


Abstract
Synonymous codon usage significantly impacts translational and transcriptional efficiency, gene expression, the secondary structure of both mRNA and proteins, and has been implicated in various diseases. However, population-specific differences in codon usage biases remain largely unexplored. Here, we present a web server, https://cubap.byu.edu, to facilitate analyses of codon usage biases across populations (CUBAP). Using the 1000 Genomes Project, we calculated and visually depict population-specific differences in codon frequencies, codon aversion, identical codon pairing, co-tRNA codon pairing, ramp sequences, and nucleotide composition in 17,634 genes. We found that codon pairing significantly differs between populations in 35.8% of genes, allowing us to successfully predict the place of origin for African and East Asian individuals with 98.8% and 100% accuracy, respectively. We also used CUBAP to identify a significant bias toward decreased CTG pairing in the immunity related GTPase M (IRGM) gene in East Asian and African populations, which may contribute to the decreased association of rs10065172 with Crohn's disease in those populations. CUBAP facilitates in-depth gene-specific and codon-specific visualization that will aid in analyzing candidate genes identified in genome-wide association studies, identifying functional implications of synonymous variants, predicting population-specific impacts of synonymous variants and categorizing genetic biases unique to certain populations.
INTRODUCTION
Ribosomes translate 61 unique codons into 20 amino acids because a natural wobble allows aminoacyl-tRNA to bind one of several different codons through non-Watson-Crick pairing that typically occurs at the third nucleotide position (1,2). Codon usage biases arise when synonymous codons occur in different frequencies across genomes, genes, or positions within genes. Codon usage biases affect translation efficiency (3–5), gene expression levels (6–9) and mRNA and protein secondary structure (10–17). Codon optimality and codon frequency directly correlate with tRNA abundance (6,18–20), and more abundant codons are generally translated faster, which increases overall protein levels (6,9,21,22). Codons with rare cognate tRNAs are translated slower than optimal codons and generally decrease gene expression because of limited cognate tRNA availability, which increases tRNA competition at that codon position (21,22). Therefore, codon usage biases that favor optimal codons can lead to complete aversion to suboptimal codons, which in turn decreases resource utilization, increases translational speed, and usually increases gene expression (6,10,21,23,24). Additionally, codon choice can alter mRNA and protein secondary structure through hydrogen bonding and co-translational folding, which also affects transcriptional and translational speed (10–17).
Recent studies show that specific locations within genes are also susceptible to codon usage biases for either optimal or suboptimal codons (3,4,11,25,26). Identical codon pairing occurs when a codon appears multiple times within a single ribosomal reading frame. Similarly, co-tRNA codon pairing occurs when multiple synonymous, but not identical, codons occur within a single reading frame. These pairings allow ribosomes to recharge tRNAs before they diffuse from the ribosome, effectively increasing translation speed and gene expression (3,11,25). A small ramp sequence of suboptimal, slowly-translated codons at the beginning of a gene surprisingly increases translational efficiency (26) by preventing downstream ribosomal ‘traffic jams’ (5,27). Additionally, ramp sequences coincide with more efficient hydrogen bonding that increases transcriptional efficiency by requiring fewer dsDNA bonds to be broken (28). More information on codon pairing and ramp sequences, including an example of each, are depicted in Supplementary Figures S1 and S2.
Since codon usage biases directly affect protein levels and protein structure, they likely have a non-trivial effect on genetic diseases and disorders. Synonymous mutations have been associated with >30 different diseases (29). In some cases, the etiological effects are attributed to the synonymous mutation altering splicing (30–34), although in many cases the biological mechanisms driving the synonymous variant association with disease remain unknown. For example, various synonymous mutations in the MDR1 gene are associated with drug resistance for a variety of diseases including cancer (35–42). Additionally, a synonymous mutation in UBE1 associated with developing X-linked infantile spinal muscular atrophy significantly lowers UBE1 expression and changes exon methylation (43). Synonymous rare variants are also associated with Alzheimer's disease biomarkers (44), and shifts in codon usage biases alone can yield high diagnostic accuracy in a variety of other diseases including cancers and multiple sclerosis (45). Furthermore, codon usage biases in rat oncogenes directly regulate tumor growth (46). Therefore, better understanding the effects of codon usage on protein expression may lead to improved protein therapeutics and better understanding the etiology of various diseases (29).
The 1000 Genomes Project contains whole genome sequencing data from 2504 individuals spanning 26 populations from five superpopulations and has been widely used to assess population-specific genetic differences. Although specific genomic inquiries of the 1000 Genomes Project can be conducted on web portals such as PopHuman (47) and CAGm (48), population-specific differences in codon usage biases remain largely unexplored and are not easily accessible. Tools such as CAICal (49) and CodonW (50) specialize in gene-specific or individual-specific calculations of codon usage biases. Codon metrics such as the Codon Adaptation Index (51), Relative Synonymous Codon Usage (52), the Effective Number of Codons (53), and GC content are often used to compare codon usage biases between small groups of individuals or species. GC content is commonly used in studies of codon usage biases because higher GC content is indicative of genic regions (54,55) and correlates with the overall nucleotide composition of the genome (56). Some databases, such as HIVE-CUT (57) and CBDB (58), enable phylogenic comparisons of species-specific codon usage biases. While these methods allow researchers to compare codon usage statistics between studies and species, the extent to which differences in specific codon usage biases (e.g. ramp sequences, codon pairing (59), codon aversion (60)) differ between human populations and their ability to predict population of origin have yet to be characterized. Additionally, a centralized database that can be used to identify conserved population-specific differences in codon usage biases that might have disease implications does not currently exist. Because codon optimality correlates with codon frequency, the effects of a synonymous mutations on protein secondary structure, expression, and disease may vary between populations where population-specific codon biases exist. A database of population-specific codon usage biases would provide an additional filter for evaluating findings from genome-wide association studies, elucidate the extent to which synonymous variants affect disease susceptibility in different populations, and provide insights into population-specific reactions to drugs in clinical trials.
Our web portal, Codon Usage Biases Across Populations (https://cubap.byu.edu), facilitates these types of analyses by providing users with annotated codon usage biases across different human populations using data from the 1000 Genomes Project. We used these biases to accurately predict the population of origin in African and East Asian populations with 98.8% and 100% accuracy, respectively. Additionally, CUBAP’s dataset can be used to better understand how synonymous variants may affect disease development in a variety of ways, and we provide a case study of how CUBAP can be used to analyze the effects of pathogenic synonymous variants. CUBAP includes various analyses of codon frequencies, codon aversion, identical codon pairing, co-tRNA codon pairing, ramp sequences, and nucleotide composition including GC content spanning 17 634 genes and 40 643 isoforms. Interactive graphics powered by Microsoft Power BI facilitate custom project-specific analyses of codon usages within specific populations, genes, or codons.
