

Abstract
Small molecule targeted therapies have demonstrated outstanding potential in the clinic. These drugs are designed to minimize adverse effects by selectively attacking cancer cells while exerting minimal damage to normal cells. Although initial response to targeted therapies may be high, yielding positive response rates and often improving survival for an important percentage of patients, resistance often limits long-term effectiveness. On the other hand, immunotherapy has demonstrated durable results, yet for a limited number of patients. Growing evidence indicates that some targeted agents can modulate different components of the anti-tumor immune response. These include immune sensitization by inhibiting tumor cell-intrinsic immune evasion programs or enhancing antigenicity, as well as direct effects on immune effector and immunosuppressive cells. The combination of these two approaches, therefore, has the potential to result in synergistic and durable outcomes for patients. In this review, we focus on the latest advances on integrating immunotherapy with small molecule targeted inhibitors. In particular, we discuss how specific oncogenic events differentially affect immune response, and the implications of these findings on the rational design of effective combinations of immunotherapy and targeted therapies.
Introduction
Over the past decade, immunotherapy has cemented its status as a vital component of cancer care. In particular, immune checkpoint blockade (ICB) agents, most notably inhibitors of the PD-1/PD-L1 and CTLA-4 pathways, have become standard of care for many solid and hematologic malignancies, leading to durable results and improved long-term protection from relapse. This latter effect is likely mediated by the induction of an adaptive immune memory capable of eradicating otherwise obstinate tumor cells. Despite their broad applicability, however, ICB benefits only a limited number of patients. Best durable responses have been observed in melanoma, where five-year survival was reported at 26% for ipilimumab (anti-CTLA-4) and 44% for nivolumab (anti-PD-1), and non-small cell lung cancer (NSCLC), where overall survival (OS) approximates 16% after five years (1,2). In an effort to harness this unique potential, the research field has seen a revamped focus on understanding how current and new therapies can influence anti-tumor immune response. In fact, multiple strategies aiming to potentiate immunotherapy are currently under preclinical and clinical investigation. Conventional cancer therapies and small molecule targeted inhibitors have been shown to modulate various components of tumor immunity and response to immunotherapy (3–5). Targeted agents in particular exert these effects by altering mechanisms of immune escape encoded by oncogenic pathways in tumor cells. Therefore, a deeper understanding of how specific oncogenic events shape tumor immunity will prove crucial to the successful development of immunomodulatory strategies. To this end, targeted therapies are ideally situated to block or enhance relevant pathways while exerting minimal damage to normal cells.
Immune evasion by cancer cells
The process by which tumor cells evade immune surveillance can be better understood through the concept of immunoediting (1,6). Initially, malignant cells are regularly detected and eliminated by the immune system through recognition of immunogenic antigens and generation of an innate and adaptive immune response. Acute inflammation activates innate immunity, leading to dendritic cell (DC) maturation and subsequent priming of T-cells, which are central to the anti-tumor response. This constant pressure, however, may act to select for tumor cells that are able to escape immune attack and remain in equilibrium until further changes promote overt tumor growth. This final process is usually accompanied by a shift from acute to chronic inflammation and the establishment of an immunosuppressive tumor microenvironment (TME) via recruitment of immune suppressive cells whose normal function is to dampen immune response, including regulatory T-cells (TRegs), pro-tumorigenic tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), which further facilitate tumor growth. As a consequence, T-cells in general become exhausted or dysfunctional, and therefore unable to mount an effective anti-tumor response (Fig. 1).
Figure 1.
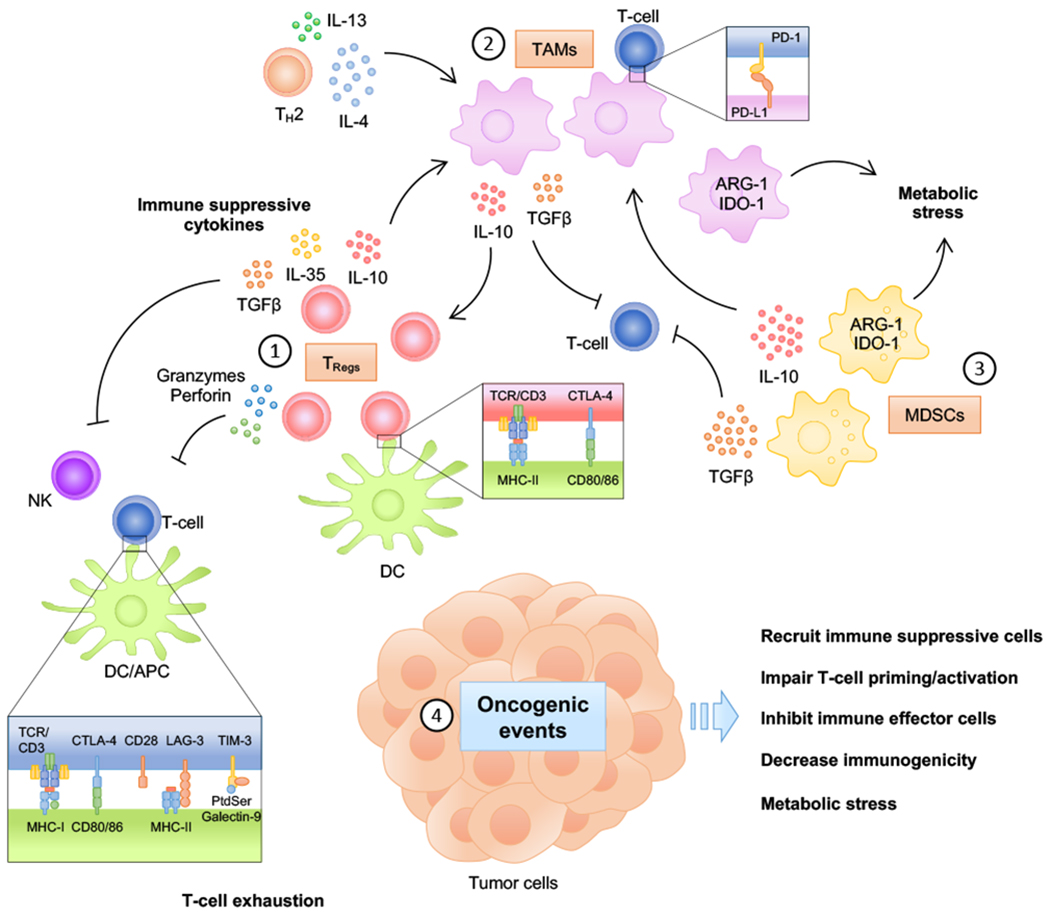
