonliu810822/CoMM; (ii) PrediXcan available at http://gene2pheno.org/; (iii) TWAS relies on the BSLMM (22) implemented in the GEMMA (22) software. All methods were used with default settings.
Simulations
In detail, we considered the following simulation settings. We first obtained 410 cis-SNPs (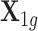 ) for the gene PARK2 on chromosome 6 from the GTEx data, and denote it as
) for the gene PARK2 on chromosome 6 from the GTEx data, and denote it as 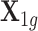 , a matrix with 491 rows representing 491 samples and 410 columns representing 410 cis-SNPs. We used PARK2 because the number of its cis-SNPs represents the median of all genes. Genotype matrix
, a matrix with 491 rows representing 491 samples and 410 columns representing 410 cis-SNPs. We used PARK2 because the number of its cis-SNPs represents the median of all genes. Genotype matrix 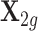 was extracted from the NFBC1966 data (see the following GWASs data section for details) with 5123 rows for individuals in this data set and 410 columns for all cis-SNPs in PARK2. To simulate the reference panel for LD calculation, another genotype matrix
was extracted from the NFBC1966 data (see the following GWASs data section for details) with 5123 rows for individuals in this data set and 410 columns for all cis-SNPs in PARK2. To simulate the reference panel for LD calculation, another genotype matrix 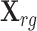 is extracted from the NFBC1966 data with 400 randomly selected individuals and the same set of 410 cis-SNPs. In Supplementary Text, we also perform additional simulations using randomly generated genotyped data.
is extracted from the NFBC1966 data with 400 randomly selected individuals and the same set of 410 cis-SNPs. In Supplementary Text, we also perform additional simulations using randomly generated genotyped data.
To generate multi-tissue gene expressions, we considered different cellular-level heritability levels ( ) and sparsity levels (s). These are key parameters to describe the genetic architecture of gene expression (23). The cellular-level heritability represents the proportion of variance of the gene expression that can be explained by genotype, while sparsity represents the proportion of genetic variants that are associated with the gene expression. First, SNP effect on gene expression was generated by
) and sparsity levels (s). These are key parameters to describe the genetic architecture of gene expression (23). The cellular-level heritability represents the proportion of variance of the gene expression that can be explained by genotype, while sparsity represents the proportion of genetic variants that are associated with the gene expression. First, SNP effect on gene expression was generated by  , where we assume the factorizable assumption (24,25) and
, where we assume the factorizable assumption (24,25) and 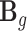 is a coefficient matrix with 410 rows for 410 cis-SNPs and 10 columns for 10 tissues. And based on our model construction,
is a coefficient matrix with 410 rows for 410 cis-SNPs and 10 columns for 10 tissues. And based on our model construction, 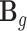 can be decomposed into two parts.
can be decomposed into two parts. 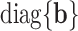 in the first part can be treated as the SNP effect shared in all the tissues, and
in the first part can be treated as the SNP effect shared in all the tissues, and 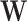 in the second part can be treated as the tissue specific effect. Under this design, we simulated SNP effect size
in the second part can be treated as the tissue specific effect. Under this design, we simulated SNP effect size 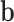 from a standard normal distribution, randomly selected 1%, 5% or 10% of the SNPs to have non-zero tissue-specific effect
from a standard normal distribution, randomly selected 1%, 5% or 10% of the SNPs to have non-zero tissue-specific effect 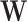 for gene expressions in all tissues, and simulated their effects from a standard normal distribution. We then simulated errors
for gene expressions in all tissues, and simulated their effects from a standard normal distribution. We then simulated errors 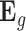 from a normal distribution, where their variances were chosen according to
from a normal distribution, where their variances were chosen according to 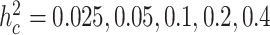 , and the covariance structure was autoregressive with ρe = 0.5, representing the shared environmental or other non-genetic factors among overlap samples in 10 tissues. Then, we simulated a multi-tissue eQTL data set assuming
, and the covariance structure was autoregressive with ρe = 0.5, representing the shared environmental or other non-genetic factors among overlap samples in 10 tissues. Then, we simulated a multi-tissue eQTL data set assuming 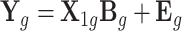 .
.
To simulate a quantitative trait, we used the equation 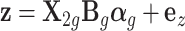 , where
, where 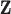 represents phenotype, αg is the vector for the effects of gene expression in 10 tissues on the phenotype, and
represents phenotype, αg is the vector for the effects of gene expression in 10 tissues on the phenotype, and 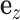 is the error term. The nonzero entries of
is the error term. The nonzero entries of 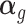 , indicating the existence of the gene expression effect on phenotype, were generated from a uniform distribution and
, indicating the existence of the gene expression effect on phenotype, were generated from a uniform distribution and 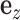 was generated from a normal distribution. The variance of
was generated from a normal distribution. The variance of 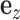 denoted by σ2 was chosen according to the tissue-level heritability
denoted by σ2 was chosen according to the tissue-level heritability 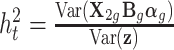 . Here, we set
. Here, we set  for null simulations and type I error control examination and
for null simulations and type I error control examination and 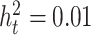 for non-null simulations and power comparisons.
for non-null simulations and power comparisons.
GWASs data
The NFBC1966 data set
The NFBC1966 data set consists of ten traits and 364 590 SNPs from 5402 individuals (26), including total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C) and triglycerides (TG), inflammatory marker C-reactive protein, markers of glucose homeostasis (glucose and insulin), body mass index (BMI) and blood pressure (BP) measurements (systolic and diastolic BP). As individual-level genotype data is available, we use this dataset to demonstrate the reliability of TisCoMM-S2. Quality control procedures are conducted following similar steps to Shi et al. (27). Specifically, individuals with missing-ness in any of the traits and with genotype missing call-rates 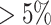 were excluded. We excluded SNPs with minor allele frequency (MAF)
were excluded. We excluded SNPs with minor allele frequency (MAF) 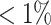 , missing call-rates
, missing call-rates 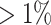 , or failed Hardy–Weinberg equilibrium. After quality control filtering, 172 412 SNPs from 5123 individuals were available for downstream analysis.
, or failed Hardy–Weinberg equilibrium. After quality control filtering, 172 412 SNPs from 5123 individuals were available for downstream analysis.
The tissues used in TisCoMM and TisCoMM-S2 were the same, and the six tissues with the largest number of overlapped individuals were used. The summary statistics for TisCoMM-S2 were calculated using PLINK (28).
Summary-level GWASs data
We obtained summary statistics from GWASs for 33 traits, including 15 traits from (19) and 18 traits from the UK Biobank. Details of these traits can be found in Supplementary Table S1. In the main text, we discussed LOAD and asthma in detail. Analysis results for other traits can be found in Supplementary Text.
GTEx eQTL data
Th GTEx data including genotype and RNA-seq data are obtained from dbGaP with accession number phs000424.v7.p2. Processed gene-expression data are available on the GTEx portal (https://gtexportal.org/home/). In the eQTL data, we removed SNPs with ambiguous alleles or MAF less 0.01. Following both S-PrediXcan (16) and UTMOST (15), we normalized gene expression data to remove potential confounding effects from sex, sequncing platform, top three principal compoments of genotye data, and top PEER factors (29). All covariates were downloaded from the GTEx portal website.
We used two different strategies to select tissues used in our real data analysis. For the NG traits, we obtained the top enriched tissues for each trait according to Supplementary Table S2 in (19), and a subset of tissues with sample sizes larger than 100 was used. For the UKB traits, we used the six tissues with the largest number of overlapped individuals.
Reference panel
Due to the absence of genotype data using summary statistics, we use reference samples to estimate the LD structures R among SNPs in the study samples. Since diseases and traits considered in our real data application are for European population cohorts, we choose to use European subsamples from the 1000 Genome Project (30) as a reference panel.
