

Abstract
Decapping is the first committed step in 5′-to-3′ RNA decay, and in the cytoplasm of human cells, multiple decapping enzymes regulate the stabilities of distinct subsets of cellular transcripts. However, the complete set of RNAs regulated by any individual decapping enzyme remain incompletely mapped, and no consensus sequence or property is currently known to unambiguously predict decapping enzyme substrates. Dcp2 was the first-identified and best-studied eukaryotic decapping enzyme, but it has been shown to regulate the stability of <400 transcripts in mammalian cells to date. Here, we globally profile changes in stability of the human transcriptome in Dcp2 knockout cells via TimeLapse-seq. We find that P-body enrichment is the strongest correlate of Dcp2-dependent decay, and that modification with m6A exhibits an additive effect with P-body enrichment for Dcp2 targeting. These results are consistent with a model in which P-bodies represent sites where translationally repressed transcripts are sorted for decay by soluble cytoplasmic decay complexes through additional molecular marks.
Keywords: TimeLapse-seq, metabolic labeling, mRNA decapping, 4-thiouridine, mRNA decay rates, m(7)G, human Dcp2, processing body, m(6)A
Graphical Abstract
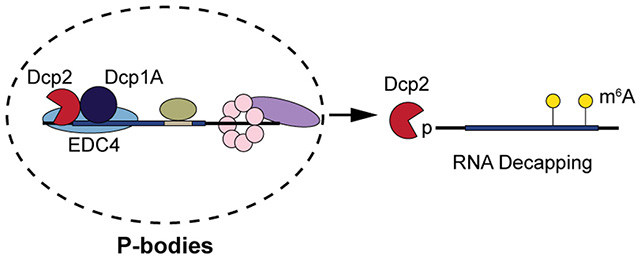
INTRODUCTION
Regulation of cytoplasmic transcript abundance by RNA degradation is a critical mechanism of post-transcriptional control of gene expression, and aberrant RNA decay has been linked to perturbed homeostasis 1–3, inflammatory responses 4, 5, and neuronal diseases 6, 7. In eukaryotes, bulk cytoplasmic RNA degradation initiates with shortening of the 3′ poly(A) tail, followed by either 5′-to-3′ or 3′-to-5′ decay 8. In the 5′-to-3′ pathway of exonucleolytic RNA decay, the 7-methylguanosine (m7G) cap is removed by a decapping enzyme in complex with regulatory proteins (the “decapping complex”), releasing m7GDP and committing 5′ phosphorylated RNA to degradation by the conserved 5′-to-3′ exonuclease Xrn1 9. Dcp2 (also referred to as hDcp2 in human), a member of the Nudix hydrolase family, was the first identified decapping enzyme in yeast and was long assumed to be the major cytoplasmic decapping factor in higher eukaryotes as well 10–14.
Recently, additional mammalian m7G decapping enzymes, Nudt16 15, 16 and Nudt3 17, were identified, and Dxo/Rai1 was shown to hydrolyze both unmethylated 18, 19 and NAD caps 20. The discovery of coexistent decapping pathways provoked the fundamental question of whether each enzyme decaps specific RNAs. Consistent with this hypothesis, mammalian Dcp2 21, 22, Nudt16 15, and Nudt3 17 have been reported to modulate the half-lives of unique, partially overlapping, subsets of cellular transcripts. For example, Dcp2 specifically controls the half-lives of several well-characterized substrates including RRP41 22, LNC13 5, and IRF7 4. However, global maps of RNA substrates of Dcp2, as well as molecular determinants that direct RNAs to Dcp2 in human cells, are only partially understood. To date, only 145 RNA have been identified as Dcp2 substrates in mice, with 216 other transcripts utilizing Dcp2 and Nudt16 redundantly 16.
Despite specific binding of one 5′ untranslated region (UTR) stem-loop structure in vitro 23, human Dcp2 is generally non-sequence-specific, instead recognizing the m7G cap as well as the backbone of ~12 nucleotides in an RNA binding channel 24. It is likely that macromolecular interactions and/or localization guide specific transcripts to Dcp2 in human cells, such as interactions with proteins that recognize specific RNA sequence motifs 2 or post-transcriptional modifications such as N6-methyladenosine (m6A) 25–27; alternatively, Dcp2 substrate specificity could in principle be controlled by dynamic exchange of Dcp2 between the cytosol and ribonucleoprotein (RNP) granules known as processing bodies (P-bodies) 28, 29. However, none of these mechanisms have yet been shown to fully define Dcp2 substrates. For example, while the Dcp2 decapping complex-associated protein tristetrapolin, or TTP, can destabilize some RNAs containing AU-rich elements (AREs) 30, 31, not all ARE-containing RNAs are Dcp2 substrates 15. Similarly, while a general destabilizing effect of m6A has been revealed 32–34, some m6A marks regulate translation rather than stability 35, 36.
Previous efforts to identify targets of RNA decapping enzymes have been hindered, at least in part, by limitations to existing experimental approaches. For example, global profiling of changes in RNA abundance at discrete timepoints after transcriptional arrest in yeast and mouse cells lacking Dcp2 was used to define Dcp2 substrates 16, 37, but these experiments were complicated by (1) incomplete penetrance of the gene-trap knockout, and (2) genetic compensation 16. Furthermore, these studies relied on transcriptional inhibition for assessment of RNA stability, which perturbs RNA decay rates due to induction of stress responses 38, 39. This suggests that detection of Dcp2 substrates is limited by challenges in differentiation of changes in RNA synthesis and processing vs. decay in genetic knockouts, as well as inadvertent alteration of RNA decay rates with transcriptional inhibitors. In this work, we apply TimeLapse-seq 40 to resolve changes in RNA synthesis (e.g. transcription and maturation) and stability in human Dcp2 knockout cells. We demonstrate that an RNA sequence motif and modification are sufficient but not necessary for decay of some Dcp2-sensitive transcripts. Instead, the portion of the transcriptome stabilized in the absence of Dcp2 best correlates with transcripts enriched in P-bodies. This correlation is improved for transcripts that both localize to P-bodies and bear m6A modifications. Our results are consistent with a model in which P-body-resident RNAs are licensed for decay by alternative pathways depending on covalent marks and sequence motifs.
MATERIALS AND METHODS
Cell culture.
HEK293T cells were purchased from ATCC, and early-passage stocks were maintained to ensure cell line provenance and sterility. Knock-out cell lines were generated as described below. Cells were maintained in DMEM (Corning) supplemented with 10% (vol/vol) FBS (Sigma-Aldrich) and 100 U/mL penicillin-streptomycin (VWR). Cells were verified to be mycoplasma-free using the ATCC Universal mycoplasma detection kit.
Antibodies.
Primary antibodies used for Western blotting and/or immunofluorescence were as follows: rabbit polyclonal anti-Dcp2 (Novus Biologicals, NBP1-41070); mouse monoclonal anti-beta actin (Invitrogen, BA3R); rabbit polyclonal anti-Xrn1 (Bethyl, A300-443A; Sigma, PLA0105); rabbit monoclonal anti-Msi2 (Abcam, ab76148); rabbit monoclonal anti-Dcp1a Alexa Fluor 647 (Abcam, ab209946). Secondary antibodies for western blotting were goat anti-rabbit peroxidase conjugate (Rockland, 611-103-122; Merck, AP132P) and goat anti-mouse peroxidase conjugate (Rockland, 610-1319). The secondary antibody for immunofluorescence was goat anti-rabbit Alexa Fluor 568 (Life Technologies, A-11011).
Generation of DCP2, MSI2 and XRN1 CRISPR knockout (KO) cells.
DCP2 knock out (KO) HEK293T cells were generated using previously reported guide RNAs (gRNAs) 41, and XRN1 KO and MSI2 KO HEK293T cells were generated using validated gRNAs 42 to target the XRN1 genomic region (gRNAs; 5′-TAAAACGCCTCCCACGCTGC-3′ and 5′-TTAAGAGAAGAAGTTCGATT-3′) or MSI2 (gRNAs; 5′-CCATGAGTTAGATTCCAAGA-3′ and 5′-ATCCCACTACGAAACGCTCC-3′), respectively. Double-stranded DNA oligonucleotides corresponding to the gRNAs were inserted into pSpCas9(BB)-2A-GFP vector (Addgene, as a gift from F. Zhang, MIT, Cambridge, MA 43). In each case, an equal mixture of the two gRNA plasmids were transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, and GFP-positive cells were sorted with flow cytometry into single clones. Loss of Dcp2 or XRN1 or Msi2 expression was confirmed by Western blot, genomic DNA PCR and sequencing. In the DCP2 KO cell line used in this study, the two alleles were disrupted by a 36-nt homozygous deletion at the catalytic site spanning E147/148. In the XRN1 KO cell line, the two alleles were disrupted with a 7-nt homozygous deletion. In the MSI2 KO cell line used in this study, the two alleles were disrupted with a 44-nt homozygous deletion. For generation of double knockout cell lines, the same DCP2 or MSI2 gRNAs were transfected into XRN1 KO cells followed by FACS as above, with confirmation by Western blotting and genomic DNA PCR/sequencing.
TimeLapse-seq.
Wild type (WT) HEK293T, DCP2 and MSI2 KO cells were treated in duplicate with 500 μM s4U for 2 h, and a single replicate control per cell line was not treated with s4U. Total RNA was isolated from cell pellets using TRIzol reagent, followed by phenol-chloroform extraction and isopropanol precipitation supplemented with 50 μM DTT. After washing twice with 75% EtOH, the RNA pellet was dried and resuspended in nuclease-free water and treated with Turbo DNase to remove genomic DNA. RNA was isolated using RNAClean beads, and was then treated with TimeLapse chemistry (2,2,2-trifluoroethylamine and sodium periodate) and purified as described in Schofield et al., 2018. Purified recoded RNA was then sequenced using the mammalian pico-input SMARTer stranded Total RNA-seq kit v2 (Takara Bio). Paired-end 100nt or 150nt RNA sequencing was performed using an Illumina Hiseq-4000 or Novaseq.
