


Abstract
The relationship between stochastic transcriptional bursts and dynamic 3D chromatin states is not well understood. Using an innovated, ultra-sensitive technique, we address here enigmatic features underlying the communications between MYC and its enhancers in relation to the transcriptional process. MYC thus interacts with its flanking enhancers in a mutually exclusive manner documenting that enhancer hubs impinging on MYC detected in large cell populations likely do not exist in single cells. Dynamic encounters with pathologically activated enhancers responsive to a range of environmental cues, involved <10% of active MYC alleles at any given time in colon cancer cells. Being the most central node of the chromatin network, MYC itself likely drives its communications with flanking enhancers, rather than vice versa. We submit that these features underlie an acquired ability of MYC to become dynamically activated in response to a diverse range of environmental cues encountered by the cell during the neoplastic process.
INTRODUCTION
Single cell studies have shown that transcriptional activation occurs in bursts in both prokaryotes and eukaryotes (1). The resulting variability in expression levels contributes to transcriptional ‘noise’, and likely depends on the probability of key limiting events, such as the accessibility to transcription factors and their on-off rates at cis-regulatory elements (2). Cis-regulatory elements, such as enhancers, are often positioned distal to the promoters they regulate. This provides another level of transcriptional control exerted by 3D chromatin conformation to influence the probability of communication between enhancers and promoters (3). The arrangement of large domains with enhancing activities, the so-called super-enhancers, has been proposed to reduce transcriptional noise and buffer against environmental perturbations in order to robustly maintain the differentiated phenotype (4,5). Super-enhancers can also be activated to drive unscheduled expression of oncogenes, such as MYC, and thus the neoplastic process (4,6–8).
When analyzing the interactomes impinging on enhancers in general and super-enhancers in particular, a common theme is that these interact with each other extensively and that such enhancer hubs collaborate to boost the transcriptional process (9–18). However, experiments attempting to resolve the stochastic character of such features have this far suffered from not being able to combine high sensitivity with high reproducibility. Although Hi-C protocols (19) have been developed to examine chromatin structure at the single cell level or in small cell populations (20,21), they are not sufficiently quantitative to robustly address specific enhancer-gene communications. We thus remain largely ignorant of the dynamics of their interactions in small cell populations to complicate any comparison to the transcriptional process.
To address these issues, we optimized the previously published Nodewalk technique (7,8) to increase sensitivity without losing the ability to quantitively measure chromatin interactions. We applied this technique to a colon cancer cell line, HCT116, with the vast majority of MYC alleles being transcriptionally active any given time (7,8). By analyzing chromatin networks with a sensitivity corresponding to 7 cells, we document that MYC stochastically screens for its distal enhancers in a mutually exclusive manner and discuss how this arrangement might benefit the fitness of cancer cells.
MATERIALS AND METHODS
Cell culture
HCT116 cells—a kind gift from Dr B. Vogelstein—human colon epithelial cells (HCEC; ScienCell, 2950), and Drosophila S2 cells (Thermo Fisher Scientific, 21720024) were cultured as previously described (7,8). Cells were routinely screened for mycoplasma contamination using EZ-PCR Mycoplasma Test Kit (Biological Industries, Cromwell, CT. 20-700-20).
Adaptation of the Nodewalk protocol for small input material
See protocol for the analyses of large input samples using S2 cells as an external control (7,8). For the adaptation of the Nodewalk protocol for small input material we used the following strategy: Following formaldehyde fixation, HCT116 cells were counted and diluted in nuclear isolation buffer to make 600 cells/μl (corresponding to ∼3 ng of genomic DNA/μl). Aliquots (0.5 μl) of the resulting cell suspension was mixed and incubated on ice for 10 min. The cell suspension was directly diluted 10 times with ×1.2 Buffer 2 (7,8). Next, the samples were digested with Hind III and ligated as described (7,8), albeit in a smaller reaction volume (20 μl for Hind III digestion; 200 μl for 3C ligation). Following reversal of the crosslink, the 3C-DNA was purified using the ChIP DNA Clean and Concentrator kit (Zymo Research, D5205). The elution buffer was pre-heated at 65°C to increase the recovery of large DNA fragments.
The digestion efficiency of crosslinked chromatin (22) was assessed by designing F1/R1 and F2/R2 PCR primers flanking the Hind III sites at the 5' and 3'ends of the MYC promoter and gene body. The amount of total DNA was quantified with the primers F3/R3 used to produce a PCR fragment lacking an internal Hind III site. The linear range of amplification was determined by serial dilution of sonicated genomic DNA. The digestion efficiency was calculated as (1- (PCRF1+R1/F2+R2/PCRF3+R3)) × 100 (%). Primer sequences and PCR condition are listed in Supplementary Table S1.