Let 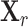 denote the genotype matrix for cis-SNPs in the reference panel. To estimate the LD matrix R, we adopt a simple shrinkage method as follows. We first calculate the empirical correlation matrix
denote the genotype matrix for cis-SNPs in the reference panel. To estimate the LD matrix R, we adopt a simple shrinkage method as follows. We first calculate the empirical correlation matrix 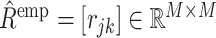 with
with 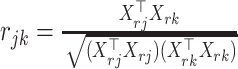 , where Xrj the jth column of
, where Xrj the jth column of 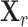 . To make the estimated correlation matrix positive definite, we apply a simple shrinkage estimator (31):
. To make the estimated correlation matrix positive definite, we apply a simple shrinkage estimator (31): 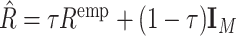 , where τ ∈ [0, 1] is the shrinkage intensity. In real data application, we fixed the shrinkage intensity at 0.95 both for simplicity and computational stability.
, where τ ∈ [0, 1] is the shrinkage intensity. In real data application, we fixed the shrinkage intensity at 0.95 both for simplicity and computational stability.
RESULTS
TisCoMM overview
Here, we provide a brief overview of the TisCoMM; more details are available in Supplementary Text. TisCoMM integrates expression regulatory information across multiple tissues by jointly considering two models. The first one is the expression prediction model, which models the relationship between genetic factors 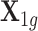 and normalized gene expressions across multiple tissues
and normalized gene expressions across multiple tissues 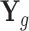 in the eQTL data set:
in the eQTL data set: 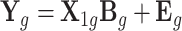 . The second one is the association model, which relates the phenotypic value
. The second one is the association model, which relates the phenotypic value 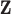 and standardized genotype
and standardized genotype  in the GWAS data:
in the GWAS data: 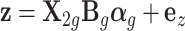 .
. 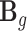 is an matrix of the corresponding effect sizes shared by the two models. And
is an matrix of the corresponding effect sizes shared by the two models. And 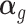 represents the effect sizes of ‘imputed’ gene expression. Our TisCoMM can be depicted as Figure 1, within which Figure 1A illustrates the TisCoMM method combing both the expression prediction model and the corresponding association model together with data input and output.
represents the effect sizes of ‘imputed’ gene expression. Our TisCoMM can be depicted as Figure 1, within which Figure 1A illustrates the TisCoMM method combing both the expression prediction model and the corresponding association model together with data input and output.
Figure 1.

TisCoMM workflow. (A) Two sets of TisCoMM input matrices are highlighted in green and blue separately (left). The probabilistic graphical model for TisCoMM is shown in the middle, which integrates gene expressions and models the co-regulation of cis-SNPs across different tissues explicitly. 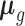 and μ
and μ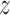 denote expectations of gene expression in eQTLs and phenotype in GWASs, respectively. The decomposition of the
denote expectations of gene expression in eQTLs and phenotype in GWASs, respectively. The decomposition of the 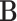 matrix is illustrated on the right-hand side of the figure. (B) The TisCoMM joint test for all genes to prioritize candidate causal genes. See more details of
matrix is illustrated on the right-hand side of the figure. (B) The TisCoMM joint test for all genes to prioritize candidate causal genes. See more details of 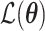 in Methods section. The example outputs (right) are shown as Manhattan plots for 33 traits. (C) The TisCoMM tissue-specific test for all candidate genes to explore the tissue-specific roles of candidate genes. The example outputs (right) are shown as heatmaps which summarize the tissue-specific effect of each gene. Significance level, effect size and heritability are converted into background color, circle color and circle size.
in Methods section. The example outputs (right) are shown as Manhattan plots for 33 traits. (C) The TisCoMM tissue-specific test for all candidate genes to explore the tissue-specific roles of candidate genes. The example outputs (right) are shown as heatmaps which summarize the tissue-specific effect of each gene. Significance level, effect size and heritability are converted into background color, circle color and circle size.
To pool expression regulatory information across relevant tissues, we assume the factorizable assumption (24,25) for 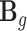 , j = 1, …, Mg, t = 1, …, T. This assumption has been empirically validated for GTEx data in an imputation study (32) and Park et al. further used this assumption in a multi-tissue TWAS (33). Here, we assume that the effect size of cis-SNP j in tissue t can be factorized by variant-dependent and tissue-dependent components: βjt = bj
, j = 1, …, Mg, t = 1, …, T. This assumption has been empirically validated for GTEx data in an imputation study (32) and Park et al. further used this assumption in a multi-tissue TWAS (33). Here, we assume that the effect size of cis-SNP j in tissue t can be factorized by variant-dependent and tissue-dependent components: βjt = bj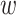 jt, where bj (variant) is the eQTL effect of cis-SNP j shared in all the T tissues, and
jt, where bj (variant) is the eQTL effect of cis-SNP j shared in all the T tissues, and 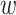 jt is the tissue-specific effect size. Thus, we have
jt is the tissue-specific effect size. Thus, we have  . This factorization allows us to model the co-regulation of cis-SNPs shared across different tissues explicitly (Figure 1A, right). To make TisCoMM identifiable, we follow the polygenic model and assume that bj independently follows a normal distribution
. This factorization allows us to model the co-regulation of cis-SNPs shared across different tissues explicitly (Figure 1A, right). To make TisCoMM identifiable, we follow the polygenic model and assume that bj independently follows a normal distribution 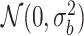 , and we adaptively assign weight
, and we adaptively assign weight 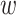 jt using the fitted coefficient from marginal regression of gene expression in tissue t on the jth genetic variant, following the adaptive weighting strategy used in (32).
jt using the fitted coefficient from marginal regression of gene expression in tissue t on the jth genetic variant, following the adaptive weighting strategy used in (32).
The parameter of our interest in TisCoMM is the vector of effect size 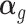 . To prioritize candidate target genes, we conduct hypothesis testing for a joint null,
. To prioritize candidate target genes, we conduct hypothesis testing for a joint null, 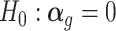 (Figure 1B). To further explore the tissue-specific roles of candidate genes, we conduct hypothesis testing for each tissue, H0: αgt = 0, t = 1, …, T (Figure 1C). We refer to the two inference tasks as the TisCoMM joint test and TisCoMM tissue-specific test, respectively. We develop an expectation-maximization (EM) algorithm for parameter estimation. A parameter expansion technique is further adopted to accelerate computational efficiency (see details in Supplementary Text). In contrast to the existing two-step TWAS methods, we perform TisCoMM analysis in a unified model by treating
(Figure 1B). To further explore the tissue-specific roles of candidate genes, we conduct hypothesis testing for each tissue, H0: αgt = 0, t = 1, …, T (Figure 1C). We refer to the two inference tasks as the TisCoMM joint test and TisCoMM tissue-specific test, respectively. We develop an expectation-maximization (EM) algorithm for parameter estimation. A parameter expansion technique is further adopted to accelerate computational efficiency (see details in Supplementary Text). In contrast to the existing two-step TWAS methods, we perform TisCoMM analysis in a unified model by treating 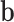 as a hidden random variable. Generally, the computational cost for the TisCoMM tissue-specific test is
as a hidden random variable. Generally, the computational cost for the TisCoMM tissue-specific test is 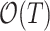 of that for the TisCoMM joint test. To enable computational efficiency, we only conduct the TisCoMM tissue-specific test for candidate genes detected in the joint test, rather than for all genes.
of that for the TisCoMM joint test. To enable computational efficiency, we only conduct the TisCoMM tissue-specific test for candidate genes detected in the joint test, rather than for all genes.
In a single-tissue analysis, it is difficult to explore the tissue-specific role of a candidate gene. The disease-associated genes will be identified in all the causal tissues as well as the tissues (possibly non-causal) highly correlated with the causal one, because there exist sharing patterns for expressions in multiple tissues. By considering all available tissues, our tissue-specific test could largely remove the spurious discoveries due to correlated expression across tissues.
