


Abstract
Horizontally acquired genes are typically regulated by ancestral regulators. This regulation enables expression of horizontally acquired genes to be coordinated with that of preexisting genes. Here, we report a singular example of the opposite regulation: a horizontally acquired gene that controls an ancestral regulator, thereby promoting bacterial virulence. We establish that the horizontally acquired regulatory gene ssrB is necessary to activate the ancestral regulatory system PhoP/PhoQ of Salmonella enterica serovar Typhimurium (S. Typhimurium) in mildly acidic pH, which S. Typhimurium experiences inside macrophages. SsrB promotes phoP transcription by binding upstream of the phoP promoter. SsrB also increases ugtL transcription by binding to the ugtL promoter region, where it overcomes gene silencing by the heat-stable nucleoid structuring protein H-NS, enhancing virulence. The largely non-pathogenic species S. bongori failed to activate PhoP/PhoQ in mildly acidic pH because it lacks both the ssrB gene and the SsrB binding site in the target promoter. Low Mg2+ activated PhoP/PhoQ in both S. bongori and ssrB-lacking S. Typhimurium, indicating that the SsrB requirement for PhoP/PhoQ activation is signal-dependent. By controlling the ancestral genome, horizontally acquired genes are responsible for more crucial abilities, including virulence, than currently thought.
INTRODUCTION
Horizontal gene transfer plays a key role in microbial evolution because it can readily endow a recipient organism with new abilities (1–4). Expression of horizontally acquired genes can be independent of the ancestral genome but is most often controlled by ancestral regulators (5–10). This control enables bacteria to coordinate expression of newly acquired genes with that of preexisting genes, thereby avoiding potential negative fitness effects resulting from their uncoordinated expression (1,8–10). Here, we provide a singular example of the antithesis: a horizontally acquired regulatory gene that controls an ancestral regulatory system required for bacterial virulence (Figure 1). This control allows bacteria to expand the environments in which a regulator operates.
Figure 1.

The horizontally acquired regulatory ssrB gene promotes S. Typhimurium virulence by activating the ancestral PhoP/PhoQ system in mildly acidic pH. The ancestral organism that gave rise to S. Typhimurium and S. bongori harbored the ancestral phoP and phoQ genes specifying the PhoP/PhoQ regulatory system and the horizontally acquired ugtL gene. S. Typhimurium acquired the spiR and ssrB genes by horizontal gene transfer, which, together with the evolution of an SsrB binding site in the ugtL promoter (purple), enables activation of PhoP/PhoQ in mildly acidic pH (by promoting transcription of ugtL and phoP genes) and virulence. The non-pathogenic species S. bongori lacks the spiR and ssrB genes and the SsrB binding site in the ugtL promoter, and is unable to activate PhoP/PhoQ in mildly acidic pH. Low Mg2+ activates the PhoP/PhoQ system in both S. Typhimurium and S. bongori. The SsrB protein is a direct transcriptional activator of the phoP gene. Thus, the evolved function of the ancestral regulatory circuit by transferred foreign gene(s) distinguishes organisms’ behavior.
The genus Salmonella consists of two species: S. enterica, which includes >2500 serovars, many of which responsible for disease conditions in humans (11), and S. bongori, which is largely non-pathogenic (12). S. enterica serovar Typhimurium (S. Typhimurium) requires a horizontally acquired gene cluster known as Salmonella pathogenicity island 2 (SPI-2) to survive inside macrophages and to cause a typhoid fever-like disease in mice (13,14). SPI-2 specifies several virulence determinants, including a regulatory system—designated SsrB/SpiR (15,16)—that controls transcription of SPI-2 genes and of other horizontally acquired genes scattered around the S. Typhimurium genome (16–19). SsrB is a DNA-binding transcriptional regulator activated by the sensor SpiR (sometimes referred to as SsrA) (15,16). Transcription of the horizontally acquired ssrB and spiR genes is regulated by several ancestral regulators, including SlyA (20), OmpR (6), and PhoP (7).
The PhoP and PhoQ proteins form a two-component regulatory system that governs virulence in S. Typhimurium (21–23) (Figure 1). The sensor PhoQ responds to inducing signals by promoting the phosphorylated state of the DNA binding regulator PhoP (PhoP-P), the active form of the PhoP protein (24). The UgtL protein increases the fraction of PhoP-P by advancing PhoQ autophosphorylation (25). PhoQ activation by mildly acidic pH is critical for S. Typhimurium virulence because inhibition of phagosome acidification impairs survival inside macrophages (26) and PhoP activation (27,28). Furthermore, amino acid substitutions in PhoQ that hinder activation by mildly acidic pH attenuate virulence even though these PhoQ variants are activated normally by low Mg2+ and antimicrobial peptides (29). Likewise, a ugtL null mutant is defective for PhoQ activation by mildly acidic pH and virulence (25).
We now report that S. Typhimurium's ability to activate the ancestral PhoP/PhoQ system in mildly acidic pH and to cause disease requires the horizontally acquired regulatory gene ssrB and an SsrB binding site in the ugtL promoter (Figure 1). Non-pathogenic S. bongori fails to activate PhoQ in mildly acidic pH because it lacks both the ssrB gene and the SsrB binding site in the ugtL promoter. These findings demonstrate that horizontally acquired genes can confer new abilities by altering the properties of an ancestral regulatory system. Moreover, they suggest that foreign genes play more critical roles in evolution than previously thought. In addition, our results imply that a horizontally acquired gene may promote different phenotypes depending on the genes pre-existing in the host genome.
MATERIALS AND METHODS
Bacterial strains, plasmids, oligodeoxynucleotides and growth conditions
Bacterial strains and plasmids used in this study are listed in Supplemental Table S1. All S. enterica serovar Typhimurium strains were derived from the wild-type strain 14028s (30) and constructed by phage P22-mediated transductions (31), and the λ-red recombineering technique (32). Bacteria were grown at 37°C in Luria-Bertani (LB) broth or N-minimal media (33) supplemented with 0.1% casamino acids, 38 mM glycerol and the indicated pH (pH 7.6 or pH 4.9) and 1 mM of MgCl2 unless specified. Escherichia coli DH5α was used as the host for preparation of plasmid DNA (34). To induce plasmid expression, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added at the indicated concentrations (0–1 mM). When necessary to select for plasmid maintenance, appropriate antibiotics were added at the following final concentrations: ampicillin at 50 μg ml−1, chloramphenicol at 20 μg ml−1, kanamycin at 50 μg ml−1 and tetracycline at 10 μg ml−1. DNA oligonucleotides used in this study are listed in Supplemental Table S2.
Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated using RNeasy Kit (Qiagen) and RNase-Free DNase Set (Qiagen) according to the manufacturer's instructions. The purified RNA was quantified using a Nanodrop machine (NanoDrop Technologies). Complementary DNA (cDNA) was synthesized from 1.5 μg RNA using High Capacity RNA-to cDNA Master Mix (Applied Biosystems) or SuperScript IV VILO master mix (Invitrogen), and then diluted 10-fold in water. The mRNA amounts of the mgtC, ugtL, pcgL, pagC, pagD, ssaG and sifB genes were determined from 2 μl of cDNA using Fast SYBR Green PCR Master Mix (Applied Biosystems) and appropriate primers (mgtC: 6962/6963; ugtL: 7295/7302; pcgL: 6627/6628; pagC: 6964/6965; pagD: 7016/7017; ssrB: 13096/13097; ssaG: 13098/13099; sifB: 17529/17530) at 0.35 μM as final concentrations, and monitored using a QuantStudio 6 machine (Applied Biosystems). Data were normalized to the levels of 16S ribosomal RNA amplified with primers 3203 and 3204 from 1000-fold diluted cDNA samples.
Determination of S. Typhimurium mRNA abundance inside macrophages
The murine-derived macrophage cell line J774A.1 was cultured in Dulbecco modified Eagle medium (DMEM; Life Technologies) supplemented with 10% FBS (Life Technologies) at 37°C under 5% CO2. Macrophages were seeded in 6-well tissue culture plates with 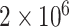 cells per well one day before infection with S. Typhimurium. Confluent monolayers were inoculated with bacterial cells that had been grown overnight in LB broth, washed with PBS and resuspended in prewarmed DMEM at a multiplicity of infection of 10. Following 30 min incubation, the wells were washed three times with prewarmed PBS to remove extracellular bacteria and then incubated with prewarmed medium supplemented with 100 µg ml−1 gentamicin for 1 h to kill extracellular bacteria. Next, the wells were washed three times with PBS and then incubated with prewarmed medium supplemented with 10 µg ml−1 gentamicin. At the indicated times, samples were harvested using TRIzol reagent (Invitrogen) solution. Total RNA was isolated and cDNA was synthesized as described above using 2 μg RNA. mRNA abundance was measured by qRT-PCR as described above. Data were normalized to the levels of 16S ribosomal RNA from twenty-fold diluted cDNA.
cells per well one day before infection with S. Typhimurium. Confluent monolayers were inoculated with bacterial cells that had been grown overnight in LB broth, washed with PBS and resuspended in prewarmed DMEM at a multiplicity of infection of 10. Following 30 min incubation, the wells were washed three times with prewarmed PBS to remove extracellular bacteria and then incubated with prewarmed medium supplemented with 100 µg ml−1 gentamicin for 1 h to kill extracellular bacteria. Next, the wells were washed three times with PBS and then incubated with prewarmed medium supplemented with 10 µg ml−1 gentamicin. At the indicated times, samples were harvested using TRIzol reagent (Invitrogen) solution. Total RNA was isolated and cDNA was synthesized as described above using 2 μg RNA. mRNA abundance was measured by qRT-PCR as described above. Data were normalized to the levels of 16S ribosomal RNA from twenty-fold diluted cDNA.
Measuring gene expression by β-galactosidase assay
Bacteria with chromosomal lacZ fusion were grown in indicated media. β-galactosidase activity was determined as described (35). Source data can be found in Supplemental Table S3.
Measuring gene expression by GFP assay
Bacterial cells expressing a gfp variant from the indicated promoter were grown in N-minimal media as described. Fluorescence and OD600 values were measured by using the multilabel plate reader VICTOR3 (PerkinElmer). The measured values of GFP expression were divided by 1000 times the OD600 values. Source data can be found in Supplemental Table S3.