Generation of an immune suppressive tumor microenvironment. (1) Regulatory T-cells (TRegs) suppress immune response via direct cell contact and humoral mechanisms. TRegs constitutively express CTLA-4, which binds to CD80 and CD86 on antigen-presenting cells, such as dendritic cells (DCs), leading to impaired DC maturation and blocking binding of CD80/CD86 to CD28 on conventional T-cells, thereby preventing co-stimulation and T-cell activation. Moreover, TRegs can directly target effector T-cells (TEff) and natural killer (NK) cells for destruction by secreting cytotoxic granzymes and perforin. Secretion of inhibitory cytokines such as TGFβ, IL-10 and IL-35 further inhibit anti-tumor immune response. (2) Tumor-associated macrophages (TAMs) are a major component of the immune infiltrate in solid tumors. Chronic inflammation within the TME and production of IL-4 and IL-13 by TH2 cells and IL-10 by TRegs induce pro-tumorigenic macrophage polarization. In turn, TAMs exacerbate immune suppression by releasing cytokines such as IL-10 and TGFβ that suppress TEff and NK cells but stimulate TRegs. Pro-tumorigenic TAMs also up-regulate metabolic enzymes such as IDO-1 and Arg-1, which can severely affect the composition of the immune infiltrate by competing for catabolism of nutrients. In addition, TAMs can directly inhibit T-cells by expressing immune checkpoint ligands PD-L1 and PD-L2. (3) Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that accumulate in response to chronic inflammation and fail to differentiate into mature cells. MDSCs secrete significant levels of IL-10 and TGFβ, thereby inducing TReg accumulation and pro-tumorigenic macrophage polarization, while simultaneously inhibiting TEff and NK cells function and activation. Furthermore, MDSCs promote metabolic stress by dramatically depleting nutrients needed for T-cell function. (4) Oncogenic events can directly and indirectly inhibit immune response by multiple mechanisms: a) Numerous cytokines secreted by tumor cells, including TGFβ, IL-10 and the pro-angiogenic molecule VEGF, promote recruitment of immune suppressive cells. b) Down-regulation of pro-inflammatory chemokines, including CCL3, CCL4 and CCL5, and CXCR3 ligands such as CXCL9 and CXCL10 result in decreased DC and T-cell recruitment and impaired T-cell priming/activation. c) Expression of PD-L1 and direct inhibition of T-cell effector function by tumor cells has been observed in numerous cancer types. PD-L1 can be induced by multiple non-exclusive mechanisms, including by cytokines such as type I and II IFNs, TNFα and IL-10, and specific oncogenic events, including chromosomal amplification and up-regulation by oncogenic signaling. d) Decreased immunogenicity may result from defects in antigen presentation and/or defective response to IFNγ, which can occur due to genomic inactivation or downregulation of class I MHC and MHC-related molecules (e.g. B2M) or of genes related to the IFNγ pathway. e) Tumor cells also exert strong metabolic stress on the immune infiltrate by competing for nutrients and secreting byproducts that negatively affect immune effector function. (Reviewed on (1,4,6).)
Oncogenic signaling pathways shape the tumor immune microenvironment
Oncogenic signaling pathways have the potential to affect every component of tumor immunity. Careful analysis of clinical studies and the development of relevant animal models are key steps to maximize translational potential. Studies using genetically engineered mouse models (GEMMs) correlated with clinical data have provided much insight into how specific oncogenic events differentially contribute to immune escape. Mechanistic studies for target prediction and biomarker discovery, as well as pre-clinical evaluation in mouse models thus provide important information for designing potentially successful clinical trials (Fig. 2). Importantly, mechanisms of immune evasion and de novo as well as acquired resistance to immunotherapy often overlap, thus underscoring the potential for targeted approaches that could simultaneously sensitize tumors to immunotherapy and prevent recurrence. In this section, we review some of the major molecular mechanisms described to date.
Figure 2.
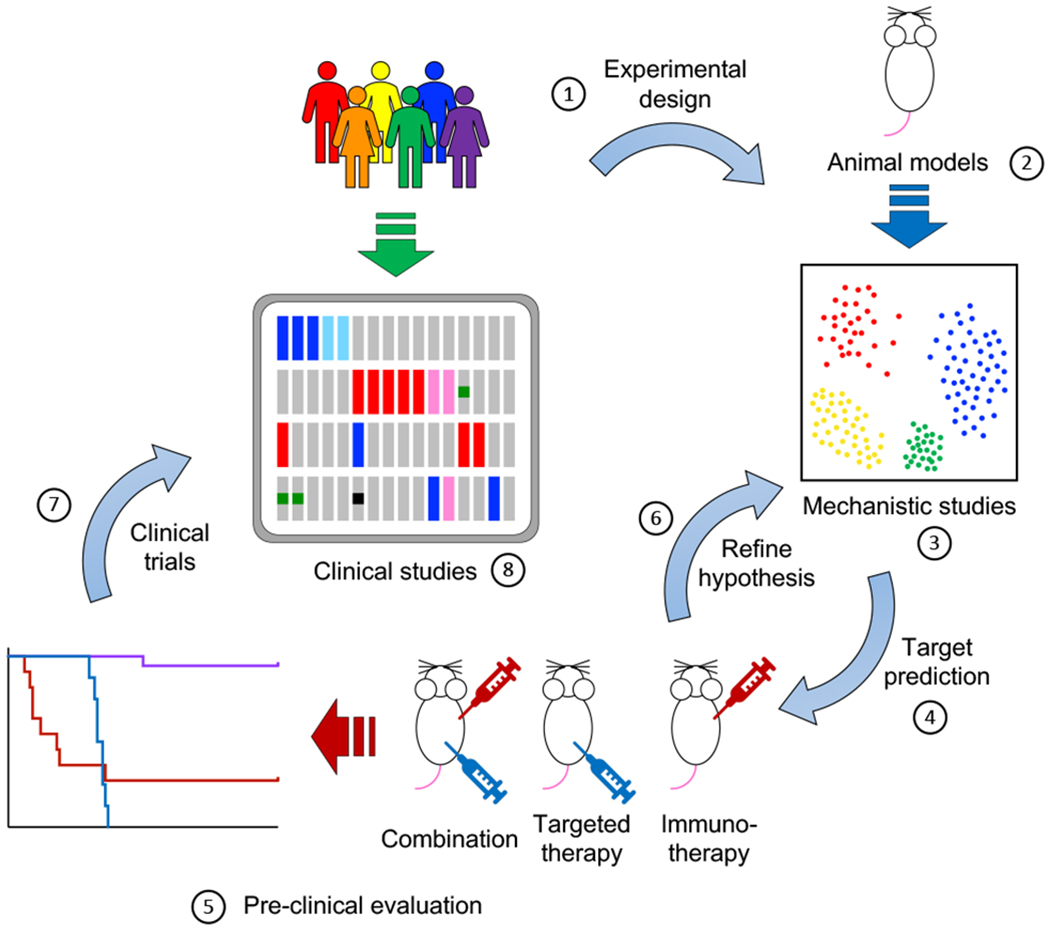
Integration of clinical and animal studies in translational immuno-oncology. (1) Experimental design informed by clinical observations to maximize translational potential. (2) Development of animal models that recapitulate genetic abnormalities found in the clinic. In this regard, immunocompetent, syngeneic mouse models provide the current gold standard. Emerging technologies, such as mouse models engrafted with humanized immune systems can improve the clinical relevance of pre-clinical studies and maximize translational feasibility. (3) Mechanistic studies are a crucial component of modern tumor immunology research. Complementing classical gain- and loss-of-function experiments, powerful technologies such as single-cell RNA sequencing (scRNA-Seq), cytometry by time-of-flight (CyTOF) and highly multiplex tissue cyclic immunofluorescence (t-CyCIF) have greatly enhanced our ability to interrogate the nature and degree of interplay between tumor and immune cells. Future work will undoubtedly entail more robust integration of single-cell expression analyses with single-cell spatial relationships within tissues. (4-6) Iterative rounds of target prediction (4), pre-clinical evaluation (5) and refining hypothesis (6) are needed to identify promising targets of clinical relevance. (7) Results from pre-clinical studies are used to inform and support the design of clinical trials for promising combinations.