The Nodewalk libraries were generated by tagmentation, as described earlier (7,8). Generally, the size distribution of the tagmented 3C DNA ranged from 200 to 300 bp (Supplementary Figure S1A). For the small inout samples we used the Nextera XT DNA sample prep kit (Illumina, San Diego, CA. FC-121–1031, FC-131–1024). The amount of the input was validated by qPCR (using primers F3 and R3, see Supplementary Table S1). Each library was sequenced on Illumina Miseq (Illumina) using Miseq reagent cartridge v2 (Illumina) that generated 140–150 bp paired-end reads.
Sequence mapping, filtering, de-duplication (Supplementary Figure S1B), identification of statistically significant interactions, and enrichment mapping of cis regulatory elements was done as previously described (7,8). The mapping efficiency was determined by the number of uniquely mapped fragments divided by the total number of fragments. The duplication rate was defined as the number of uniquely mapped fragments with duplication versus the sum of uniquely mapped fragments with and without duplication. The read depth was approximated by the number of uniquely mapped fragments without duplication versus the number of HindIII fragments.
Calculation of the recovery of ligation events and quantification of the input cells
The recovery of the ligation events was defined as the amount of all observed detected ligation events compared to the expected amount of all possible ligation events (LE) using the number of the de-duplicated reads. For this calculation, we included the LE of non-significant interactions. The sum of LE was therefore divided by the number of the possible LE estimated as follows: Calculating with 5 pg genomic DNA / cell, we estimated the number of cells in the input sample based on the quantity of 3C DNA measured by either Nanodrop (for the input of 10 000 cells) or qPCR (using primers F3 and R3, see Supplementary Table S1). As HCT116 cells harbor 3 MYC alleles, the estimated number of alleles was therefore multiplied with 3 for HCT116 and 2 for HCEC cells. These values indicate the potential number of ligation events at one end of the bait fragment, including self-ligation as well as interactions with neighboring and distant fragments.
Mapping the interactors with increased window size
Ligation events of nine Nodewalk libraries derived from 300 cells or 10 libraries derived from 0.88 ng of 3C DNA were summed up at each window size (either 25, 50 or 100 kbps). The windows were shifted each 10 kbps and visualized on WashU Epigenome Browser (23) (summary method: Average).
Comparison between expected and observed number of enhancers within the MYC interactome
A permutation-based approach was adopted to test whether the observed number of enhancers impinging on MYC in the small input protocols, namely Nodewalk with 0.88 ng and with 34.8 pg inputs, significantly differs from the corresponding expected numbers if the interactome was formed stochastically. Specifically, a null distribution was formed by randomly selecting interacting regions 10 000 times from the MYC network with 10k cells input keeping the numbers of regions as of the corresponding observed low input interactomes. The method assigned the sampled interactors with probability weights which were based on their observed number of ligation events (LE) in the higher cell count network. In each cycle, the total number of interacting regions that overlapped with H3K27ac peaks was counted. The approach was repeated separately for each analysis.
Network visualization
All the network figures were generated by Gephi 0.9.1 (24) (layout: Force Atlas 2).
Bootstrapping network generation
From the reads computed after Nodewalk data processing we generated a set of networks that represent the family of possible networks that can be originated from the sequencing data by bootstrapping. To this end, we first selected a number of reads to be included (n = 500, 1500, 5000) and randomly selected with replacement a number of reads equal to n from the pool of reads. The number of bootstrapped networks computed was 1000. We then calculated 95% CI (Confidence Interval) of the median of influence index and entropy on these 1000 weighted networks (weights are number of reads) using bootstrap technique.
3D DNA FISH analyses
DNA FISH probes covering the MYC promoter and gene body (chr8:128 746 000–128 756 177), the oncogenic super-enhancer (positioned at chr8:128 216 526–128 225 855), and an in-between enhancer (EnhD, positioned at chr8:128413009–128414109), were prepared and labeled with 496-dUTP (Enzo, 42831), Cy3-dCTP (PA53021, GE Healthcare) and Cy5-dCTP (PA55021, GE Healthcare), respectively, as previously described (8). The genomic regions surrounding the OSE and MYC loci were visualized using the bacterial artificial chromosome (BAC) clone CTD-3066D1. DNA FISH analyses were performed, as previously described (8).
Grid wide-field microscopy
Cell imaging and the generation of optical sections in 3D were carried out on a Leica DMi8 microscope using the Thunder Imaging System (Leica Microsystems). Stacks were taken at 0.3 μm intervals in the z-axis. Pictures were analyzed with the use of Leica Application Suite X (LasX) software. The Epi-Fluorescence microscopic images were generated using a 63× objective with a 1.4 numerical aperture and camera DFC9000. Due to the limitations in the resolution of the fluorophores (with CY3 at 239.6 nm), the distance data were stratified using 240 nm as the first cut-off. In total, 940 alleles of the MYC, EnhD and OSE regions were analyzed in distance measurements in two independent experiments.
Dynamic importance index
We used Eigenvalue centrality that quantifies the role of each node in propagating signal through the whole network to estimate the node influence metric using igraph package (25) of R.