MATERIALS AND METHODS
Data availability
All analyses were performed on 2504 human samples from the 1000 Genomes Project database. This dataset was selected because of its credibility, accuracy, and large number of samples spanning many different populations. Individuals are labeled as belonging to one of 26 different subpopulations spanning five superpopulations (see Table 1). After excluding genes that had annotated translational exceptions, partial genes, or potential errors, we analyzed 17 634 complete gene sequences and 40 643 isoforms using human reference assembly hg19. All scripts used to analyze these data are publicly available at https://github.com/kauwelab/cubap. CUBAP’s documentation can be viewed at https://cubap.readthedocs.io/.
Table 1.
Population data used from 1000 Genomes Project
| Superpopulation | Subpopulation | Description | Number of Samples |
|---|---|---|---|
| Africa | ASW | African Ancestry in Southwest US | 61 |
| ACB | African Caribbean in Barbados | 96 | |
| ESN | Esan in Nigeria | 99 | |
| GWD | Gambian in Western Division, The Gambia | 113 | |
| LWK | Luhya in Webuye, Kenya | 99 | |
| MSL | Mende in Sierra Leone | 85 | |
| YRI | Yoruba in Ibadan, Nigeria | 108 | |
| Total: | 661 | ||
| America | CLM | Colombian in Medellin, Colombia | 94 |
| MXL | Mexican Ancestry in Los Angeles, California | 64 | |
| PEL | Peruvian in Lima, Peru | 85 | |
| PUR | Puerto Rican in Puerto Rico | 104 | |
| Total: | 347 | ||
| East Asia | CDX | Chinese Dai in Xishuangbanna, China | 93 |
| CHB | Han Chinese in Bejing, China | 103 | |
| JPT | Japanese in Tokyo, Japan | 104 | |
| KHV | Kinh in Ho Chi Minh City, Vietnam | 99 | |
| CHS | Southern Han Chinese, China | 105 | |
| Total: | 504 | ||
| Europe | GBR | British in England and Scotland | 91 |
| FIN | Finnish in Finland | 99 | |
| IBS | Iberian populations in Spain | 107 | |
| TSI | Toscani in Italy | 107 | |
| CEU | Utah residents with Northern and Western European ancestry | 99 | |
| Total: | 503 | ||
| South Asia | BEB | Bengali in Bangladesh | 86 |
| GIH | Gujarati Indian in Houston, TX | 103 | |
| ITU | Indian Telugu in the UK | 102 | |
| PJL | Punjabi in Lahore, Pakistan | 96 | |
| STU | Sri Lankan Tamil in the UK | 102 | |
| Total: | 489 | ||
| Grand Total: | 2,504 |
The distribution of samples and populations within the 1000 Genomes Project.
Calculating codon usage biases
We calculated various codon usage biases for each individual and population in our dataset. Codon frequency biases were calculated for each person by incrementing a counter for each codon and writing the resulting values for each gene to a comma separated values (CSV) file. Using the codon frequency CSV file, we calculated codon aversion data for each gene by isolating only codons that had a frequency of zero in any given gene or isoform.
We calculated identical and co-tRNA codon pairing frequencies for each gene using a modified script from Miller et al. (59). Identical codon pairing was calculated for each of the 61 amino-acid encoding codons by counting the number of times the same codon occurred within a ribosomal window of nine codons, which encompasses the average length of a ribosome (61). Similarly, co-tRNA codon pairing was calculated for each of 20 amino acids by counting the number of times non-identical codons that encode the same amino acid occurred multiple times within a ribosomal window. The resulting identical codon pairing and co-tRNA codon pairing data were also written to separate CSV files that can be downloaded from the website.
We used ExtRamp (27) to calculate the relative synonymous codon usage of each codon within each gene, and we determined the length of a ramp sequence (if it existed) within that gene for an individual. A ramp sequence is a short segment of suboptimal, slowly-translated codons at the beginning of a gene that is predicted to increase gene expression by preventing downstream ribosome collisions. ExtRamp (27) uses the relative synonymous codon usages calculated from all longest isoforms in human reference assembly hg19. Those relative synonymous codon usage values are used to calculate the harmonic mean translational speed within a sliding ribosomal window (e.g. nine codons), calculated for all ribosomal windows across a gene sequence. If an outlier local minimum occurs within the first percentile of the gene (i.e. the beginning of the gene), then a ramp sequence is written to an output file including the nucleotides at the beginning of the gene sequence through the end of the outlier region. We computed and report the harmonic mean of the relative synonymous codon usages in the ramp sequence as well as the harmonic mean of all codons within the gene sequence for each gene containing a predicted ramp sequence. We used the harmonic mean instead of the arithmetic or geometric mean because it is most appropriate for averaging ratios such as the relative synonymous codon usage, it is not significantly affected by outliers that may exist within the ramp sequence, and it was used by ExtRamp to calculate the existence of a ramp sequence (27). Results were written to a CSV file that can also be downloaded from https://cubap.byu.edu.
Nucleotide frequencies and GC content were also calculated by counting the occurrences of each nucleotide in each gene. GC content was computed for each gene within Microsoft Power BI.
Supplementary Figure S3 depicts how data were generated for the CUBAP server and details the format of the CSVs of each codon usage bias file.
Population identification
Population-specific differences in codon usage biases
We performed an analysis of variance (ANOVA) on population-specific differences in codon frequency, co-tRNA codon pairs, identical codon pairs, codon aversion, and ramp sequences across the longest isoforms of 17 634 genes. The purpose of an ANOVA is to determine the significance of a relationship between a numeric variable (e.g. mean number of codon pairings) and categorical groups (e.g. population). Since codon frequencies, codon aversion, codon pairing, and ramp sequences were discrete analyses, a Bonferroni significance threshold was established for each of those biases independently depending on the number of genes and codons analyzed for each bias (see Supplementary Table S1). Pairwise t-tests were performed on all significant ANOVAs to determine which populations significantly differed from each other. We established a Bonferroni significance threshold for each set of t-tests by using the total number of tests for each codon bias (see Supplementary Table S1). Additionally, we calculated the Cohen's d (62) effect size for all significant t-tests. The ANOVA and t-tests are well-justified for eliciting significant population-specific differences in codon usages because the superpopulations and subpopulations were well-represented and consisted of large sample sizes (see Table 1).