MYC
The MYC oncogene was shown to directly up-regulate expression of the innate immune inhibitor receptor CD47, a so called “don’t eat me” signal, and of the adaptive immune checkpoint ligand PD-L1 in lymphoma/leukemia models of conditional MYC over-expression (Fig. 3A) (7). These results were subsequently corroborated by multiple groups in different cancer models (8). Furthermore, conditional MYC activation in a KRASG12D-driven model of lung cancer showed that MYC drives tumor progression and recruitment of an immunosuppressive TME characterized by a marked influx of macrophages and depletion of T-cells, B-cells and natural killer (NK) cells (9). These effects were mediated by tumor-secreted CCL9 and IL-23, which enhanced recruitment of PD-L1+ macrophages and promoted lymphocyte exclusion, respectively (Fig. 3A) (9). In turn, MYC deactivation reversed these changes and led to tumor regression, which was dependent on NK cells but not on T-cells (9). Notably, CCL9/IL-23 co-blockade inhibited tumor progression, while PD-L1 blockade restored T-cell infiltration but did not measurably affect tumor growth (9). More recently, newly developed small molecule MYC inhibitors that disrupt MYC/MAX dimerization were shown to promote anti-tumor immune response, and to synergistically inhibit tumor growth of MyC-Cap mouse prostate cancer allografts when combined with PD-1 blockade (10).
Figure 3.
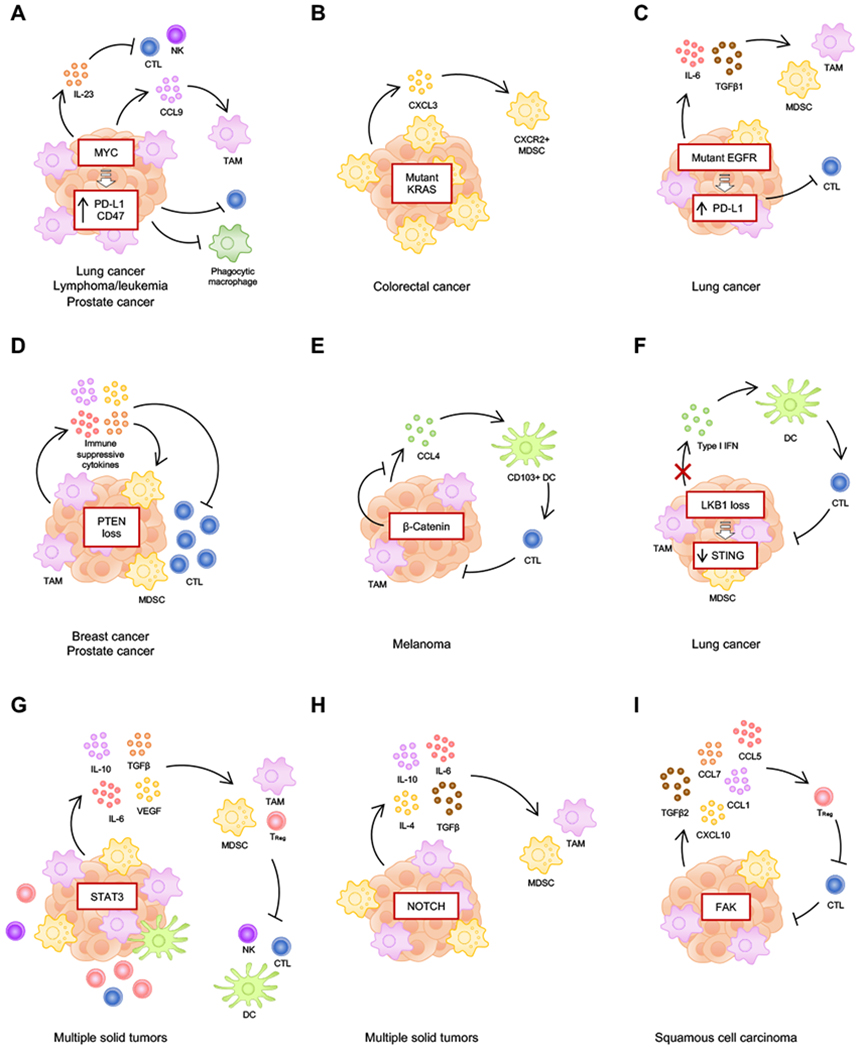
Tumor-intrinsic molecular mechanisms of immune suppression driven by specific oncogenic events. Selected examples are depicted based on mechanistic studies on animal models. Of note, co-occurring oncogenic events affect immune suppressive mechanisms, thus increasing immune heterogeneity between cancer cases. Additional mechanisms linked to each example have also been described but could not be included in this diagram due to space constraints. A, MYC has been shown to promote T-cell and natural killer (NK) cell exclusion, and infiltration of tumor-associated macrophages (TAMs), while also directly inhibiting T-cells and phagocytic macrophages via upregulation of PD-L1 and CD47. B, Mutant KRas has been shown to promote recruitment of myeloid-derived suppressor cells (MDSCs) to the TME through upregulation of CXCL3. C, Mutant EGFR has been shown to up-regulate PD-L1 in tumor cells and to induce recruitment of TAMs and MDSCs. D, Loss of PTEN is associated with increased production of immune suppressive cytokines, which promote the establishment of an immune suppressive tumor microenvironment (TME) and inhibit T-cell infiltration. E, β-Catenin has been shown to inhibit secretion of CCL4 by tumor cells, hence preventing activation of CD103+ dendritic cells (DCs) and subsequent cytotoxic T lymphocyte (CTL) activation. F, Loss of LKB1 down-regulates the STING pathway in tumor cells, thereby preventing release of type I IFNs in response to cytoplasmic double-stranded DNA (dsDNA), which would otherwise stimulate immune response. G, STAT3 signaling in tumor cells induces upregulation of multiple cytokines that contribute to the establishment of an immune suppressive TME by stimulating suppressive immune cells and inhibiting effector cells. H, Dysregulated NOTCH promotes an immune suppressive TME via multiple anti-inflammatory cytokines. I, FAK has been shown to stimulate regulatory T-cells (TRegs) by upregulating numerous cytokines.