Inferring TisCoMM results from GWASs summary statistics
To make our method widely applicable, we extend TisCoMM to use summary-level GWASs data, denoted as TisCoMM-S2. The model details are given in Supplementary Text.
We observe high concordance between TisCoMM and TisCoMM-S2 results. Figure 2 shows the comparison of TisCoMM and TisCoMM-S2 test statistics for ten traits from the Northern Finland Birth Cohorts program 1966 (NFBC1966) data set (26) (see Methods section). The reference panel was 400 subsamples from the NFBC1966 data set. The high correlation between TisCoMM and TisCoMM-S2 suggests the reliability of detections for trait-associated genes using summary-level GWASs data.
Figure 2.

Comparison of TisCoMM and TisCoMM-S2 results in NFBC1966 traits. The reference panel is subsamples from the NFBC1966 data set. The summary-based method shows similar results to the individual-based method. The blue rectangle indicates the null region.
Simulations: testing gene-trait associations
We focused on the detection of trait-associated genes in the first set of simulations. Here, we compared TisCoMM and TisCoMM-S2 with three different multi-tissue methods, including MultiXcan, S-MultiXcan, and UTMOST. We set all tissues to be causal. For each scenario, we ran 5000 replicates. We first examined type I error control of different methods under the null. Results are shown in Supplementary Figure S1. By comparing the distribution of P-values with the expected uniform distribution, we observe that all methods provide well-controlled type I errors.
Next, we examined the power of different methods under the alternative hypothesis, as shown in Figure 3. We observe that the performance of all five methods improves with the increment of cellular heritability. In general, the summary-level methods (TisCoMM-S2 and S-MultiXcan) perform similarly to their counterparts in individual-level data. Moreover, the power of TisCoMM and TisCoMM-S2 is robust to sparsity level s but the power of alternative methods favors settings with smaller sparsity level s. When sparsity level s is small and cellular heritability becomes large, the power of all methods become comparable.
Figure 3.

TisCoMM joint test outperforms the other multi-tissue methods. The number of replicates is 5000. In each subplot, the x-axis stands for the SNP heritability level, and the y-axis stands for the proportion of significant genes within 5000 replicates.
Simulations: testing tissue-specific effects
We focused on the detection of tissue-specific effects in the second set of simulations. Here, we compared the TisCoMM tissue-specific test with the single-tissue methods including CoMM (10), PrediXcan (7), and TWAS (8) under the alternative hypothesis with fixed tissue heritability 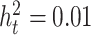 and fixed sparsity s = 0.05. We considered ten tissues and varied the number of causal tissues to simulate different levels of tissue specificity of a trait. Specifically, we considered settings with one and two causal tissues, respectively. To allow correlated gene expression in the GWASs, the nonzero of tissue-specific effect
and fixed sparsity s = 0.05. We considered ten tissues and varied the number of causal tissues to simulate different levels of tissue specificity of a trait. Specifically, we considered settings with one and two causal tissues, respectively. To allow correlated gene expression in the GWASs, the nonzero of tissue-specific effect  was generated with rows drawn from a multivariate normal distribution, with AR correlation parameter ρW = 0.2, 0.5, 0.8. A large value of ρW implied a higher correlation among columns of
was generated with rows drawn from a multivariate normal distribution, with AR correlation parameter ρW = 0.2, 0.5, 0.8. A large value of ρW implied a higher correlation among columns of 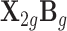 . Other sittings were similar to Simulation I.
. Other sittings were similar to Simulation I.
We repeated the whole process 1000 times and calculated the statistical power and false-positive rate (FPR) as the proportion of P-values reaching the significance level in causal tissues and non-causal tissues, respectively. Specifically, we set the significance level at 0.05/10 for all considered methods. Figure 4 shows simulation results for the case that one tissue is causal. We observe that in all settings, the TisCoMM tissue-specific test has slightly inferior power, compared to the single-tissue methods, but much smaller FPR. As expected, the statistical power of all methods increases with cellular heritability (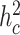 ). However, the FPR of single-tissue methods substantially inflates while that of TisCoMM tissue-specific test remains at the same level. Furthermore, the FPR of TisCoMM tissue-specific test does not vary with correlations among expressions across multiple tissues (ρW) while that of single-tissue methods increase with ρW. The similar pattern can be observed for the case that two tissues are causal (Supplementary Figure S2). These results demonstrate the usefulness of TisCoMM tissue specific test in exploring the tissue-specific role of genes with controlled FPR.
). However, the FPR of single-tissue methods substantially inflates while that of TisCoMM tissue-specific test remains at the same level. Furthermore, the FPR of TisCoMM tissue-specific test does not vary with correlations among expressions across multiple tissues (ρW) while that of single-tissue methods increase with ρW. The similar pattern can be observed for the case that two tissues are causal (Supplementary Figure S2). These results demonstrate the usefulness of TisCoMM tissue specific test in exploring the tissue-specific role of genes with controlled FPR.
Figure 4.

The comparison of the TisCoMM tissue-specific test and the single-tissue association tests under the alternative hypothesis with one causal tissue. Upper panel: the power of TisCoMM tissue-specific test and the single tissue methods with Bonferroni correction applied. Lower panel: the corresponding false-positive rates under each setting.
Additional simulations with randomly generated genotype data are presented in Supplementary Figures S3–S10. All these results are consistent with the above observations. To better mimic the real data, we further conducted the simulations by adjusting for covariates or confounding factors; results are shown in Supplementary Text (Supplementary Figures S11–S22).
Real data applications
We performed multi-tissue TWAS analysis for summary-level GWASs data in 33 complex traits (see Supplementary Table S1 for details), including 15 traits from the Nature Genetics paper (NG traits) (19) and 18 traits from the UK Biobank (UKB traits). These traits can be roughly divided into four categories, including metabolites (e.g. HDL-C, LDL-C and fasting glucose), autoimmune diseases (e.g. asthma, Crohn’s disease and macular degeneration), psychiatric/neurodegenerative disorders (e.g. Alzheimer’s disease, major depression disorder, and psychiatric disorder), and cardiovascular disorders (e.g. coronary artery disease and peripheral vascular disease). The Genotype-Tissue Expression (GTEx) Project (6) reported eQTL in 48 tissues, where the number of genes in each tissue ranges from 16 333 to 27 378. In the analysis, we extracted cis-SNP that are within either 500 kb upstream of the transcription start site or 500 kb downstream of the transcription end site.
Similar to a single-tissue analysis, there are two different strategies to select tissues for TWASs: the first strategy uses expressions from the most biologically related tissues while the second strategy selects top tissues with the largest number of available individuals (9). In Supplementary Table S2 of (19), it provides the most biologically related tissues and thus we could use trait-relevant tissues for the NG traits. In detail, for each trait, a set of tissues with significant enrichment P-values (after Bonferroni correction) was identified, and a subset with >100 overlapped samples (34) was chosen for further analysis in TisCoMM-S2. On the other hand, although methods like LD score regression (17) can be used for the UKB traits, it is difficult to balance the tissue relevance and sample size for each tissue. To make efficient use of the GTEx data set, we used six tissues with the largest number of overlapped samples for the UKB traits.