TimeLapse-seq analysis.
TimeLapse-seq read trimming and alignment to mature RNA isoforms were performed as described previously in Schofield et al., 2018. Mutation calling was performed as described previously with the following modifications: for bases with overlapping coverage in a read pair, only the mutation with higher base quality is used for analysis. Reads were assigned as new or old based on a per-read 3.6% T-to-C mutation rate. The per-read mutation cutoff was determined through maximization of sensitivity and specificity for varying mutation rates, determined using the following equations:
For this determination of a mutation rate cutoff, “new” reads are intronic reads in highly expressed transcripts with s4U and TimeLapse chemistry treatment, and “old” reads are in matched transcripts without s4U treatment to reflect the background T-to-C mutation rate.
Old and new read counts per gene were normalized per sample based on total RNA counts using EdgeR. Log2-fold change analysis was performed using DEseq2 on total, new, and old RNA counts, with scale factors from total RNA input into new and old RNA analyses. Change in synthesis and degradation rates for mRNAs were determined using the following equations:
where fnew is the fraction of new RNA in exonic features and [RNA] is the normalized total exonic RNA read count per gene.
Cell-based mRNA decay assay by quantitative real-time RT-PCR.
DCP2 KO, MSI2 KO or WT HEK293T cells were cultured in 6-well plates to 70% confluency. Cells were harvested on ice at the indicated time points after transcriptional arrest by actinomycin D (5 μg/mL). Total RNA was isolated with TRIzol (Invitrogen) following the manufacturer’s instructions, followed by DNase I (NEB) treatment. RNA levels were quantified from reverse-transcribed cDNA by real-time PCR using iTaq Universal SYBR Green Supermix (Bio-Rad). RNA amounts remaining at individual time points relative to time = 0 hour were calculated from Ct values as described previously 44. Half-lives were derived from linear regression fit of ln(relative RNA remaining) as a function of time using the following equation: t1/2 = ln(2)/slope 45, 46. Primer sequences are listed in Table S1.
Cell-based mRNA decay assay after siRNA knockdown of METTL3.
WT or DCP2 KO HEK293T cells were transfected with METTL3 siRNA (Qiagen, SI04317096) or control siRNA (Qiagen, 1027281) at 30% confluency. After 6 h, each flask was re-seeded into separate 6-well plates. After 72 h, actinomycin D (5 μg/mL) was added to stop transcription, and cells were harvested at indicated time points. Total RNA was extracted with TRIzol and treated with DNase I. Selected m6A-containing mRNAs were then quantified with qRT-PCR based on a comparative Ct method 47 using previously reported primers 32. Relative RNA levels were normalized to beta actin 32, and half-lives were derived from linear regression fit.
P-body imaging using confocal microscopy.
HEK293T cells were grown on fibronectin-coated glass coverslips in a 48-well plate to 70% confluency. Cells were fixed with 10% neutral buffered formalin (Fisher Scientific), permeabilized with methanol at −20 °C, and blocked with blocking buffer (3% BSA in DPBS) for 1 h at room temperature. Cells were stained with rabbit anti-Msi2 at a 1:1000 dilution in blocking buffer overnight at 4 °C, followed by 3x PBS washes. Goat anti-rabbit Alexa Fluor 568 was applied at a 1:1000 dilution in blocking buffer for 1 to 4 h at room temperature in dark, with 5 subsequent PBS washes. Anti-Dcp1a Alexa Fluor 647 was then added at 1:1000 dilution and incubated for 1 h, followed by 3 PBS washes. Cells were post-fixed with 10% buffered formalin, stained with DAPI, and imaged by laser scanning confocal microscopy (Leica TCS SP8) with PL (field planarity) APO (apochromatic) 63x/1.40 oil, CS2 and PL APO 100x/1.44 oil, CORR (correction collar) CS (confocal scanning).
Quantitative splinted ligation RT-PCR (qSL-RT-PCR).
The detection of decapped mRNA by qSL-RT-PCR was performed in WT, XRN1 KO, and DCP2/XRN1 or MSI2/XRN1 double knockout (DKO) HEK293T cells following a procedure previously developed in yeast lacking XRN1 44, 48. HEK293T WT, XRN1 KO, DCP2/XRN1 DKO or MSI2/XRN1 DKO cells were cultured in 6-well plates to 70% confluency. Actinomycin D (5 μg/mL) was added, and cells were harvested on ice at the indicated time points. Total RNA was isolated with TRIzol, and 10 μg total RNA was mixed with 20 pmol of each splint oligonucleotide and 30 pmol of anchor RNA (5′-GCUGAUGGCGAUGAAUGAACACUGCGUUUGCUGGCUUUGAUG-3′). The oligos were annealed by sequential incubation for 5 min from 70 °C to 60 °C to 42 °C and to 25 °C, followed by the ligation step with 2 μL T4 DNA ligase (NEB) at 16 °C overnight. 20 U RNasin Plus (Promega) was included to prevent RNA degradation. 2 μL DNase I (NEB) was then added to digest genomic DNA for 1 h at 37 °C. The RNA was precipitated with 0.3 M sodium acetate (pH 5.2) and 2.5 volume of ethanol at −20 °C overnight, washed with 70% ethanol and resuspended in 30 μL of DEPC-treated water. cDNA was synthesized from 1 μg of the ligated RNA with GoScript kit (Promega) using gene specific reverse primers (Rev-2, see Table S1). Total mRNA and splinted ligation product were then quantified by real-time PCR using gene specific Fwd-2/Rev-2 primers and Anchor Fwd/Rev-2 primers, respectively (Supplementary Table 1). A ΔΔCt method was used to determine the relative level of decapped mRNA as previously described 47, and the amounts of total mRNA remaining at each time point relative to time = 0 h were calculated from 2−ΔΔCt using ΔCt (t = 0) as the reference value.
CDF analysis and density volcano plot.
Datasets were filtered for genes enriched or depleted in P-bodies (enrichment > 2.5 or < −2.5, Hubstenberger et al., 2017), and/or containing m6A or m6Am modifications (Wei et al., 2018). Cumulative distributions of the change in decay rate between KO and WT were plotted using R with package ggplot2. For volcano and density plots, transcripts were plotted using R with package ggplot2 according to P-body enrichment and significance 49, and those identified as stabilized by DCP2 KO were colored differentially from remaining transcripts.
Quantification and statistical analysis.
Two-tailed and equal-variance Student’s t-test, Mann Whitney U test, Kruskal-Wallis test and analysis of variance (ANOVA) with Dunnett’s test were performed using Excel, GraphPad Prism 7 or R for statistical significance with significance defined as a P value<0.05. Equal variance between samples was established using an F-test. All data obtained represent at least three biological replicates unless otherwise stated. Data are presented as the mean ± standard deviation.
Data and software availability.
The accession number for the TimeLapse-seq data reported in this paper is NCBI Gene Expression Omnibus: GSE143662. All software and parameters are described above, and custom scripts are available upon request.
RESULTS
Measurement of RNA dynamics in human DCP2 knockout (KO) cells using TimeLapse-seq
TimeLapse-seq utilizes metabolic RNA labeling with 4-thiouridine (s4U) followed by oxidative nucleophilic aromatic substitution, which recodes s4U to cytidine analogs identifiable as T-to-C mutations in RNA sequencing 50. Several studies have demonstrated application of these metabolic RNA labeling-based methods in improved measurement of RNA turnover in different organisms 51, 52. Utilizing a 2-hour s4U treatment combined with TimeLapse-seq, we observe the proportion of mature mRNA that is newly generated over the metabolic labeling period. The content of T-to-C mutations defines mature transcripts synthesized during metabolic labeling (high mutation content, referred to herein as “new” RNA) vs. pre-existing (background/low mutations, referred to herein as “old” RNA) mature RNA populations. From these observed mutations, rates of transcript synthesis and turnover can be differentiated in the processed mRNA population (by considering reads aligning to exonic features) at steady state, contrasting approaches that utilize intronic reads to estimate rates of transcription53. Furthermore, the observed proportion of new and old RNAs allows us to directly measure transcript half-lives at steady state, and to interrogate the effect of a genetic knockout on transcript half-lives. Importantly, at the s4U concentrations applied in TimeLapse-seq, no toxicity has been observed in HEK293T cells, in contrast to transcriptional inhibition-based methods 38. We therefore applied TimeLapse-seq using optimized treatment conditions for studying mRNA turnover in wild type (WT) and a DCP2 KO HEK293T cell line generated using CRISPR-Cas9 technology as previously reported 41 (Figure 1a and Figure S1a). Per-gene total RNA counts correlate well between biological replicates, and s4U-specific recoding is evident in sequencing tracks (Figure 1b) and in high-turnover intronic mutation rates (ranging between 6.3-9.2% per T).
Figure 1.