RESULTS
MYC as a driver of chromatin networks
We have earlier identified an enhancer hub organized around an active MYC and involving primarily local sequences (7,8) distributed in the neighboring topologically associated domains (TADs), which define regions with preferred physical interactions (26). To identify the most important nodes in the ensemble network, we first constructed virtual chromatin networks in both cancer (HCT116) and primary cultures of human colon epithelial cells (HCEC) and determined the nodes with the highest connectivity (regions 1–9, Figure 1A, B). To compensate for any bias in network comparisons arising from the fact that MYC is triploid in HCT116 (27) and diploid in HCECs, we randomly sampled two thirds of the interactions from the HCT116 network. Although the un-stratified network displayed only minor differences between the normal and cancerous counterparts, the most connected nodes were considerably more prominent in colon cancer cells than in normal cells (Figure 1B, Supplementary Figure S2A). Next, we determined their cohesion by stratifying the nodes according to their increasing k-core values (28) (Figure 1B, Supplementary Figure S2B). This is calculated by sequentially identifying the most connected nodes. Examining the chromatin marks associated with higher connectivity, we found that primed or active enhancers were strongly enriched with increasing k-core values specifically in cancer cells (Supplementary Figure S2C). This information was next used to identify the node with the highest dynamic index, which is one of the best proxies for the identification of the most influential spreaders of information in a complex network (29). Thus, removing a node with high dynamic index changes the structure of the network significantly and globally (30). In contrast to the most connected enhancer nodes, which preferentially localized within the two flanking TADs, the MYC bait itself showed the highest dynamic index in both HCEC and HCT116 cells (Figure 1C). Taken together, this information suggests that the MYC-enhancer interactions are very dynamic and that these processes are driven by MYC screening for enhancer regions rather than the other way around.
Figure 1.

The generation of a hierarchical chromatin network impinging on MYC. (A) The position of the 9 Nodewalk baits around the MYC locus with bait nr 10 that originated from chromosome 5. Vertical lines indicate the interactors and their ligation events (LE) impinging on MYC. (B) The network structures from 20 000 HCT116 cells and 20 000 human normal colon epithelial cells (HCEC) stratified by their k- values (8). The red and green nodes identify regions overlapping with H3K27ac and H3K4me1 peaks, respectively. The size of each node reflects the number of the interactors. The size of each node reflects the number of the interactors with small nodes reflecting non-bait regions common to two or more baits. (C) Dynamic importance index (D.I. Index) analysis of the nine enhancer baits in HCEC and HCT-116 cells.
The MYC chromatin network is the sum of stochastic interactions
To document that such enhancer networks represented the sum of stochastic interactions detected only in a large input cell population faced a significant challenge: the smaller the sample, the more variable the responses will be (31). Under this scenario, high biological variability representing stochastic interactions in very small cell populations would be expected to compromise attempts to evaluate reproducibility between such aliquots. To resolve this enigma, we first modified the initial Nodewalk protocol (7,8) to enable analyses of very low input samples (Supplementary Figure S3A). Next, we prepared three types of samples: One set of technical replicas with large input material were derived from the same pool of 3C cDNA sample to assess technical reproducibility in the ensuing steps. Ten samples representing technical replicas with small input material were then derived from a large pool of crosslinked and ligated chimeric DNA representing one million cells. Each of the ten replicas contained 0.88 ng of 3C DNA aliquot corresponding to 176 cells. These were compared to another set of nine biological replicas derived from small cell populations, each corresponding to 177 cells to represent biological variability (Figure 2A). In all cases, we were able to quantitatively restriction digest and ligate (Supplementary Figure S3B, C) and recover the total DNA (Supplementary Figure S3D) as well as the bait alleles (Supplementary Figure S3E) to enable quantitative analyses of chromatin interactions in small samples.
Figure 2.

The strategy to discriminate between virtual and real enhancer hubs. (A) The principle of analysis of stochastic chromatin networks displayed in libraries with smaller amount of input. The frequency of the distribution of interactors in the panel showing the 0.88 ng aliquots are predicted to closely follow a normalized distribution profile representative of the initial 3C sample derived from one million cells. Under the assumption that the network represents stochastic interactions, the biological variability is expected to be higher in the 177-cell sample than in the 0.88 ng sample with the highest variability represented by the 34.8 pg/21 alleles samples. (B) Sampling of technical and biological replicas. ‘Tech.’ represents technical whereas ‘Biol.’ represents biological replicas. (C) Venn diagram showing the overlap in interactors in cis between three different technical replicas. (D) Chromatin networks detected by the MYC bait in 10 samples of 0.88 ng 3C DNA aliquots (average recovery was 74.4%) or 9 different samples each containing 177 cells. The size of each node reflects its connectivity. (E) Bar diagram showing the relationship between the number of interactors (in cis) versus the number of the replicates showing the specific interaction. The bait was omitted from this analysis. (F) The stratification of interactors based on the presence or absence of enhancer marks in HCT116 cells. (G) Venn diagram demonstrating the overlap between pooled small input samples and the 50 ng ‘ensemble’ library. (H) Heatmap of enhancer-MYC interactions in the flanking TADs within the libraries of the 177 cell aliquots. Black indicates absence of interacting sequences. (I) The observed frequencies of enhancers impinging on the MYC bait compared to the expected frequencies, generated by random resampling of interactors in TADs with enhancer marks impinging on the MYC bait scaled from 50 ng 3C DNA input to 177 cells. (J) The observed frequencies of enhancers impinging on the MYC bait compared to the expected frequencies, generated by random resampling of interactors in TADs scaled from 50 ng 3C DNA input to 0.88 ng. Values were calculated from interactors in the whole genome. A statistical test showed that the P-values for panels I and J were found to be 0.3274 and 0.6373, respectively. In both instances it thus failed to reject the null hypothesis at the 95% confidence level.