Since codon aversion and codon pairing have previously been used to recover phylogenies, we also performed a single one-way ANOVA on the overall codon pairing frequencies of the five superpopulations and a second ANOVA on the overall codon aversion frequencies between each superpopulation to determine if overall codon pairing or codon aversion biases might be used in an alignment-free clustering algorithm to predict the population of origin of each individual in the 1000 Genomes Project. Since both ANOVAs were significant (P-values = 2.56 × 10−189 and 5.03 × 10−31, respectively), we proceeded to cluster individuals in the 1000 Genomes Project based on codon pairing or codon aversion across the genome.
Clustering
We assessed the predictive power of codon usage biases at identifying the superpopulation of origin for each person in the 1000 Genomes Project, as well as the hg19 human reference genome, by using two alignment-free phylogenomic algorithms to classify individuals based on codon aversion (60) and codon pairing (59). These phylogenomic algorithms perform similar calculations by first calculating a motif of codons that are either averted or pair at least once within a single gene. This process is repeated for all genes, and each motif is added to a set for a genome. Next, a distance is calculated between genomes using a set union of the motifs between individuals, without regard to gene names. Finally, a phylogeny is recovered using neighbor-joining. These phylogenies were used as pedigrees to assess the effectiveness of population clustering.
Evaluation of population identification
After recovering the proposed phylogeny using only codon pairing or codon aversion, we counted the unique clusters (i.e. clades) of individuals originating from the same superpopulation using the 1000 Genomes Project population annotations. Finally, we calculated the percent predictive accuracy for each of the five superpopulations as follows, where a cluster was defined as the largest discrete grouping that consisted of only individuals from the same superpopulation:
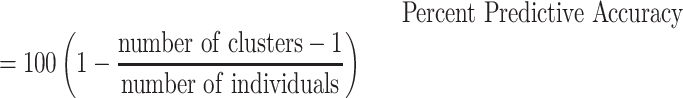 |
The scripts used to perform these calculations are available at https://github.com/kauwelab/cubap/population_stratification.
Data analysis & visualization
We opted to use Microsoft Power BI (Microsoft, 2019, Power BI Desktop) to visualize codon usage biases on a server (https://cubap.byu.edu) because of its rich library of interactive graphics that allow for optimized, state-of-the-art subsetting and visualization. Power BI allows users to interact with bar charts, violin plots, and box plots for each of the codon usage biases, genes, and populations. However, Power BI does not allow users to upload their own data to reports published on websites. Since the purpose of CUBAP is to provide a tool to query results from the 1000 Genomes Project, Power BI was optimally suited to allow user-specific queries of these data. We used the Power BI custom visual Violin Plot (Daniel Marsh-Patrick, version 1.3.0.4, 2019) that calculates the maximum, minimum, median, mean, and standard deviation for the number of codons or codon pairs for each population group. We used the Box and Whisker Chart custom visual (Jan Pieter Posthuma, version 2.5.3.0, 2017) to depict the relative synonymous codon usage for ramp sequences and the whole gene sequence. Users can interact with most of the visualizations on CUBAP by selecting specific genes, populations, and codons. We also implemented custom visual Smart Filters (OKViz, version 1.2.5.0, 2020) to improve the performance time of gene queries.
Case study: evaluating population-specific biases in pathogenic synonymous variants
The extent to which population-specific codon usage biases affect disease susceptibility or synonymous variant pathogenicity within specific populations remains largely unknown. We mapped the minor allele frequencies (MAF) of 15 synonymous variants labeled ‘Pathogenic’ and supported by multiple submitters in ClinVar (62) across the 1000 Genomes Project to identify synonymous variants with a MAF that significantly differed between populations. Supplementary Table S2 shows the ANOVA P-values of comparing population-specific MAFs of these 15 synonymous pathogenic variants. We found that rs10065172 (313C>T; p.Leu105Leu) in the immunity related GTPase M (IRGM) gene had a significantly different MAF between superpopulations (P-value = 1.06 × 10−10), after correcting for multiple tests using a Bonferroni correction (0.05/15 variants; α = 0.003333). By performing a Tukey test on rs10065172 population-specific MAFs, we found that both Africa and East Asia were significantly different from all other superpopulations (P-values ≤ 6.096 × 10−4) except each other (P-value = 0.764) (see Supplementary Table S3 for complete comparisons). We then used CUBAP to identify population-specific codon usage biases (e.g., codon pairing, codon aversion, ramp sequences, and codon frequency) affecting IRGM, and evaluated the effects of rs10065172 on changing these biases. We performed a literature search on rs10065172 to identify population-specific differences in pathogenicity and compared those reported differences to the population-specific codon usage biases in IRGM reported by CUBAP.
RESULTS
CUBAP features six interactive Power BI visuals for analyzing codon frequency, identical codon pairing, co-tRNA codon pairing, codon aversion, ramp sequences, and nucleotide composition (see Supplementary Figures S4–S9 for example screenshots). Apart from the ramp sequences visual, which includes only genes that have a ramp sequence, each of those visuals allows users to analyze codon usage biases for 40 643 isoforms spanning 17 634 genes in the human genome from all 2504 individuals encompassing 26 subpopulations and five superpopulations included in the 1000 Genomes Project. All raw CSV data files are available for download online by navigating to the ‘Results’ page of https://cubap.byu.edu.
Interactive bar charts for codon frequency, identical codon pairing, and co-tRNA codon pairing allow users to analyze these codon usage biases in detail (see Supplementary Figures S4-S6 for example screenshots). Each user query displays the mean and standard deviation for the user-selected gene or isoform and allows users to easily subset the results or query different genes. When analyzing multiple genes or isoforms, users can view codon usage data either as an average across all isoforms or for each isoform individually. Users can compare codon usage frequencies across all sub- and superpopulations using violin plots by selecting specific codons or amino acids.
Codon aversion plots allow users to analyze codon aversion frequencies in different genes, populations, and across the genome. Codon aversion is the absence of specific codons in a gene sequence. Often, averted codons correlate to rare tRNAs that are expected to slow translation. Supplementary Figure S7 shows an example of a codon aversion query on the website. Results from Power BI allow users to view and subset the total number of alleles in each subpopulation or superpopulation that use specific codons.
In the ramp sequence report, users can compare population frequencies of ramp sequences in specific genes (see Supplementary Figure S8 for an example screenshot). These sequences of slowly-translated codons at the beginning of a gene evenly space ribosomes to increase translation efficiency and can provide insight into gene and population-specific expression levels. Box and whisker plots show the harmonic mean of the relative synonymous codon usages of codons in both the ramp sequence and the entire gene. These data are then plotted across all subpopulations. Additionally, users may compare the average length of the ramp sequence to the length of the selected gene.