KRAS
KRASG12D was shown to mediate immune suppression in a GEMM of colorectal carcinoma (CRC) with inducible KRASG12D and additional APC and p53 double deletion (11). In this case, KRASG12D repressed expression of IRF2, thus alleviating repression of CXCL3 expression by CRC tumor cells and promoting recruitment of CXCR2+ MDSCs to the TME (Fig. 3B) (11). While single agents against PD-1 or CXCR2 did not affect tumor growth or survival, combined treatment significantly increased survival and inhibited tumor growth (11). Furthermore, a novel KRASG12C-specific inhibitor, AMG510, strongly promoted a pro-inflammatory TME and synergized with anti-PD-1 to inhibit mouse syngeneic CT-26 CRC tumors with enforced KRASG12C expression, which led to complete regression in 90% of cases (9/10) and immunological memory, as shown by the ability to reject a second challenge of CT-26 tumor cells (12).
EGFR and HER2
Mutant EGFR in lung cancer mouse models has been shown to promote the establishment of an immunosuppressive TME characterized by low levels of cytotoxic T lymphocytes (CTLs) and increased markers of T-cell exhaustion (Fig. 3C) (13). Ectopic mutant EGFR expression in bronchial epithelial BEAS2B cells up-regulates PD-L1 expression, while small molecule EGFR inhibition in NSCLC cell lines down-regulates PD-L1 (13). Consistently, mouse lung adenocarcinoma tumors driven by EgfrL858R display high myeloid cells infiltration, reduced CD4+ T-helper response and blunted CD8+ T-cell expansion, compared to tumors driven by KrasG12D or concomitant KrasG12D and p53 deletion (14). In the case of HER2, HER2-positive breast cancers predominantly exhibit immune subtypes consistent with ongoing immune activity, including IFNγ-dominant phenotype (~50% of cases; characterized by strong CD8+ and anti-tumorigenic macrophage signals) and wound healing phenotype (~44% of cases; characterized by high expression of angiogenic genes, high proliferation and TH2-type responses) (15,16). In addition to inhibiting oncogenic HER2 signaling in tumor cells, anti-HER2 targeted monoclonal antibodies stimulate innate and adaptive immune responses critical for clinical efficacy (17). These effects are mediated primarily via antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis, and by inducing antigen cross-presentation and T-cell priming (17). Considering the aggressive nature of HER2+ breast cancers and the outstanding therapeutic effect of anti-HER2 monoclonal antibodies, these observations underscore the power of the immune system to subdue highly malignant tumor cells.
PTEN
Genetic loss of PTEN is associated with reduced anti-tumor immunity in multiple cancers (18–20). In melanoma, PTEN deficiency correlated with decreased response to ICB in a cohort of patients (n = 39), and with decreased immune activation scores in melanoma samples from TCGA (20). Interestingly, PTEN deficiency and WNT/β-catenin pathway activation were largely non-overlapping (20). Using a BRAF-mutant melanoma xenograft model with ectopic expression of melanoma antigen gp100 and MHC class I H2-Db, which is specifically recognized by CD8+ T-cells from transgenic PMEL-1 mice, it was shown that PTEN silencing in tumor cells reduced T-cell infiltration and cytotoxic response (Fig. 3D) (20). Moreover, since PTEN-deficient tumors preferentially signal through PI3Kβ (21), treatment with the PI3Kβ isoform-specific inhibitor GSK2636771 improved response to PD-1 blockade in a GEMM of BRAFV600E/PTEN-null melanoma (20). Similarly, a novel chimeric GEMM of metastatic castration-resistant prostate cancer (mCRPC) with triple deletion of PTEN, p53 and Smad4 showed markedly enhanced response to combined PD-1/CTLA-4 blockade when combined with GSK2636771 (22). These mCRPC tumors were highly infiltrated by Gr-MDSCs, which contributed to primary resistance to immunotherapy, and showed synergistic response to ICB in combination with targeted agents that preferentially affect Gr-MDSCs, such as the tyrosine kinase inhibitor cabozantinib, the PI3K/mTOR dual inhibitor BEZ235 and the CXCR1/2 inhibitor SX-682 (22). Of note, the same group had previously reported that additional loss of Smad4 in a PTEN-null prostate cancer GEMM dramatically enhances tumor progression, metastatic spread and lethality (23), and up-regulates CXCL5 expression in tumor cells via HIPPO-YAP1 signaling, which enhances recruitment of immune suppressive CXCR2+ MDSCs (22).
WNT/β-catenin
Analysis of human melanoma samples revealed a correlation between T-cell exclusion and WNT/β-catenin signaling, including gain-of-function mutations on the β-catenin gene (CTNNB1) and up-regulated expression of β-catenin target genes (24). To further investigate these findings, the authors compared a GEMM of metastatic melanoma driven by BRAFV600E and PTEN loss with a syngeneic model harboring additional constitutively active β-catenin, thus showing that β-catenin inhibits production of CCL4 by tumors cells, which leads to impaired recruitment of CD103+ DCs and consequently to impaired T-cell activation (Fig. 3E) (24). Of note, while BRAFV600E/PTEN-null tumors responded to combined PD-L1/CTLA-4 blockade and exhibited significant growth inhibition, tumors with additional β-catenin activation failed to respond to this immunotherapy (24). Consistently, WNT/β-catenin signaling was found to inversely correlate with T-cell infiltration in colorectal cancer (25), and across multiple cancer types compiled from The Cancer Genome Atlas (TCGA) (26).
LKB1
Loss of LKB1 in a mouse model of NSCLC driven by mutant KRAS results in neutrophil accumulation and increased T-cell exhaustion (27). Interestingly, LKB1 loss is associated with decreased PD-L1 expression and resistance to PD-1 blockade in mouse models and patient tumors (27). Indeed, retrospective analyses of clinical response in patients with KRAS-mutant lung adenocarcinoma identified genomic mutations on LKB1 as a significant biomarker for primary resistance to anti-PD-1/PD-L1 immunotherapy, as well as in another cohort of NSCLC irrespective of KRAS status (28). Further work demonstrated that LKB1 deficiency in KRAS-mutant lung cancer results in down-regulation of STING and, consequently, an inability to respond to cytoplasmic double-stranded DNA (dsDNA) (29). STING down-regulation facilitates immune escape by preventing STING-mediated expression of type I interferons (IFNs) and pro-inflammatory cytokines, which are necessary for proper engagement and activation of anti-tumor immune response (Fig. 3F) (30).