Focusing on interactors in cis, which dominates the MYC chromatin network (7,8), we observed that the overlap in interactors present in three different technical replicas taken from the same initial RNA library, which was prepared from 0.88 ng of 3C DNA (Figure 2B), generated a technical reproducibility of >90% (Figure 2C). Next, we compared the reproducibility of chromatin fibre interactions among the 0.88 ng technical replicas to the reproducibility of chromatin fibre interactions among the 177-cell samples representing biological replicas, an approach that was simplified by a low variability in the quantity of input and the quality of 3C DNA (Supplementary Figure S3B–E). As could be expected from the hypothetical scheme in Figure 2A, >70% of interactors impinging on the MYC bait were detected in only one library among the libraries of the nine 177-cell samples (Figure 2D, E). In contrast, the libraries of the ten 0.88 ng 3C DNA technical replicas showed that >85% of the interactors were reproduced in two or more libraries (Figure 2D, E). Irrespective of this difference, both sets of samples recapitulated a similar proportion of interactor categories (Figure 2F). Importantly, the overlap between the pooled 0.88 ng technical aliquots or the pooled 177-cell samples and the large ensemble of interactome generated from 3C DNA aliquots corresponding to 10 000 cells exceeded 91%, highlighting that Nodewalk using small input material reliably recapitulated a subset of the interactors already present in the ensemble network (Figure 2G). As predicted in Figure 2A, the overlap between interactomes present in the libraries that were generated from small input material was only limited (Figure 2H). Importantly, there was no significant difference between an expected stochastic interactome (generated by 10 000 iterations of random sampling from ensemble network) and the actual observed interactome of MYC in the libraries derived from the nine 177 cell samples (Figure 2I). Moreover, this conclusion was reinforced when comparing expected to the observed interactome (Figure 2J) in re-sampled networks correcting for the MYC copy number (Supplementary Figure S2).
MYC-enhancer interactions are mutually exclusive
To examine the stochastic features of the MYC chromatin network, we pushed the limits of the Nodewalk technique by further reducing the input sample size to 34.8 pg (Figure 2A) corresponding to 21 alleles in seven cells (Supplementary Figure S3F). For a more robust analysis, only interactors within the MYC TADs, which were found to be reproducible across the ensemble network, were retained. This strategy revealed that 6 out of 8 different TAD1/2-specific interactors overlapped entirely with the parental libraries containing a total of 42 TAD1/2-specific interactors, which included non-enhancer as well as enhancer regions in both cases (Figure 3A). When assessing the distribution of the above-mentioned reproducible interactors among the twenty-three different 34.8 pg aliquots, we found that the observed number of different enhancers interacting with MYC in each aliquot ranged from 0 to 1, which closely agreed with the permutated number of interactions of a stochastic interactome, i.e. between 0 and 3 different enhancers expected to bind MYC in each aliquot (Figure 3B). Note that as there is no interaction between the enhancers labeled as ‘a’ and ‘b’ in the heatmap of Figure 3B within the ensemble network (Figure 3C), these likely occurred in different cells, or between different alleles within the same cell. Although the number of enhancers impinging on MYC increased somewhat when including all cis and trans enhancers in the ensemble network, there was still no significant difference between an expected stochastic interactome and the observed number of enhancers impinging on MYC either locally (Figure 3D) or genome wide (Figure 3E). Based on all of these considerations and that the average recovery of the bait was 36.2% (Figure 3B), we conclude that an average of 0.7 enhancer regions per 34.8 ng aliquot interacts with an average of 7.6 different MYC alleles. As the vast majority of MYC alleles are transcriptionally active in HCT116 cells (7,8), we conclude that enhancers only transiently engage with active MYC. Moreover, by binning the interactome data generated from small samples in successively larger windows, the overall patterns of the ensemble network could be reproduced to demonstrate the stochastic, but preferential patterns of interactions between MYC and its enhancers (Figure 4). We conclude that MYC likely interacts with its flanking enhancers in a dynamic and stochastic manner.
Figure 3.