In vivo detection of phosphorylated PhoP
PhoP and PhoP-P were separated on 12.5% polyacrylamide gels containing acrylamide-Phos-tag™ ligand (Phos-tag™ Consortium) as described by the manufacturer. Gels were copolymerized with 50 μM Phos-tag™ acrylamide and 100 μM MnCl2. Whole-cell extracts were prepared as described (36) and normalized by OD600. The samples were electrophoresed on Phos-tag™ gels with standard running buffer [0.4% (w/v) SDS, 25 mM Tris, 192 mM glycine] at 4°C under 20 mAmp for 3.5 h, transferred to nitrocellulose membranes, and analyzed by immunoblotting using polyclonal rabbit antibodies recognizing PhoP (1:2000) and polyclonal mouse antibodies recognizing AtpB (F0F1 ATP synthase subunit A) (1:5000). Secondary horseradish peroxidase-conjugated antisera recognizing rabbit and mouse antibodies (GE healthcare) were used at 1:5000 dilution. The blots were developed with the Amersham ECL Western Blotting Detection Reagents (GE Healthcare) or SuperSignal West Femto Chemiluminescent system (Pierce), and were visualized using LAS-4000 (Fuji Film). The density of protein bands was determined by quantification using ImageJ software version 1.52 (NIH).
Western blot analysis
Bacterial cells were grown as described and crude extracts were prepared in B-PER bacterial protein extraction reagent (Pierce) with 100 μg ml–1 lysozyme and EDTA-free protease inhibitor (Roche). Samples were separated in 4–12% NuPAGE gels (Life Technologies). Then, samples were analyzed by Western blotting using antibodies recognizing FLAG (Sigma; 1:2000) and AtpB (Abcam; 1:5000). Secondary horseradish peroxidase-conjugated antisera recognizing mouse antibodies (GE healthcare) were used at 1:5000 dilution. The blots were developed with the Amersham ECL Western Blotting Detection Reagents (GE Healthcare) or SuperSignal West Femto Chemiluminescent system (Pierce), and were visualized using LAS-4000 (Fuji Film). The density of protein bands was determined by quantification using ImageJ software version 1.52 (NIH).
Chromatin immunoprecipitation
hns-FLAG-expressing wild-type (JC805) and ugtL-sifBmu (JC1625) S. Typhimurium were grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9. Bacterial cells were cross-linked with 1% formaldehyde at room temperature for 15 min, quenched with 200 mM glycine at room temperature for 10 min and washed three times with cold phosphate-buffered saline (PBS). Then, cells were lysed in cell lysis solution A [10 mM Tris, pH 8.0, 50 mM NaCl, 10 mM EDTA, 20% sucrose, 10 mg ml−1 lysozyme] and 10× RIPA solution (Millipore). DNA was fragmented to average size of 500 bp by sonication (VirTis) and a 50 μl aliquot was taken as input DNA. Immunoprecipitation of complexes between the heat stable nucleoid-structuring (H-NS) protein and DNA were performed using antibodies recognizing FLAG, and using MagnaChip protein A/G magnetic beads (Millipore). Samples were then washed twice with 1× RIPA solution, twice with LiCl immune complex wash buffer (Millipore), twice with TE buffer [20 mM Tris (pH 8.0), 1 mM EDTA], and eluted in elution buffer [50 mM Tris (pH 8.0), 10 mM EDTA, 1% SDS] with incubation at 65°C for 15 min. Both immunoprecipitated (IP) and input DNA samples were incubated at 65°C for 9 h to reverse crosslinks, purified using Qiagen PCR purification column, and were quantified using qRT-PCR using primers (mgtA: 7225/7226; ugtL: 7295/7302; sifB: 17529/17530; STM14_3310: 16879/16880; rpoD: 4149/4150). Binding of H-NS protein to DNA were calculated as ratio of ‘IP DNA/input DNA’ and normalized to that of rpoD.
Purification of the SsrBc protein
To purify the SsrBc protein, E. coli BL21 (DE3) harboring a plasmid expressing His6-tagged SsrB C-terminus portion (pH6-SsrBc) was grown in LB at 37°C for 3 h and 0.7 mM of IPTG was added to induce gene expression and further incubated at 30°C for 3 h. Cells were collected and washed twice with a solution containing 50 mM Tris–HCl (pH 8.0) and 150 mM NaCl. Washed cells were resuspended in solution A [50 mM Tris–HCl (pH 8.0), 150 mM NaCl] containing 150 μg ml−1 lysozyme, 1 mM MgCl2, DNase I (Promega) and protease EDTA-free protease inhibitor (Roche), and incubated at 4°C for 30 min. Cells were broken using Cell Disruptor (Constant Systems Ltd). After adding imidazole to 20 mM as final concentration, cell debris was removed by centrifugation (12 000 × g, 30 min) and the supernatant was applied to Ni-Nta agarose (Qiagen) column. The column was washed with solution A containing 25 mM imidazole, and proteins were eluted with solution A containing 100–300 mM imidazole and dialyzed with the same solution without imidazole. The purified SsrBc protein was separated in 4–12% NuPAGE gels (Life Technologies) and visualized by coomassie G-250 staining (Thermo Scientific) (Supplemental Figure S1), demonstrating purity of ∼94%.
Electrophoretic mobility shift assay
DNA fragments corresponding the ugtL-sifB intergenic region and the 3′ end of purB coding region with the purB-phoP intergenic region were generated by PCR from wild-type S. Typhimurium (14028s) using primers 17222/17223 and 14244/14217, respectively. The DNA fragments were gel purified with QIAquick column (Qiagen). Purified probe DNA (2 or 4 nM) and purified SsrBc protein at indicated concentrations (0–2 μM) were mixed with binding buffer [20 mM HEPES (pH 8.0), 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 0.1 mM DTT, 50 μg ml−1 BSA, 10% (vol/vol) glycerol, and 10 ng μl−1 of poly(dI-dC) (Sigma)] in a total volume of 20 μl and incubated for 15 min at room temperature. Samples were then electrophoresed on 6% Tris-borate-EDTA gels (Life Technologies) and stained using EMSA kit (Invitrogen) according to the manufacturer's instructions.
DNase I footprint assay
DNA fragments corresponding to the 3′ end of the purB coding region with the purB-phoP intergenic region were generated by PCR from wild-type S. Typhimurium (14028s) using primers 14244 and 32P-labeled 14217. The DNA fragments were gel purified with QIAquick column (Qiagen). A total of ∼105 cpm of labeled DNA probe (∼2 nM) and purified SsrBc (0, 0.25 and 1 μM) were mixed with the same binding buffer used in the electrophoretic mobility shift assay including 50 ng μl−1 of poly(dI-dC) (Sigma) in a total volume of 20 μl and incubated for 15 min at room temperature. DNase I (Promega) (0.01 units), 10 mM CaCl2, and 10 mM MgCl2 were added and incubated for 3 min at room temperature. The reaction was stopped by the addition of 100 μl of phenol chloroform, and the aqueous phase was precipitated with ethanol. The precipitate was dissolved in sequence-loading buffer and electrophoresed on a 6% acrylamide/7 M urea gel together with a sequence ladder initiated with the labeled primer by using the T7 Sequenase 2.0 DNA-sequencing kit (Amersham Biosciences), and the gels were dried and autoradiographed (Fujifilm).
Mouse virulence assay
Six-week-old female BALB/c or C3H/HeN mice were purchased from Charles River Laboratories. Five mice in each group were infected intraperitoneally with 0.1 ml of PBS containing ∼102 (for BALB/c) or ∼2 × 104 (for C3H/HeN) S. Typhimurium that had been grown overnight in LB broth and resuspended and diluted in PBS. Mouse survival was monitored every 12 h for 3 weeks. Virulence assays were conducted twice with similar outcomes, and data for each experimental group correspond to groups of five mice. All animals were housed in temperature- and humidity-controlled rooms and maintained on a 12 h light/12 h dark cycle. All procedures complied with regulations of the Institutional Animal Care and Use Committee of the Yale School of Medicine.
Construction of chromosomal mutant strains
To generate a mutant strain expressing the ugtL gene from the heterologous promoter plac1–6, a cat cassette with plac1–6 was introduced upstream of the ugtL leader region to replace the ugtL promoter. That is, the cat fragment with the plac1–6 was amplified from plasmid pKD3 using primers 16655/16658, then introduced into wild-type S. Typhimurium (14028s; S. Typhimurium) harboring plasmid pKD46 (32). The cat cassette was removed using plasmid pCP20 (32).
To generate strains deleted for SPI-2 genes, a kan cassette was introduced in the indicated region of SPI-2: the kan fragment was amplified from plasmid pKD13 using primers 13160/13161 (for deleting sseA–sseG genes), 13160/13165 (for deleting sseA-ssaU genes), 13164/13165 (for deleting ssaG-ssaU genes) then introduced into wild-type S. Typhimurium (14028s) harboring plasmid pKD46 (32).
To generate a strain harboring ugtL-FLAG, a kan cassette was introduced in the 3’end of the ugtL gene as follows: the kan fragment with the FLAG coding sequence was amplified from plasmid pKD4 using primers 16686/16687, then introduced into wild-type S. Typhimurium (14028s) harboring plasmid pKD46 (32).
To generate a strain with mutated SsrB binding region in the ugtL-sifB intergenic region, a cassette with cat and PrhaB-relE was introduced into the region containing a putative SsrB binding site: the cat PrhaB-relE fragment was amplified from plasmid pSLC-242 (37) using primers 17220/17221, then introduced into wild-type S. Typhimurium (14028s) harboring plasmid pKD46 (32). Then, the cat PrhaB-relE cassette was replaced by annealed oligonucleotides 17234/17235 to remove SsrB binding site. The resulting strain was obtained following selection against RelE-mediated toxicity on media containing 0.2% rhamnose as described (37). Mutation was confirmed by DNA sequencing.
To generate S. bongori expressing the phoQ gene from S. Typhimurium (phoQST), a tetRA cassette was amplified using primers 14810/14811 from transposon Tn10 in strain MS7953s and was introduced into the phoQ gene of wild-type S. bongori (S3041) harboring pKD46 (32). The phoQ gene of S. Typhimurium was amplified using primers 14821/14822 and genomic DNA of strain 14028s. The tetAR cassette was replaced by a PCR product to create S. bongori with phoQST. The resulting strain was selected against tetracycline resistance on media containing fusaric acid (38), and the gene replacement was confirmed by DNA sequencing.
To generate a strain with mutated SsrB binding site in the region upstream of the phoP gene (located in the purB gene), the SsrB binding sequence was mutated and a copy of intact purB gene and upstream gene promoter (PycfC) were integrated into the attachment Tn7 site. The normal purB gene was deleted except the portion containing the SsrB binding site. The engineering of these chromosomal mutations was carried out as follows: a tetRA cassette was amplified by PCR using primers 14950/14951 from transposon Tn10 in strain MS7953s, then introduced into wild-type S. Typhimurium (14028s) harboring plasmid pKD46 (32). The resulting strain was grown in media containing 0.5 mM adenine. The tetRA cassette was then replaced with annealed oligonucleotides 14952/14953 to mutate SsrB binding site. The resulting strain was obtained following selection against tetracycline resistance on media containing fusaric acid (38), and the mutation was confirmed by DNA sequencing. Then, the PycfC and purB gene were introduced into the attachment Tn7 site: the PycfC and purB gene were amplified by PCR from wild-type S. Typhimurium (14028s) genomic DNA using primers 16540/16541, 16542/16543, then introduced into pGRG36 (digested with XmaI and XhoI) by Gibson assembly. The resulting plasmid was verified by DNA sequencing and introduced into the attachment Tn7 site as described (39). Integration of the PycfC-purB gene and its upstream gene promoter was verified by DNA sequencing. Then, a kan cassette was introduced into the purB gene at normal chromosomal location: the kan cassette was amplified from plasmid pKD4 using primers 16869/16870, then introduced into S. Typhimurium strains with the PycfC-purB fragment at the attachment Tn7 site and wild-type or mutated SsrB binding site in the upstream region of the phoP gene harboring plasmid pKD46 (32). The kan cassette was removed using plasmid pCP20 (32).