Users can view nucleotide frequencies and calculate GC content using the interactive visual for nucleotide composition (see Supplementary Figure S9). Interactive bar charts show the average frequency and standard deviation of each nucleotide for user-selected genes. Users can select a specific nucleotide to view population-specific frequencies on both bar and violin plots. Lines depicting mean GC content for the gene and across all populations are also plotted on the violin plot.
CUBAP is a resource for analyzing population differences in codon usage bias and does not allow users to upload data. However, users can calculate codon usage biases in their own data using the python scripts provided at https://github.com/kauwelab/cubap. These scripts require input data in a FASTA file format, which necessitates including both the reference sequence and the mutated sequence for variant analyses. These data can be compared directly to the 1000 Genomes Project codon usage bias data available at https://cubap.byu.edu/Results.html#data.
A single ANOVA test conducted on the total number of codon pairs in each genome significantly differed between superpopulations (P-value = 2.56 × 10−189). A second ANOVA test found that codon aversion also significantly differed between superpopulations (P-value = 5.03 × 10−31), indicating that these biases may be used as a metric to stratify populations. We performed 10 756 740 additional pairwise t-tests for each protein-encoding codon in each gene in each superpopulation, which resulted in 35.8% of all genes having at least one codon with a statistically significant difference in the average number of codon pairs between at least two superpopulations after correcting for multiple tests using a Bonferroni correction (P-value < 4.64 × 10−9). Aversion to any of the 64 codons, including the stop codons, significantly differed between at least two populations in at least one codon in 9.9% of genes after a Bonferroni correction (P-value < 4.43 × 10−9). The striking population differences in codon pairing and codon aversion between populations allowed us to use alignment-free phylogenetic algorithms to predict the superpopulation of origin for people in the 1000 Genomes Project. We also included the hg19 reference genome, which may have caused additional splits between clusters of individuals originating from the same population. Using codon pairing alone, we successfully identified the East Asian population (n = 504) with 100% accuracy and the African population (n = 661) with 98.8% accuracy. Table 2 shows the percent predictive accuracy for all populations. Codon aversion has similarly high predictive power within these populations, and accurately recovered 95.0% of the East Asian population and 85.9% of the African population (see Table 3 for all population comparisons). Although codon usage biases in American and European populations are difficult to differentiate, when stratified as a single group, 91.6% of individuals clustered in a single cluster using only codon pairing. Gene-specific analyses of the most statistically significant differences between populations are found at https://cubap.byu.edu/population_stratification.html.
Table 2.
Population identification accuracy for codon pairing
| Population | Total individuals | Number of clusters | Percent accuracy |
|---|---|---|---|
| East Asia | 504 | 1 | 100 |
| Africa | 661 | 9 | 98.7897 |
| South Asia | 489 | 56 | 88.7526 |
| Europe | 503 | 91 | 82.1074 |
| America | 347 | 100 | 71.4697 |
Using an alignment-free phylogenomic algorithm that analyzes only codon pairing usages across a genome, individuals in the 1000 Genomes Projects were clustered in a phylogeny (i.e. similar to a pedigree). Cluster accuracy was determined based on the number of clusters (i.e. clades) of individuals belonging the same superpopulation, where a new cluster was formed when an individual from a different population was added to the cluster. The table shows the superpopulation, the number of individuals in that population, the number of clusters identified using the phylogenomic algorithm, and the percent accuracy of the classification.
Table 3.
Population identification accuracy for codon aversion
| Population | Total individuals | Number of clusters | Percent accuracy |
|---|---|---|---|
| East Asia | 504 | 26 | 95.0397 |
| Africa | 661 | 94 | 85.9304 |
| Europe | 503 | 216 | 57.2565 |
| South Asia | 489 | 214 | 56.4417 |
| America | 347 | 165 | 52.7378 |
Using an alignment-free phylogenomic algorithm that analyzes only codon aversion usages across a genome, individuals in the 1000 Genomes Projects were clustered in a phylogeny (i.e. similar to a pedigree). Cluster accuracy was determined based on the number of clusters (i.e. clades) of individuals belonging the same superpopulation, where a new cluster was formed when an individual from a different population was added to the cluster. The table shows the superpopulation, the number of individuals in that population, the number of clusters identified using the phylogenomic algorithm and the percent accuracy of the classification.
Case study: Crohn's disease and rs10065172
Several genome-wide association studies suggest rs10065172 in the IRGM gene is strongly associated with Crohn's disease in European (odds ratio (OR) = 1.284; 95% confidence interval (CI) = 1.055–1.564) and Korean individuals (OR = 1.42; 95% CI = 1.12–1.80), but not in Japanese (OR = 0.94; 95% CI = 0.81–1.11) nor African American individuals (OR = 0.91 95% CI = 0.67–1.33) (63–69). When accounting for population stratification, the association between rs10065172 and Crohn's disease increases in Europeans, but not Asians (69). Notably, Korean individuals are not included in the 1000 Genomes Project and thus not included in our analyses. While other variants are in near perfect linkage disequilibrium with rs10065172, rs10065172 specifically increases Crohn's disease susceptibility in individuals of European ancestry (70).
Both African and East Asian populations exhibit a significantly higher MAF of rs10065172 (MAF = 0.502 and MAF = 0.434 respectively), while the European population has the lowest (MAF = 0.105). Despite not altering the amino acid sequence, rs10065172 removes a CTG pair, which may alter the efficiency of translation and mRNA secondary structure. Using CUBAP, we found that CTG pairing in IRGM varies significantly between populations (see Figure 1). When considering only individuals without synonymous variant rs10065172, East Asian and African populations having fewer instances of CTG codon pairing (P-value = 2.52 × 10−41) than all other populations.
Figure 1.

Identical Codon Pairing Screenshot of IRGM. A screenshot of the Identical Codon Pairing visual with the IRGM gene selected. The lower graphs show population differences in the frequency of CTG pairing, which is decreased by rs10065172.
DISCUSSION
CUBAP enables users to easily visualize cross-population analyses of codon frequencies, codon aversion, identical codon pairing, co-tRNA codon pairing, ramp sequence, and nucleotide composition, including GC content for 17 634 genes and 40 643 isoforms. CUBAP can be used to identify gene-specific and population-specific biases at a codon-by-codon level to help users better understand the extent to which synonymous codon usage biases affect a gene or region of interest. We anticipate that this resource will be used to inspect candidate genes identified in genome-wide association studies, determine the extent to which synonymous codon usage biases are fixed in a population, and predict the effects that changing a codon usage bias will have on different populations. Additionally, our population identification analyses show that biases in codon pairing alone are enough to determine the population of origin in East Asian and African populations, indicating that codon usage might be effectively used in genealogy. CUBAP’s large dataset and interactive, dynamic visuals provide users with the most comprehensive view of codon usage biases across human populations to date.