STAT3 and NF-κB
Signaling pathways that regulate expression of inflammatory cytokines, such as STAT3 and NF-κB, have the potential to dramatically affect immune response. STAT3 can promote immune escape by up-regulating immune suppressive genes, including IL-6, IL-10, TGFβ and VEGF, while simultaneously down-regulating immune effector genes such as type I and II IFNs, IL-12, CD80, CD86, MHC class II molecules, CCL5 and CXCL10 (31). Tumor cell-intrinsic STAT3 promotes paracrine activation of STAT3 in various populations of immune cells, thereby reducing NK and T-cell cytotoxicity, inhibiting DC maturation and TH1-type response, and stimulating immunosuppressive cells such as MDSCs, TRegs and TAMs (Fig. 3G) (32,33). In a breast cancer GEMM driven by the polyoma virus middle T antigen (PyMT), which is characterized by aggressive and metastatic tumors with latencies around three to four weeks and 80% penetrance, genetic ablation of Stat3 resulted in early hyperplastic lesions that were readily cleared by the immune system, although after a latency averaging 40 weeks, 30% of these mice developed non-metastatic tumors that escaped immune surveillance and markedly lacked immune infiltration (34). In addition, STAT3 inhibits expression of numerous immunostimulatory genes downstream of NF-κB (31). The NF-κB pathway plays an important role in activating programs of immune response; however, aberrant NF-κB signaling has been shown to exert strong oncogenic effects by up-regulating genes that promote cell proliferation and survival (35). STAT3 binding to NF-κB promotes transactivation of oncogenic genes and prevents binding to genes involved in immune response (31,36). Furthermore, multiple upstream events, including growth factor and cytokine receptors, non-receptor tyrosine kinases like Src and Abl, and Toll-like receptors (TLRs) induce STAT3 and NF-κB activation either directly, or indirectly via autocrine and paracrine signaling (31).
NOTCH
Dysregulated NOTCH signaling in tumor cells can up-regulate expression of anti-inflammatory cytokines, including TGFβ, IL-4, IL-6 and IL-10, thereby promoting an immunosuppressive TME (Fig. 3H) (37).
FAK
Focal Adhesion Kinase (FAK) was shown to induce CD8+ T-cell exhaustion and promote TReg recruitment via regulation of multiple cytokines, including CCL1/5/7, CXCL10 and TGFβ2, in a mouse model of squamous cell carcinoma (SCC) (Fig. 3I), and these effects could be reversed by pharmacological targeting of FAK by VS-4718 (38). Similar findings were described in pancreatic ductal adenocarcinoma (PDAC), where FAK inhibition with VS-4718 renders KrasG12D; Trp53L/+ PDAC tumors sensitive to adoptive cell transfer (ACT) or PD-1 blockade immunotherapy (39).
Integrating small molecule targeted therapy and immunotherapy to improve therapeutic outcomes
Distinct small molecule targeted therapies have been shown to exert specific effects on anti-tumor immune response in mouse models and in the clinic (Fig. 4A). Inhibitors of BRAF, Cyclin-dependent kinase 4 and 6 (CDK4/6) and Poly (ADP-ribose) polymerase 1/2 (PARP) are currently being tested in combination with ICB in clinical trials and have thus far shown promising potential. In this section we discuss these three kinds of inhibitors as examples of targeted agents with immune modulatory properties.
Figure 4.
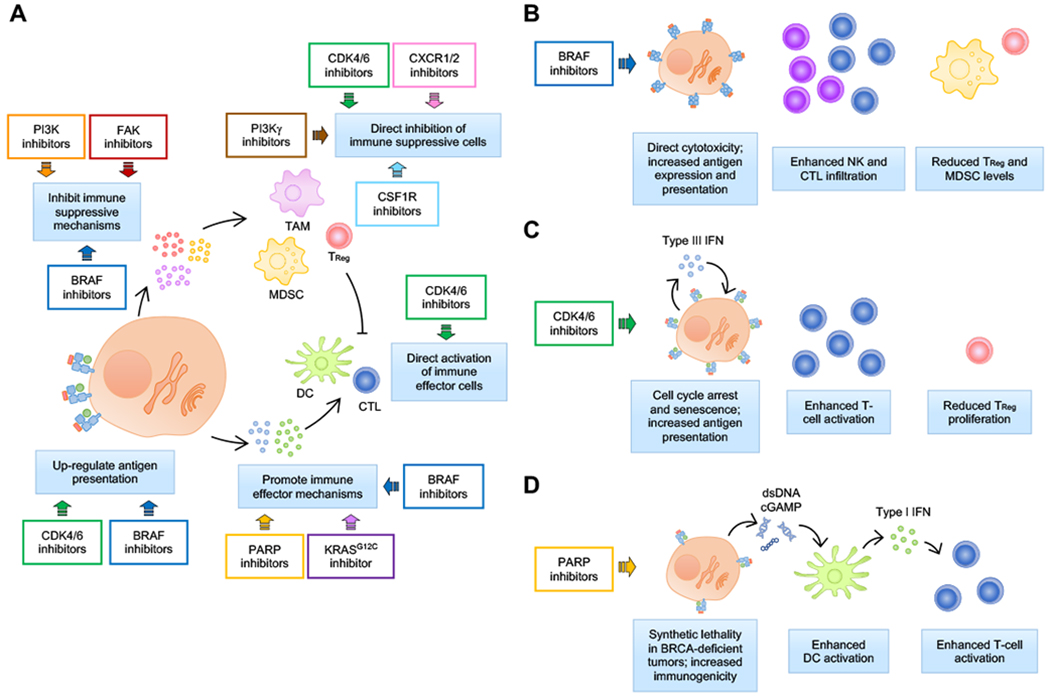
Immune modulation by small molecule targeted therapies. A, Targeted therapies have been shown to affect multiple aspects of cancer immunity, including inhibition of anti-inflammatory mechanisms and promotion of pro-inflammatory mechanisms, up-regulation of antigen presentation, and direct modulatory effects on immune cells. Some targeted agents have been designed to specifically target immune sub-populations. For example, PI3Kγ and CSFR1 inhibitors are used to deplete tumor-associated macrophages (TAMs), and CXCR1/2 inhibitors are used to inhibit myeloid-derived suppressor cells (MDSCs). Other drugs, such as BRAF, PI3K, FAK and KRASG12C inhibitors, were found to affect immune-related mechanisms in addition to their intended cytotoxic effect on tumor cells largely because oncogenic signaling from tumor cells modulates immune response. In the case of PARP inhibitors, enhanced immunogenicity seems to be a corollary of its primary effect on inducing irreparable DNA damage; however, engagement of a robust immune response is required for effective response. And in the case of CDK4/6 inhibitors, unexpected effects in tumor antigenicity as well as direct effects on immune suppressive and immune effector cells have been reported by independent research groups. B-D, Summary of immune modulatory effect of selected examples of targeted therapies currently under clinical investigation in combination with immune checkpoint blockade immunotherapy.