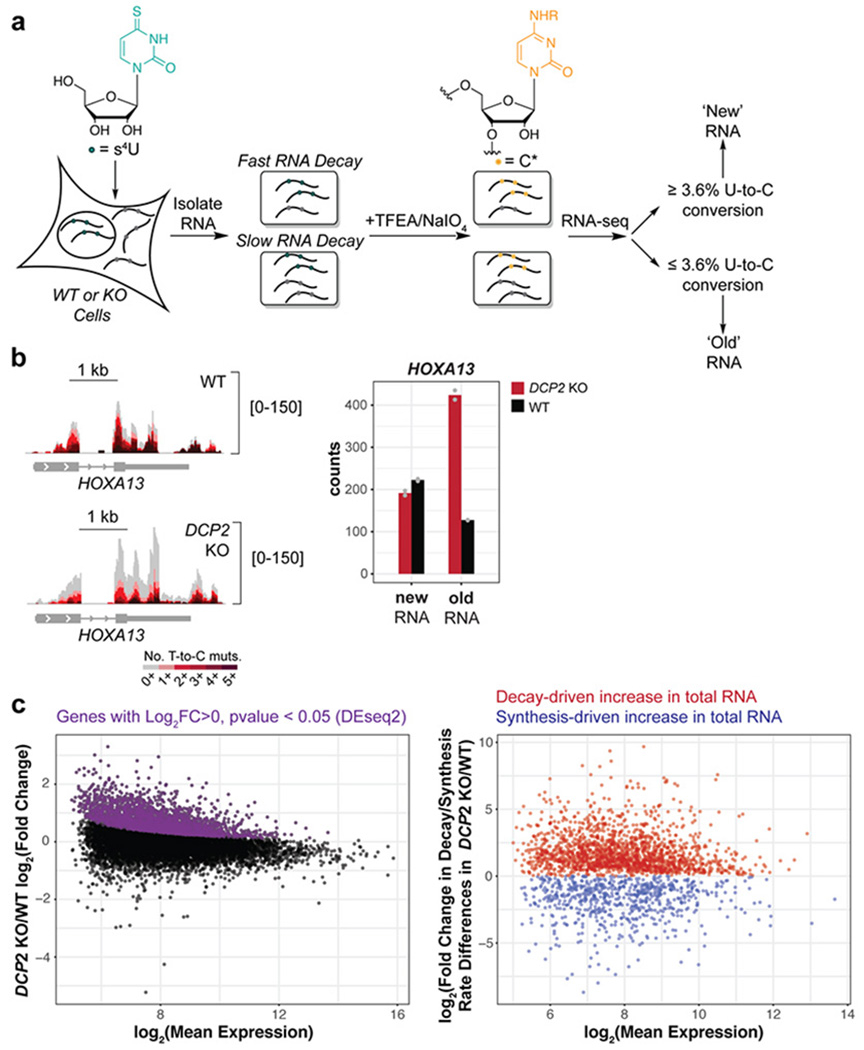
TimeLapse-seq identifies and differentiates global changes in RNA stability and transcription in human cells lacking the cytoplasmic RNA decapping enzyme Dcp2. (a) Experimental scheme of RNA stability measurement by TimeLapse-seq. (b) TimeLapse-seq tracks of HOXA13 in wild type (WT) (top left) and DCP2 knockout (KO) HEK293T cells (bottom left). Reads with no T-to-C mutations represent old RNA and increasing mutational content corresponds to newly synthesized RNA containing recoded s4U incorporated over the 2 h period of metabolic labeling. The bar plot (right) displayed inferred new and old reads from corresponding tracks. (c) Determination of post-transcriptionally stabilized RNAs in DCP2 KO versus WT HEK293T cells. Left, the MA plot indicates significantly upregulated genes (purple) derived from DESeq2 gene-level analysis of total RNAs. Decay- vs. synthesis-driven changes were then separated based on differences in the changes of the estimated rates of degradation and synthesis in DCP2 KO versus WT HEK293T cells (right). Dcp2 targets or substrates are defined as RNAs stabilized in DCP2 KO vs. WT.
In order to evaluate changes in transcript decay rate attributed to DCP2 KO, we estimated the fraction of new mature RNAs (fnew) aligning to exonic features using a thresholding approach to separate new and old reads based on T-to-C mutation content. We note that “new” reads are defined as reads mapping to those RNAs, including mature, processed RNAs, that were synthesized during the labeling window and “synthesis” includes both transcription and post-transcriptional processing of mature transcripts. Sensitivity and specificity analyses were used to determine an optimal T-to-C mutation rate per read cutoff (3.6%, see methods) to assign individual reads as new or old, and we found that this simple binomial approach correlates well with more complex modeling approaches (Figure S2a). New reads aligning anywhere to the gene correlate well between replicates but only moderately with intronic reads (Figure S2b), consistent with the long s4U treatment relative to intronic half-lives, and underscoring the need to assess RNA dynamics on the mature transcript. Next, we quantified and compared relative stability and synthesis rate changes using TimeLapse-seq fraction new estimates and read counts, which allows us to distinguish between stability-driven and synthesis-driven changes in transcripts found to be significantly upregulated in total RNA in the DCP2 KO. To be specific, we assumed first order kinetics at steady state and modeled the rate of RNA decay (kdeg) as a function of fraction new reads (), and the rate of RNA synthesis (ksyn) as a function of kdeg (Ntotal*kdeg), similar to a prior study 54. To further distinguish between decay- and synthesis-driven changes, we combined the calculated changes in the rate of degradation and synthesis in DCP2 KO versus WT cells for transcripts that are significantly up- and down-regulated in DCP2 KO cells from the DESeq2 analysis of the total RNA level (Figure 1c).
After computationally modeling the changes in rates of decay and synthesis in the DCP2 KO cells, we defined classes of dominantly synthesis vs. decay regulated transcripts using an arbitrary cutoff with subsequent experimental validation ( and , respectively) (Figure 1c and Figure S2c). Using this cutoff, 1803 RNAs were identified as stabilized, 514 RNAs were destabilized, and 700 and 2105 were up- or down-regulated by changes in synthesis, respectively, in DCP2 KO versus WT cells. To verify the transcript class definitions, we selected 34 genes identified as stabilized/destabilized in the DCP2 KO, 5 genes that change neither in synthesis or stability (“controls”), and 11 genes that exhibit synthesis changes only (Figure S2c), for validation by quantitative real-time RT-PCR (qRT-PCR) after transcriptional inhibition (Figure 2a and Figure S3a–e). We chose both genes near and away from the class boundary (i.e. or ). 29 out of 33 total tested stabilized genes were validated as true positives stabilized in the DCP2 KO by qRT-PCR (Figure 2a and Figure S3a), while all control genes were validated to have no significant difference in RNA lifetime by qRT-PCR (Figure S3c). A de-stabilized gene, MRPL40, decreased in RNA lifetime by qRT-PCR in DCP2 KO as expected (Figure S3d). Half of predominantly synthesis regulated genes exhibited no significant changes in RNA lifetime in the DCP2 KO by qRT-PCR, also as expected (Figure S3b, e). We interpret changes in synthesis as well as destabilization in the DCP2 KO relative to wild type cells as evidence of compensatory responses to the loss of Dcp2 expression.
Figure 2. Validation of transcript stabilization in DCP2 KO cells.
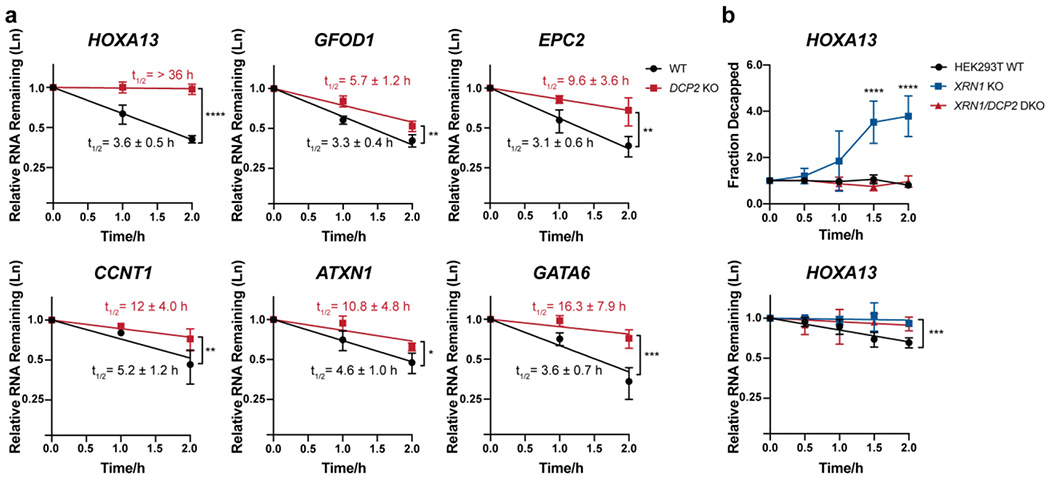
(a) The stabilities of selected Dcp2 targets were measured using qRT-PCR in DCP2 KO versus WT HEK293T cells treated with actinomycin D for the indicated times. Half-lives (95% CI) were obtained from linear regression analysis, and significance was determined using two-tailed t-test of the slope of regression lines. Error bars represent mean ± s.d. n=4 biological replicates for HOXA13, GATA6 and CCNT1 and n=3 biological replicates for all other genes. (b) Splinted ligation RT-PCR (qSL-RT-PCR) 45 in WT, XRN1 KO and XRN1/DCP2 double knockout (DKO) HEK293T cell lines was performed to assay decapping of selected Dcp2 targets. Significance was analyzed by ANOVA linear regression (total RNA) or at individual time points (splint ligation). Error bars represent mean ± s.d. n=4 biological replicates. P-values are denoted by asterisks; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Prior reports have defined Dcp2 substrates as RNAs stabilized in the absence of Dcp2 relative to wild type cells 16. In order to validate transcripts stabilized in the DCP2 KO cells as Dcp2 substrates, we confirmed that decay of selected representatives of this class is inhibited at the decapping step. We utilized a splinted ligation assay (qSL-RT-PCR) previously developed in yeast 44, 48. In this assay, sequence-specific enzymatic ligation of decapped RNAs, which bear a 5′ phosphate group, was mediated by a DNA splint bridging an anchor RNA oligonucleotide of known sequence and 5′ UTR of the target transcript. Semi-quantitative analysis was then carried out via qRT-PCR using primers recognizing the splinted ligation product vs. total RNA (decapped + capped) for each target gene with a ΔΔCt method47. These measurements are made on an XRN1 KO background, which allows the decapped product to accumulate sufficiently for detection 55. Three DCP2 KO-stabilized transcripts, HOXA13 (Figure 2b), CCNT1 and GATA6 (Figure S3f), were selected for analysis. We generated human XRN1 KO cells using CRISPR-Cas9 technology as previously reported 43 (Figure S1b). We measured the decapping kinetics of each target after transcriptional block in WT HEK293T and XRN1 KO cells. For all 3 RNAs, decapped product increased over 2 h in the XRN1 KO, but was undetectable in wild type HEK293T as expected (Figure 2b and Figure S3f, blue versus black trace). We then generated XRN1/DCP2 double knock out (DKO) cells (Figure S1b). In comparison to the XRN1 KO, no decapped product was detectable for any of the three target genes in the DKO cells (Figure 2b and Figure S3f, red trace), consistent with a requirement of Dcp2 activity for their decapping. Taken together, these data suggest that RNAs stabilized in DCP2 KO cells are cellular decapping substrates of Dcp2.