Since codon usage biases play an integral role in regulating translation rates (3,4,11,25,26), gene expression (7), mRNA and protein secondary structure (12), and can be used in phylogenetics (59,71,72), we anticipate that this resource will be widely used for cross-population analyses and gene-specific inquiries. For instance, traditional genetic analyses of complex diseases rarely evaluate the effects of synonymous codon usage biases because they are generally thought to not contribute significantly to disease. However, recent studies have shown that synonymous codon usage alone significantly alters protein levels and can have a causal effect on Alzheimer's disease (44), cancer (45,46) and multiple sclerosis (45). CUBAP enables researchers to query synonymous codon usage biases in genes identified in case-control studies to determine the extent to which codon usage biases already exist within various populations. Since the values are pre-computed and users can dynamically subtype the data, the web portal is designed to facilitate creative and personalized analyses of specific genes and populations of interest.
We also propose that population-specific codon usage biases can be used as an additional filter in genome-wide association studies. Adjusting for population stratification in genome-wide association studies is difficult (73,74) because variants that are identified as highly associated with disease risk within one population may not be as highly associated in another population (75). We propose that one mechanism that might affect disease association within different populations is the underlying codon usage biases distinct to those populations. For instance, a variant that introduces a codon that was previously averted in one population may not affect the same codon usage dynamic in a different population. Therefore, additional studies might determine if candidate synonymous variants that alter codon dynamics differently between populations are less likely to be generally associated with disease across all populations. Conversely, future studies may find that changing codon usage biases that are uniform across all populations are more likely to have similar pathogenic effects across all populations.
Understanding population-specific codon usage biases may also impact the results of clinical trials of new drugs because drugs can induce different responses depending on the recipient's ethnicity (76–78). Since codon usage dynamics directly affect protein expression levels, population-specific differences in these biases may directly affect a drug's response or dosage requirement within a specific population. Therefore, CUBAP provides a resource for researchers designing clinical trials to visually determine the extent to which codon usage biases might lead to confounding effects between populations.
Case study: Crohn's disease and rs10065172
Despite being in linkage disequilibrium with several other variants (64–66,70,79), in vitro experiments show that rs10065172 alone promotes Crohn's disease development (70). Altered IRGM mRNA structure caused by removing a CTG pair in IRGM could significantly affect gene expression because rs10065172 occurs in the seed region of the IRGM mRNA, a short sequence vital for microRNA (miRNA) binding and subsequent gene regulation. In vitro data reveals that the presence of rs10065172 in this seed region prevents the binding of MiR-196 microRNAs to IRGM mRNA. Without MiR-196 regulation, IRGM is overexpressed and alters xenophagy of gut bacteria, thereby increasing the likelihood of developing Crohn's disease (70).
While rs10065172 increases Crohn's disease susceptibility in Europeans, it is not strongly associated with disease in Africans and East Asians. Additionally, despite having a significantly higher MAF in individuals of African and East Asian descent, Crohn's disease prevalence is relatively low in these populations, especially in Africa, compared to North America and Europe (80), suggesting other population-specific biases may limit the deleterious effects of rs10065172. African and East Asian individuals without rs10065172 have a significantly lower frequency of CTG pairing than individuals without rs10065172 in other populations (P-value = 2.52 × 10−41). Because the translation efficiency or optimality of codons correlates with their frequency, the loss of a CTG pair caused by rs10065172 may have a more pronounced effect on translation in European populations that have significantly higher average CTG pairing in IRGM. Conversely, since baseline CTG pairing in IRGM is less common in African and East Asian populations, rs10065172 may have a more muted effect on translational dynamics, which would explain its lower association with Crohn's disease in those populations. CUBAP provides a platform for analyzing these population-specific codon preferences that provide contextual support to explain a likely mechanism by which rs10065172 promotes Crohn's disease development in certain populations. The same process can be applied to any variant by using CUBAP to determine the extent to which population-specific codon usage biases affect a specific gene, which may contribute to differential disease association between populations.
Although 35.8% of genes have at least one codon that exhibits a significantly different frequency of codon pairing between populations, each gene averages only 3.044 codons with a significantly different number of pairings between at least two populations. Therefore, only 1.75% of codons have significantly different codon pairings between populations. Similar to the global average, IRGM has only three codons that significantly differ between populations: CTG, GAG and ACA. Additionally, the likelihood of correctly choosing at random two out of five superpopulations that were previously implicated as having a decreased risk for Crohn's disease would be 10%. Therefore, the probability of correctly identifying a specific codon pairing as affecting only two populations is represented by 0.0175 × 0.1 = 1.75 × 10−3 = 0.175%. Given that the probability of randomly identifying CTG pairing in IRGM as affecting Crohn's disease in only African and East Asian populations is very low (P-value = 1.75 × 10−3), our case study illustrates the utility of CUBAP in adding additional support to previous studies, as well as narrowing the search space for potential impacts of population-specific codon usage biases on disease. Additionally, CUBAP provides a starting point for researchers who may not know the effects of a variant a priori to identify codons that have significant population-specific differences in various codon usage biases that are predicted to affect gene expression.
CONCLUSION
Analytical codon usage bias tools have played an important role in understanding the dynamics and effects of codon usage bias in genes and species. While many codon usage bias calculators and databases exist, CUBAP offers the most comprehensive and interactive analysis of codon usage biases across human populations to date. CUBAP is a powerful tool that allows researchers to easily compare codon frequency, codon aversion, identical codon pairing, co-tRNA codon pairing, ramp sequences, and nucleotide composition across 26 populations and five superpopulations. We anticipate that this tool will allow researchers to better visualize codon usage biases within specific genes, identify population differences in codon usages, and better predict potential implications of synonymous variants associated with genetic diseases.
Supplementary Material
ACKNOWLEDGEMENTS
We appreciate the contributions of Brigham Young University for supporting this research. We also recognize the Office of Research Computing and the Life Sciences Office of Information Technology at Brigham Young University for their technical assistance.
Contributor Information
Matthew W Hodgman, Department of Biology, Brigham Young University, Provo, UT 84602, USA.
Justin B Miller, Department of Biology, Brigham Young University, Provo, UT 84602, USA.
Taylor E Meurs, Department of Biology, Brigham Young University, Provo, UT 84602, USA.