The analysis for each trait based on its GWAS summary statistics together with the eQTL data from 4–6 tissues can be done around 100 min on a Linux platform with 2.6 GHz Intel Xeon CPU E5-2690 with 30 720KB cache and 96GB RAM (only 10–12GB RAM used) on 24 cores. To prioritize trait-associated genes, we compared TisCoMM-S2 with other two multi-tissue TWAS methods, i.e. S-MultiXcan and UTMOST. Both alternative methods take advantage of prediction models to impute gene expressions across multiple tissues. The prediction models used here were Elastic Net models trained on 48 GTEx tissues. See Table 1 for the summary of detections across different approaches for the 15 NG and 18 UKB traits. Generally, TisCoMM-S2 identifies more genome-wide associations than S-MultiXcan and UTMOST in most traits. In detail, TisCoMM-S2/S-MultiXcan/UTMOST identified 3058/2008/1769 and 443/338/277 genome-wide significant genes in all the NG traits and UKB traits, respectively. Their qq-plots of P-values are shown in Supplementary Figures S23–S26 and plots for their genomic inflation factors are shown in Supplementary Figure S27. As case study examples, we carefully examined the results for late-onset Alzheimer’s disease (LOAD) and asthma. Additional results for other traits are shown in Supplementary Figures S31–S36.
Table 1.
Numbers of significant gene-trait associations across 15 NG traits and 18 UKB traits. The reference penal is European subsamples from 1000 Genome. The number in the parenthesis denoted genes reported on the GWASs catalog. The full names of traits can be found in Supplementary Table S1
| TisCoMM-S2 | S-MultiXcan | UTMOST | |
|---|---|---|---|
| NG traits | |||
| 2hrGlu | 1(0) | 0(0) | 5(0) |
| LOAD | 92(24) | 71(20) | 70(19) |
| BMI | 82(30) | 59(21) | 68(25) |
| CAD | 69(9) | 28(5) | 36(11) |
| CD | 468(52) | 339(50) | 291(55) |
| FG | 94(11) | 66(11) | 54(8) |
| FI | 3(0) | 1(0) | 2(0) |
| HDL-C | 464(14) | 268(13) | 237(12) |
| HOMAB | 10(0) | 7(0) | 4(0) |
| HOMAIR | 1(0) | 1(0) | 1(0) |
| LDL-C | 498(5) | 273(5) | 228(5) |
| TC | 603(88) | 376(77) | 330(86) |
| TG | 360(58) | 250(48) | 192(40) |
| UC | 301(30) | 262(31) | 243(41) |
| WHR | 12(3) | 7(1) | 8(1) |
| UKB traits | |||
| ALLERGIC_RHINITIS | 25(3) | 23(2) | 12(4) |
| ASTHMA | 200(31) | 157(29) | 140(36) |
| CARD | 2(0) | 2(0) | 4(0) |
| DEPRESS | 2(0) | 1(0) | 0(0) |
| DYSLIPID | 166(0) | 120(0) | 91(0) |
| HEMORRHOIDS | 0 | 1(0) | 0 |
| HERNIA_ABDOMINOPELVIC | 2(1) | 2(1) | 1(1) |
| INSOMNIA | 0 | 0 | 0 |
| IRON_DEFICIENCY | 0 | 0 | 0 |
| IRRITABLE_BOWEL | 0 | 1(0) | 0 |
| MACDEGEN | 0 | 0 | 0 |
| OSTIOA | 1(0) | 2(0) | 2(1) |
| OSTIOP | 0 | 1(0) | 0 |
| PEPTIC_ULCERS | 1(0) | 1(0) | 0 |
| PSYCHIATRIC | 1 | 1 | 3 |
| PVD | 2(0) | 3(0) | 3(0) |
| STRESS | 1(0) | 1(0) | 1(0) |
| VARICOSE_VEINS | 40(2) | 22(2) | 20(2) |
To examine the effect of tissue selection strategy on the performance of gene prioritization, we also used the six tissues for the NG traits. Results are shown in Supplementary Table S2. We can observe that the two different strategies lead to different sets of significant candidate genes. However, there is a large number of overlaps between the two sets. This result demonstrates the usefulness of the second strategy to identify gene-trait associations in joint tests.
Real Data analysis for LOAD
TisCoMM-S2 Joint test for LOAD and validation
Based on TisCoMM-S2 joint test and two other methods (S-MultiXcan and UTMOST), 92/71/70 genome-wide significant genes were identified respectively after Bonferroni correction. The list of significant gene-LOAD associations returned from TisCoMM-S2, S-MultiXcan and UTMOST can be found in Supplementary Table S3. The qq-plots for associations tested in these three approaches are shown in Figure 5A. To validate our findings in another independent data set, we used the summary statistics from a GWAS by proxy (GWAX (35), the sample size is 114 564). Our replication rate was the highest (Supplementary Table S4, Figure 6) among all the three methods, where 31 out of 92 (33.7%) genes were successfully replicated under the Bonferroni-corrected significance threshold and the numbers of replicated genes raised to 189 under a relaxed P-value cutoff of 0.05. On the other hand, the replication rates of S-MultiXcan (32.4%) and UTMOST (32.9%) were lower. Moreover, TisCoMM-S2 had the highest replication rate in either the uniquely detected gene sets (genes uniquely detected by single method) or commonly detected gene sets (genes detected by multiple methods).
Figure 5.

TisCoMM-S2 results for LOAD and asthma. The reference panel is European subsamples from 1000 Genome. In each row, the two panels show the qq-plot (left) and Manhatton plot (right).
Figure 6.

Overlap of detected genes from three methods and replication rates. Circles represent the detected gene sets from each method (TisCoMM-S2, S-MultiXcan and UTMOST), and the size of the overlapped regions are proportional to the number of genes inside. The replication rates are shown in two ways as ratio and heatmap. In the ratio form, denominator indicates the number of detected genes in our first test data set, and numerator indicates the number of validated genes in the second validation data set. To visualize the replication rates, the ratios are then converted to heatmaps. RRT, RRM and RRU correspond to the replication rate of TisCoMM-S2, S-MultiXcan and UTMOST.
TisCoMM-S2 tissue-specific test infers tissue-specific roles of candidate genes for LOAD
To demonstrate the utility of the TisCoMM-S2 tissue-specific test, we applied the tissue-specific test to all 92 candidate genes of LOAD identified by the TisCoMM-S2 joint test, and compared analysis results with those from CoMM (10,11). Table 2 shows the distributions of identified tissues in which candidate genes are associated with LOAD (see details in Supplementary Table S7). Among all identified candidate genes for LOAD, 76.1% were significant in no more than two tissues using TisCoMM-S2 while 70.7% were significant in all four tissues using CoMM-S2. The most plausible explanation is that compared to the multivariate perspective of our TisCoMM-S2 tissue-specific test, single-tissue approaches, e.g. CoMM-S2, tend to have larger tissue bias and more inflation in significant findings (9). Suppose a gene is causal in tissue A but not in tissue B, and its expressions in tissues A and B are correlated. In a single-tissue test, the association can be spuriously significant for tissue B because of the similar gene expression pattern observed in both tissues. By performing a tissue-specific test for this gene in tissue B conditioned on tissue A, the significant spurious association will be largely excluded. We also illustrate the relationship between effect size and P-value as well as the cellular-heritability in each tissue using volcano plots (Supplementary Figure S29A).
Table 2.
Distributions of tissues in which the candidate genes’ associations arise in LOAD and asthma
| Trait |
 tissues tissues |
0 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|---|
| LOAD | TisCoMM-S2 | 5 | 28 | 37 | 17 | 5 | - | - |
| CoMM-S | 6 | 5 | 7 | 9 | 65 | - | - | |
| Asthma | TisCoMM-S2 | 37 | 68 | 58 | 28 | 6 | 3 | 0 |
| CoMM-S | 20 | 11 | 5 | 5 | 9 | 30 | 120 |
We further performed TisCoMM-S2 tissue-specific test on genes identified by all three methods (TisCoMM-S2, S-MultiXcan and UTMOST, see details in Supplementary Table S7). The detailed overlap of genes returned from these three methods is illustrated in Figure 7. We notice that the tissue-specific rate is different in each method and genes uniquely identified by TisCoMM-S2 has the largest tissue-specific rate (100%, Figure 7). Genes uniquely identified by UTMOST has the smallest tissue-specific rate. Only 3 out of 14 genes uniquely identified by UTMOST have tissue-specific effect.