Comparing our list of 1803 Dcp2 substrates to prior reports 16, we have expanded the number of known Dcp2 substrates by more than tenfold. 29 previously reported Dcp2 substrates are re-identified in our TimeLapse-seq analysis. We note that overlap with previously reported datasets is low (20% of reported targets), likely due to (1) different cells and species utilized for analysis (e.g. mouse16 vs. human cells in this study), (2) insignificant changes at the total RNA level (and hence exclusion by our statistical cutoff) as a result of altered synthesis of some Dcp2 targets, or (3) omission of substrates with half-lives significantly longer than the chosen time point for the TimeLapse-seq experiment. Our list is therefore likely incomplete as well as cell type- and/or organism-specific. Despite the low overlap for specific gene targets, gene ontology (GO) analysis using PANTHER 56 revealed that RNA groups that were previously reported to be regulated by Dcp2, such as innate immune response 4, were enriched among stabilized genes in our dataset (FDR=4.6 e-3) (Figure S2d), providing additional evidence that we have identified an expanded list of bona fide Dcp2 substrates.
Human Musashi-2 (Msi2) regulates stability of a subset of human transcripts partially distinct from Dcp2 targets
While Dcp2 has previously been shown to be redundant for decapping of RNAs bearing AU-rich element (ARE) motifs 15, we sought to determine if another motif, the Musashi binding element (MBE), could direct transcripts to Dcp2-mediated decapping. Recently, Musashi-2 (Msi2), an RNA-binding protein that regulates cell proliferation 57–61, was shown to promote decay of its RNA targets without significantly altering their translational efficiency, in contrast to its paralog Msi1 57, 61. In 2014, Park et al. showed that previously reported Dcp2-regulated mRNAs 22 are also upregulated in Msi2-deficient mouse cells 62. It is, however, presently unclear whether human Msi2 targets are selectively degraded through the 5′-to-3′ decay pathway.
We therefore profiled RNA dynamics in a MSI2 KO HEK293T cell line relative to wild type cells (Figure S1c and Figure 3a) via TimeLapse-seq. The validity of the statistical cutoff for significant synthesis and stability changes was confirmed by qRT-PCR (Figure 3b–c). Based on this analysis, we observed stabilization (1171 transcripts) and, to a much lesser degree, destabilization (107 transcripts) in the MSI2 KO, consistent with prior reports that Msi2 destabilizes specific RNA targets. Msi2 targets tend to be translationally repressed (Figure 3d). However, only a small number of genes sensitive to Msi2-mediated decay overlapped with Dcp2 substrates (20%, or 240 total genes stabilized in both the MSI2 KO and DCP2 KO), and poor correlation was found between the differences in decay rate in the DCP2 KO vs WT and that in the MSI2 KO vs WT TimeLapse-seq experiment (Figure 3e), consistent with regulation of largely orthogonal subsets of cellular transcripts by these two proteins. GO analysis of Msi2 targets revealed enrichment for different functions than those regulated by Dcp2, as well as genes related to humoral immune response, as previously reported 59 (Figure S4a). However, for a small number of specific genes, co-regulation by Msi2 and Dcp2 does occur. For example, qSL-RT-PCR of an MBE-containing Dcp2 substrate, HOXA13, in a MSI2/XRN1 DKO cell line (Figure S1d), revealed inhibition of HOXA13 decapping in the absence of Msi2 (Figure S4b), and Msi2 protein partially colocalizes with P-bodies in HEK293T cells (Figure S4c). These observations are consistent with interaction of Msi2 with multiple decay factors, only one of which is Dcp2, for regulation of its bound RNAs, and indicate that, while a small number of specific transcripts require both Msi2 and Dcp2 for their decay, most Dcp2 substrates are not Msi2 targets.
Figure 3.
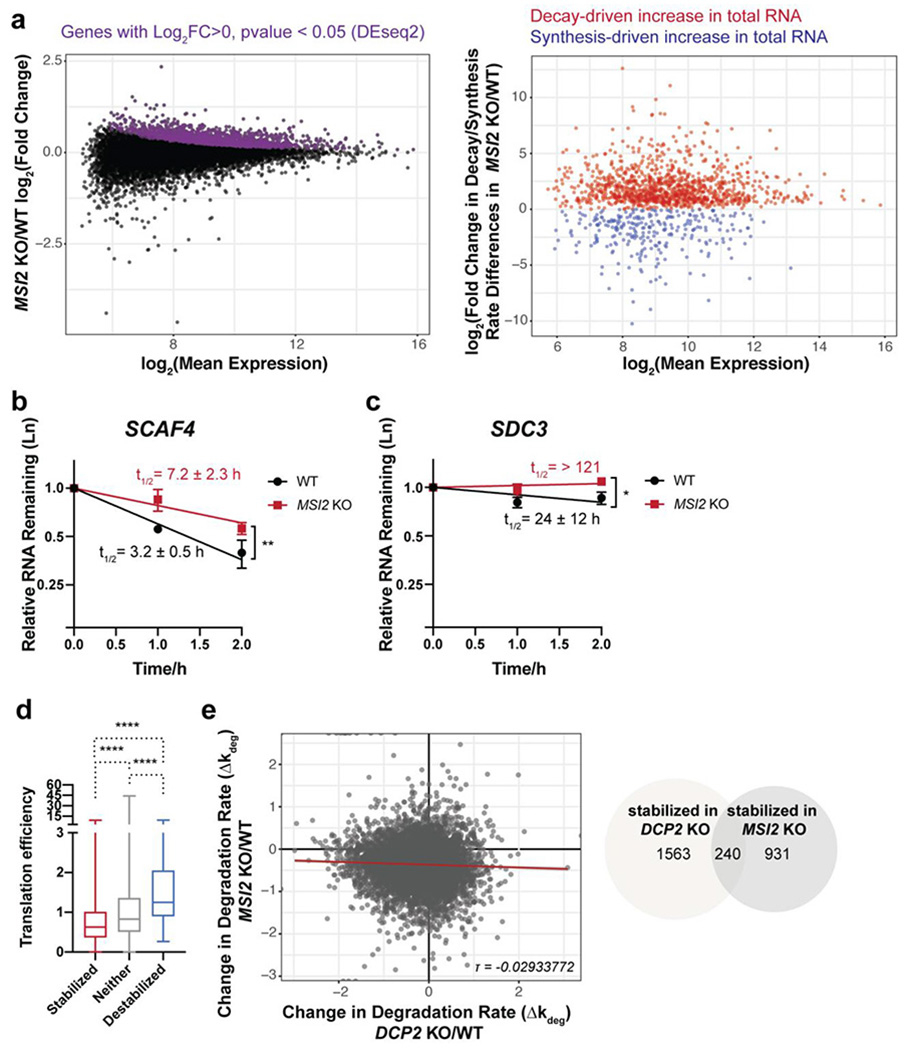
Human Msi2 and Dcp2 regulate distinct subsets of transcripts. (a) TimeLapse-seq analysis of changes in RNA stability and synthesis in MSI2 KO cells. (b-c) qPCR validation in MSI2 KO cells after transcriptional arrest of transcripts stabilized by both MSI2 and DCP2 KO (b), or exclusively by MSI2 KO (c). Error bars represent mean ± s.d. n=3 biological replicates. Half-lives (95% CI) were calculated from linear model, and significance was analyzed by linear regression t-test; *P<0.05; **P < 0.01. (d) Boxplots depicting translation efficiency (data from Sidrauski et al., 2015) for each of the three classes of mRNA stability changes in MSI2 KO versus WT HEK293T cells. Statistical significance is calculated using Mann-Whitney U test; ****P < 0.0001. (e) Msi2 and Dcp2 globally regulate the stability of distinct subsets of RNAs, as shown by low correlation between the changes in estimated RNA degradation rate in each KO relative to WT cells (left, Kendall tau rank correlation coefficient τ=−0.02933772) and low overlap of post-transcriptionally stabilized genes in each KO (right).
The decay of a subset of m6A-modified RNAs is accelerated by Dcp2
m6A is a dynamic internal RNA modification with multiple regulatory functions including modulation of transcription, translation and RNA stability 25–27. m6A regulates RNA stability through reader proteins 32, 63, which can either stabilize or destabilize RNA. For example, the m6A reader protein YTHDF2 has been reported to accelerate decay of its targets partly by recruiting the CCR4-NOT deadenylase complex 64 and localizing to P-bodies 32, while IGF2BP1-3 increases the stability of m6A-modified targets 65. Both of these protein classes have been reported to localize to P-bodies, either transiently or stably, suggesting proximity to Dcp2 32, 49, 65, though YTHDF2 has also been reported to accelerate RNA decay through another pathway, RNase P/MRP 66. In contrast to m6A, m6Am (N6,2-O-dimethyladenosine) has been shown to stabilize RNAs 41, 67. We therefore sought to determine whether m6A and m6Am modifications are enriched or de-enriched in Dcp2 target RNAs, and/or whether these modifications modulate RNA stability through multiple decay pathways.