Construction of plasmids
Plasmid pSsrB was constructed as follows: the ssrB gene was amplified from wild-type S. Typhimurium (14028s) using primers 14225/14226, then introduced between the BamHI and HindIII sites of pUHE21–2lacIq (40).
Plasmid pSsrBV197A was constructed by site-directed mutagenesis using the Quikchange lightning site-directed mutagenesis kit with primers 15755/15756 and plasmid pSsrB as a template following manufacturer's instructions.
Plasmid pH6-SsrBc was constructed as follows: the ssrB gene was amplified from wild-type S. Typhimurium (14028s) using primers 14224/14225, then introduced between the BamHI and HindIII sites of pUHE21–2lacIq (40).
Plasmid pUgtLSB was constructed as follows: the ugtL gene was amplified from wild-type S. bongori (S3041) using primers 16058/16059, then introduced between the BamHI and HindIII sites of pUHE21–2lacIq(40).
Plasmid pFPV25::PpmrD-GFPaav, pFPV25::Pmig-14-GFPaav were constructed as follows: the pmrD and mig-14 promoters were amplified from wild-type S. Typhimurium (14028s) using primers 5900/4740 and 4803/6044, then introduced between the EcoRI and BamHI sites of pFPV25AAV (41).
Protein and nucleotide sequence comparisons
Amino acid sequence and the upstream region of DNA in the indicated strains were compared to those of S. Typhimurium 14028s using TBLASTN/BLASTN and Clustal Omega (EMBL-EBI). A phylogenetic tree was generated by interactive Tree of Life software (v5) based on the analysis of the ugtL gene and its upstream region containing the SsrB binding site (720 nt upstream from the start codon (AUG) of the ugtL gene and whole coding region).
Statistical analyses
Sample sizes (biological replicates) for each experimental group or condition are described in each figure legend. For comparisons of two groups, t-tests were applied. Two-sided analysis provides P-values for each comparison. For comparisons of more than two groups, one-way ANOVA with Brown-Forsythe and Welch tests were applied. Each group was compared with a control group (wild-type unless specified).
RESULTS
Non-pathogenic S. bongori harbors a functional ugtL virulence gene
The PhoP/PhoQ systems of S. bongori and S. Typhimurium differ in that the former is activated by low Mg2+ and antimicrobial peptides, but not mildly acidic pH (29), whereas the latter is activated by all three signals (29,42–44). This disparity is due, in part, to differences between their PhoQ proteins because a S. Typhimurium strain expressing the S. bongori phoQ gene instead of its own was defective for activation by mildly acidic pH but not by low Mg2+ and antimicrobial peptides (29).
Unexpectedly, we found that a S. bongori strain expressing the S. Typhimurium phoQ gene instead of its own was also defective for activation by mildly acidic pH but not by low Mg2+ (Supplemental Figure S2). This result indicates that an additional factor(s) is responsible for the disparate abilities of the two Salmonella species to activate the PhoP/PhoQ system in mildly acidic pH.
We reasoned that the ugtL gene might be that factor given that the deduced amino acid sequences of the ugtL genes share only 55% identity (Supplemental Figure S3), which is much lower than most proteins present in both species (e.g. the PhoQ proteins are 99% identical). However, a plasmid expressing the S. bongori ugtL gene from a heterologous promoter increased the fraction of PhoP-P in a S. Typhimurium ugtL mutant as much as the isogenic plasmid with the S. Typhimurium ugtL gene (Figure 2A). A 10-fold increase in the inducer concentration used to activate the heterologous promoter driving ugtL transcription resulted in the quasi-complete conversion of PhoP into PhoP-P (Figure 2A). By contrast, the plasmid vector had no effect (Figure 2A). These results demonstrate that the S. bongori ugtL gene is competent for activation of the S. Typhimurium PhoP/PhoQ system in mildly acidic pH. Why, then, does mildly acidic pH activate PhoP/PhoQ in S. Typhimurium but not in S. bongori even though both species have functional ugtL genes?
Figure 2.

SsrB is necessary for PhoP activation, which requires UgtL, in mildly acidic pH. (A, B) Phos-tag western blot analysis of crude extracts prepared from (A) wild-type (JC805) and ugtL (JC925) S. Typhimurium strains with indicated plasmids (pVec, empty vector; pUgtLST, plasmid expressing S. Typhimurium ugtL; pUgtLSB, plasmid expressing S. bongori ugtL) grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9 supplemented with indicated concentrations of IPTG, and (B) wild-type (14028s) and ssrB (EG14411) S. Typhimurium strains grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9 (acidic pH), 10 μM Mg2+ at pH 7.6 (low Mg2+), or 1 mM Mg2+ at pH 7.6 (non-inducing) using antibodies recognizing PhoP or the loading control AtpB. Representatives of at least three independent experiments are shown. Numbers under the blots indicate % phosphorylated PhoP (PhoP-P). (C) β-galactosidase activity produced from a chromosomal pcgL-lacZ fusion in wild-type (EG9331) and ssrB (JC201) S. Typhimurium with an empty vector (pVec) or a plasmid expressing SsrB (pSsrB) grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9 (acidic pH), 10 μM Mg2+ at pH 7.6 (low Mg2+), or 1mM Mg2+ at pH 7.6 (non-inducing) supplemented with 0.2 mM IPTG. The mean and SD from four independent experiments are shown (n = 4). Each dot represents individual biological sample. One-way ANOVA with Brown-Forsythe and Welch tests (wild-type vs. others); ns, not significant; ***P < 0.001, ****P < 0.0001.
PhoP activation in mildly acidic pH requires the regulatory gene ssrB
S. bongori lacks the horizontally acquired SPI-2 (12) and is unable to activate PhoP/PhoQ in mildly acidic pH (Supplemental Figure S2) (29). Because the SPI-2-encoded SsrB protein is activated in mildly acidic pH (16,45), like PhoP (27,29,44), we investigated the possibility of the ssrB gene being required for activation of the PhoP/PhoQ system when S. Typhimurium experiences mildly acidic pH.
We determined that the fraction of PhoP-P was much lower in the ssrB mutant than in wild-type S. Typhimurium when bacteria were grown in mildly acidic pH (pH 4.9, 1 mM Mg2+) (Figure 2B), mimicking the behavior of the ugtL null mutant (Figure 2A) (25). By contrast, the fraction of PhoP-P was almost the same in isogenic wild-type and ssrB S. Typhimurium when grown in low Mg2+ (pH 7.6, 10 μM Mg2+) (Figure 2B), an inducing condition that activates PhoP as much as mildly acidic pH in a ugtL-independent manner (25,29). Under non-inducing conditions, PhoP-P was not detected in wild-type or ssrB S. Typhimurium (Figure 2B). These results demonstrate that ssrB is necessary to activate PhoP when the PhoQ-activating condition is mildly acidic pH.
The ssrB-dependent activation of PhoP taking place in mildly acidic pH is necessary for PhoP-dependent gene transcription because the β-galactosidase activity from the PhoP-activated pcgL-lac transcriptional fusion was much lower in the ssrB mutant than in the isogenic ssrB+ strain (Figure 2C), reflecting the abundance of PhoP-P (Figure 2B) (46). By contrast, the two strains had similarly high β-galactosidase activity when grown in low Mg2+ (Figure 2C) and similarly low β-galactosidase activity when grown in non-inducing conditions (Figure 2C). A plasmid with a wild-type copy of the ssrB gene complemented the ssrB mutant, whereas the plasmid vector did not (Figure 2C). The ssrB gene is required for normal transcription of other PhoP-activated genes because the ssrB mutant exhibited lower expression of PhoP-activated genes in strains harboring a lac transcriptional fusion to the ugtL gene (Supplemental Figure S4A), gfp transcriptional fusions to the phoP, pmrD, and mig-14 genes (Supplemental Figure S4B), and also when examining the mRNA amounts of the mgtC, ugtL, pcgL, pagC and pagD genes (Supplemental Figure S4C), than the isogenic ssrB+ strain (40,42,46). Thus, the defect of the ssrB mutant is neither specific to a particular PhoP-activated gene nor to the reporter used.
SsrB appears to activate PhoP by regulating expression of a gene(s) outside of SPI-2 because the fluorescence from a phoP-gfp fusion was not altered upon deletion of 26 of 30 genes in SPI-2 (Supplemental Figure S4D). Taken together, the results in this section indicate that when S. Typhimurium experiences mildly acidic pH, the horizontally acquired ssrB gene activates the ancestral regulator PhoP, resulting in transcription of PhoP-activated genes.
SsrB activates PhoP in a phoQ-dependent manner
We hypothesized that PhoP activation by SsrB requires PhoQ because this sensor is the only known PhoP phosphodonor (47) and SsrB promotes the phosphorylated state of PhoP (Figure 2B). In agreement with this notion, fluorescence from phoP-gfp was largely similar (i.e. 2-fold difference) in a strain with a phoQ null allele and specifying a PhoP variant capable of autophosphorylation from acetyl phosphate (47) and the isogenic ssrB null mutant (Figure 3A). By contrast, fluorescence was 10 times higher in wild-type S. Typhimurium than in the ssrB mutant (Figure 3A and Supplemental Figure S4B). Thus, SsrB activation of PhoP is largely PhoQ-dependent. (The two-fold phoQ-independent effect is addressed below under SsrB promotes phoP transcription by binding to the purB coding region upstream of the phoP promoter.)
Figure 3.