BRAF inhibitors
Treatment with BRAF inhibitors has been shown to increase melanoma differentiation antigen (MDA) expression and presentation by tumor cells, increase NK cell infiltration, and reduce TReg and MDSC levels in cell and mouse models of BRAF-mutant melanoma (Fig. 4B) (40–42). Using the SM1 model of BRAFV600E mouse melanoma and SM1 cells stably expressing the chicken ovalbumin (OVA) antigen (SM1-OVA), treatment with the BRAF inhibitor vemurafenib improved ACT immunotherapy with T-cells specific against OVA as well as with PMLE-1 T-cells recognizing endogenous gp100 in SM1 cells (43). Furthermore, BRAF inhibition with dabrafenib in combination with the MEK inhibitor trametinib enhanced PMLE-1 ACT, leading to increased CD8+ T-cell infiltration and cytotoxicity, and complete tumor regressions (44). Combined dabrafenib and trametinib also improved response to PD-1 blockade in this model (44). Analysis of biopsy samples from patients with metastatic melanoma also revealed an association between treatment with combined BRAF and MEK inhibition, and increased MDA expression and CD8+ T-cell infiltration (45). More recently, results from a randomized phase 2 clinical trial of combined dabrafenib, trametinib and PD-1 blockade by pembrolizumab compared to dabrafenib, trametinib and placebo showed encouraging results, including improved progression-free survival and enhanced response, although the triple combination also resulted in increased adverse effects (46,47).
CDK4/6 inhibitors
CDK4/6 inhibitors exert direct immune-stimulatory effects on both tumor and immune cells (Fig. 4C). In tumor cells, the CDK4/6 inhibitors palbociclib and abemaciclib were shown to down-regulate expression of the DNA methyltransferase DNMT1, leading to decreased methylation and subsequently increased expression of endogenous retrovirus (ERV) elements, thus stimulating production of type III IFNs, and a consequent increase in antigen presentation and enhanced CD8+ T-cell effector function (48). Moreover, CDK4/6 inhibition specifically inhibited ex vivo proliferation of CD4+ CD25+ TRegs, but did not affect proliferation of CD4+ CD25− and CD8+ T-cells (48). Splenic CD4+ FOXP3+ TReg levels were also decreased upon treatment in vivo independently of the presence of a tumor (48). PD-L1 inhibition significantly improved response to abemaciclib in an inducible GEMM of HER2+ breast cancer, and resulted in complete tumor regression of CT-26 CRC tumors in all cases, as well as the ability to reject a second challenge with CT-26 tumor cells (48). In addition, an in vitro small molecule screen identified CDK4/6 inhibitors as capable of directly enhancing T-cell activation via up-regulation of NFAT signaling, a family of transcription factors that are required for proper activation and function of T-cells (49). Consistently, CDK4/6 inhibition by palbociclib or trilaciclib potentiated PD-1 blockade to stimulate anti-tumor T-cell function and inhibit tumor growth in the MC38 and CT-26 CRC models (49). Interestingly, Cyclin D-CDK4 was shown to promote PD-L1 proteasomal degradation (50). In vivo treatment with CDK4/6 inhibitors increased tumor PD-L1 levels and sensitized CT-26 tumors to ICB, resulting in complete tumor regression in 67% (8/12) of mice receiving combined palbociclib and anti-PD-1 (50). A study of 348 ER+/HER2− tumor samples collected from patients prior to start of CDK4/6 inhibitor treatment with palbociclib, ribociclib or abemaciclib revealed FAT1 deletion as a mechanism of therapeutic resistance (51). Mechanistically, FAT1 loss resulted in engagement of the Hippo pathway, leading to YAP/TAZ translocation to the nucleus and up-regulation of CDK6 expression (51). In the clinic, preliminary results from a phase Ib clinical trial of combined abemaciclib and pembrolizumab in ER-positive/HER2-negative metastatic breast cancer have shown safety profiles similar to single agents and an initial objective response rate (ORR) of 14.3% (52).
PARP inhibitors
Recent studies have demonstrated that, in addition to direct cytotoxicity, the therapeutic efficacy of PARP inhibitors (PARPi) requires coordinated activation of robust local and systemic anti-tumor immune response, such as increased infiltration of effector CD4+ and CD8+ T-cells into the TME, increased intratumoral DCs with potent antigen-presentation capacity, and systemic reduction of MDSCs in tumor, spleen and blood (53,54). Mechanistically, dsDNA derived from homologous recombination (HR)-deficient tumor cells upon PARP inhibition activates cGAS/STING in tumor cells and/or DCs to drive a cGAS/STING-dependent type I IFN signal that mediates antitumor immunity (Fig. 4D) (53). This mechanism of PARPi-triggered STING-dependent antitumor immunity has been demonstrated in several cancer types, including ovarian cancer, triple-negative breast cancer (TNBC), and lung cancer (53–57). Interestingly, PARP inhibitors have also been shown to induce expression of PD-L1 in tumor cells via multiple mechanisms, including as a response to interferon expression, inactivation of GSK3β, reduced poly(ADP-ribosyl)ation with concomitantly increased phosphorylation of STAT3, and STING activation (56,58–62). While PARPi-mediated PD-L1 up-regulation can promote adaptative immune suppression, it can be overcome by ICB. Indeed, pre-clinical studies have shown that PD-1/PD-L1 blockade further augments PARPi-triggered immune response, leading to more durable suppression of tumor growth and prolonged survival (53–56). Combined PARP inhibition and ICB is being evaluated by numerous clinical trials in first-line, maintenance, and recurrent settings of both HR-deficient and HR-proficient solid tumors (63–68). In general, these trials have found combination therapies are well-tolerated, with safety concerns consistent with individual agent profiles, and have produced encouraging initial results. While PARP inhibition and PD-1/PD-L1 monotherapy exhibit low efficacy for patients with platinum-resistant ovarian cancer who lack a BRCA mutation, with ORRs approximately 5% and 4-10%, respectively (69–74), in the ongoing phase I/II TOPACIO/KEYNOTE-162 trial, combined niraparib plus pembrolizumab demonstrated improved efficacy (ORR, 19%) in BRCA wild type patients with recurrent platinum-resistant ovarian cancer (75).