John S K Kauwe, Department of Biology, Brigham Young University, Provo, UT 84602, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute on Aging [RF1 AG054052]. Funding for open access charge: National Institute on Aging [RF1 AG054052]; Brigham Young University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Rozov A., Demeshkina N., Khusainov I., Westhof E., Yusupov M., Yusupova G.. Novel base-pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat. Commun. 2016; 7:10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crick F.H. Codon–anticodon pairing: the wobble hypothesis. J. Mol. Biol. 1966; 19:548–555. [DOI] [PubMed] [Google Scholar]
- 3. Shao Z.Q., Zhang Y.M., Feng X.Y., Wang B., Chen J.Q.. Synonymous codon ordering: a subtle but prevalent strategy of bacteria to improve translational efficiency. PLoS One. 2012; 7:e33547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Irwin B., Heck J.D., Hatfield G.W.. Codon pair utilization biases influence translational elongation step times. J. Biol. Chem. 1995; 270:22801–22806. [DOI] [PubMed] [Google Scholar]
- 5. Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y.. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010; 141:344–354. [DOI] [PubMed] [Google Scholar]
- 6. Quax T.E., Claassens N.J., Soll D., van der Oost J.. Codon bias as a means to Fine-Tune gene expression. Mol. Cell. 2015; 59:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gutman G.A., Hatfield G.W.. Nonrandom utilization of codon pairs in Escherichia coli. Proc. Natl Acad. Sci. USA. 1989; 86:3699–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985; 2:13–34. [DOI] [PubMed] [Google Scholar]
- 9. Duret L. tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet. 2000; 16:287–289. [DOI] [PubMed] [Google Scholar]
- 10. Zhou M., Guo J., Cha J., Chae M., Chen S., Barral J.M., Sachs M.S., Liu Y.. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature. 2013; 495:111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buchan J.R., Aucott L.S., Stansfield I.. tRNA properties help shape codon pair preferences in open reading frames. Nucleic Acids Res. 2006; 34:1015–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Purvis I.J., Bettany A.J., Santiago T.C., Coggins J.R., Duncan K., Eason R., Brown A.J.. The efficiency of folding of some proteins is increased by controlled rates of translation in vivo. A hypothesis. J. Mol. Biol. 1987; 193:413–417. [DOI] [PubMed] [Google Scholar]
- 13. Goodman D.B., Church G.M., Kosuri S.. Causes and effects of N-terminal codon bias in bacterial genes. Science. 2013; 342:475–479. [DOI] [PubMed] [Google Scholar]
- 14. Chamary J.V., Hurst L.D.. Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol. 2005; 6:R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mukhopadhyay P., Basak S., Ghosh T.C.. Synonymous codon usage in different protein secondary structural classes of human genes: implication for increased non-randomness of GC(3) rich genes towards protein stability. J. Biosci. 2007; 32:947–963. [DOI] [PubMed] [Google Scholar]
- 16. Gu W., Zhou T., Ma J., Sun X., Lu Z.. The relationship between synonymous codon usage and protein structure in Escherichia coli and Homo sapiens. Biosystems. 2004; 73:89–97. [DOI] [PubMed] [Google Scholar]
- 17. Hia F., Yang S.F., Shichino Y., Yoshinaga M., Murakawa Y., Vandenbon A., Fukao A., Fujiwara T., Landthaler M., Natsume T. et al.. Codon bias confers stability to human mRNAs. EMBO Rep. 2019; 20:e48220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berg O.G., Kurland C.G.. Growth rate-optimised tRNA abundance and codon usage. J. Mol. Biol. 1997; 270:544–550. [DOI] [PubMed] [Google Scholar]
- 19. Dong H., Nilsson L., Kurland C.G.. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996; 260:649–663. [DOI] [PubMed] [Google Scholar]
- 20. Hanson G., Coller J.. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018; 19:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dana A., Tuller T.. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. 2014; 42:9171–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gardin J., Yeasmin R., Yurovsky A., Cai Y., Skiena S., Futcher B.. Measurement of average decoding rates of the 61 sense codons in vivo. Elife. 2014; 3:e03735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Plotkin J.B., Kudla G.. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 2011; 12:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Navon S., Pilpel Y.. The role of codon selection in regulation of translation efficiency deduced from synthetic libraries. Genome Biol. 2011; 12:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang F.P., Li H.. Codon-pair usage and genome evolution. Gene. 2009; 433:8–15. [DOI] [PubMed] [Google Scholar]
- 26. Tuller T., Zur H.. Multiple roles of the coding sequence 5′ end in gene expression regulation. Nucleic Acids Res. 2015; 43:13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miller J.B., Brase L.R., Ridge P.G.. ExtRamp: a novel algorithm for extracting the ramp sequence based on the tRNA adaptation index or relative codon adaptiveness. Nucleic Acids Res. 2019; 47:1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Villada J.C., Duran M.F., Lee P.K.H.. Interplay between position-dependent codon usage bias and hydrogen bonding at the 5' end of ORFeomes. mSystems. 2020; 5:doi:10.1128/mSystems.00613-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sauna Z.E., Kimchi-Sarfaty C.. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011; 12:683–691. [DOI] [PubMed] [Google Scholar]
- 30. Richard P., Gaudon K., Fournier E., Jackson C., Bauché S., Haddad H., Koenig J., Echenne B., Hantaï D., Eymard B.. A synonymous CHRNE mutation responsible for an aberrant splicing leading to congenital myasthenic syndrome. Neuromuscul. Disord. 2007; 17:409–414. [DOI] [PubMed] [Google Scholar]
- 31. Del Gatto F., Breathnach R.. A Crouzon syndrome synonymous mutation activates a 5′ splice site within the IIIc exon of the FGFR2 gene. Genomics. 1995; 27:558–559. [DOI] [PubMed] [Google Scholar]
- 32. Vidal C., Cachia A., Xuereb-Anastasi A.. Effects of a synonymous variant in exon 9 of the CD44 gene on pre-mRNA splicing in a family with osteoporosis. Bone. 2009; 45:736–742. [DOI] [PubMed] [Google Scholar]
- 33. Macaya D., Katsanis S.H., Hefferon T.W., Audlin S., Mendelsohn N.J., Roggenbuck J., Cutting G.R.. A synonymous mutation in TCOF1 causes Treacher Collins syndrome due to mis-splicing of a constitutive exon. Am. J. Med. Genet. A. 2009; 149A:1624–1627. [DOI] [PubMed] [Google Scholar]