SsrB promotes UgtL expression, thereby enhancing PhoP activation in mildly acidic pH. (A, C) Fluorescence produced from a PphoP-gfp transcriptional fusion displayed by (A) wild-type (14028s), ssrB (EG14411), phoP*phoQ (EG10232), and phoP*phoQ ssrB (JC449) or (C) wild-type (14028s), ssrB (EG14411), plac1–6-ugtL (JC1360), plac1–6-ugtL ssrB (JC1449) S. Typhimurium. Bacteria were grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9. (B) Schematics of the intergenic region between the ugtL and sifB genes in wild-type, plac1–6-ugtL and ugtL-sifBmuS. Typhimurium strains. (D) In vitro binding of SsrBc to the wild-type or mutant (mu) ugtL-sifB intergenic region DNA. Four nM of the ugtL-sifB intergenic region DNA (wild-type or mu) was incubated with purified SsrBc (0, 0.5, 1 and 2 μM) proteins. A representative of at least three independent experiments is shown. (E) mRNA abundance of the ugtL, pagC and phoP genes produced by wild-type (14028s), ugtL-sifBmu (JC1547), sifB (JC1567), ugtL-sifBmussrB (JC1548), and ssrB (EG14411) S. Typhimurium grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9. (F) In vivo binding of H-NS to the promoter regions of the sifB, ugtL, STM14_3310 and mgtA genes were determined in hns-FLAG (JC805) and hns-FLAG ugtL-sifBmu (JC1625) S. Typhimurium strains grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9 using chromatin immunoprecipitation. (G) mRNA abundance of the ugtL and ssaG genes produced by wild-type (14028s) and ssrB (EG14411) S. Typhimurium with plasmids expressing wild-type SsrB or its variant (V197A) grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9 (acidic pH) supplemented with 5 μM IPTG. The mean and SD from at least four independent experiments are shown (A, n = 6; C, E–G, n = 4). Each dot represents individual biological sample (A, C, E–G). Two-tailed t-test with (A and C) ssrB+ versusssrB− or (F) wild-type versus ugtL-sifBmu. One-way ANOVA with Brown-Forsythe and Welch tests (wild-type versus others) (E and G); ns, not significant; **P < 0.01, ***P < 0.001, ****P < 0.0001.
SsrB activates PhoP by promoting transcription of the ugtL gene
We posit that SsrB activates PhoP in mildly acidic pH by promoting ugtL transcription because ugtL is necessary for PhoQ-dependent phosphorylation of PhoP in mildly acidic pH (25), and also because SsrB promotes transcription of horizontally acquired genes outside of SPI-2 (17–19) and ugtL is a horizontally acquired gene (48). As proposed, the amounts of the ugtL mRNA and UgtL protein were higher in wild-type S. Typhimurium than in the ssrB mutant (Supplemental Figures S4C and S5). Moreover, when ugtL transcription was driven from the constitutive plac1–6 promoter (49) (Figure 3B), isogenic ssrB strains displayed similar fluorescence from phoP-gfp (Figure 3C) (i.e., ∼2-fold difference. The two-fold ugtL promoter-independent effect is addressed below under SsrB promotes phoP transcription by binding to the purB coding region upstream of the phoP promoter). In other words, ugtL transcription from a constitutive promoter largely bypassed the SsrB requirement for PhoP activation. How, then, does SsrB promote ugtL transcription in wild-type S. Typhimurium?
A previous study showed SsrB binding to the intergenic region that separates the divergently transcribed ugtL and sifB genes (Figure 3B) (17). This region is absent from the equivalent region of the S. bongori genome (Supplemental Figure S6), which has neither sifB (Supplemental Figure S6) nor ssrB (12). The identified region has a bona fide SsrB binding site because the purified DNA binding domain of SsrB (19) bound to a DNA fragment harboring this region but not to one deleted for it (Figure 3D). Moreover, the site is necessary for PhoP activation in mildly acidic pH because the mRNA abundances of the PhoP-activated pagC and phoP genes were lower in an engineered strain deleted for the SsrB binding site (Figure 3B) than in the isogenic wild-type strain (Figure 3E). Independent support for this notion was obtained when examining bacteria carrying the phoP-gfp fusion: fluorescence was ∼5-fold lower in the SsrB binding site mutant than in the isogenic wild-type strain (Supplemental Figure S7A).
The defective PhoP activation exhibited by the SsrB binding site mutant is due to the inability of the SsrB protein to promote ugtL expression. This is because the SsrB binding site mutant produced the same low ugtL mRNA amounts as a mutant lacking the ssrB gene and a double mutant lacking both ssrB and the SsrB binding site (Figure 3E). By contrast, a strain deleted for the sifB gene retained wild-type ugtL mRNA amounts (Figure 3E), indicating that the phenotype of the SsrB binding site mutant is not due to compromised sifB expression.
When low Mg2+ was the activating signal for PhoQ, the SsrB binding site mutant retained wild-type expression of the phoP and ugtL genes (Supplemental Figure S7). This result is congruent with ssrB being dispensable for PhoP activation during growth in low Mg2+ (Figure 2B). Critically, ugtL transcription from a heterologous promoter bypasses the SsrB requirement (Supplemental Figure S8) but not the need for a mildly acidic pH (Supplemental Figure S8) for activation of the PhoP protein. Therefore, PhoP activation in mildly acidic pH requires PhoQ’s intrinsic ability to respond to a mildly acidic pH. In sum, the SsrB binding site in the ugtL promoter region is specifically required for PhoP activation in mildly acidic pH.
SsrB antagonizes the gene silencer H-NS in the ugtL promoter region
The xenogeneic silencer H-NS binds to AT-rich horizontally acquired DNA, preventing expression of the corresponding genes (50,51), including ugtL and sifB. Given that SsrB overcomes the silencing effects of H-NS at certain horizontally acquired genes (52), we wondered whether SsrB promotes ugtL transcription by binding to the SsrB binding site in the ugtL-sifB intergenic region that is bound by H-NS. Using chromatin immunoprecipitation, we determined that H-NS bound to the ugtL-sifB region more strongly in the strain with the mutant SsrB binding site than in the isogenic wild-type strain (Figure 3F). Control experiments showed that the SsrB binding site mutant retained normal H-NS binding to the STM14_3310 DNA (Figure 3F), one of the genes most tightly bound by H-NS (50), and no detectable binding to the DNA region upstream of mgtA (Figure 3F), a PhoP-activated gene not bound by H-NS (50).
SsrB appears to promote ugtL transcription solely by antagonizing silencing by H-NS (as opposed to being required to recruit RNA polymerase) because ugtL mRNA abundance was similar in isogenic ssrB mutant strains harboring plasmids expressing either the wild-type SsrB protein or the SsrB (V197A) variant (Figure 3G), which retains wild-type DNA binding ability but is impaired in RNA polymerase recruitment (53). Control experiments showed defective ssaG transcription in the strain expressing the SsrB (V197A) variant (Figure 3G), in agreement with previous results demonstrating that ssaG transcription by SsrB requires RNA polymerase recruitment (53). (Please note that it is not possible to genetically test the participation of hns in ugtL expression because hns is an essential gene in S. Typhimurium (50), and also because suppressors that allow growth of an hns mutant fail to grow under the virulence-relevant conditions necessary to activate the SsrB protein.) Taken together, the results presented in this section suggest that SsrB promotes ugtL transcription by displacing H-NS from the ugtL-sifB intergenic region.
SsrB promotes phoP transcription by binding to the purB coding region upstream of the phoP promoter
SsrB appears to activate PhoP by means other than promoting ugtL transcription because inactivation of the ssrB gene decreases expression of the PhoP-activated phoP promoter in a strain in which the ugtL gene is transcribed from a heterologous promoter (Figure 3C), and also because the ssrB mutant exhibited defective phoP expression in a phoP* phoQ strain (Figure 3A) even though UgtL activates PhoP in a PhoQ-dependent manner (25). As detailed below, we have now established that SsrB is a direct transcriptional activator of the phoP gene.
DNase I footprinting analysis of the phoP promoter region (∼300 bp) demonstrated that the purified DNA binding domain of SsrB protects a region 145–167 nt upstream of the PhoP-dependent transcription start site (P1) of the phoP gene (Figure 4A and 4B). This region is located within the purB coding region (Figure 4B) and resembles other SsrB binding sites (17). Nucleotide substitutions in the identified SsrB binding site (Figure 4C and 4D) hampered SsrB binding in vitro (Figure 4C), and reduced phoP transcription in vivo in bacteria experiencing a mildly acidic pH (Figure 4E). (Because the nucleotide substitutions in the SsrB binding site disrupt the purB coding region, these experiments were carried out using isogenic strains harboring a wild-type copy of the purB gene integrated into the attachment Tn7 site (Figure 4D).) A double mutant with nucleotide substitutions in the SsrB binding site in the purB coding region upstream of the phoP promoter and the deletion of the SsrB binding region in the ugtL promoter was as defective in PhoP-dependent transcription as the ssrB null mutant (Figure 4E). The effect of the nucleotide substitutions in the SsrB binding sites and ssrB gene are specific to mildly acidic pH because the corresponding mutants behaved like the isogenic wild-type strain when experiencing low Mg2+ or non-inducing conditions (Figure 4E). Cumulatively, these results indicate that SsrB activates PhoP by promoting transcription of both the phoP and ugtL genes.
Figure 4.

SsrB promotes phoP transcription by binding to the purB coding region upstream of the phoP promoter. (A) DNase I footprinting analysis of the phoP promoter using purified SsrBc-His6 protein (0, 0.25 and 1 μM, triangle; 1 μM, square). Lanes G, A and T correspond to dideoxy chain-termination sequences for the phoP promoter DNA. The black bar indicates the region protected by the SsrBc-His6 protein. (B) Schematic of the purB-phoP chromosomal region. The SsrB binding site is located in the coding region of the purB gene. Numbers under the black box indicate distance from the +1 (P1, PhoP-activated transcriptional start site). (C) In vitro binding of SsrBc-His6 to the wild-type or mutant SsrB binding site DNAs. DNA (2 nM) was incubated with SsrBc-His6 proteins (0, 0.06, 0.13, 0.25, 0.5 and 1 μM). A representative of at least three independent experiments is shown. Numbers underneath the nucleotides indicate distance from P1. (D) Schematic of strains with wild-type (PphoPWT) or mutated (PphoPmu) SsrB binding site in the purB coding region (on the purB gene which is inactivated) and a copy of purB gene integrated into attachment Tn7 site. (E) Fluorescence produced from a PphoP-gfp transcriptional fusion displayed by PphoPWT (JC1482), PphoPmu (JC1458), PphoPWTugtL-sifBmu (JC1582), PphoPmuugtL-sifBmu (JC1583), PphoPWTssrB (JC1463), PphoPmussrB (JC1464), and PphoPWTsifB (JC1586) Salmonella grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9 (acidic pH), 10 μM Mg2+ at pH 7.6 (low Mg2+), or 1 mM Mg2+ at pH 7.6 (non-inducing). The mean and SD from four independent experiments are shown. One-way ANOVA with Brown-Forsythe and Welch tests (PphoPWT vs. others); ns, not significant; ***P < 0.001, ****P < 0.0001.