MYC interacts with flanking enhancers in a mutually exclusive manner. (A) Overlap between pooled libraries from 34.8 pg input and pooled libraries from 177 cells within TADs. (B) The overall recovery of MYC bait alleles in 23 × 34.8 pg aliquots, each of which corresponds to 21 alleles input material is shown on top of a heatmap of the MYC network. The interactors were organized in cis keeping their relative position on the physical map. ‘a’ and ‘b’ indicates the location of the interactors represented in C). (C) The virtual enhancer hubs observed within TAD1/2 in the ensemble libraries. The right-most network image identifies the lack of physical and direct interaction between nodes ‘a’ and ‘b’. (D) The observed frequencies of enhancers interacting with MYC bait compared to the expected frequencies scaled down from 50 ng 3C DNA input to 21 alleles. (E) The observed frequencies of enhancers interacting with MYC bait compared to the expected frequencies scaled down from 50 ng 3C DNA input to 21 alleles. Values were calculated from genome wide interactors. A statistical test showed that the P-values were found to be 0.5648 and 0.4523 for panels D and E, respectively. In both instances it thus failed to reject the null hypothesis at the 95% confidence level.
Figure 4.

The stochastic character of the MYC network. Comparison of interaction profiles in the TADs flanking MYC with interaction profiles binned into larger windows resulting from Nodewalk analyses using 50 ng 3C DNA/10 000 cells input (green), 0.88 ng 3C DNA aliquots (red) and 177 cell aliquots (blue).
It could be argued that the Nodewalk technique might have approached a technical limitation for being able to pick up multiple interactions from such small aliquots provided by the 34.8 ng samples. However, not only is the number of total alleles recovered in the 23 aliquots exceeding the recovered alleles from each aliquot of the 177 cell samples, but the total number of enhancers within TADs impinging on MYC are comparable. Moreover, using 3D DNA FISH analyses comparing the proximities between two major MYC interactors, the oncogenic super-enhancer and EnhD (7,8), and MYC (Figure 5A) we could document that these three regions rarely, if at all, occupied the same space within the limits of the microscopic resolution (Figure 5B, Supplementary Figure S4). Taken together the data strongly implies that the enhancer hubs observed in the ensemble network are only virtual, which is consistent with previous results on the dynamics of 3D interactomes (32–35). Moreover, the observation that enhancer hubs were formed more readily in the absence of a functional cohesin complex (36) reinforces the conclusion that enhancer hubs are only indirectly associated with transcription. We thus conclude that although the identity of the interacting regions is specific and highly reproducible in large cell populations, these represent the sum of stochastic events in small populations.
Figure 5.
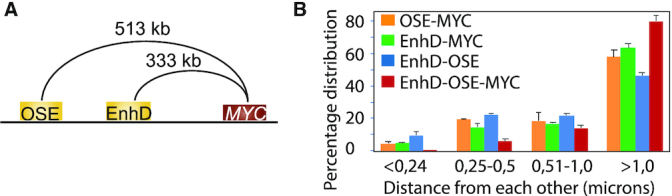
3D DNA FISH analyses to discriminate between ‘dating versus partying’ interactions (35). (A) The regions examined for simultaneous DNA FISH analyses. (B) shows the percentage of the proximities for the different combination of alleles, as indicated in the image representing two independent experiments. OSE = oncogenic super-enhancer. In the three-way analyses between EnhD-OSE-MYC, the distribution was determined by the most distant partner. See text and (8) for additional information.
DISCUSSION
We have here addressed the dynamic nature of a chromatin interactome characterized by MYC likely screening for neighboring enhancers in both normal and cancerous cells. If such networks represented dynamic and stochastic events, the interaction patterns would also be expected to display increased variability between the aliquots, as the size of a statistical sample affects the standard error for that sample to render such studies technically difficult. We have solved this enigma by enhancing the sensitivity of the Nodewalk technique while retaining its ability to quantitatively determine network structures in numerous aliquots of very small samples. The results are compatible with the conclusion that the chromatin networks impinging on MYC in cancer cells are stochastic and have likely evolved to facilitate redundant mechanisms of MYC activation. In this regard, we find it interesting that <10% of MYC alleles interact with an enhancer within the chromatin network at any given time. Due to that >95% of MYC alleles are transcriptionally active in HCT-116 cells, as estimated by RNA FISH analyses (8), this result also suggests that the regional enhancers do not generally associate with MYC once transcription has been initiated.
In contrast to the most connected enhancer nodes, which preferentially localized within the two flanking TADs, the MYC bait itself showed the highest dynamic index in both HCEC and HCT116 cells. This observation likely reflects the position of MYC itself at or close to the inter-TAD boundary to facilitate its ability to explore the neighboring two TADs equally well in both HCT116 cells and HCECs (7,8). We therefore submit that the mobility of MYC juxtaposed to the inter-TAD boundary generally drives its communications with its neighboring TADs in both normal and cancerous cells (Figure 6). Under this scenario, the unscheduled emergence of a enhancer would enhance the probability of pathological MYC expression. However, this is likely a simplified scenario that does not explain the higher dynamic index for the oncogenic super-enhancer in the colon cancer cells (regions 2 and 3 in Figure 2A, C). We therefore argue that this cancer cell-specific feature relates to its ability to recruit MYC to nuclear pores to increase its expression post-transcriptionally (8). We also predict that the plasticity underlying this process provides the cancer cell with a selective advantage, as multiple signaling pathways converge on different sets of MYC enhancers in cancer cells (37) (Figure 6) to ensure excessive cell proliferation in response to a changing microenvironment during the neoplastic process.