- 34. Ho P.Y., Huang M.Z., Fwu V.T., Lin S.C., Hsiao K.J., Su T.S.. Simultaneous assessment of the effects of exonic mutations on RNA splicing and protein functions. Biochem. Biophys. Res. Commun. 2008; 373:515–520. [DOI] [PubMed] [Google Scholar]
- 35. Alenius M., Wadelius M., Dahl M.L., Hartvig P., Lindström L., Hammarlund-Udenaes M.. Gene polymorphism influencing treatment response in psychotic patients in a naturalistic setting. J. Psychiatr. Res. 2008; 42:884–893. [DOI] [PubMed] [Google Scholar]
- 36. Fung K.L., Gottesman M.M.. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim. Biophys. Acta. 2009; 1794:860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Herrlinger K.R., Koc H., Winter S., Teml A., Stange E.F., Fellermann K., Fritz P., Schwab M., Schaeffeler E.. ABCB1 single-nucleotide polymorphisms determine tacrolimus response in patients with ulcerative colitis. Clin. Pharmacol. Ther. 2011; 89:422–428. [DOI] [PubMed] [Google Scholar]
- 38. Komar A.A. Silent SNPs: impact on gene function and phenotype. Pharmacogenomics. 2007; 8:1075–1080. [DOI] [PubMed] [Google Scholar]
- 39. Kwon W.S., Rha S.Y., Jeung H.C., Ahn J.B., Jung J.J., Noh S.H., Chung H.C.. G-T haplotype (2677G>T/A and 3435C>T) of ABCB1 gene polymorphisms is associated with ethnic differences to paclitaxel sensitivity in cancer cells with different gene expression pattern. Cancer Lett. 2009; 277:155–163. [DOI] [PubMed] [Google Scholar]
- 40. Ni L.N., Li J.Y., Miao K.R., Qiao C., Zhang S.J., Qiu H.R., Qian S.X.. Multidrug resistance gene (MDR1) polymorphisms correlate with imatinib response in chronic myeloid leukemia. Med. Oncol. 2011; 28:265–269. [DOI] [PubMed] [Google Scholar]
- 41. Tsai C.J., Sauna Z.E., Kimchi-Sarfaty C., Ambudkar S.V., Gottesman M.M., Nussinov R.. Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J. Mol. Biol. 2008; 383:281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van der Veldt A.A., Eechoute K., Gelderblom H., Gietema J., Guchelaar H.J., van Erp N.P., van den Eertwegh A.J., Haanen J.B., Mathijssen R.H., Wessels J.A.. Genetic polymorphisms associated with a prolonged progression-free survival in patients with metastatic renal cell cancer treated with sunitinib. Clin. Cancer Res. 2011; 17:620–629. [DOI] [PubMed] [Google Scholar]
- 43. Ramser J., Ahearn M.E., Lenski C., Yariz K.O., Hellebrand H., von Rhein M., Clark R.D., Schmutzler R.K., Lichtner P., Hoffman E.P. et al.. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am. J. Hum. Genet. 2008; 82:188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miller J.E., Shivakumar M.K., Risacher S.L., Saykin A.J., Lee S., Nho K., Kim D.. Codon bias among synonymous rare variants is associated with Alzheimer's disease imaging biomarker. Pac. Symp. Biocomput. 2018; 23:365–376. [PMC free article] [PubMed] [Google Scholar]
- 45. Fornasiero E.F., Rizzoli S.O.. Pathological changes are associated with shifts in the employment of synonymous codons at the transcriptome level. BMC Genomics. 2019; 20:566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lampson B.L., Pershing N.L., Prinz J.A., Lacsina J.R., Marzluff W.F., Nicchitta C.V., MacAlpine D.M., Counter C.M.. Rare codons regulate KRas oncogenesis. Curr. Biol. 2013; 23:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Casillas S., Mulet R., Villegas-Mirón P., Hervas S., Sanz E., Velasco D., Bertranpetit J., Laayouni H., Barbadilla A.. PopHuman: the human population genomics browser. Nucleic Acids Res. 2018; 46:D1003–D1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kinney N., Titus-Glover K., Wren J.D., Varghese R.T., Michalak P., Liao H., Anandakrishnan R., Pulenthiran A., Kang L., Garner H.R.. CAGm: a repository of germline microsatellite variations in the 1000 genomes project. Nucleic Acids Res. 2019; 47:D39–D45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Puigbo P., Bravo I.G., Garcia-Vallve S.. CAIcal: A combined set of tools to assess codon usage adaptation. Biol. Direct. 2008; 3:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peden J. Analysis of codon usage. 1999; Ph.D. Thesis. University of Nottingham.
- 51. Sharp P.M., Li W.H.. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987; 15:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sharp P.M., Tuohy T.M., Mosurski K.R.. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986; 14:5125–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990; 87:23–29. [DOI] [PubMed] [Google Scholar]
- 54. Galtier N., Piganeau G., Mouchiroud D., Duret L.. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics. 2001; 159:907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Qi W.H., Yan C.C., Li W.J., Jiang X.M., Li G.Z., Zhang X.Y., Hu T.Z., Li J., Yue B.S.. Distinct patterns of simple sequence repeats and GC distribution in intragenic and intergenic regions of primate genomes. Aging (Albany NY). 2016; 8:2635–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou H.Q., Ning L.W., Zhang H.X., Guo F.B.. Analysis of the relationship between genomic GC Content and patterns of base usage, codon usage and amino acid usage in prokaryotes: similar GC content adopts similar compositional frequencies regardless of the phylogenetic lineages. PLoS One. 2014; 9:e107319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Athey J., Alexaki A., Osipova E., Rostovtsev A., Santana-Quintero L.V., Katneni U., Simonyan V., Kimchi-Sarfaty C.. A new and updated resource for codon usage tables. BMC Bioinformatics. 2017; 18:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hilterbrand A., Saelens J., Putonti C.. CBDB: the codon bias database. BMC Bioinformatics. 2012; 13:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Miller J.B., Mckinnon L.M., Whiting M.F., Kauwe J.S.K., Ridge P.G.. Codon pairs are phylogenetically conserved: A comprehensive analysis of codon pairing conservation across the Tree of Life. PLoS One. 2020; 15:e0232260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miller J.B., Hippen A.A., Belyeu J.R., Whiting M.F., Ridge P.G.. Missing something? Codon aversion as a new character system in phylogenetics. Cladistics. 2017; 33:545–556. [DOI] [PubMed] [Google Scholar]