SsrB-dependent activation of the ugtL gene is necessary for virulence
S. Typhimurium's ability to cause disease in a mouse model of infection requires functional phoP, phoQ (21,22), ugtL (25) and ssrB (54) genes, as well as PhoQ’s ability to respond to mildly acidic pH (27,29,55). To determine whether the SsrB-dependent activation of PhoP in mildly acidic pH is necessary for virulence, we compared a set of isogenic strains with a wild-type or defective SsrB binding site in the ugtL-sifB intergenic region (ugtL-sifBmu; Figure 3B), and, as controls, mutants lacking the ssrB or sifB genes.
When inoculated via the intraperitoneal route, the SsrB binding site mutant was attenuated in both C3H/HeN and Balb/C mice (Figure 5A and Supplemental Figure S9). (A salient difference between these mouse strains is that the former has two functional copies of the Slc11a1 gene, whereas the latter has two defective copies of it. Although Slc11a1 confers resistance to S. Typhimurium (56), phoP and phoQ single mutants are attenuated for virulence in both C3H/HeN and Balb/C mice (21–23,25,29).) The virulence attenuation of the SsrB binding site mutant (ugtL-sifBmu) is due to impaired expression of ugtL (as opposed to sifB) because the sifB null mutant displayed wild-type virulence (Figure 5A and Supplemental Figure S9), as reported (57). The SsrB binding site mutant (ugtL-sifBmu) was not as attenuated as the ssrB null mutant (Figure 5A and Supplemental Figure S9), in agreement with SsrB being required for expression of virulence genes in addition to ugtL (16,17,19).
Figure 5.
SsrB-dependent activation of ugtL transcription is required for S. Typhimurium virulence in mice and for transcription of PhoP-activated genes inside macrophages. (A) Survival of BALB/c mice inoculated intraperitoneally with ∼102 wild-type (14028s), ugtL-sifBmu (JC1547), sifB (JC1567) and ssrB (EG14411) S. Typhimurium. Data are representative of two independent experiments, which produced similar results, n = 5 mice per each experimental group. Mantel-Cox test was performed between wild-type and isogenic mutant Salmonella infected mice; ns, not significant, ****P < 0.0001. (B) mRNA abundance of the ugtL, pagC, ssrB and ssaG genes produced by wild-type (14028s), phoP (MS7953s), ssrB (EG14411) and ugtL-sifBmu (JC1547) Salmonella harvested from the macrophage-like cell line J774A.1 at the indicated times. The mean and SD from three independent experiments are shown. One-way ANOVA with Brown-Forsythe and Welch tests (wild-type vs. others) were applied at each time point; no *, not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
PhoP and SsrB exhibit different activation kinetics when S. Typhimurium is inside macrophages
We hypothesized that the virulence defect of the SsrB binding site mutant (ugtL-sifBmu) (Figure 5A and Supplemental Figure S9) is due to diminished PhoP activation inside macrophages because PhoP is activated when S. Typhimurium is in a mildly acidic macrophage phagosome (27,28) and also because PhoP activation in mildly acidic pH is necessary for S. Typhimurium virulence (25,27,29). To test this hypothesis, we examined the mRNA abundance of PhoP-activated genes at different times after S. Typhimurium internalization by a murine macrophage-like cell line.
The mRNA abundance of the PhoP-activated ugtL, pagC and ssrB genes was similar among wild-type, ugtL-sifBmu mutant and ssrB null strains at 1 h post bacterial internalization by macrophages (Figure 5B). By contrast, the mRNA abundance of ugtL and pagC genes was lower in both the ugtL-sifBmu mutant and the ssrB null strain than in wild-type S. Typhimurium at 6 h post bacterial internalization by macrophages (Figure 5B). (ssrB mRNA amounts were similar in the ugtL-sifBmu mutant and wild-type strain (Figure 5B).) The phoP null mutant had low mRNA amounts for all three genes at both times (Figure 5B). Therefore, PhoP activation is ssrB-dependent at 6 h but -independent at 1 h inside macrophages.
The behavior of the ugtL and pagC genes is in contrast to that of ssrB and the SsrB-activated ssaG gene (58). That is, there was little ssaG expression at 1 h (Figure 5B), and the mRNA at 6 h accumulated in a ssrB- and phoP-dependent manner (Figure 5B). The ugtL-sifBmu mutant was minimally defective at 6 h post bacterial internalization (Figure 5B). Although the phoP mutant is defective in ssaG expression at 6 h inside macrophages (Figure 5B), it behaves like the wild-type strain in bacteria grown in defined laboratory media of mildly acidic pH (Supplemental Figure S10). These results indicate that PhoP activation occurs early on, and SsrB activation takes place later when S. Typhimurium is inside macrophages. Moreover, they support the notion of SsrB promoting S. Typhimurium virulence, in part, by activating the PhoP protein.
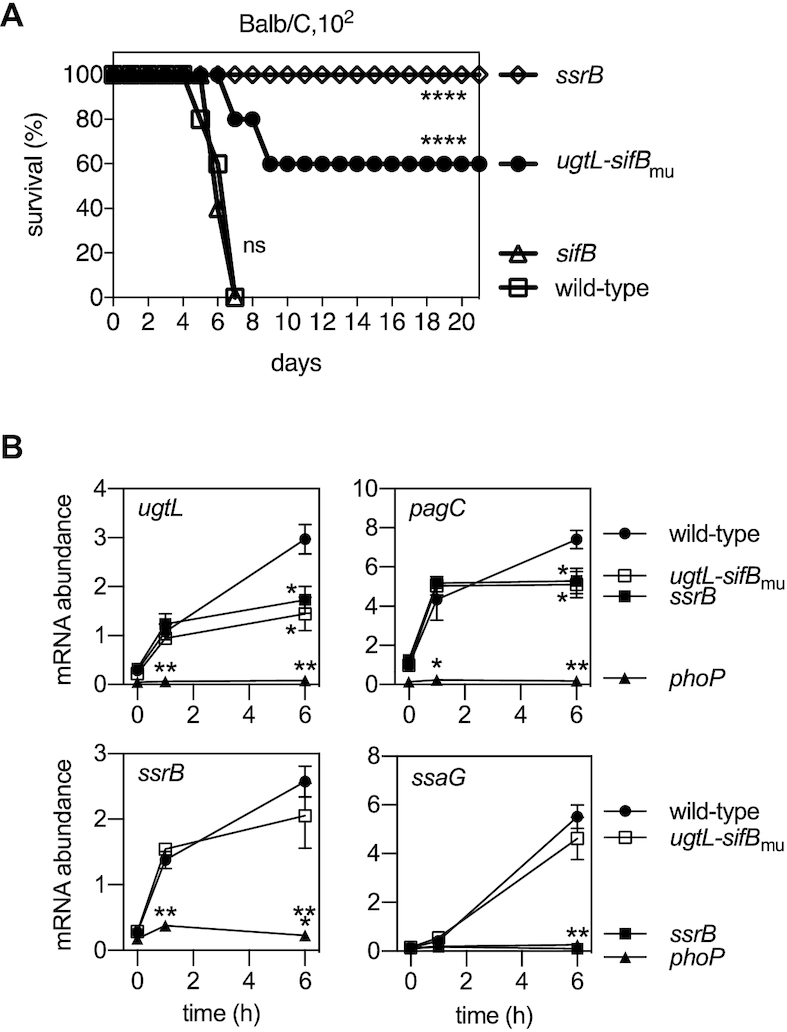
The SsrB binding site in the ugtL promoter is conserved among S. enterica serovars that infect warm-blooded animals
The phoP gene is required for virulence in S. enterica serovars other than Typhimurium that infect a variety of warm-blooded animals. That is, inactivation of the phoP gene in S. Gallinarum, S. Choleraesuis, and S. Typhi attenuates virulence in chickens, pigs, and humans, respectively (59–61). Thus, we wondered whether the SsrB activation of ugtL is retained in S. enterica serovars with different host ranges.
The nucleotide sequences corresponding to the ssrB coding region and the SsrB binding region in the ugtL promoter are highly conserved (>99% and >85%, respectively) among the Salmonella serovars that infect warm-blooded animals (Figure 6A). By contrast, the ugtL promoter, leader, and coding regions are much less conserved in ssrB-containing S. enterica subspecies that infect cold-blooded animals. For example, sequences resembling the SsrB binding region are not found in the ugtL promoter of S. enterica subspecies diarizonae, despite this bacterium harboring ssrB (Figure 6A and 6B). This and other Salmonella subspecies that infect cold-blooded animals have different serovar-specific gene sets in the region occupied by the sifB gene in S. Typhimurium (Figure 6B). These results suggest that the SsrB binding site in the ugtL promoter contributes to the different habitats of individual S. enterica serovars.
Figure 6.
The nucleotide sequences upstream of the ugtL gene distinguishes phenotypic properties of Salmonella species. (A) A heat map of sequence identity among Salmonella species and serovars. Sequence identity of the deduced amino acid sequences of the phoP, phoQ, ugtL and ssrB genes analyzed using TBLASTN. DNA sequence identity of the SsrB binding region in the ugtL promoter (SsrB binding site) was analyzed using BLASTN. Sequences of indicated elements from S. bongori (NCTC12419), S. enterica subsp. diarizonae (SA20044251; S. diarizonae), S. enterica subsp. arizonae (RKS2983; S. arizonae), S. enterica subsp. houtenae (CFSAN000552; S. houtenae), S. enterica subsp. salamae (RSE42; S. salamae), S. enterica subsp. indica (NCTC12420; S. indica), S. enterica subsp. enterica serovar Parayphi A (ATCC11511; S. Paratyphi A), S. enterica subsp. enterica serovar Typhi (Ty2; S. Typhi), S. enterica subsp. enterica serovar Dublin (ATCC39184; S. Dublin), S. enterica subsp. enterica serovar Gallinarum (1984; S. Gallinarum), S. enterica subsp. enterica serovar Paratyphi B (SPB7; S. Paratyphi B), S. enterica subsp. enterica serovar Cholerasuis (SC-B67; S. Cholerasuis), S. enterica subsp. enterica serovar Heidelberg (41578; S. Heidelberg), and S. enterica subsp. enterica serovar Enteritidis (92–0392; S. Enteritidis) were compared to those of wild-type S. Typhimurium (14028s). % identity values are displayed in color map. The phylogenetic tree was made by the interactive Tree of Life software (v5) based on analysis of the ugtL gene and its upstream region containing the SsrB binding site (720 nt upstream from the start codon (AUG) of the ugtL gene and whole coding region) using Clustal Omega. (B) Schematic of the ugtL gene and its neighboring genes in S. Typhimurium, S. salamae, S. diarizonae, and S. bongori. The conserved regions are indicated with gray color.