Generally, m6A modified transcripts decayed slower in the DCP2 KO cells (Figure 4a). Consistent with this trend, a weak but significant positive correlation between genes stabilized by silencing of YTHDF2 expression with Dcp2 substrates defined in this study was also observed, as well as negative correlation of stabilization by YTHDF2 silencing with DCP2 KO destabilization (Figure 4b). Conversely, m6Am modified RNAs were degraded faster and enriched among transcripts destabilized in the DCP2 KO (Figure 4c and Figure S5a). We noted that two previously reported m6A-containing, YTHDF2-associated RNAs, LDLR and CREBBP32, were stabilized in the DCP2 KO. We therefore tested whether m6A is required for their recognition by Dcp2 by silencing expression of the m6A methyltransferase METTL3 in wild type and DCP2 KO cells (Figure 4d–e). Both transcripts increased in stability in wild type HEK293T cells after METTL3 knockdown, as expected, but METTL3 silencing conferred no stabilization in DCP2 KO cells relative to a non-silencing control, indicating that decay of these m6A modified transcripts requires Dcp2 and vice versa. However, only 21% of m6A-modified transcripts are Dcp2 substrates, and conversely only 52% of Dcp2 substrates are modified with m6A (Figure S5a). We therefore conclude that m6A modifications define a subset of Dcp2 substrates, but likely direct RNAs to decay via multiple pathways; conversely, m6A modification is not necessary, but in a subset of modified transcripts may be sufficient, to define a Dcp2 substrate.
Figure 4.
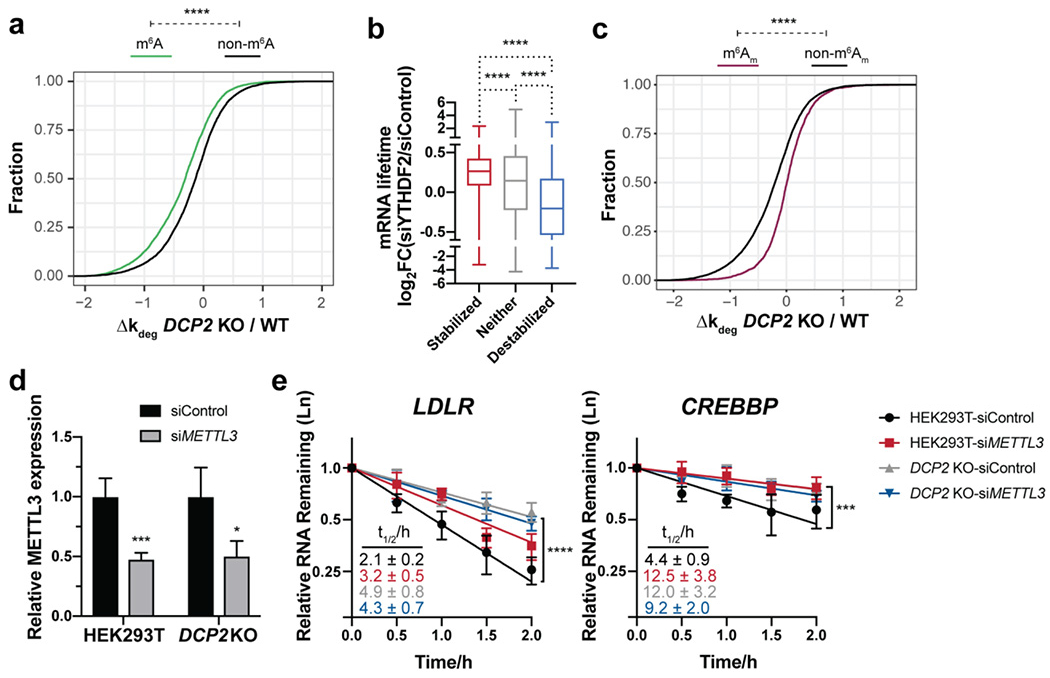
m6A modifications are present in many Dcp2 targets and required for their decay, but are not sufficient to define Dcp2 substrates globally. (a) Cumulative distribution function (CDF) plots of the relative changes in decay rate of transcripts among m6A-containing (data from Jaffrey and co-workers, 2012) and non-target RNAs in DCP2 KO vs. WT. P-values were calculated using two-sided Mann-Whitney test; ****P < 0.0001. (b) Boxplots depicting RNA half-life changes after YTHDF2 silencing in HeLa cells (data from Wang et al., 2014) for each of the three classes of RNA stability changes in DCP2 KO cells. Statistical significance is derived from Mann-Whitney U test; ****P < 0.0001. (c) CDF plots of the relative changes in decay rate of transcripts among m6Am-containing (data from Mauer et al., 2016) and non-target RNAs in DCP2 KO vs. WT. P-values were calculated using two-sided Mann-Whitney test; ****P < 0.0001. (d) Silencing of METTL3 mRNA expression determined by qRT-PCR with reference to β-actin in WT or DCP2 KO HEK293T cells treated with non-targeting siRNA (siControl) or siMETTL3. (e) Relative RNA decay rates of two m6A-containing YTHDF2 targets were quantified by qRT-PCR after transcriptional inhibition in DCP2 KO or WT HEK293T cells transfected with either siMETTL3 or siControl. Error bars shown are mean ± s.d., n=4 biological replicates. P-values are calculated from ANOVA linear regression; ***P < 0.001; ****P < 0.0001.
Dcp2 targets regulatory RNAs enriched in macroscopically observable P-bodies
P-bodies are phase separated liquid droplets composed primarily of ribonucleoprotein complexes associated with translational repression and 5′-to-3′ mRNA decay 68, 69. P-body formation is dependent on RNA and intrinsically disordered regions of Dcp2-associated proteins such as Dhh1/DDX670 and EDC471. Early work showed that, while inhibition of RNA decapping caused an increase in P-body size and number72, nucleation of macroscopically observable P-bodies is unnecessary for RNA decay28. Recently, reports from Chao, Singer, Parker and co-workers demonstrated that 5′-to-3′ decay does not occur within P-bodies, but rather on soluble cytoplasmic protein complexes or small, macroscopically invisible P-bodies29, 73, 74. Macroscopically detectable P-bodies therefore most likely represent sites of RNA sequestration rather than decay, and their involvement in 5′-to-3′ RNA decay is unclear. We therefore sought to determine whether bona fide Dcp2 targets are enriched in large P-bodies. We queried the transcripts stabilized in the DCP2 KO cells for sequence properties and/or RNA-protein interactions associated with 5′-to-3′ decay and P-body enrichment via comparison to published datasets. Stabilization by silencing of DDX675, a P-body-associated protein that couples translational repression to 5′-to-3′ RNA decay76–81, correlated significantly with stabilization in the DCP2 KO (Figure 5a). Correspondingly, decreased translational efficiency and long 5′ and 3′ UTRs were associated with Dcp2 substrates, as expected 82–84 (Figure 5b–d). While transcript length and coding sequence length correlate positively with Dcp2 substrates (Figure S5b–c), as expected due to its correlation with translational efficiency 85, they also exhibit more complex correlations with DCP2 KO destabilized transcripts. Surprisingly, high GC content did not correlate with Dcp2 substrates, despite its impact on translational repression through DDX6 75 (Figure S5d). We finally observed a significant positive correlation of DCP2 KO-stabilized RNAs with RNAs stabilized by knockout of the Dcp2- and P-body-associated exonuclease XRN1 71, 86 (Figure 5e). Perhaps most importantly, an accounting of transcripts enriched in macroscopically observable P-bodies partially purified from human cells via flow cytometry has recently been published 49. A clear separation of the DCP2 KO stabilized and destabilized transcripts according to P-body enrichment vs. de-enrichment, respectively, was observed (Figure 5f). Overall, these trends suggest that Dcp2 substrates accumulate in macroscopically observable P-bodies due to sensing of their translational repression by DDX6; their decapping and 5′-to-3′ decay by Dcp2 and XRN1 may occur after release from large P-bodies.
Figure 5.