- 61. Martens A.T., Taylor J., Hilser V.J.. Ribosome A and P sites revealed by length analysis of ribosome profiling data. Nucleic Acids Res. 2015; 43:3680–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R.. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McCarroll S.A., Huett A., Kuballa P., Chilewski S.D., Landry A., Goyette P., Zody M.C., Hall J.L., Brant S.R., Cho J.H. et al.. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat. Genet. 2008; 40:1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Glas J., Seiderer J., Bues S., Stallhofer J., Fries C., Olszak T., Tsekeri E., Wetzke M., Beigel F., Steib C. et al.. IRGM variants and susceptibility to inflammatory bowel disease in the German population. PLoS One. 2013; 8:e54338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Parkes M., Barrett J.C., Prescott N.J., Tremelling M., Anderson C.A., Fisher S.A., Roberts R.G., Nimmo E.R., Cummings F.R., Soars D. et al.. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat. Genet. 2007; 39:830–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prescott N.J., Dominy K.M., Kubo M., Lewis C.M., Fisher S.A., Redon R., Huang N., Stranger B.E., Blaszczyk K., Hudspith B. et al.. Independent and population-specific association of risk variants at the IRGM locus with Crohn's disease. Hum. Mol. Genet. 2010; 19:1828–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moon C.M., Shin D.J., Kim S.W., Son N.H., Park A., Park B., Jung E.S., Kim E.S., Hong S.P., Kim T.I. et al.. Associations between genetic variants in the IRGM gene and inflammatory bowel diseases in the Korean population. Inflamm. Bowel Dis. 2013; 19:106–114. [DOI] [PubMed] [Google Scholar]
- 68. Wang M.H., Okazaki T., Kugathasan S., Cho J.H., Isaacs K.L., Lewis J.D., Smoot D.T., Valentine J.F., Kader H.A., Ford J.G. et al.. Contribution of higher risk genes and European admixture to Crohn's disease in African Americans. Inflamm. Bowel Dis. 2012; 18:2277–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lu X.C., Tao Y., Wu C., Zhao P.L., Li K., Zheng J.Y., Li L.X.. Association between variants of the autophagy related gene–IRGM and susceptibility to Crohn's disease and ulcerative colitis: a meta-analysis. PLoS One. 2013; 8:e80602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brest P., Lapaquette P., Souidi M., Lebrigand K., Cesaro A., Vouret-Craviari V., Mari B., Barbry P., Mosnier J.F., Hébuterne X. et al.. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat. Genet. 2011; 43:242–245. [DOI] [PubMed] [Google Scholar]
- 71. Miller J., McKinnon L., Whiting M., Ridge P.. Codon use and aversion is largely phylogenetically conserved across the tree of life. Mol. Phylogenet. Evol. 2020; 144:106697. [DOI] [PubMed] [Google Scholar]
- 72. Miller J.B., McKinnon M.M., Whiting M.F., Ridge P.G.. CAM: an alignment-free method to recover phylogenies using codon aversion motifs. PeerJ. 2019; 7:e6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Peterson R.E., Kuchenbaecker K., Walters R.K., Chen C.Y., Popejoy A.B., Periyasamy S., Lam M., Iyegbe C., Strawbridge R.J., Brick L. et al.. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell. 2019; 179:589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim M.S., Patel K.P., Teng A.K., Berens A.J., Lachance J.. Genetic disease risks can be misestimated across global populations. Genome Biol. 2018; 19:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rosenberg N.A., Huang L., Jewett E.M., Szpiech Z.A., Jankovic I., Boehnke M.. Genome-wide association studies in diverse populations. Nat. Rev. Genet. 2010; 11:356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Burroughs V.J., Maxey R.W., Levy R.A.. Racial and ethnic differences in response to medicines: towards individualized pharmaceutical treatment. J. Natl. Med. Assoc. 2002; 94:1–26. [PMC free article] [PubMed] [Google Scholar]
- 77. Muñoz C., Hilgenberg C.. Ethnopharmacology: understanding how ethnicity can affect drug response is essential to providing culturally competent care. Holist. Nurs. Pract. 2006; 20:227–234. [DOI] [PubMed] [Google Scholar]
- 78. Yasuda S.U., Zhang L., Huang S.M.. The role of ethnicity in variability in response to drugs: focus on clinical pharmacology studies. Clin. Pharmacol. Ther. 2008; 84:417–423. [DOI] [PubMed] [Google Scholar]
- 79. Ajayi T.A., Innes C.L., Grimm S.A., Rai P., Finethy R., Coers J., Wang X., Bell D.A., McGrath J.A., Schurman S.H. et al.. Crohn's disease IRGM risk alleles are associated with altered gene expression in human tissues. Am. J. Physiol. Gastrointest. Liver Physiol. 2019; 316:G95–G105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Collaborators G.I.B.D. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020; 5:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All analyses were performed on 2504 human samples from the 1000 Genomes Project database. This dataset was selected because of its credibility, accuracy, and large number of samples spanning many different populations. Individuals are labeled as belonging to one of 26 different subpopulations spanning five superpopulations (see Table 1). After excluding genes that had annotated translational exceptions, partial genes, or potential errors, we analyzed 17 634 complete gene sequences and 40 643 isoforms using human reference assembly hg19. All scripts used to analyze these data are publicly available at https://github.com/kauwelab/cubap. CUBAP’s documentation can be viewed at https://cubap.readthedocs.io/.
Table 1.
Population data used from 1000 Genomes Project
| Superpopulation | Subpopulation | Description | Number of Samples |
|---|---|---|---|
| Africa | ASW | African Ancestry in Southwest US | 61 |
| ACB | African Caribbean in Barbados | 96 | |
| ESN | Esan in Nigeria | 99 | |
| GWD | Gambian in Western Division, The Gambia | 113 | |
| LWK | Luhya in Webuye, Kenya | 99 | |
| MSL | Mende in Sierra Leone | 85 | |
| YRI | Yoruba in Ibadan, Nigeria | 108 | |
| Total: | 661 | ||
| America | CLM | Colombian in Medellin, Colombia | 94 |
| MXL | Mexican Ancestry in Los Angeles, California | 64 | |
| PEL | Peruvian in Lima, Peru | 85 | |
| PUR | Puerto Rican in Puerto Rico | 104 | |
| Total: | 347 | ||
| East Asia | CDX | Chinese Dai in Xishuangbanna, China | 93 |
| CHB | Han Chinese in Bejing, China | 103 | |
| JPT | Japanese in Tokyo, Japan | 104 | |
| KHV | Kinh in Ho Chi Minh City, Vietnam | 99 | |
| CHS | Southern Han Chinese, China | 105 | |
| Total: | 504 | ||
| Europe | GBR | British in England and Scotland | 91 |
| FIN | Finnish in Finland | 99 | |
| IBS | Iberian populations in Spain | 107 | |
| TSI | Toscani in Italy | 107 | |
| CEU | Utah residents with Northern and Western European ancestry | 99 | |
| Total: | 503 | ||
| South Asia | BEB | Bengali in Bangladesh | 86 |
| GIH | Gujarati Indian in Houston, TX | 103 | |
| ITU | Indian Telugu in the UK | 102 | |
| PJL | Punjabi in Lahore, Pakistan | 96 | |
| STU | Sri Lankan Tamil in the UK | 102 | |
| Total: | 489 | ||
| Grand Total: | 2,504 |
The distribution of samples and populations within the 1000 Genomes Project.