Mildly acidic pH activates PhoP in S. bongori if ugtL is transcribed from a heterologous promoter
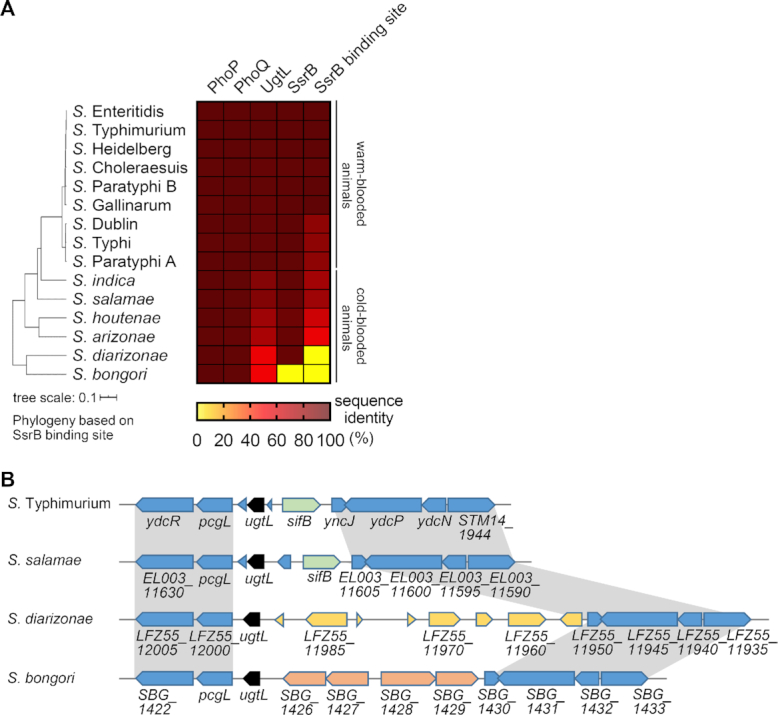
The data presented above argue that S. bongori fails to activate PhoP/PhoQ in mildly acidic pH because it lacks the ssrB gene and the SsrB binding region in the ugtL promoter region (Supplemental Figure S6). If this is correct, it should be possible to bypass the requirement for both ssrB and the SsrB binding site in the ugtL promoter by expressing the ugtL gene from a heterologous promoter. As hypothesized, ugtL-expressing plasmids increased the PhoP-P-to-PhoP ratio in both wild-type S. bongori and in an S. bongori derivative expressing the S. Typhimurium phoQ gene instead of its own (Figure 7A and B). This was true whether the ugtL gene originated from S. Typhimurium or S. bongori (Figure 7A and B). (We note that the S. bongori UgtL had higher activity than the S. Typhimurium UgtL when investigated in wild-type S. bongori (Figure 7A).) By contrast, the vector control had no effect (Figure 7A and B). Furthermore, a plasmid expressing the ssrB gene from a heterologous promoter complemented the ssrB S. Typhimurium mutant (Figure 2A) but failed to promote ugtL transcription in S. bongori (Figure 7C) because S. bongori lacks the SsrB binding site in the ugtL-sifB intergenic region.
Figure 7.
Expression of the ugtL gene from a heterologous promoter increases the fraction of PhoP-P in S. bongori grown in mildly acidic pH. (A, B) Phos-tag Western blot analysis of crude extracts prepared from wild-type S. bongori (A) or S. bongori harboring S. Typhimurium phoQ (phoQST) (JC325) (B) strains harboring a plasmid expressing the S. Typhimurium ugtL (pUgtLST) or S. bongori ugtL (pUgtLSB) gene from a heterologous promoter or an empty vector (pVec), or with no plasmid (–) grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9 grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9 (acidic pH) with 500 μM of IPTG using antibodies recognizing PhoP or the loading control AtpB. Representatives of at least three independent experiments are shown. Numbers under the blots indicate % phosphorylated PhoP (PhoP-P). (C) mRNA abundance of the ugtL gene in wild-type S. bongori harboring a plasmid expressing the ssrB gene (pSsrB) from a heterologous promoter or an empty vector (pVec) grown to mid-log phase in N-minimal media with 1 mM of Mg2+ at pH 4.9. The mean and SD from three independent experiments are shown (n = 3). Each dot represents individual biological sample. Two-tailed t-test with strain with pVec vs. the one with pSsrB; ns, not significant.
DISCUSSION
The phenotypic properties that distinguish closely related bacterial species are often ascribed to differences in gene content. These differences usually result from horizontal gene transfer, whereby foreign genes confer new abilities upon a recipient organism. Foreign genes, such as those conferring resistance to antibiotics, can operate independently of the ancestral genome, and this is why virtually identical resistance determinants are found in distantly related bacterial species (1,3). By contrast, our results now establish that a horizontally acquired gene can act on the ancestral genome by providing abilities that are realized only by the simultaneous presence of horizontally acquired and ancestral genes in the same organism (Figure 1). Therefore, the phenotypic properties that distinguish closely related bacterial species are more accurately ascribed to both the intrinsic properties of species-specific genes and their effects on ancestral genes.
Control of the virulence regulator PhoP by the SsrB protein
We suggest the following model of how the horizontally acquired ssrB gene confers upon S. Typhimurium the ability to activate the ancestral PhoP/PhoQ system in mildly acidic pH (Figure 1). The SsrB protein binds to the SsrB binding site in the ugtL-sifB intergenic region (Figure 3D), which displaces the foreign gene silencer H-NS and results in ugtL transcription (Figure 3EF). The UgtL protein stimulates autophosphorylation of PhoQ (25), which increases the fraction of PhoP-P, thereby promoting transcription of PhoP-activated genes (Figure 3E and Supplemental Figure S7). In addition, SsrB furthers PhoP amounts by increasing phoP transcription (Figure 4). In this way, activation of the ancestral PhoP/PhoQ system by the horizontally acquired ssrB gene enables S. Typhimurium to cause disease (Figures 1 and 8).
Figure 8.

The horizontally acquired ssrB gene activates the ancestral regulator PhoP at late times inside macrophages. (Left) At early times following bacterial internalization by macrophages, phagosomal signals, such as mildly acidic pH, stimulate the ancestral sensor PhoQ to promote the phosphorylated state of PhoP. PhoP-P promotes transcription of its target genes, including the phoP, ugtL, and ssrB genes. The SsrB protein shows little activity at this time. The little ugtL expression taking place is not sufficient for full PhoP activation. (Right) At later times following bacterial internalization by macrophages, SpiR activates SsrB in response to signals yet to be identified, thereby promoting transcription of the ugtL gene by antagonizing the xenogeneic silencer H-NS. UgtL enhances PhoQ autophosphorylation, further activating PhoP. SsrB also increases phoP transcription by directly binding to the purB coding region upstream of the phoP promoter region. As a consequence, PhoP further increases transcription of its activated genes. The SsrB-mediated PhoP activation is necessary for normal Salmonella virulence.
The mildly acidic pH of the macrophage phagosome activates the PhoP protein in an ssrB-independent manner shortly after bacterial entry (Figure 5B and 8). At 6 h, however, active SsrB promotes ugtL expression, thereby enhancing PhoP activation (Figure 5B and 8). Given that the phagosome acidifies less than 1 h after phagocytosis (26,27) and that SsrB shows close to wild-type activity in laboratory media of mildly acidic pH (Supplemental Figure S10), SsrB may be activated by another phagosomal signal(s) at 6 h inside a phagosome.
The DNA binding proteins PhoP, SsrB, and SlyA have been implicated in ugtL transcription. PhoP binds at two different sites in the ugtL promoter (Supplemental Figure S6) (62) and is necessary to recruit RNA polymerase (63). By contrast, SsrB and SlyA operate as anti-silencers that antagonize silencing by H-NS (Figure 3 and 8) (63). Expression of both SsrB and SlyA is transcriptionally controlled by PhoP (7,64). However, SsrB and SlyA differ in several properties: (i) SsrB is necessary for ugtL transcription in mildly acidic pH but not in low Mg2+ (Figure 2B, 4E and Supplemental Figure S7), whereas SlyA is necessary for activation in low Mg2+ (62,63); (ii) the SlyA binding site is proximal to the transcription start sites of the ugtL gene (62), whereas the SsrB binding site is located further upstream (Supplemental Figure S6) and (iii) SlyA is present in both S. enterica and S. bongori, whereas SsrB is present only in S. enterica. Cumulatively, these findings suggest that S. Typhimurium employs different DNA binding proteins to aid PhoP transcribe the ugtL gene, depending on the identity of the PhoQ inducing signal.
Our findings have implications regarding PhoP/PhoQ-mediated bacterial physiology. First, they raise the possibility of signals altering UgtL abundance impacting PhoP activation status. For example, SsrB responds to redox changes (65). Likewise, the Salmonella-specific regulators HilD and SprB (50,66) have been implicated in ugtL expression (67). And second, SsrB activation of PhoP is likely to have genome-wide effects, beyond the genes directly controlled by PhoP because: (i) PhoP is a direct transcriptional activator of the rstA and slyA genes, which specify DNA binding regulatory proteins (63,64,68,69); (ii) PhoP activates the transcriptional regulator PmrA post-translationally (70,71); (iii) PhoP promotes degradation of the gene silencer H-NS (72); and (iv) PhoP reduces proteolysis by different proteases that target pleiotropic regulators (73–75). The PhoP-dependent effects may account, in part, for SsrB controlling expression of ∼5% of the S. Typhimurium genome (17,18).
Our findings suggest that the SsrB-mediated induction of the ugtL gene taking place inside macrophages (Figure 5B) contributes to virulence in an animal host (Figure 5A and Supplemental Figure S9) by generating active PhoP protein (Figure 5B). Given that PhoP-activated genes, including ugtL, are highly induced inside the vacuolar compartment of epithelial cells (76), the PhoP activation mechanisms described in this paper may also take place in such cells during infection, and thus, promote Salmonella virulence.
The genetic basis for phenotypic differences among Salmonella species and serovars
The two species that comprise the Salmonella genus differ in hundreds of genes (12,13,77). Some of these genes are directly responsible for S. Typhimurium being pathogenic and S. bongori being largely non-pathogenic. We have now established that this phenotypic difference also results from a S. Typhimurium-specific gene acting on genes that are shared between the two species (Figure 1). That is, the S. Typhimurium-specific ssrB gene is necessary for activation of the widespread PhoP protein in mildly acidic pH (Figure 2). S. bongori fails to activate PhoP in mildly acidic pH (Figure 7) (29) because it lacks both the ssrB gene (12) and the SsrB binding site in the ugtL promoter region (Figure 1 and Supplemental Figure S6), and also because of differences in the phoQ gene (29). That nucleotide differences in bacterial promoter sequences play a critical role in bacterial evolution is further supported by the dramatic increase in virulence displayed by an African clade of S. Typhimurium with a single nucleotide difference in the promoter region of the pgtE gene (78).