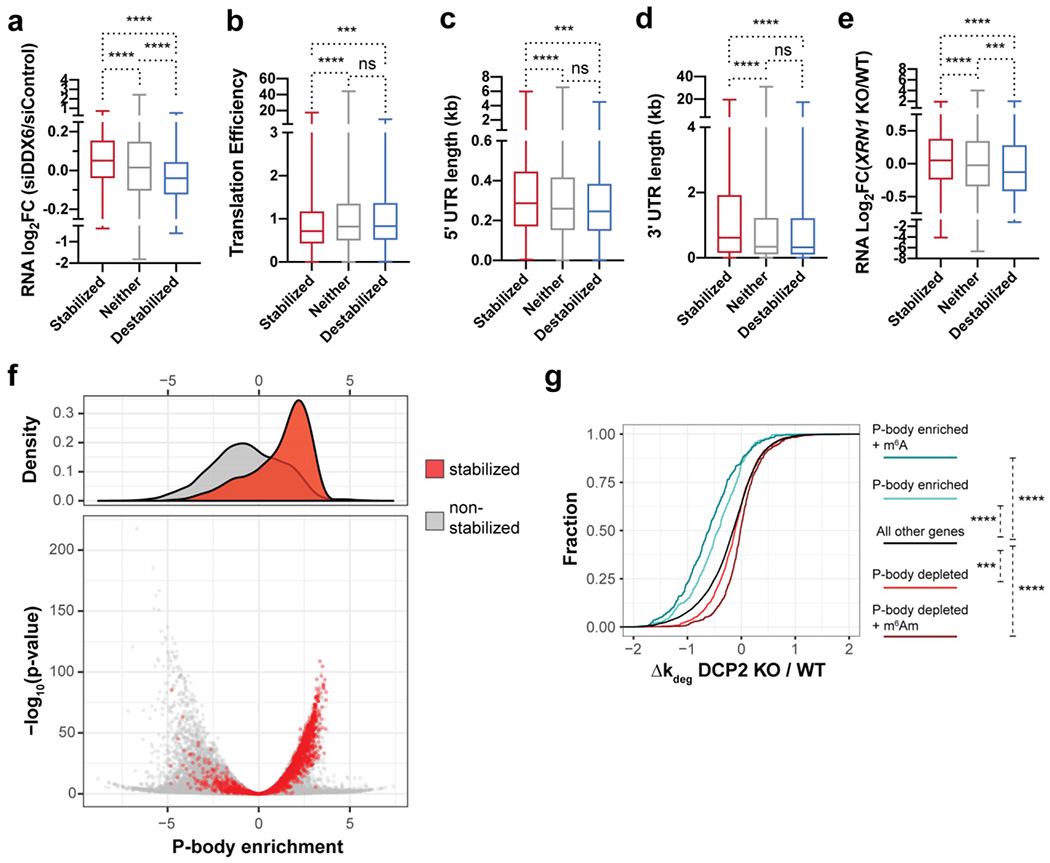
Dcp2 targeting of RNAs correlates with P-body enrichment and is enhanced by m6A modification. (a-e) Boxplots quantifying differential expression of RNAs after DDX6 silencing in HEK293T cells relative to control (data from Courel et al., 2019) (a), translation efficiency (data from Sidrauski et al., 2015) (b), 5′ UTR length (c), 3′ UTR length (reference datasets from Khong et al., 2017) (d), and RNA enrichment after XRN1 knockout in HEK293T cells (data from Chang et al., 2019) (e) for each of the three classes of mRNA stability changes in DCP2 KO versus WT HEK293T cells. Statistical significance is calculated using Mann-Whitney U test. Ns, not significant (p>0.05); ***P<0.001; ****P < 0.0001. (f) P-body-enriched and -depleted RNAs were defined based on data from Hubstenberger et al., 2017 ((log2(fold change RNA enrichment in sorted P-body/pre-sorted fraction)>2.5 and <−2.5, respectively). In the density volcano plot of P-body association status, the red and grey spots represent transcripts stabilized in DCP2 KO cells and all other genes, respectively. (g) CDF plots of the relative changes in decay rate of transcripts among significantly P-body-enriched (defined as in c), P-body depleted, P-body enriched and m6A-containing (m6A data from Jaffrey and co-workers, 2012), P-body depleted and m6Am-containing (m6Am data from Mauer et al., 2017), and non-target RNAs in DCP2 KO relative to WT. P-values were calculated using two-sided Kruskal-Wallis test; ***P<0.001; ****P < 0.0001.
Approximately 70% of Dcp2 substrates are P-body enriched, while only 14% of Dcp2 substrates identified in this study are de-enriched in P-bodies; only 19% of non-Dcp2 substrates are P-body enriched (Figure S5a), all supporting a strong correlation between Dcp2 targets and P-body enrichment. However, only approximately 20% of P-body-enriched transcripts are identified as Dcp2 targets in this study (Figure S5a), suggesting that a mechanism may exist to target specific P-body-enriched transcripts to Dcp2. We asked whether additional RNA marks are required to promote Dcp2-dependent decay of P-body-enriched transcripts. Indeed, P-body enriched and m6A-modified RNAs showed markedly increased stability in DCP2 KO cells compared to P-body-enrichment alone, while P-body depleted and m6Am containing RNAs were significantly destabilized in DCP2 KO relative to RNAs exhibiting only P-body depletion (Figure 5g). Taken together, these results suggest that Dcp2-dependent decay is specified by both the localization to macroscopically observable P-bodies as well as modification by m6A.
DISCUSSION
TimeLapse-seq enables identification of RNAs post-transcriptionally regulated by Dcp2
In this work, we adapted TimeLapse-seq to identify RNA synthesis and stability changes in DCP2 KO human cells, interpreting the set of RNAs post-transcriptionally stabilized in the DCP2 KO as substrates of the human decapping enzyme Dcp2. While overlap with a previous study identifying Dcp2 substrates in mouse cells is low (~20%) 16, similar trends in regulation of pathways such as innate immunity are reproduced, and two key experiments argue that we are effectively identifying the true Dcp2 substrates. First, qRT-PCR validation confirmed significantly increased lifetimes of putative substrates in DCP2 KO cells; second, a splinted ligation assay showed that the observed changes were caused by accumulation of capped intermediates. Overall, we have expanded the list of known Dcp2 substrates in human cells by ~10-fold.
The global nature of TimeLapse-seq, and the ability to distinguish regulation at the synthesis or degradation stage, unexpectedly revealed significant indirect effects of the DCP2 KO. Similar to a previous study in yeast 37, 514 transcripts were destabilized in the DCP2 KO cells - about 30% as many as were stabilized. One explanation could be compensatory upregulation of alternative RNA decay pathways that destabilize these targets. In support of this hypothesis, various other proteins related to RNA destabilization are upregulated in the DCP2 KO, including exosome components (Table S2). Furthermore, 49% of the destabilized RNAs contain m6Am (Figure S5a), consistent with previous reports on the protective role of this modification from Dcp2 activity 41 and an indirect effect of the DCP2 KO on their stability. Combined with the observed changes in synthesis of >2800 transcripts in the DCP2 KO, these results demonstrate significant genetic compensation for loss of DCP2 activity, and suggest that acute inhibition or silencing, rather than constitutive genetic deletion, may be necessary to more fully uncouple the effects of DCP2 enzymatic activity and substrate specificity from secondary changes to the transcriptome.
P-body enrichment is the strongest correlate of Dcp2 substrate specificity and has an additive effect with m6A modification
One key observation from our analysis of the physical basis of Dcp2 specificity is its correlation with P-body enrichment of corresponding RNAs. Although recent evidence has demonstrated that RNAs within P-bodies were protected from decapping 29, 73, 87, 70% of human Dcp2 substrates defined in this study are, in fact, P-body enriched (adjusted P-value<0.05, log2Fold enrichment >0) (Figure S5a), indicating a functional link between P-body enrichment and Dcp2 activity. However, conversely, Dcp2 targets account for only a small fraction of all P-body enriched transcripts (20%), which is consistent with previous studies showing coexisting, orthogonal pathways regulating P-body localized RNAs75. Therefore, P-body localization is necessary, but not sufficient, to direct RNAs to Dcp2-dependent decapping and decay. Our data indicate that no single explanatory factor – sequence motif, modification, or P-body localization - fully determines human Dcp2 substrate selectivity. Instead, we showed that P-body localization and m6A modification exert additive effects on the magnitude of stabilization in the DCP2 KO. Dcp2 substrate selectivity is, therefore, multifactorial.
Our data are consistent with a model for Dcp2 target selection that involves multiple regulatory elements (Figure 6). In this model, a translationally repressed mRNA first undergoes phase separation into macroscopic P-bodies, then is released from the P-body and sorted to different decay pathways based on additional properties; m6A contributes to Dcp2 targeting, while other marks and motifs may sort RNAs for decay by other pathways. We note that our data do not permit direct testing of the order of steps proposed in this model, and that alternative models and explanations for our observed data cannot be excluded - including the possibility that macroscopic P-bodies form spontaneously via phase separation of inactive decapping complexes, and RNAs simply escape from P-bodies via mass action after decay on active, cytoplasmic complexes. In summary, this study defines the Dcp2 targetome in human cells, reveals the complexity of genetic compensation for loss of Dcp2 activity, and identifies an interplay between P-body localization and post-transcriptional modification state in RNA decay pathway selectivity.
Figure 6.
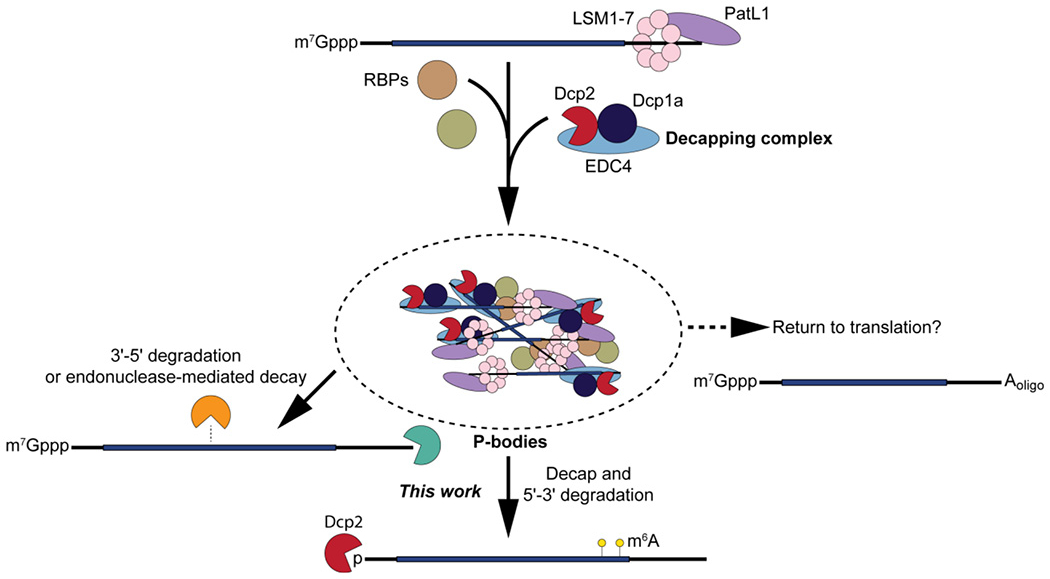
Model: Dcp2 regulation of P-body-associated RNA stability.