Future prospects
It is clear that tumor cell-intrinsic signaling mechanisms strongly affect immune composition and function. A deeper understanding of these molecular and cellular mechanisms will not only help in the design of potentially promising clinical trials of combination therapies targeted to specific groups of patients, but will also help discover new therapeutic targets with previously unknown functions in tumor immunity. Nevertheless, clinical development may still be limited by lack of significant benefit and compounding adverse effects. Careful pre-clinical and clinical studies are needed to improve the efficacy and tolerability of targeted therapy and immunotherapy combinations. Some areas of focus should include the need to: 1) better understand tissue-specific oncogene-related immune effects; 2) identify and validate biomarkers to predict response and resistance to oncogene targeting; 3) develop high fidelity animal models incorporating patient-derived tumors and humanized immune systems to better identify effective combinations without causing increased toxicity to patients; and 4) use multiplexed assays to integrate immune and tumor intrinsic molecular changes in response to combination therapy. Still, current evidence from pre-clinical and clinical trials is in aggregate promising and encouraging. The notion that specific targeted agents can sensitize tumor cells to immunotherapy, thereby leading to durable and effective responses in patients that would otherwise not respond is worth pursuing. Continued basic and pre-clinical research integrated with careful clinical trial planning of combination therapies will likely continue to yield meaningful treatment options for patients afflicted by cancer.
Acknowledgements
We thank Drs. Harvey Cantor and Hye-Jung Kim for scientific discussions. We thank Elizabeth Cahn and the Breast Cancer Advocacy Group at Dana-Farber/Harvard Cancer Center for discussions on patient issues and needs. We thank the Dale Family Foundation for their charitable contributions. This work was supported in part by grants from The Susan G. Komen Foundation PDF16376814 (J.S.B.), Friends of Dana-Farber Cancer Institute (J.S.B.), Cancer Research Institute (Q.W.), Terri Brodeur Breast Cancer Foundation (S.K.), Breast Cancer Research Foundation (J.J.Z.), DoD CDMRP BC171657 (J.J.Z.) and NIH P50 CA168504 (J.J.Z), P50 CA165962 (J.J.Z), and R35 CA210057 (J.J.Z).
Footnotes
Disclosure of potential conflicts of interest
J.S.B. is a scientific consultant for Geode Therapeutics. J.S.B. and J.J.Z. are co-inventors of DFCI 2180.001 (DFS-166.25). Q.W. is a scientific consultant for Crimson Biopharm. Q.W. and J.J.Z. are co-inventors of DFCI 2409.001 (DFS-203.60). J.J.Z. is a founder and director of Crimson Biopharm and Geode Therapeutics.
All figures are original artwork by the authors and thus permission is not required
References
- 1.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nature reviews Clinical oncology 2019;16(3):151–67 doi 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 2.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. The New England journal of medicine 2019;381(16):1535–46 doi 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 3.Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nature reviews Cancer 2017;17(5):286–301 doi 10.1038/nrc.2017.17. [DOI] [PubMed] [Google Scholar]
- 4.Wellenstein MD, de Visser KE. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018;48(3):399–416 doi 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer immunology research 2015;3(5):436–43 doi 10.1158/2326-6066.CIR-15-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168(4):707–23 doi 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (New York, NY) 2016;352(6282):227–31 doi 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casey SC, Baylot V, Felsher DW. The MYC oncogene is a global regulator of the immune response. Blood 2018;131(18):2007–15 doi 10.1182/blood-2017-11-742577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017;171(6):1301–15.e14 doi 10.1016/j.cell.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer cell 2019;36(5):483–97.e15 doi 10.1016/j.ccell.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer cell 2019;35(4):559–72.e7 doi 10.1016/j.ccell.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575(7781):217–23 doi 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 13.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer discovery 2013;3(12):1355–63 doi 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung Cancer Subtypes Generate Unique Immune Responses. Journal of immunology (Baltimore, Md : 1950) 2016;197(11):4493–503 doi 10.4049/jimmunol.1600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatti-Mays ME, Balko JM, Gameiro SR, Bear HD, Prabhakaran S, Fukui J, et al. If we build it they will come: targeting the immune response to breast cancer. NPJ breast cancer 2019;5(1):37–13 doi 10.1038/s41523-019-0133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, et al. The Immune Landscape of Cancer. Immunity 2018;48(4):812–30.e14 doi 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianchini G, Gianni L. The immune system and response to HER2-targeted treatment in breast cancer. The Lancet Oncology 2014;15(2):e58–68 doi 10.1016/S1470-2045(13)70477-7. [DOI] [PubMed] [Google Scholar]
- 18.George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017;46(2):197–204 doi 10.1016/j.immuni.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature medicine 2007;13(1):84–8 doi 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 20.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer discovery 2016;6(2):202–16 doi 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nature reviews Cancer 2015;15(1):7–24 doi 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017;543(7647):728–32 doi 10.1038/nature21676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding Z, Wu C-J, Chu GC, Xiao Y, Ho D, Zhang J, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011;470(7333):269–73 doi 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015;523(7559):231–5 doi 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 25.Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer discovery 2018;8(6):730–49 doi 10.1158/2159-8290.CD-17-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 2019. doi 10.1158/1078-0432.CCR-18-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer research 2016;76(5):999–1008 doi 10.1158/0008-5472.CAN-15-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer discovery 2018;8(7):822–35 doi 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer discovery 2019;9(1):34–45 doi 10.1158/2159-8290.CD-18-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barber GN. STING: infection, inflammation and cancer. Nature reviews Immunology 2015;15(12):760–70 doi 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nature reviews Cancer 2009;9(11):798–809 doi 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature reviews Immunology 2007;7(1):41–51 doi 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 33.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nature medicine 2004;10(1):48–54 doi 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 34.Jones LM, Broz ML, Ranger JJ, Ozcelik J, Ahn R, Zuo D, et al. STAT3 Establishes an Immunosuppressive Microenvironment during the Early Stages of Breast Carcinogenesis to Promote Tumor Growth and Metastasis. Cancer research 2016;76(6):1416–28 doi 10.1158/0008-5472.CAN-15-2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia Y, Shen S, Verma IM. NF-κB, an active player in human cancers. Cancer immunology research 2014;2(9):823–30 doi 10.1158/2326-6066.CIR-14-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee H, Deng J, Xin H, Liu Y, Pardoll D, Yu H. A requirement of STAT3 DNA binding precludes Th-1 immunostimulatory gene expression by NF-κB in tumors. Cancer research 2011;71(11):3772–80 doi 10.1158/0008-5472.CAN-10-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colombo M, Mirandola L, Chiriva-Internati M, Basile A, Locati M, Lesma E, et al. Cancer Cells Exploit Notch Signaling to Redefine a Supportive Cytokine Milieu. Frontiers in immunology 2018;9:1823 doi 10.3389/fimmu.2018.01823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 2015;163(1):160–73 doi 10.1016/j.cell.