The PhoP-P-to-PhoP ratio was higher in wild-type S. bongori when harboring a plasmid expressing the S. bongori ugtL gene than when harboring an isogenic plasmid expressing the S. Typhimurium ugtL gene (Figure 7A). By contrast, the ratio was similar when the plasmids were present in a S. bongori variant expressing the S. enterica phoQ gene in place of S. bongori’s own phoQ gene (Figure 7B). These results suggest co-evolution of the horizontally acquired ugtL and the ancestral phoQ genes in Salmonella species.
A phylogeny based on the ∼2.6-Mb core Salmonella sequence (79) resembles that based on the ugtL promoter, leader, and coding regions (∼1.2 Kb) (Figure 6A). This analysis suggests that the PhoP activation resulting from the anti-silencing effects of SsrB on the ugtL promoter is a key feature of Salmonella serovars that infect warm-blooded animals. Moreover, it implies that PhoP activation by mildly acidic pH is dispensable in Salmonella serovars that infect cold-blooded animals.
Different horizontally acquired genes enable Escherichia coli to activate PhoP/PhoQ in mildly acidic pH
E. coli can activate the PhoP/PhoQ system in mildly acidic pH (80) despite lacking the ssrB and ugtL genes. This activation requires the regulatory system encoded by the evgA and evgS genes (81), which have features of horizontally acquired DNA, including an unusually low GC content, absence from closely related species, and being bound by the gene silencer H-NS (82). It is also dependent on EvgA-activated safA (83), a horizontally acquired gene that specifies a post-translational activator of the PhoQ protein (84). There is no shared sequence identity between the SafA and UgtL proteins or between the EvgA and SsrB proteins, suggesting that E. coli and S. enterica independently acquired the ability to activate the ancestral regulatory system PhoP/PhoQ in mildly acidic pH.
CONCLUDING REMARKS
Finally, the mobile nature of some horizontally acquired genes suggests that, as long as such genes are expressed, they operate regardless of the host genetic background. For example, a gene specifying an antibiotic-inactivating protein typically confers antibiotic resistance to a wide variety of bacterial species. However, our findings present a new paradigm: a horizontally acquired gene regulating an ancestral regulatory system. This paradigm indicates that the activity of conserved ancestral genes can vary across species, depending on which horizontally acquired genes are present in a given organism.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author on reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jennifer Aronson for comments on the manuscript.
Author contributions: J.C. and E.A.G. designed research; J.C. performed research; J.C. and E.A.G. analyzed data; and J.C. and E.A.G. wrote the paper.
Contributor Information
Jeongjoon Choi, Department of Microbial Pathogenesis, Yale School of Medicine, 295 Congress Avenue, New Haven, CT 06536, USA.
Eduardo A Groisman, Department of Microbial Pathogenesis, Yale School of Medicine, 295 Congress Avenue, New Haven, CT 06536, USA; Yale Microbial Sciences Institute, P.O. Box 27389, West Haven, CT 06516, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health of the USA [AI120558]. Funding for open access charge: NIH [AI120558].
Conflict of interest statement. None declared.
REFERENCES
- 1. Soucy S.M., Huang J., Gogarten J.P.. Horizontal gene transfer: building the web of life. Nat. Rev. Genet. 2015; 16:472–482. [DOI] [PubMed] [Google Scholar]
- 2. Daubin V., Szollosi G.J.. Horizontal gene transfer and the history of life. Cold Spring Harb. Perspect. Biol. 2016; 8:a018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ochman H., Lawrence J.G., Groisman E.A.. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000; 405:299–304. [DOI] [PubMed] [Google Scholar]
- 4. McDaniel T.K., Kaper J.B.. A cloned pathogenicity island from enteropathogenic Escherichia coli confers the attaching and effacing phenotype on E. coli K-12. Mol. Microbiol. 1997; 23:399–407. [DOI] [PubMed] [Google Scholar]
- 5. Furniss R.C.D., Clements A.. Regulation of the locus of enterocyte effacement in attaching and effacing pathogens. J. Bacteriol. 2018; 200:e00336-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee A.K., Detweiler C.S., Falkow S.. OmpR regulates the two-component system SsrA-SsrB in Salmonella pathogenicity island 2. J. Bacteriol. 2000; 182:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bijlsma J.J., Groisman E.A.. The PhoP/PhoQ system controls the intramacrophage type three secretion system of Salmonella enterica. Mol. Microbiol. 2005; 57:85–96. [DOI] [PubMed] [Google Scholar]
- 8. Dobrindt U., Hochhut B., Hentschel U., Hacker J.. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2004; 2:414–424. [DOI] [PubMed] [Google Scholar]
- 9. Lercher M.J., Pal C.. Integration of horizontally transferred genes into regulatory interaction networks takes many million years. Mol. Biol. Evol. 2008; 25:559–567. [DOI] [PubMed] [Google Scholar]
- 10. Dorman C.J. Regulatory integration of horizontally-transferred genes in bacteria. Front. Biosci. (Landmark Ed.). 2009; 14:4103–4112. [DOI] [PubMed] [Google Scholar]
- 11. Coburn B., Grassl G.A., Finlay B.B.. Salmonella, the host and disease: a brief review. Immunol. Cell Biol. 2007; 85:112–118. [DOI] [PubMed] [Google Scholar]
- 12. Fookes M., Schroeder G.N., Langridge G.C., Blondel C.J., Mammina C., Connor T.R., Seth-Smith H., Vernikos G.S., Robinson K.S., Sanders M. et al.. Salmonella bongori provides insights into the evolution of the Salmonellae. PLoS Pathog. 2011; 7:e1002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ochman H., Groisman E.A.. Distribution of pathogenicity islands in Salmonella spp. Infect. Immun. 1996; 64:5410–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shea J.E., Hensel M., Gleeson C., Holden D.W.. Identification of a virulence locus encoding a second type III secretion system in Salmonella Typhimurium. Proc. Natl. Acad. Sci. USA. 1996; 93:2593–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fass E., Groisman E.A.. Control of Salmonella pathogenicity island-2 gene expression. Curr. Opin. Microbiol. 2009; 12:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deiwick J., Nikolaus T., Erdogan S., Hensel M.. Environmental regulation of Salmonella pathogenicity island 2 gene expression. Mol. Microbiol. 1999; 31:1759–1773. [DOI] [PubMed] [Google Scholar]
- 17. Tomljenovic-Berube A.M., Mulder D.T., Whiteside M.D., Brinkman F.S., Coombes B.K.. Identification of the regulatory logic controlling Salmonella pathoadaptation by the SsrA-SsrB two-component system. PLoS Genet. 2010; 6:e1000875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colgan A.M., Kroger C., Diard M., Hardt W.D., Puente J.L., Sivasankaran S.K., Hokamp K., Hinton J.C.. The impact of 18 ancestral and Horizontally-Acquired regulatory proteins upon the transcriptome and sRNA landscape of Salmonella enterica serovar Typhimurium. PLoS Genet. 2016; 12:e1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng X., Walthers D., Oropeza R., Kenney L.J.. The response regulator SsrB activates transcription and binds to a region overlapping OmpR binding sites at Salmonella pathogenicity island 2. Mol. Microbiol. 2004; 54:823–835. [DOI] [PubMed] [Google Scholar]
- 20. Banda M.M., Zavala-Alvarado C., Perez-Morales D., Bustamante V.H.. SlyA and HilD counteract H-NS-Mediated repression on the ssrAB virulence operon of Salmonella enterica serovar Typhimurium and thus promote its activation by OmpR. J. Bacteriol. 2019; 201:e00530-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fields P.I., Groisman E.A., Heffron F.. A Salmonella locus that controls resistance to microbicidal proteins from phagocytic cells. Science. 1989; 243:1059–1062. [DOI] [PubMed] [Google Scholar]
- 22. Miller S.I., Kukral A.M., Mekalanos J.J.. A two-component regulatory system (phoP phoQ) controls Salmonella Typhimurium virulence. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:5054–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Groisman E.A., Chiao E., Lipps C.J., Heffron F.. Salmonella Typhimurium phoP virulence gene is a transcriptional regulator. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:7077–7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shin D., Groisman E.A.. Signal-dependent binding of the response regulators PhoP and PmrA to their target promoters in vivo. J. Biol. Chem. 2005; 280:4089–4094. [DOI] [PubMed] [Google Scholar]
- 25. Choi J., Groisman E.A.. Activation of master virulence regulator PhoP in acidic pH requires the Salmonella-specific protein UgtL. Sci. Signal. 2017; 10:eaan6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rathman M., Sjaastad M.D., Falkow S.. Acidification of phagosomes containing Salmonella Typhimurium in murine macrophages. Infect. Immun. 1996; 64:2765–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alpuche Aranda C.M., Swanson J.A., Loomis W.P., Miller S.I.. Salmonella Typhimurium activates virulence gene transcription within acidified macrophage phagosomes. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:10079–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin-Orozco N., Touret N., Zaharik M.L., Park E., Kopelman R., Miller S., Finlay B.B., Gros P., Grinstein S.. Visualization of vacuolar acidification-induced transcription of genes of pathogens inside macrophages. Mol. Biol. Cell. 2006; 17:498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi J., Groisman E.A.. Acidic pH sensing in the bacterial cytoplasm is required for Salmonella virulence. Mol. Microbiol. 2016; 101:1024–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fields P.I., Swanson R.V., Haidaris C.G., Heffron F.. Mutants of Salmonella Typhimurium that cannot survive within the macrophage are avirulent. Proc. Natl. Acad. Sci. U.S.A. 1986; 83:5189–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ronald W., Davis D.B., John R.R.. Advanced Bacterial Genetics. 1980; NY: Cold Spring Harbor Lab Press. [Google Scholar]
- 32. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Snavely M.D., Miller C.G., Maguire M.E.. The mgtB Mg2+ transport locus of Salmonella Typhimurium encodes a P-type ATPase. J. Biol. Chem. 1991; 266:815–823. [PubMed] [Google Scholar]
- 34. Hanahan D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983; 166:557–580. [DOI] [PubMed] [Google Scholar]
- 35. Camp A.H., Losick R.. A feeding tube model for activation of a cell-specific transcription factor during sporulation in Bacillus subtilis. Genes Dev. 2009; 23:1014–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barbieri C.M., Stock A.M.. Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using Phos-tag-based reagents. Anal. Biochem. 2008; 376:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khetrapal V., Mehershahi K., Rafee S., Chen S., Lim C.L., Chen S.L.. A set of powerful negative selection systems for unmodified Enterobacteriaceae. Nucleic Acids Res. 2015; 43:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maloy S.R., Nunn W.D.. Selection for loss of tetracycline resistance by Escherichia coli. J. Bacteriol. 1981; 145:1110–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McKenzie G.J., Craig N.L.. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol. 2006; 6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soncini F.C., Vescovi E.G., Groisman E.A.. Transcriptional autoregulation of the Salmonella Typhimurium phoPQ operon. J. Bacteriol. 1995; 177:4364–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Choi J., Groisman E.A.. The lipopolysaccharide modification regulator PmrA limits Salmonella virulence by repressing the type three-secretion system Spi/Ssa. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:9499–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garcia Vescovi E., Soncini F.C., Groisman E.A.. Mg2+ as an extracellular signal: environmental regulation of Salmonella virulence. Cell. 1996; 84:165–174. [DOI] [PubMed] [Google Scholar]