CONCLUSION
We have used TimeLapse-seq to investigate global RNA stability changes in Dcp2 knockout vs wild type HEK293T and address the important question of Dcp2 selectivity in human cells. We extensively analyzed RNA features and found that P-body enrichment is the major determinants of Dcp2 targeting, and that m6A modifications have additive effects with P-body enrichment. We posit that translationally repressed RNAs are recruited to P-bodies before being selected for decay by specialized soluble decay complexes via additional molecular modalities.
Supplementary Material
ACKNOWLEDGMENT
We thank all members of the Slavoff and Simon lab for helpful discussion.
Funding Sources
This work was supported in part by the NIH (R01GM122984) and Yale University West Campus start-up funds (to S.A.S.) and by NIH DP2 HD083992-01 (to M.D.S).
ABBREVIATIONS
- m7G
7-methylguanosine
- Dcp2
human m7GpppN-mRNA hydrolase
- UTR
untranslated region
- m6A
N6-methyladenosine
- RNP
ribonucleoprotein
- P-bodies
processing bodies
- TTP
tristetrapolin
- AREs
AU-rich elements
- WT
wild type
- KO
knockout
- DKO
double knockout
- s4U
4-thiouridine
- qRT-PCR
quantitative real-time reverse transcription polymerase chain reaction
- GO
gene ontology
- qSL-RT-PCR
splinted ligation reverse transcription polymerase chain reaction
- fnew
fraction new
- kdeg
rate of RNA decay
- ksyn
rate of RNA synthesis
- MBE
Musashi binding element
- Msi2
Musashi-2
- m6Am
N6,2’-O-dimethyladenosine
- CDF
cumulative distribution function
Footnotes
ACCESSION CODES
Dcp2: Q8IU60
Msi2: Q96DH6
Dcp1a: Q9NPI6
XRN1: Q8IZH2
DDX6: P26196
YTHDF2: Q9Y5A9
METTL3: Q86U44
ASSOCIATED CONTENT
Supporting Information. The following files are available free of charge.
Supplementary figures and list of primers (PDF)
Supplementary table of gene classes described in this work (XLSX)
REFERENCES
- [1].Lykke-Andersen S, and Jensen TH (2015) Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes, Nat. Rev. Mol. Cell Biol 16, 665–677. [DOI] [PubMed] [Google Scholar]
- [2].Schoenberg DR, and Maquat LE (2012) Regulation of cytoplasmic mRNA decay, Nat. Rev. Genet 13, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Popp MW, and Maquat LE (2018) Nonsense-mediated mRNA Decay and Cancer, Curr. Opin. Genet. Dev 48, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Li Y, Dai J, Song M, Fitzgerald-Bocarsly P, and Kiledjian M (2012) Dcp2 decapping protein modulates mRNA stability of the critical interferon regulatory factor (IRF) IRF-7, Mol. Cell. Biol 32, 1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Castellanos-Rubio A, Fernandez-Jimenez N, Kratchmarov R, Luo X, Bhagat G, Green PH, Schneider R, Kiledjian M, Bilbao JR, and Ghosh S (2016) A long noncoding RNA associated with susceptibility to celiac disease, Science 352, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shukla S, and Parker R (2014) Quality control of assembly-defective U1 snRNAs by decapping and 5′-to-3′ exonucleolytic digestion, Proc. Natl. Acad. Sci. U. S. A 111, E3277–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jiao X, Wang Z, and Kiledjian M (2006) Identification of an mRNA-decapping regulator implicated in X-linked mental retardation, Mol. Cell 24, 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen CY, and Shyu AB (2011) Mechanisms of deadenylation-dependent decay, Wiley. Interdiscip. Rev. RNA 2, 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li Y, and Kiledjian M (2010) Regulation of mRNA decapping, Wiley. Interdiscip. Rev. RNA 1, 253–265. [DOI] [PubMed] [Google Scholar]
- [10].Dunckley T, and Parker R (1999) The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif, EMBO J. 18, 5411–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang Z, Jiao X, Carr-Schmid A, and Kiledjian M (2002) The hDcp2 protein is a mammalian mRNA decapping enzyme, Proc. Natl. Acad. Sci. U. S. A 99, 12663–12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xu J, Yang JY, Niu QW, and Chua NH (2006) Arabidopsis DCP2, DCP1, and VARICOSE form a decapping complex required for postembryonic development, Plant Cell 18, 3386–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, and Izaurralde E (2006) mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes, Genes Dev. 20, 1885–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lykke-Andersen J (2002) Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay, Mol. Cell. Biol 22, 8114–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Y, Song M, and Kiledjian M (2011) Differential utilization of decapping enzymes in mammalian mRNA decay pathways, RNA 17, 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Song MG, Li Y, and Kiledjian M (2010) Multiple mRNA decapping enzymes in mammalian cells, Mol. Cell 40, 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Grudzien-Nogalska E, Jiao X, Song MG, Hart RP, and Kiledjian M (2016) Nudt3 is an mRNA decapping enzyme that modulates cell migration, RNA 22, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jiao X, Chang JH, Kilic T, Tong L, and Kiledjian M (2013) A mammalian pre-mRNA 5′ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing, Mol. Cell 50, 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chang JH, Jiao X, Chiba K, Oh C, Martin CE, Kiledjian M, and Tong L (2012) Dxo1 is a new type of eukaryotic enzyme with both decapping and 5′-3′ exoribonuclease activity, Nat. Struct. Mol. Biol 19, 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jiao X, Doamekpor SK, Bird JG, Nickels BE, Tong L, Hart RP, and Kiledjian M (2017) 5′ End Nicotinamide Adenine Dinucleotide Cap in Human Cells Promotes RNA Decay through DXO-Mediated deNADding, Cell 168, 1015–1027.e1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cohen LS, Mikhli C, Jiao X, Kiledjian M, Kunkel G, and Davis RE (2005) Dcp2 Decaps m2,2,7GpppN-capped RNAs, and its activity is sequence and context dependent, Mol. Cell. Biol 25, 8779–8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li Y, Song MG, and Kiledjian M (2008) Transcript-specific decapping and regulated stability by the human Dcp2 decapping protein, Mol. Cell. Biol 28, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li Y, Ho ES, Gunderson SI, and Kiledjian M (2009) Mutational analysis of a Dcp2-binding element reveals general enhancement of decapping by 5′-end stem-loop structures, Nucleic Acids Res. 37, 2227–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Arribas-Layton M, Wu D, Lykke-Andersen J, and Song H (2013) Structural and functional control of the eukaryotic mRNA decapping machinery, Biochim. Biophys. Acta 1829, 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shi H, Wei J, and He C (2019) Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers, Mol. Cell 74, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Patil D, Pickering B, and Jaffrey S (2018) Reading m(6)A in the Transcriptome: m(6)A-Binding Proteins, Trends Cell Biol. 28, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen C, and Shyu A (2017) Emerging Themes in Regulation of Global mRNA Turnover in cis, Trends Biochem. Sci. 42, 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Eulalio A, Behm-Ansmant I, Schweizer D, and Izaurralde E (2007) P-body formation is a consequence, not the cause, of RNA-mediated gene silencing, Mol. Cell. Biol 27, 3970–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Horvathova I, Voigt F, Kotrys AV, Zhan Y, Artus-Revel CG, Eglinger J, Stadler MB, Giorgetti L, and Chao JA (2017) The Dynamics of mRNA Turnover Revealed by Single-Molecule Imaging in Single Cells, Mol. Cell 68, 615–625.e619. [DOI] [PubMed] [Google Scholar]
- [30].Clement SL, Scheckel C, Stoecklin G, and Lykke-Andersen J (2011) Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment, Mol. Cell. Biol 31, 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Franks TM, and Lykke-Andersen J (2007) TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements, Genes Dev. 21, 719–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, Ren B, Pan T, and He C (2014) N6-methyladenosine-dependent regulation of messenger RNA stability, Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, and Wu L (2016) YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex, Nat. Commun 7, 12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Park OH, Ha H, Lee Y, Boo SH, Kwon DH, Song HK, and Kim YK (2019) Endoribonucleolytic Cleavage of m, Mol. Cell 74, 494–507.e498. [DOI] [PubMed] [Google Scholar]
- [35].Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015) N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency, Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li A, Chen Y, Ping X, Yang X, Xiao W, Yang Y, Sun H, Zhu Q, Baidya P, Wang X, Bhattarai D, Zhao Y, Sun B, and Yang Y (2017) Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation, Cell Res. 27, 444–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].He F, Celik A, Wu C, and Jacobson A (2018) General decapping activators target different subsets of inefficiently translated mRNAs, Elife 7, e34409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lugowski A, Nicholson B, and Rissland O (2018) Determining mRNA half-lives on a transcriptome-wide scale, Methods 137, 90–98. [DOI] [PubMed] [Google Scholar]