Figure 6.
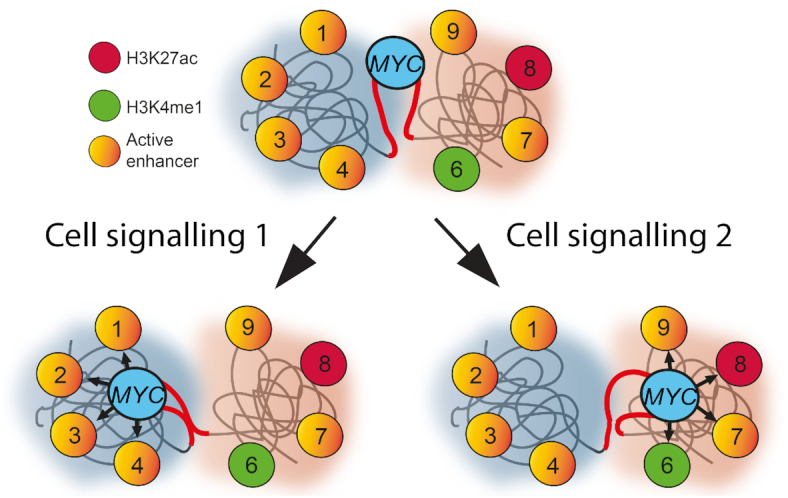
Schematic representation of MYC and its flanking enhancers. The MYC locus is at or close to an inter-TAD boundary with the flanking TADs marked in the background as blue or red spheres. From its default state it will upon transcriptional activation connect with the enhancer regions in both of its flanks switching between the TADs in a mutually exclusive manner. It is also envisaged that these processes are controlled by external factors produced in the microenvironment of the evolving cancer cells.
To summarize, by optimizing the Nodewalk technique to very small input cell populations we have here documented that the MYC chromatin networks stochastically impinging on flanking enhancers have likely evolved to facilitate redundant mechanisms of MYC activation in cancer cells. The major advantage of this decentralized network topology is that there might be no ‘single point of failure’ within the network to ensure the potential for continuous, yet variable activation of MYC to increase the fitness of cancer cells by promoting their adaptability to changing microenvironments (38). The extreme sensitivity and versatility of the optimized Nodewalk technique open up the possibility to decipher cellular heterogeneity in 3D chromatin structures for specific regions within solid and liquid tumor biopsies as well as circulating tumor cells. Such an approach might be essential to determine the precise order of events underlying enhancer-mediated, stochastic transcriptional activation and gene gating (8) fueling cancer evolution.
DATA AVAILABILITY
All the Nodewalk sequence data presented here have been deposited to the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession number of GSE76049. The ChIP-seq data were retrieved from GEO as follows: CTCF (accession nr GSM749690); H3K27ac (accession nr GSM946854); H3K4me1 (accession nr GSM1240111).
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge initial assistance from Drs Alejandro Woodbridge and Peter J Svensson as well as support from Science for Life Laboratory, the National Genomics Infrastructure, NGI, and Uppmax for providing assistance in massive parallel sequencing and computational infrastructure as well as the extensive data sets from ENCODE.
Author contributions: N.S.: Developed the Nodewalk protocol, made all Nodewalk analyses, and performed some of the bioinformatic analyses. E.G.S.: Co-supervised and performed parts of the bioinformatic analyses. N.A.K.: Made parts of the enrichment analyses as well as the dynamic importance index analyses. A.L.R. and B.A.S contributed to the DNA FISH analyses. J.V.: Contributed to the Nodewalk protocol. D.G.C.: Contributed to the network analysis. J.T.: Supervised the design of the bioinformatics analyses. A.G. and R.O.: Contributed conceptually and to the design of the experiments as well as wrote the manuscript.
Notes
Present address: Noriyuki Sumida, Bio Systems Design Department, Bio Analytical Systems Product Division, Science & Medical Systems Business Group, Hitachi High-Tech, Ichige 882, Hitachinaka, Ibaraki 312-8504, Japan.
Contributor Information
Noriyuki Sumida, Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital, Z1:00, SE-171 76 Stockholm, Sweden.
Emmanouil G Sifakis, Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital, Z1:00, SE-171 76 Stockholm, Sweden.
Narsis A Kiani, Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital, Z1:00, SE-171 76 Stockholm, Sweden.
Anna Lewandowska Ronnegren, Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital, Z1:00, SE-171 76 Stockholm, Sweden.
Barbara A Scholz, Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital, Z1:00, SE-171 76 Stockholm, Sweden.