Figure 7.

Overlap of detected genes from three methods and tissue-specific rates. Circles represent the detected gene sets from each method (TisCoMM-S2, S- MultiXcan and UTMOST), and the size of the overlapped regions are proportional to the number of genes inside. The tissue-specific rates are shown in two ways as ratio and heatmap. In the ratio form, denominator indicates the number of detected genes in each method, and numerator indicates the number of tissue-specific genes. To visualize the tissue-specific rates, the ratios are then converted to heatmaps, so that the intensity of the color is proportional to the tissue-specific rate.
Lastly, we investigate the molecular functions of LOAD associated genes in each tissue. In each of tested tissues in LOAD, there are ∼40 tissue-specific genes. It is difficult to carry out a proper pathway analysis with such limited gene sets. So we classified the genes into seven functional groups based on which molecular functions they belong to. As shown in Figure 8, majority (>62%) of LOAD-associated genes belonged to binding and catalytic activity, and a small portion of significant LOAD genes were transcription factors suggesting that many regulation processes are going on at both protein and mRNA levels in different tissues.
Figure 8.

(A) Each pie chart corresponding to a different tissue shows the percentage of LOAD-associated genes in each molecular function group (from gene ontology). (B) The x-axis of the heatmap represents the union of LOAD-associated genes in 3 function groups (binding, catalytic activity, and transcription factor). The y-axis represents different tissue types. In each cell, the background color (shades of gray) indicates the significance level, the circle size indicates the heritability, and the color inside each circle indicates the effect size. (C) Pathway analysis of asthma-associated genes in thyroid and lung. Pathway analysis was done using a web-based software DAVID, testing the enrichments of asthma-associated genes in biological processes (from gene ontology). Significant pathways were selected if gene count ≥ 5 and Benjamini-Hochberg (BH) corrected P-value ≤0.05. The asthma-associated genes are highlighted in blue. (D) The x-axis of the heatmap represents the asthma-associated genes in the immune response pathway. And all the other settings are the same as the one used in part B.
Known LOAD GWAS genes captured by TisCoMM-S2
Among the 92/71/70 LOAD associated genes identified by the three methods (TisCoMM-S2 joint test, S-MultiXcan and UTMOST), 17 out of the 45 overlapping genes are known LOAD GWAS genes. Here, we define known LOAD GWAS gene as the ones reported in GWAS Catalog. Among the 92 candidate target genes identified by TisCoMM-S2 joint test, 24 of them are previously known LOAD GWAS genes, which are annotated in the Manhattan plot in Figure 5A. These include genes on the chromosome (CHR) 2 (BIN1), CHR 6 (CD2AP), CHR 7 (EPHA1), CHR 8 (CLU), CHR 11 (PICALM, CCDC89, MS4A2, MS4A6A), CHR 16 (IL34) , and CHR 19 (STK11 and APOE region). Moreover, TisCoMM-S2 also identified 35 genes that were not significant in neither S-MultiXcan nor UTMOST, and four of them are known LOAD GWAS genes, including IL34 (P-value = 1 × 10−6), PTK2b (P-value = 1.4 × 10−9), EPHX (P-value = 4.7 × 10−8) and STK11 (P-value = 7.2 × 10−7).
The well-replicated risk gene APOE (36) and its 50Kb downstream CLPTM1 have been identified by the TisCoMM-S2 joint test. Moreover, the TisCoMM-S2 tissue-specific test identified CLPTM1 to be significantly associated with LOAD in all four tissues (artery aorta, esophagus mucosa, nerve tibial and skin sun-exposed lower leg with tissue-specific P-values < 4.9 × 10−7), but APOE to be only significantly associated with LOAD in artery aorta (tissue-specific P-value = 8.3 × 10−9) and nerve tibial (tissue-specific P-value = 1.2 × 10−8). On the other hand, CoMM-S2 significantly identified both APOE and CLPTM1 in all four tissues (P-values ≤ 1 × 10−10) but failed to identify the difference of tissue-specific role for these two genes.
Novel LOAD genes captured by TisCoMM-S2
Among all novel genes for LOAD identified by TisCoMM-S2 joint test, some of them were identified to be LOAD-related genes based on other computational models (e.g. MAP3K2) while some of them have not been directly linked to LOAD yet, but have been proven to be important regulators in different regions of the neuron system (e.g. STMN4, EED and APC2). MAP3K2 is 200 kb downstream of B1N1, a reported LOAD risk gene (37) that was also genome-wide significant in our joint test (P-values for both B1N1 and MAP3K2 ≤1 × 10−10). MAP3K2 belongs to the serine/threonine protein kinase family and has been previously identified as a member of the Alzheimer’s disease susceptibility network (38). In Supplementary Figure S28, regional plots based on GWAS, TWAS and eQTL results are shown for the LOAD associated locus near the MAP3K2 gene. It can be seen that although the GWAS signals are weak and moderate, the enrichment of eQTL signals near the MAP3K2 gene have led to improved TWAS results. STMN4 (P-value ≤ 1 × 10−10 ) encodes the known protein that exhibits microtubule-destabilizing activity. The expression levels of this gene in mouse neurons have been shown to change significantly after different exposure of cortical nerve cells to the Aβ peptide (39). The expression of STMN4 in zebrafish has also been shown to have an important role in regulating neurogenesis in the neural keel stage (40). EED (P-value = 5.7 × 10−7 ) encodes a Polycomb protein, which plays a starring role as an important modulator of hippocampal development (41). APC2 (P-value = 1.3 × 10−6) is preferentially expressed in postmitotic neurons and involved in brain development through its regulation of neuronal migration and axon guidance (42). We annotate these four genes in red in Figure 5A.
LOAD associated genes in brain tissues
According to our tissue selection strategy, above LOAD genes were tested in four non-brain tissues (enriched tissues). To further investigate the gene expression changes in the well-studied disease tissues, three more brain regions (hippocampus, frontal cortex, and cerebellar hemisphere) were selected for another tissue-specific analysis for LOAD. Because it is known that hippocampus is one of the first brain regions to be affected by Alzheimer’s disease and related to the memory lost (43), markers such as Aβ in frontal cortex can be used to predict future Alzheimer’s disease (44), and cerebellum is affected in the final stage of the disease and related to cognitive decline (45). The joint test conducted on brain regions revealed 105 LOAD associated genes, of which 73 were identified in the enriched tissues (Supplementary Figure S30A), and the other 32 genes were uniquely identified in brain regions (Supplementary Figure S30B). According to the joint test, the most significant gene uniquely identified in brain regions is KLC3 (P-value ≤1 × 10−10 ), which is within 50 kb downstream of APOE. Moreover, it is significantly associated with LOAD in hippocampus region only, but not the other two brain regions according to the tissue-specific test (Supplementary Figure S30B and Table S8). Thus, we propose KLC3 as one of the potential novel targets for LOAD in hippocampus.