- [39].Russo J, Heck AM, Wilusz J, and Wilusz CJ (2017) Metabolic labeling and recovery of nascent RNA to accurately quantify mRNA stability, Methods 120, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schofield JA, Duffy EE, Kiefer L, Sullivan MC, and Simon MD (2018) TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding, Nat. Methods 15, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik A, Patil D, Linder B, Pickering B, Vasseur J, Chen Q, Gross S, Elemento O, Debart F, Kiledjian M, and Jaffrey S (2017) Reversible methylation of m(6)A(m) in the 5 ′ cap controls mRNA stability, Nature 541, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sanjana NE, Shalem O, and Zhang F (2014) Improved vectors and genome-wide libraries for CRISPR screening, Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013) Genome engineering using the CRISPR-Cas9 system, Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Blewett N, Coller J, and Goldstrohm A (2011) A quantitative assay for measuring mRNA decapping by splinted ligation reverse transcription polymerase chain reaction: qSL-RT-PCR, RNA 17, 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen CY, Ezzeddine N, and Shyu AB (2008) Messenger RNA half-life measurements in mammalian cells, Methods Enzymol. 448, 335–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Trcek T, Sato H, Singer RH, and Maquat LE (2013) Temporal and spatial characterization of nonsense-mediated mRNA decay, Genes Dev. 27, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schmittgen TD, and Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method, Nat. Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- [48].Hu W, Sweet TJ, Chamnongpol S, Baker KE, and Coller J (2009) Co-translational mRNA decay in Saccharomyces cerevisiae, Nature 461, 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hubstenberger A, Courel M, Bénard M, Souquere S, Ernoult-Lange M, Chouaib R, Yi Z, Morlot JB, Munier A, Fradet M, Daunesse M, Bertrand E, Pierron G, Mozziconacci J, Kress M, and Weil D (2017) P-Body Purification Reveals the Condensation of Repressed mRNA Regulons, Mol. Cell 68, 144–157.e145. [DOI] [PubMed] [Google Scholar]
- [50].Schofield J, Duffy E, Kiefer L, Sullivan M, and Simon M (2018) TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding, Nat. Methods 15, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chan L, Mugler C, Heinrich S, Vallotton P, and Weis K (2018) Non-invasive measurement of mRNA decay reveals translation initiation as the major determinant of mRNA stability, Elife 7, e32536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wu Q, Medina S, Kushawah G, DeVore M, Castellano L, Hand J, Wright M, and Bazzini A (2019) Translation affects mRNA stability in a codon-dependent manner in human cells, Elife 8, e45396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gaidatzis D, Burger L, Florescu M, and Stadler MB (2015) Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation, Nat. Biotechnol 33, 722–729. [DOI] [PubMed] [Google Scholar]
- [54].Neymotin B, Athanasiadou R, and Gresham D (2014) Determination of in vivo RNA kinetics using RATE-seq, RNA 20, 1645–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Baudrimont A, Voegeli S, Viloria E, Stritt F, Lenon M, Wada T, Jaquet V, and Becskei A (2017) Multiplexed gene control reveals rapid mRNA turnover, Sci. Adv 3, e1700006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, and Thomas P (2019) Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0), Nat. Protoc 14, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bennett CG, Riemondy K, Chapnick DA, Bunker E, Liu X, Kuersten S, and Yi R (2016) Genome-wide analysis of Musashi-2 targets reveals novel functions in governing epithelial cell migration, Nucleic Acids Res. 44, 3788–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Katz Y, Li F, Lambert N, Sokol E, Tam W, Cheng A, Airoldi E, Lengner C, Gupta P, Yu Z, Jaenisch R, and Burge C (2014) Musashi Proteins are Post-transcriptional Regulators of the Epithelial-Luminal Cell State, Elife 3, e03915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rentas S, Holzapfel N, Belew M, Pratt G, Voisin V, Wilhelm B, Bader G, Yeo G, and Hope K (2016) Musashi-2 attenuates AHR signalling to expand human haematopoietic stem cells, Nature 532, 508–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sheng W, Dong M, Chen C, Li Y, Liu Q, and Dong Q (2017) Musashi2 promotes the development and progression of pancreatic cancer by down-regulating Numb protein, Oncotarget 8, 14359–14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Duggimpudi S, Kloetgen A, Maney S, Munch P, Hezaveh K, Shaykhalishahi H, Hoyer W, McHardy A, Lang P, Borkhardt A, and Hoell J (2018) Transcriptome-wide analysis uncovers the targets of the RNA-binding protein MSI2 and effects of MSI2’s RNA-binding activity on IL-6 signaling, J. Biol. Chem 293, 15359–15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Park S, Deering R, Lu Y, Tivnan P, Lianoglou S, Al-Shahrour F, Ebert B, Hacohen N, Leslie C, Daley G, Lengner C, and Kharas M (2014) Musashi-2 controls cell fate, lineage bias, and TGF-beta signaling in HSCs, J. Exp. Med 211, 71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shi H, Wang X, Lu Z, Zhao B, Ma H, Hsu P, Liu C, and He C (2017) YTHDF3 facilitates translation and decay of N-6-methyladenosine-modified RNA, Cell Res. 27, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, and Wu L (2016) YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex, Nat. Commun 7, 12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao B, Mesquita A, Liu C, Yuan C, Hu Y, Huttelmaier S, Skibbe J, Su R, Deng X, Dong L, Sun M, Li C, Nachtergaele S, Wang Y, Hu C, Ferchen K, Greis K, Jiang X, Wei M, Qu L, Guan J, He C, Yang J, and Chen J (2018) Recognition of RNA N-6- methyladenosine by IGF2BP proteins enhances mRNA stability and translation, Nat. Cell Biol. 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Park O, Ha H, Lee Y, Boo S, Kwon D, Song H, and Kim Y (2019) Endoribonucleolytic Cleavage of m(6)A-Containing RNAs by RNase P/MRP Complex, Mol. Cell 74, 494–507. [DOI] [PubMed] [Google Scholar]
- [67].Boulias K, Toczydłowska-Socha D, Hawley BR, Liberman N, Takashima K, Zaccara S, Guez T, Vasseur JJ, Debart F, Aravind L, Jaffrey SR, and Greer EL (2019) Identification of the m, Mol. Cell 75, 631–643.e638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Decker CJ, and Parker R (2012) P-bodies and stress granules: possible roles in the control of translation and mRNA degradation, Cold Spring Harb. Perspect. Biol. 4, a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Luo Y, Na Z, and Slavoff S (2018) P-Bodies: Composition, Properties, and Functions, Biochemistry 57, 2424–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kamenska A, Simpson C, Vindry C, Broomhead H, Benard M, Ernoult-Lange M, Lee BP, Harries LW, Weil D, and Standart N (2016) The DDX6-4E-T interaction mediates translational repression and P-body assembly, Nucleic Acids Res. 44, 6318–6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Eulalio A, Behm-Ansmant I, Schweizer D, and Izaurralde E (2007) P-body formation is a consequence, not the cause, of RNA-mediated gene silencing, Mol. Cell. Biol 27, 3970–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Parker R (2003) Decapping and Decay of messenger rna occur in cytoplasmic processing bodies, Science 300, 805–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tutucci E, Vera M, Biswas J, Garcia J, Parker R, and Singer RH (2018) An improved MS2 system for accurate reporting of the mRNA life cycle, Nat. Methods 15, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Pitchiaya S, Mourao M, Jalihal A, Xiao L, Jiang X, Chinnaiyan A, Schnell S, and Walter N (2019) Dynamic Recruitment of Single RNAs to Processing Bodies Depends on RNA Functionality, Mol. Cell 74, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Courel M, Clément Y, Bossevain C, Foretek D, Vidal Cruchez O, Yi Z, Bénard M, Benassy MN, Kress M, Vindry C, Ernoult-Lange M, Antoniewski C, Morillon A, Brest P, Hubstenberger A, Roest Crollius H, Standart N, and Weil D (2019) GC content shapes mRNA storage and decay in human cells, Elife 8, e49708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Coller J, and Parker R (2005) General translational repression by activators of mRNA decapping, Cell 122, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zeidan Q, He F, Zhang F, Zhang H, Jacobson A, and Hinnebusch AG (2018) Conserved mRNA-granule component Scd6 targets Dhh1 to repress translation initiation and activates Dcp2-mediated mRNA decay in vivo, PLoS Genet. 14, e1007806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Radhakrishnan A, Chen YH, Martin S, Alhusaini N, Green R, and Coller J (2016) The DEAD-Box Protein Dhh1p Couples mRNA Decay and Translation by Monitoring Codon Optimality, Cell 167, 122–132.e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Rouya C, Siddiqui N, Morita M, Duchaine TF, Fabian MR, and Sonenberg N (2014) Human DDX6 effects miRNA-mediated gene silencing via direct binding to CNOT1, RNA 20, 1398–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ozgur S, Basquin J, Kamenska A, Filipowicz W, Standart N, and Conti E (2015) Structure of a Human 4E-T/DDX6/CNOT1 Complex Reveals the Different Interplay of DDX6-Binding Proteins with the CCR4-NOT Complex, Cell Rep. 13, 703–711. [DOI] [PubMed] [Google Scholar]
- [81].Carroll JS, Munchel SE, and Weis K (2011) The DExD/H box ATPase Dhh1 functions in translational repression, mRNA decay, and processing body dynamics, J. Cell Biol 194, 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Roy B, and Jacobson A (2013) The intimate relationships of mRNA decay and translation, Trends Genet. 29, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Mishima Y, and Tomari Y (2016) Codon Usage and 3′ UTR Length Determine Maternal mRNA Stability in Zebrafish, Mol. Cell 61, 874–885. [DOI] [PubMed] [Google Scholar]
- [84].Mugridge JS, Coller J, and Gross JD (2018) Structural and molecular mechanisms for the control of eukaryotic 5′-3′ mRNA decay, Nat. Struct. Mol. Biol 25, 1077–1085. [DOI] [PubMed] [Google Scholar]
- [85].Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, and Parker R (2017) The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules, Mol. Cell 68, 808–820.e805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Chang CT, Muthukumar S, Weber R, Levdansky Y, Chen Y, Bhandari D, Igreja C, Wohlbold L, Valkov E, and Izaurralde E (2019) A low-complexity region in human XRN1 directly recruits deadenylation and decapping factors in 5′-3′ messenger RNA decay, Nucleic Acids Res. 47, 9282–9295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Schütz S, Nöldeke ER, and Sprangers R (2017) A synergistic network of interactions promotes the formation of in vitro processing bodies and protects mRNA against decapping, Nucleic Acids Res. 45, 6911–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.