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nature medicine 2016;22(8):851–60 doi 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw C-NJ, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer research 2010;70(13):5213–9 doi 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 41.Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. The Journal of clinical investigation 2013;123(3):1371–81 doi 10.1172/JCI66236. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Steinberg SM, Zhang P, Malik BT, Boni A, Shabaneh TB, Byrne KT, et al. BRAF inhibition alleviates immune suppression in murine autochthonous melanoma. Cancer immunology research 2014;2(11):1044–50 doi 10.1158/2326-6066.CIR-14-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer research 2012;72(16):3928–37 doi 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Science translational medicine 2015;7(279):279ra41–ra41 doi 10.1126/scitranslmed.aaa4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2013;19(5):1225–31 doi 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nature medicine 2019;25(6):941–6 doi 10.1038/s41591-019-0448-9. [DOI] [PubMed] [Google Scholar]
- 47.Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH, Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nature medicine 2019;25(6):936–40 doi 10.1038/s41591-019-0476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017;548(7668):471–5 doi 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 Inhibition Augments Anti-Tumor Immunity by Enhancing T Cell Activation. Cancer discovery 2017:CD-17-0915 doi 10.1158/2159-8290.CD-17-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via Cul3SPOP to control cancer immune surveillance. Nature 2017. doi 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer cell 2018;34(6):893–905.e8 doi 10.1016/j.ccell.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tolaney SM, Kabos P, Dickler MN, Gianni L, Jansen V, Lu Y, et al. Updated efficacy, safety, & PD-L1 status of patients with HR+, HER2− metastatic breast cancer administered abemaciclib plus pembrolizumab. Journal of Clinical Oncology 2018;36(15_suppl):1059- doi 10.1200/JCO.2018.36.15_suppl.1059. [DOI] [Google Scholar]
- 53.Ding L, Kim H-J, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell reports 2018;25(11):2972–80.e5 doi 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer research 2019;79(2):311–9 doi 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. The Journal of clinical investigation 2019;129(3):1211–28 doi 10.1172/JCI123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer discovery 2019;9(5):646–61 doi 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pantelidou C, Sonzogni O, de Oliveira Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8+ T cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer discovery 2019:CD-18-1218 doi 10.1158/2159-8290.CD-18-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding L, Chen X, Xu X, Qian Y, Liang G, Yao F, et al. PARP1 Suppresses the Transcription of PD-L1 by Poly(ADP-Ribosyl)ating STAT3. Cancer immunology research 2019;7(1):136–49 doi 10.1158/2326-6066.CIR-18-0071. [DOI] [PubMed] [Google Scholar]
- 59.Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, et al. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer immunology research 2015;3(11):1257–68 doi 10.1158/2326-6066.CIR-15-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen M-K, Hsu J-M, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(14):3711–20 doi 10.1158/1078-0432.CCR-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. Journal of the National Cancer Institute 2017;109(1):djw199 doi 10.1093/jnci/djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Sun K, Xiao Y, Feng B, Mikule K, Ma X, et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Scientific reports 2019;9(1):1853–12 doi 10.1038/s41598-019-38534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drew Y, de Jonge M, Hong SH, Park YH, Wolfer A, Brown J, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecologic oncology 2018;149:246–7 doi 10.1016/j.ygyno.2018.04.555. [DOI] [Google Scholar]
- 64.Friedlander M, Meniawy T, Markman B, Mileshkin L, Harnett P, Millward M, et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. The Lancet Oncology 2019;20(9):1306–15 doi 10.1016/S1470-2045(19)30396-1. [DOI] [PubMed] [Google Scholar]
- 65.Friedlander M, Meniawy T, Markman B, Mileshkin LR, Harnett PR, Millward M, et al. A phase 1b study of the anti-PD-1 monoclonal antibody BGB-A317 (A317) in combination with the PARP inhibitor BGB-290 (290) in advanced solid tumors. Journal of Clinical Oncology 2017;35(15_suppl):3013- doi 10.1200/JCO.2017.35.15_suppl.3013. [DOI] [Google Scholar]
- 66.Karzai F, Madan RA, Owens H, Hankin A, Couvillon A, Houston ND, et al. A phase II study of the anti-programmed death ligand-1 antibody durvalumab (D; MEDI4736) in combination with PARP inhibitor, olaparib (O), in metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology 2017;35(6_suppl):162- doi 10.1200/JCO.2017.35.6_suppl.162. [DOI] [Google Scholar]
- 67.Konstantinopoulos PA, Waggoner SE, Vidal GA, Mita MM, Fleming GF, Holloway RW, et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. Journal of Clinical Oncology 2018;36(15_suppl):106- doi 10.1200/JCO.2018.36.15_suppl.106. [DOI] [Google Scholar]
- 68.Vinayak S, Tolaney SM, Schwartzberg LS, Mita MM, McCann GA-L, Tan AR, et al. TOPACIO/Keynote-162: Niraparib + pembrolizumab in patients (pts) with metastatic triple-negative breast cancer (TNBC), a phase 2 trial. Journal of Clinical Oncology 2018;36(15_suppl):1011- doi 10.1200/JCO.2018.36.15_suppl.1011. [DOI] [Google Scholar]
- 69.Disis ML, Patel MR, Pant S, Hamilton EP, Lockhart AC, Kelly K, et al. Avelumab (MSB0010718C; anti-PD-L1) in patients with recurrent/refractory ovarian cancer from the JAVELIN Solid Tumor phase Ib trial: Safety and clinical activity. Journal of Clinical Oncology 2017;34(15_suppl):5533- doi 10.1200/JCO.2016.34.15_suppl.5533. [DOI] [Google Scholar]
- 70.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England journal of medicine 2009;361(2):123–34 doi 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 71.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. The Lancet Oncology 2011;12(9):852–61 doi 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 72.Matulonis UA, Shapira-Frommer R, Santin A, Lisyanskaya AS, Pignata S, Vergote I, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Interim results from the phase 2 KEYNOTE-100 study. Journal of Clinical Oncology 2018;36(15_suppl):5511- doi 10.1200/JCO.2018.36.15_suppl.5511. [DOI] [PubMed] [Google Scholar]
- 73.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. The Lancet Oncology 2013;14(9):882–92 doi 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 74.Varga A, Piha-Paul SA, Ott PA, Mehnert JM, Berton-Rigaud D, Morosky A, et al. Pembrolizumab in patients (pts) with PD-L1–positive (PD-L1+) advanced ovarian cancer: Updated analysis of KEYNOTE-028. Journal of Clinical Oncology 2017;35(15_suppl):5513- doi 10.1200/JCO.2017.35.15_suppl.5513. [DOI] [Google Scholar]
- 75.Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA oncology 2019;5(8):1141–9 doi 10.1001/jamaoncol.2019.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]