Real data analysis for asthma
TisCoMM-S2 Joint test for asthma and validation
After Bonferroni correction, TisCoMM-S2/S-MultiXcan/UTMOST identified 200/157/140 genome-wide significant genes, respectively. The list of significant gene-asthma associations returned from TisCoMM-S2 , S-MultiXcan, and UTMOST can be found in Supplementary Table S5. The qq-plots for associations in these three approaches are shown in Figure 5B. To replicate our findings in another independent data set, we used the summary statistics from TAGC European-ancestry GWAS (46) (the sample size is 127 669). Our replication rate was the second highest (Supplementary Table S6, Figure 6) among all the three methods, where 179 out of 200 (89.5%) genes were successfully replicated under the Bonferroni-corrected significance threshold and the numbers of replicated genes raised to 44 under a relaxed P-value cutoff of 0.05. The replication rate of S-MultiXcan was the highest (89.8%), and the replication rate of UTMOST (80%) was much lower than the other two methods. Moreover, TisCoMM-S2 had the highest replication rate in either the uniquely detected gene sets (genes uniquely detected by single method) or commonly detected gene sets (genes detected by multiple methods).
TisCoMM-S2 tissue-specific test infers tissue-specific roles of candidate genes for asthma
Similar to the tissue-specific test conducted on LOAD, 200 candidate genes of asthma identified by TisCoMM-S2 joint test were then subjected to TisCoMM-S2 tissue-specific test. As we can see in Table 2, 81.5% asthma candidate genes were significant in less than three tissues using TisCoMM-S2 while 60.0% were significant in all six tissues using CoMM-S2. The relationship between effect size and P-value as well as the cellular-heritability in each tissue are shown in Supplementary Figure S29B. We further conducted tissue-specific test on genes identified by all three methods (TisCoMM-S2, S-MultiXcan and UTMOST, see details in Supplementary Table S9). As shown in Figure 7, we can observe a similar pattern as those for LOAD. Compared to TisCoMM-S2, genes uniquely identified by S-MultiXcan and UTMOST have much lower chance to be tissue-specific genes.
We further conducted pathway analysis using DAVID (47) on six sets of asthma-associated genes in all six tissues (thyroid, lung, artery tibial, muscle skeletal, adipose subcutaneous, and skin sun-exposed lower leg), respectively. As listed in Figure 8B, all three significant pathways in thyroid tissue belonged to the immune system, and the only significant pathway in lung tissue was immune response. However, no significant pathways were detected in the other four tissues. Among asthma-associated genes in immune response (first row in Figure 8C and D), the majority of them were shared between thyroid and lung, and located in the MHC region on CHR 6 including several HLA genes and LST1. Our pathway analysis suggests that nearly the same set of immune genes in thyroid and lung are responsible for asthma development.
Known asthma GWAS genes captured by TisCoMM-S2
Among the 200/157/140 asthma associated genes identified by the three methods (TisCoMM-S2 joint test, S-MultiXcan and UTMOST), 21 out of the 98 overlapping genes are known asthma GWAS genes. Among all 200 candidate target genes identified by TisCoMM-S2, 31 of them are known asthma GWAS genes, which is annotated in the Manhattan plot in Figure 5B, including genes on CHR 2 (IL1RL1/IL18R1), CHR 5 (TSLP/WDR36, RAD50), CHR 6 (HLA-DR/DQ regions, MAP3K7), CHR 9 (IL33), CHR 11 (C11orf30, LRRC32), CHR 15 (SMAD3) and CHR 17 (genes from the 17q21 asthma locus). Also, TisCoMM-S2 identified 56 genes that were not significant in neither S-MultiXcan nor UTMOST, and two of them are known asthma GWAS genes, which are PSORS1C1 (P-value = 2.2 × 10−7), and MAP3K7 (P-value =3 × 10−7).
A lot of known HLA genes in the MHC region were successfully identified as asthma related genes using our method, including HLA-DOA, HLA-DOB, HLA-DPB1, HLA-DQB1, and HLA-DRA. Another example of known asthma locus is 17q21 locus at CHR 17. ORMDL3 and GSDMB were identified to be asthma associated genes in this locus, have been mentioned as asthma susceptibility genes by many studies, a comprehensive review was written by Stein et al. (48) The original finding of ORMDL3 was observed in one GWAS study, and have been further validated in a mouse model (49). The TisCoMM-S2 tissue-specific test identified both ORMDL3 and GSDMB to be significantly associated with asthma only in lung tissue (see the volcano plot in Supplementary Figure S29B, tissue-specific P-values for these two genes are 1.7 × 10−3 and 7.1 × 10−7 , respectively). However, CoMM-S2 identified both ORMDL3 and GSDMB in all six tissues (P-values ≤ 1 × 10−10) but failed to identify the relevant tissues with which these two genes are causally related to asthma. Besides, LTA is a reported GWAS hit for asthma in GWAS Catalog, and was also identified to be specifically regulated in thyroid tissue based on our tissue-specific test. It is a cytokine produced by lymphocytes, and known as a regulator of lipid metabolism (50).
Novel asthma genes captured by TisCoMM-S2
Among all novel loci for asthma identified by TisCoMM-S2 joint test, PDCD1LG2 was shown to have essential roles in modulating and polarizing T-cell functions in airway hyperreactivity (51). Based on our tissue-specific test, TNF which is a well-studied asthma gene (52,53) was explicitly identified to be associated with asthma in lung tissue. The positive correlation between TNF expression and asthma in lung confirmed our previous understanding of TNF activation in asthma, promoting airway inflammation and airway hyperresponsiveness. However, this gene was not reported as an asthma associated gene in GWAS Catalog. Another novel asthma gene regulated individually in thyroid tissue is NCR3, which mediates the crosstalk between natural killer cells and dendritic cells (54). However, it remains unclear how the alteration of NCR3 in thyroid could lead to asthma development. Validating causal role of these gene in asthma requires further investigation. We annotate them in red in Figure 5B.
DISCUSSION
Despite the substantial successes of TWASs and its variants, the existing multi-tissue methods have several limitations, e.g., incapability to identify the tissue-specific effect of a gene, ignorance of imputation uncertainty, and failure to efficiently use tissue-shared patterns in eQTLs. To overcome these limitations and provide additional perspectives over tissue-specific roles of identified genes, we have proposed a powerful multi-tissue TWAS model, together with a computationally efficient inference method and software implementation in TisCoMM. Specifically, we have developed a joint test for prioritizing gene-trait associations and a tissue-specific test for identifying the tissue-specific role of candidate genes. Conditioned on the inclusion of trait-relevant tissues, the tissue-specific test in TisCoMM can mostly remove the spurious associations in a single-tissue test due to high correlations among gene expression across tissues. We have also developed a summary-statistic-based model, TisCoMM-S2, extending the applicability of TisCoMM to publicly available GWAS summary data. Using both simulations and real data, we examined the relationship between TisCoMM and TisCoMM-S2. Our results, as shown in Figure 2, show that the test statistics from TisCoMM and TisCoMM-S2 are highly correlated (R2 > 0.95). We further analyzed summary-level GWAS data from 33 traits with replication data for Alzheimer’s disease and asthma. Overall, the findings from TisCoMM-S2 are around 30% more than those from S-MultiXcan or UTMOST while qq-plots from these studies show that there are no apparent inflations. To replicate our findings for Alzheimer’s disease and asthma, we applied TisCoMM-S2 to independent data sets for each disease. Results show that replication rates for Alzheimer’s disease and asthma are high.