Johanna Vestlund, Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital, Z1:00, SE-171 76 Stockholm, Sweden; Unit of Computational Medicine, Department of Medicine, Center for Molecular Medicine, Karolinska Institutet, Karolinska University Hospital, L8:05, SE-171 76, Stockholm, Sweden.
David Gomez-Cabrero, Unit of Computational Medicine, Department of Medicine, Center for Molecular Medicine, Karolinska Institutet, Karolinska University Hospital, L8:05, SE-171 76, Stockholm, Sweden; Mucosal and Salivary Biology Division, King's College London Dental Institute, London SE1 9RT, UK.
Jesper Tegner, Unit of Computational Medicine, Department of Medicine, Center for Molecular Medicine, Karolinska Institutet, Karolinska University Hospital, L8:05, SE-171 76, Stockholm, Sweden; Science for Life Laboratory, Tomtebodavägen 23A, SE-17165, Solna, Sweden; Biological and Environmental Sciences and Engineering Division, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia.
Anita Göndör, Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital, Z1:00, SE-171 76 Stockholm, Sweden.
Rolf Ohlsson, Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital, Z1:00, SE-171 76 Stockholm, Sweden.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swedish Research Council [VR 2017-04670 and VR 2016-03108]; Swedish Childhood Cancer Fund [PR2017-0132]; Swedish Cancer Society [CAN2017/515, CAN 2016/708]; Lundberg Foundation [2018-0138]; Karolinska Institutet; Novo Nordisk Foundation [NNF16OC0021512]; Cancer Society in Stockholm (2018–2021 and 2019–2021); KA Wallenberg Foundation [KAW 2017.0077]. Funding for open access charge: KA Wallenberg Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Sanchez A., Golding I.. Genetic determinants and cellular constraints in noisy gene expression. Science. 2013; 342:1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hager G.L., McNally J.G., Misteli T.. Transcription dynamics. Mol. Cell. 2009; 35:741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fullwood M.J., Liu M.H., Pan Y.F., Liu J., Xu H., Mohamed Y.B., Orlov Y.L., Velkov S., Ho A., Mei P.H. et al.. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009; 462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A., Young R.A.. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hay D., Hughes J.R., Babbs C., Davies J.O., Graham B.J., Hanssen L.L., Kassouf M.T., Oudelaar A.M., Sharpe J.A., Suciu M.C. et al.. Genetic dissection of the alpha-globin super-enhancer in vivo. Nat. Genet. 2016; 48:895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Loven J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R., Bradner J.E., Lee T.I., Young R.A.. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013; 153:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sumida N., Sifakis E., Scholz B., Fernandes A., Kiani N., Gomez-Cabrero D., Svensson J., Tegner J., Göndör A., Ohlsson R.. The ultra-sensitive Nodewalk technique identifies stochastic from virtual, population-based enhancer hubs regulating MYC in 3D: implications for the fitness of cancer cells. 2018; bioRxiv doi:27 March 2018, preprint: not peer reviewed 10.1101/286583. [DOI]
- 8. Scholz B.A., Sumida N., de Lima C.D.M., Chachoua I., Martino M., Tzelepis I., Nikoshkov A., Zhao H., Mehmood R., Sifakis E.G. et al.. WNT signaling and AHCTF1 promote oncogenic MYC expression through super-enhancer-mediated gene gating. Nat. Genet. 2019; 51:1723–1731. [DOI] [PubMed] [Google Scholar]
- 9. Ing-Simmons E., Seitan V.C., Faure A.J., Flicek P., Carroll T., Dekker J., Fisher A.G., Lenhard B., Merkenschlager M.. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res. 2015; 25:504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Z., Legant W.R., Chen B.C., Li L., Grimm J.B., Lavis L.D., Betzig E., Tjian R.. 3D imaging of Sox2 enhancer clusters in embryonic stem cells. Elife. 2014; 3:e04236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berlivet S., Paquette D., Dumouchel A., Langlais D., Dostie J., Kmita M.. Clustering of tissue-specific sub-TADs accompanies the regulation of HoxA genes in developing limbs. PLos Genet. 2013; 9:e1004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gavrilov A.A., Razin S.V.. Spatial configuration of the chicken alpha-globin gene domain: immature and active chromatin hubs. Nucleic Acids Res. 2008; 36:4629–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kieffer-Kwon K.R., Tang Z., Mathe E., Qian J., Sung M.H., Li G., Resch W., Baek S., Pruett N., Grontved L. et al.. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell. 2013; 155:1507–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Markenscoff-Papadimitriou E., Allen W.E., Colquitt B.M., Goh T., Murphy K.K., Monahan K., Mosley C.P., Ahituv N., Lomvardas S.. Enhancer interaction networks as a means for singular olfactory receptor expression. Cell. 2014; 159:543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patrinos G.P., de Krom M., de Boer E., Langeveld A., Imam A.M., Strouboulis J., de Laat W., Grosveld F.G.. Multiple interactions between regulatory regions are required to stabilize an active chromatin hub. Genes Dev. 2004; 18:1495–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiang J.F., Yin Q.F., Chen T., Zhang Y., Zhang X.O., Wu Z., Zhang S., Wang H.B., Ge J., Lu X. et al.. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014; 24:513–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim T., Cui R., Jeon Y.J., Lee J.H., Lee J.H., Sim H., Park J.K., Fadda P., Tili E., Nakanishi H. et al.. Long-range interaction and correlation between MYC enhancer and oncogenic long noncoding RNA CARLo-5. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:4173–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schuijers J., Lee T.I., Zhao K. et al.. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014; 159:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagano T., Lubling Y., Stevens T.J., Schoenfelder S., Yaffe E., Dean W., Laue E.D., Tanay A., Fraser P.. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013; 502:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stevens T.J., Lando D., Basu S., Atkinson L.P., Cao Y., Lee S.F., Leeb M., Wohlfahrt K.J., Boucher W., O'Shaughnessy-Kirwan A et al.. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature. 2017; 544:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gondor A., Rougier C., Ohlsson R.. High-resolution circular chromosome conformation capture assay. Nat. Protoc. 2008; 3:303–313. [DOI] [PubMed] [Google Scholar]
- 23. Zhou X., Maricque B., Xie M., Li D., Sundaram V., Martin E.A., Koebbe B.C., Nielsen C., Hirst M., Farnham P. et al.. The Human Epigenome Browser at Washington University. Nat. Methods. 2011; 8:989–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bastian M., Heymann S., Jacomy M.. Gephi: an open source software for exploring and manipulating networks. Int. AAAI Conf. Weblogs Social Media. 2009; 8:361–362. [Google Scholar]
- 25. Csardi G., Nepusz T.. The igraph software package for complex network research. Inter J. of Comp. Syst. 2006; 5:1695. [Google Scholar]
- 26. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langer S., Geigl J.B., Ehnle S., Gangnus R., Speicher M.R.. Live cell catapulting and recultivation does not change the karyotype of HCT116 tumor cells. Cancer Genet. Cytogenet. 2005; 161:174–177. [DOI] [PubMed] [Google Scholar]
- 28. Seidman S.B. Network structure and minimum degree. Social Netw. 1983; 5:269–287. [Google Scholar]
- 29. Rodriguez F. Macau E. Network Centrality: An Introduction. 2019; 22:Springer International Publishing; 177–196. [Google Scholar]
- 30. Lohmann G., Margulies D.S., Horstmann A., Pleger B., Lepsien J., Goldhahn D., Schloegl H., Stumvoll M., Villringer A., Turner R.. Eigenvector centrality mapping for analyzing connectivity patterns in fMRI data of the human brain. PLoS One. 2010; 5:e10232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kenney J.F. Mathematics of Statistics. 1951; Chapman & Hall LTD. [Google Scholar]
- 32. Olivares-Chauvet P., Mukamel Z., Lifshitz A., Schwartzman O., Elkayam N.O., Lubling Y., Deikus G., Sebra R.P., Tanay A.. Capturing pairwise and multi-way chromosomal conformations using chromosomal walks. Nature. 2016; 540:296–300. [DOI] [PubMed] [Google Scholar]
- 33. Ay F., Vu T.H., Zeitz M.J., Varoquaux N., Carette J.E., Vert J.P., Hoffman A.R., Noble W.S.. Identifying multi-locus chromatin contacts in human cells using tethered multiple 3C. BMC Genomics. 2015; 16:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gondor A., Woodbridge A.F., Shi C., Aurell E., Imreh M., Ohlsson R.. Window into the complexities of chromosome interactomes. Cold Spring Harb. Symp. Quant. Biol. 2010; 75:493–500. [DOI] [PubMed] [Google Scholar]
- 35. Sandhu K.S., Shi C., Sjolinder M., Zhao Z., Gondor A., Liu L., Tiwari V.K., Guibert S., Emilsson L., Imreh M.P. et al.. Nonallelic transvection of multiple imprinted loci is organized by the H19 imprinting control region during germline development. Genes Dev. 2009; 23:2598–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rao S.S.P., Huang S.C., Glenn St Hilaire B., Engreitz J.M., Perez E.M., Kieffer-Kwon K.R., Sanborn A.L., Johnstone S.E., Bascom G.D., Bochkov I.D. et al.. Cohesin loss eliminates all loop domains. Cell. 2017; 171:305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hnisz D., Schuijers J., Lin C.Y., Weintraub A.S., Abraham B.J., Lee T.I., Bradner J.E., Young R.A.. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell. 2015; 58:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mautner J., Joos S., Werner T., Eick D., Bornkamm G.W., Polack A.. Identification of two enhancer elements downstream of the human c-myc gene. Nucleic Acids Res. 1995; 23:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the Nodewalk sequence data presented here have been deposited to the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession number of GSE76049. The ChIP-seq data were retrieved from GEO as follows: CTCF (accession nr GSM749690); H3K27ac (accession nr GSM946854); H3K4me1 (accession nr GSM1240111).