- 43. Bader M.W., Sanowar S., Daley M.E., Schneider A.R., Cho U., Xu W., Klevit R.E., Le Moual H., Miller S.I.. Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell. 2005; 122:461–472. [DOI] [PubMed] [Google Scholar]
- 44. Prost L.R., Daley M.E., Le Sage V., Bader M.W., Le Moual H., Klevit R.E., Miller S.I.. Activation of the bacterial sensor kinase PhoQ by acidic pH. Mol. Cell. 2007; 26:165–174. [DOI] [PubMed] [Google Scholar]
- 45. Mulder D.T., McPhee J.B., Reid-Yu S.A., Stogios P.J., Savchenko A., Coombes B.K.. Multiple histidines in the periplasmic domain of the Salmonella enterica sensor kinase SsrA enhance signaling in response to extracellular acidification. Mol. Microbiol. 2015; 95:678–691. [DOI] [PubMed] [Google Scholar]
- 46. Soncini F.C., Garcia Vescovi E., Solomon F., Groisman E.A.. Molecular basis of the magnesium deprivation response in Salmonella Typhimurium: identification of PhoP-regulated genes. J. Bacteriol. 1996; 178:5092–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chamnongpol S., Groisman E.A.. Acetyl phosphate-dependent activation of a mutant PhoP response regulator that functions independently of its cognate sensor kinase. J. Mol. Biol. 2000; 300:291–305. [DOI] [PubMed] [Google Scholar]
- 48. Hilbert F., Garcia-del Portillo F., Groisman E.A.. A periplasmic D-alanyl-D-alanine dipeptidase in the gram-negative bacterium Salmonella enterica. J. Bacteriol. 1999; 181:2158–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu M., Tolstorukov M., Zhurkin V., Garges S., Adhya S.. A mutant spacer sequence between -35 and -10 elements makes the Plac promoter hyperactive and cAMP receptor protein-independent. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:6911–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Navarre W.W., Porwollik S., Wang Y., McClelland M., Rosen H., Libby S.J., Fang F.C.. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science. 2006; 313:236–238. [DOI] [PubMed] [Google Scholar]
- 51. Lucchini S., Rowley G., Goldberg M.D., Hurd D., Harrison M., Hinton J.C.. H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2006; 2:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Walthers D., Li Y., Liu Y., Anand G., Yan J., Kenney L.J.. Salmonella enterica response regulator SsrB relieves H-NS silencing by displacing H-NS bound in polymerization mode and directly activates transcription. J. Biol. Chem. 2011; 286:1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Carroll R.K., Liao X., Morgan L.K., Cicirelli E.M., Li Y., Sheng W., Feng X., Kenney L.J.. Structural and functional analysis of the C-terminal DNA binding domain of the Salmonella Typhimurium SPI-2 response regulator SsrB. J. Biol. Chem. 2009; 284:12008–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Coombes B.K., Wickham M.E., Lowden M.J., Brown N.F., Finlay B.B.. Negative regulation of Salmonella pathogenicity island 2 is required for contextual control of virulence during typhoid. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:17460–17465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Arpaia N., Godec J., Lau L., Sivick K.E., McLaughlin L.M., Jones M.B., Dracheva T., Peterson S.N., Monack D.M., Barton G.M.. TLR signaling is required for Salmonella Typhimurium virulence. Cell. 2011; 144:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Plant J., Glynn A.A.. Natural resistance to Salmonella infection, delayed hypersensitivity and Ir genes in different strains of mice. Nature. 1974; 248:345–347. [DOI] [PubMed] [Google Scholar]
- 57. Freeman J.A., Ohl M.E., Miller S.I.. The Salmonella enterica serovar Typhimurium translocated effectors SseJ and SifB are targeted to the Salmonella-containing vacuole. Infect. Immun. 2003; 71:418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walthers D., Carroll R.K., Navarre W.W., Libby S.J., Fang F.C., Kenney L.J.. The response regulator SsrB activates expression of diverse Salmonella pathogenicity island 2 promoters and counters silencing by the nucleoid-associated protein H-NS. Mol. Microbiol. 2007; 65:477–493. [DOI] [PubMed] [Google Scholar]
- 59. Rodrigues Alves L.B., Neto O.C.F., Batista D.F.A., Barbosa F.O., Rubio M.D.S., de Souza A.I.S., Almeida A.M., Barrow P.A., Junior A.B.. Inactivation of phoPQ genes attenuates Salmonella Gallinarum biovar Gallinarum to susceptible chickens. Braz. J. Microbiol. 2018; 49:601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dominguez-Bernal G., Tierrez A., Bartolome A., Martinez-Pulgarin S., Salguero F.J., Orden J.A., de la Fuente R.. Salmonella enterica serovar Choleraesuis derivatives harbouring deletions in rpoS and phoP regulatory genes are attenuated in pigs, and survive and multiply in porcine intestinal macrophages and fibroblasts, respectively. Vet. Microbiol. 2008; 130:298–311. [DOI] [PubMed] [Google Scholar]
- 61. Hohmann E.L., Oletta C.A., Killeen K.P., Miller S.I.. phoP/phoQ-deleted Salmonella Typhi (Ty800) is a safe and immunogenic single-dose typhoid fever vaccine in volunteers. J. Infect. Dis. 1996; 173:1408–1414. [DOI] [PubMed] [Google Scholar]
- 62. Shi Y., Latifi T., Cromie M.J., Groisman E.A.. Transcriptional control of the antimicrobial peptide resistance ugtL gene by the Salmonella PhoP and SlyA regulatory proteins. J. Biol. Chem. 2004; 279:38618–38625. [DOI] [PubMed] [Google Scholar]
- 63. Perez J.C., Latifi T., Groisman E.A.. Overcoming H-NS-mediated transcriptional silencing of horizontally acquired genes by the PhoP and SlyA proteins in Salmonella enterica. J. Biol. Chem. 2008; 283:10773–10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Norte V.A., Stapleton M.R., Green J.. PhoP-responsive expression of the Salmonella enterica serovar Typhimurium slyA gene. J. Bacteriol. 2003; 185:3508–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Husain M., Jones-Carson J., Song M., McCollister B.D., Bourret T.J., Vazquez-Torres A.. Redox sensor SsrB Cys203 enhances Salmonella fitness against nitric oxide generated in the host immune response to oral infection. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:14396–14401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Eichelberg K., Hardt W.D., Galan J.E.. Characterization of SprA, an AraC-like transcriptional regulator encoded within the Salmonella Typhimurium pathogenicity island 1. Mol. Microbiol. 1999; 33:139–152. [DOI] [PubMed] [Google Scholar]
- 67. Banda M.M., Manzo R., Bustamante V.H.. HilD induces expression of a novel Salmonella Typhimurium invasion factor, YobH, through a regulatory cascade involving SprB. Sci. Rep. 2019; 9:12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Minagawa S., Ogasawara H., Kato A., Yamamoto K., Eguchi Y., Oshima T., Mori H., Ishihama A., Utsumi R.. Identification and molecular characterization of the Mg2+ stimulon of Escherichia coli. J. Bacteriol. 2003; 185:3696–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jeon J., Kim H., Yun J., Ryu S., Groisman E.A., Shin D.. RstA-promoted expression of the ferrous iron transporter FeoB under iron-replete conditions enhances Fur activity in Salmonella enterica. J. Bacteriol. 2008; 190:7326–7334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kox L.F., Wosten M.M., Groisman E.A.. A small protein that mediates the activation of a two-component system by another two-component system. EMBO J. 2000; 19:1861–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kato A., Groisman E.A.. Connecting two-component regulatory systems by a protein that protects a response regulator from dephosphorylation by its cognate sensor. Genes Dev. 2004; 18:2302–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Choi J., Groisman E.A.. Salmonella expresses foreign genes during infection by degrading their silencer. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:8074–8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tu X., Latifi T., Bougdour A., Gottesman S., Groisman E.A.. The PhoP/PhoQ two-component system stabilizes the alternative sigma factor RpoS in Salmonella enterica. Proc. Natl. Acad. Sci. USA. 2006; 103:13503–13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yeom J., Gao X., Groisman E.A.. Reduction in adaptor amounts establishes degradation hierarchy among protease substrates. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E4483–E4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gao X., Yeom J., Groisman E.A.. The expanded specificity and physiological role of a widespread N-degron recognin. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:18629–18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Westermann A.J., Forstner K.U., Amman F., Barquist L., Chao Y., Schulte L.N., Muller L., Reinhardt R., Stadler P.F., Vogel J.. Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature. 2016; 529:496–501. [DOI] [PubMed] [Google Scholar]
- 77. Baumler A.J. The record of horizontal gene transfer in Salmonella. Trends Microbiol. 1997; 5:318–322. [DOI] [PubMed] [Google Scholar]
- 78. Hammarlof D.L., Kroger C., Owen S.V., Canals R., Lacharme-Lora L., Wenner N., Schager A.E., Wells T.J., Henderson I.R., Wigley P. et al.. Role of a single noncoding nucleotide in the evolution of an epidemic African clade of Salmonella. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E2614–E2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Desai P.T., Porwollik S., Long F., Cheng P., Wollam A., Bhonagiri-Palsikar V., Hallsworth-Pepin K., Clifton S.W., Weinstock G.M., McClelland M.. Evolutionary genomics of Salmonella enterica subspecies. mBio. 2013; 4:e00579-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lippa A.M., Goulian M.. Feedback inhibition in the PhoQ/PhoP signaling system by a membrane peptide. PLoS Genet. 2009; 5:e1000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Burton N.A., Johnson M.D., Antczak P., Robinson A., Lund P.A.. Novel aspects of the acid response network of E. coli K-12 are revealed by a study of transcriptional dynamics. J. Mol. Biol. 2010; 401:726–742. [DOI] [PubMed] [Google Scholar]
- 82. Grainger D.C., Hurd D., Goldberg M.D., Busby S.J.. Association of nucleoid proteins with coding and non-coding segments of the Escherichia coli genome. Nucleic Acids Res. 2006; 34:4642–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Eguchi Y., Itou J., Yamane M., Demizu R., Yamato F., Okada A., Mori H., Kato A., Utsumi R.. B1500, a small membrane protein, connects the two-component systems EvgS/EvgA and PhoQ/PhoP in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:18712–18717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ishii E., Eguchi Y., Utsumi R.. Mechanism of activation of PhoQ/PhoP two-component signal transduction by SafA, an auxiliary protein of PhoQ histidine kinase in Escherichia coli. Biosci. Biotechnol. Biochem. 2013; 77:814–819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.