We further inferred the tissue-specific effects of identified genes using the TisCoMM-S2 tissue-specific test. By classifying these genes into seven functional groups, we observed that majority (62%) of LOAD-associated genes were related to binding and catalytic activity while a small portion was from transcription factors suggesting active regulation processes at both protein and mRNA level in different tissues. We also observed about 40 LOAD-associated genes in each non-brain tissues. The significance of these genes could be due to the exclusion of LOAD-relevant tissues, e.g. brain tissues. To fill this gap, we further conducted one more analysis on three brain regions, and identified 32 brain specific genes. For asthma, genes ORMDL3 and GSDMB were identified to be significantly associated with asthma only in lung tissue using TisCoMM-S2 tissue-specific test. However, single-tissue analysis (CoMM-S2) identified both genes significant in all six tested tissues. Further pathway analysis shows that all three significant pathways for thyroid tissue belong to the immune system and the only significant pathway for lung tissue was immune response. The majority of shared genes between thyroid and lung tissues are located in the MHC region on CHR 6, including several HLA genes and LST1. The proteins encoded by HLA genes are known as antigens. In combination with antigen-presenting cells (e.g. macrophages and dendritic cells), they play an essential role in the activation of immune cells as well as airway inflammation in response to asthma-related allergens (55,56). Despite the utility of TisCoMM to perform gene-trait association analysis in a tissue-specific manner, it is primarily designed to test genes with direct effects from cis-eQTL. Recently, an omnigenic model was proposed to better understand the underlying mechanism of so-called polygenicity in complex traits (57). Liu et al. (58) further provided a theoretical model to understand complex trait architecture by partitioning genetic contributions into direct effects from core genes and indirect effects from peripheral genes acting in trans. Most works from TWASs identify core genes with direct effects. How to effectively interrogate peripheral genes with indirect effects essentially remains an open question. Furthermore, we restricted to common variants (SNPs with MAF  ) in real data applications due to the limited sample sizes of the multiple eQTL data. As the sample size from the reference data set becomes large, combining effects from both common and rare variants may increase the statistical power for finding gene-trait associations. As high-throughput data are continuously generating for a much larger sample size with more precision, TisCoMM sheds light on how to integrate useful data for the desired analysis effectively.
) in real data applications due to the limited sample sizes of the multiple eQTL data. As the sample size from the reference data set becomes large, combining effects from both common and rare variants may increase the statistical power for finding gene-trait associations. As high-throughput data are continuously generating for a much larger sample size with more precision, TisCoMM sheds light on how to integrate useful data for the desired analysis effectively.
URLs
The software implementation of TisCoMM with a User Manual is freely available at: https://github.com/XingjieShi/TisCoMM/. The code to reproduce all the analyses presented in the paper is also included.
Supplementary Material
ACKNOWLEDGEMENTS
The computational work for this article was partially performed using resources from the National Supercomputing Centre, Singapore (https://www.nscc.sg).
Contributor Information
Xingjie Shi, Department of Statistics, Nanjing University of Finance and Economics, Nanjing, China; Centre for Quantitative Medicine, Health Services & Systems Research, Duke-NUS Medical School, Singapore.
Xiaoran Chai, Beijing Advanced Innovation Center for Genomics (ICG) & Biomedical Pioneering Innovation Center (BIOPIC), Peking University, Beijing, China; School of Medicine, National University of Singapore, Singapore.
Yi Yang, Centre for Quantitative Medicine, Health Services & Systems Research, Duke-NUS Medical School, Singapore.
Qing Cheng, Centre for Quantitative Medicine, Health Services & Systems Research, Duke-NUS Medical School, Singapore.
Yuling Jiao, School of Mathematics and Statistics, and Hubei Key Laboratory of Computational Science, Wuhan University, Wuhan, China.
Haoyue Chen, School of International Studies, Zhejiang University, Hangzhou, China.
Jian Huang, Department of Statistics and Actuarial Science, University of Iowa, USA.
Can Yang, Department of Mathematics, The Hong Kong University of Science and Technology, Hong Kong, China.
Jin Liu, Centre for Quantitative Medicine, Health Services & Systems Research, Duke-NUS Medical School, Singapore.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Duke-NUS Medical School [R-913-200-098-263]; AcRF Tier 2 [MOE2016-T2-2-029, MOE2018-T2-1-046, MOE2018-T2-2-006] from the Ministry of Education, Singapore; Grant [11871474, 61701547] from National Natural Science Foundation of China; Hong Kong Research Grant Council [12301417, 16307818, 16301419]; Grant DMS-1916199 from the National Science Foundation of USA. Funding for open access charge: Ministry of Education, Singapore [MOE2018-T2-2-006].
Conflict of interest statement. None declared.
REFERENCES
- 1. Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J.. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 2017; 101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. et al.. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012; 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cookson W., Liang L., Abecasis G., Moffatt M., Lathrop M.. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009; 10:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J.. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010; 6:e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Albert F.W., Kruglyak L.. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 2015; 16:197. [DOI] [PubMed] [Google Scholar]
- 6. Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N. et al.. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013; 45:580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gamazon E.R., Wheeler H.E., Shah K.P., Mozaffari S.V., Aquino-Michaels K., Carroll R.J., Eyler A.E., Denny J.C., Nicolae D.L., Cox N.J. et al.. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015; 47:1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W., Jansen R., De Geus E.J., Boomsma D.I., Wright F.A. et al.. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016; 48:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wainberg M., Sinnott-Armstrong N., Mancuso N., Barbeira A.N., Knowles D.A., Golan D., Ermel R., Ruusalepp A., Quertermous T., Hao K. et al.. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 2019; 51:592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang C., Wan X., Lin X., Chen M., Zhou X., Liu J.. CoMM: a collaborative mixed model to dissecting genetic contributions to complex traits by leveraging regulatory information. Bioinformatics. 2018; 35:1644–1652. [DOI] [PubMed] [Google Scholar]
- 11. Yang Y., Shi X., Jiao Y., Huang J., Chen M., Zhou X., Sun L., Lin X., Yang C., Liu J.. CoMM-S2: a collaborative mixed model using summary statistics in transcriptome-wide association studies. Bioinformatics. 2020; 36:2009–2016. [DOI] [PubMed] [Google Scholar]
- 12. Nagpal S., Meng X., Epstein M.P., Tsoi L.C., Patrick M., Gibson G., De Jager P.L., Bennett D.A., Wingo A.P., Wingo T.S. et al.. TIGAR: an improved Bayesian tool for transcriptomic data imputation enhances gene mapping of complex traits. Am. J. Hum. Genet. 2019; 105:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y.I., Wong G., Humphrey J., Raj T.. Prioritizing Parkinson’s Disease genes using population-scale transcriptomic data. Nat. Commun. 2019; 10:994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barbeira A.N., Pividori M.D., Zheng J., Wheeler H.E., Nicolae D.L., Im H.K.. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 2019; 15:e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu Y., Li M., Lu Q., Weng H., Wang J., Zekavat S.M., Yu Z., Li B., Gu J., Muchnik S. et al.. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet. 2019; 51:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barbeira A.N., Dickinson S.P., Bonazzola R., Zheng J., Wheeler H.E., Torres J.M., Torstenson E.S., Shah K.P., Garcia T., Edwards T.L., et al.. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018; 9:1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Finucane H.K., Reshef Y.A., Anttila V., Slowikowski K., Gusev A., Byrnes A., Gazal S., Loh P.-R., Lareau C., Shoresh N. et al.. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 2018; 50:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cai M., Chen L., Liu J., Yang C.. IGREX for quantifying the impact of genetically regulated expression on phenotypes. NARGenom. Bioinform. 2020; 2:lqaa010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gamazon E.R., Segrè A.V., van de Bunt M., Wen X., Xi H.S., Hormozdiari F., Ongen H., Konkashbaev A., Derks E.M., Aguet F. et al.. Using an atlas of gene regulation across 44 human tissues to inform complex disease-and trait-associated variation. Nat. Genet. 2018; 50:956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Consortium G. et al.. Genetic effects on gene expression across human tissues. Nature. 2017; 550:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Urbut S.M., Wang G., Carbonetto P., Stephens M.. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nature Genetics. 2018; 51:187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou X., Stephens M.. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012; 44:821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wheeler H.E., Shah K.P., Brenner J., Garcia T., Aquino-Michaels K., Cox N.J., Nicolae D.L., Im H.K., Consortium G. et al.. Survey of the heritability and sparse architecture of gene expression traits across human tissues. PLoS Genet. 2016; 12:e1006423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tukey J.W. One degree of freedom for non-additivity. Biometrics. 1949; 5:232–242. [Google Scholar]
- 25. Chatterjee N., Kalaylioglu Z., Moslehi R., Peters U., Wacholder S.. Powerful multilocus tests of genetic association in the presence of gene-gene and gene-environment interactions. Am. J. Hum. Genet. 2006; 79:1002–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sabatti C., Service S.K., Hartikainen A.-L., Pouta A., Ripatti S., Brodsky J., Jones C.G., Zaitlen N.A., Varilo T., Kaakinen M. et al.. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 2009; 41:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shi X., Jiao Y., Yang Y., Cheng C. Y., Yang C., Lin X., Liu J.. VIMCO: variational inference for multiple correlated outcomes in genome-wide association studies. Bioinformatics. 2019; 35:3693–3700. [DOI] [PubMed] [Google Scholar]
- 28. Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., De Bakker P.I., Daly M.J. et al.. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007; 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stegle O., Parts L., Durbin R., Winn J.. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput. Biol. 2010; 6:e1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schäfer J., Strimmer K.. A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat. Appl. Genet. Mol. 2005; 4:32. [DOI] [PubMed] [Google Scholar]
- 32. Wang J., Gamazon E.R., Pierce B.L., Stranger B.E., Im H.K., Gibbons R.D., Cox N.J., Nicolae D.L., Chen L.S.. Imputing gene expression in uncollected tissues within and beyond GTEx. Am. J. Hum. Genet. 2016; 98:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park Y., Sarkar A.K., Bhutani K., Kellis M.. Multi-tissue polygenic models for transcriptome-wide association studies. 2017; bioRxiv doi:10 February 2017, preprint: not peer reviewed 10.1101/107623. [DOI]
- 34. Mancuso N., Shi H., Goddard P., Kichaev G., Gusev A., Pasaniuc B.. Integrating gene expression with summary association statistics to identify genes associated with 30 complex traits. Am. J. Hum. Genet. 2017; 473–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu J.Z., Erlich Y., Pickrell J.K.. Case–control association mapping by proxy using family history of disease. Nat. Genet. 2017; 49:325. [DOI] [PubMed] [Google Scholar]
- 36. Yu C.-E., Seltman H., Peskind E.R., Galloway N., Zhou P.X., Rosenthal E., Wijsman E.M., Tsuang D.W., Devlin B., Schellenberg G.D.. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: patterns of linkage disequilibrium and disease/marker association. Genomics. 2007; 89:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jager Philip L D., Gyan S., Katie L., Jeremy B., Schalkwyk L.C., Lei Y., Eaton M.L., Keenan B.T., Jason E., Cristin M.C.. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014; 17:1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kurakin A., Bredesen D.E.. Dynamic self-guiding analysis of Alzheimer’s disease. Oncotarget. 2015; 6:14092–14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Romito-Digiacomo R.R., Harry M., Cicero S.A., Karl H.. Effects of Alzheimer’s disease on different cortical layers: the role of intrinsic differences in Abeta susceptibility. J. Neurosci. Off. J. Soc. Neurosci. 2007; 27:8496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin M.J., Lee S.J.. Stathmin-like 4 is critical for the maintenance of neural progenitor cells in dorsal midbrain of zebrafish larvae. Sci. Rep.-UK. 2016; 6:36188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu P.-P., Xu Y.-J., Dai S.-K., Du H.-Z., Wang Y.-Y., Li X.-G., Teng Z.-Q., Liu C.-M.. Polycomb protein EED regulates neuronal differentiation through targeting SOX11 in hippocampal dentate gyrus. Stem Cell Rep. 2019; 13:115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Almuriekhi M., Shintani T., Fahiminiya S., Fujikawa A., Kuboyama K., Takeuchi Y., Nawaz Z., Nadaf J., Kamel H., Kitam A.K. et al.. Loss-of-function mutation in APC2 causes sotos syndrome features. Cell Rep. 2015; 10:1585–1598. [DOI] [PubMed] [Google Scholar]
- 43. Maruszak A., Thuret S.. Why looking at the whole hippocampus is not enough—a critical role for anteroposterior axis, subfield and activation analyses to enhance predictive value of hippocampal changes for Alzheimer’s disease diagnosis. Front. Cell. Neurosci. 2014; 8:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leinonen V., Koivisto A.M., Savolainen S., Rummukainen J., Tamminen J.N., Tillgren T., Vainikka S., Pyykkö O.T., Mölsä J., Fraunberg M. et al.. Amyloid and tau proteins in cortical brain biopsy and Alzheimer’s disease. Ann. Neurol. 2010; 68:446–453. [DOI] [PubMed] [Google Scholar]
- 45. Jacobs H.I., Hopkins D.A., Mayrhofer H.C., Bruner E., van Leeuwen F.W., Raaijmakers W., Schmahmann J.D.. The cerebellum in Alzheimer’s disease: evaluating its role in cognitive decline. Brain. 2017; 141:37–47. [DOI] [PubMed] [Google Scholar]
- 46. Demenais F., Margaritte-Jeannin P., Barnes K.C., Cookson W.O., Altmüller J., Ang W., Barr R.G., Beaty T.H., Becker A.B., Beilby J. et al.. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat. Genet. 2018; 50:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang D.W., Sherman B.T., Lempicki R.A.. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stein M.M., Thompson E.E., Schoettler N., Helling B.A., Magnaye K.M., Stanhope C., Igartua C., Morin A., Washington C. III, Nicolae D. et al.. A decade of research on the 17q12-21 asthma locus: piecing together the puzzle. J. Allergy. Clin. Immun. 2018; 142:749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen J., Miller M., Unno H., Rosenthal P., Sanderson M.J., Broide D.H.. Orosomucoid-like 3 (ORMDL3) upregulates airway smooth muscle proliferation, contraction, and Ca2+ oscillations in asthma. J. Allergy Clin. Immun. 2018; 142:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lo J.C., Wang Y., Tumanov A.V., Bamji M., Yao Z., Reardon C.A., Getz G.S., Fu Y.-X.. Lymphotoxin ß receptor–dependent control of lipid homeostasis. Science. 2007; 316:285–288. [DOI] [PubMed] [Google Scholar]
- 51. Singh A.K., Stock P., Akbari O.. Role of PD-L1 and PD-L2 in allergic diseases and asthma. Allergy. 2010; 66:155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Berry M., Brightling C., Pavord I., Wardlaw A.J.. TNF-α in asthma. Curr. Opin. Pharmacol. 2007; 7:279–282. [DOI] [PubMed] [Google Scholar]
- 53. Brightling C., Berry M., Amrani Y.. Targeting TNF-α: a novel therapeutic approach for asthma. J. Allergy. Clin. Immun. 2008; 121:5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mulcahy H., O’rourke K., Adams C., Molloy M., O’gara F.. LST1 and NCR3 expression in autoimmune inflammation and in response to IFN-γ, LPS and microbial infection. Immunogenetics. 2006; 57:893–903. [DOI] [PubMed] [Google Scholar]
- 55. Anderson G., Morrison J.. Molecular biology and genetics of allergy and asthma. Arch. Dis. Child. 1998; 78:488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gandhi N.A., Bennett B.L., Graham N.M., Pirozzi G., Stahl N., Yancopoulos G.D.. Targeting key proximal drivers of type 2 inflammation in disease. Nat. Rev. Drug Discov. 2016; 15:35. [DOI] [PubMed] [Google Scholar]
- 57. Boyle E.A., Li Y.I., Pritchard J.K.. An expanded view of complex traits: from polygenic to omnigenic. Cell. 2017; 169:1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu X., Li Y.I., Pritchard J.K.. Trans effects on gene expression can drive omnigenic inheritance. Cell. 2019; 177:1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.