

Abstract
Cancers that arise from BRCA1 germline mutations are deficient for homologous recombination (HR) DNA repair and are sensitive to DNA damaging agents such as platinum and PARP inhibitors (PARPi). In vertebrate organisms, knockout of critical HR genes including BRCA1 and BRCA2 is lethal because HR is required for genome replication. Thus, cancers must develop strategies to cope with loss of HR activity. Furthermore, as established tumors respond to chemotherapy selection pressure, additional genetic adaptations transition cancers to an HR-proficient state. In this review, we discuss biological mechanisms that influence the ability of BRCA1-mutant cancers to perform HR. Furthermore, we consider how the HR status fluctuates throughout the cancer life course, from tumor initiation to the development of therapy refractory disease.
Keywords: BRCA1, BRCA2, RNF168, homologous recombination, breast cancer, ovarian cancer, DNA damage, DNA repair, 53BP1, RNF8
Introduction
BRCA1/2 gene mutations account for the majority of hereditary forms of breast and ovarian cancer (1). It is broadly accepted that both BRCA1 and BRCA2 proteins suppress the formation of tumors by facilitating homologous recombination (HR) DNA repair, which in turn, ensures genomic stability (2–4). Breaks frequently arise during DNA replication, and HR is required for DNA replication fork restart (5). Because HR is critical for genome duplication, it is obligatory for the viability of vertebrate organisms. Indeed, genetic disruption of critical HR factors, including Brca1, Brca2 and Rad51, can induce early embryonic lethality in mice (6–12). In striking contrast, BRCA1/2 mutant cancers are highly proliferative and malignant, despite being defective for HR. This apparent paradox raises several questions; including, why are cancers, but not normal cells, able to thrive without HR? Alternatively, do BRCA1/2 mutant cancers retain residual HR activity? And, what are the biological mechanisms that provide HR in the absence of BRCA proteins?
The HR status of tumors is highly consequential for cancer patients. In particular, PARP inhibitors (PARPi) and platinum generate DNA damage during replication, which is usually repaired by the HR machinery (13–16). Patients with BRCA1/2 mutation-containing tumors gain most benefit from PARPi therapy. In a portion of patients, PARPi provides long-term anti-tumor activity. However, in other cases, PARPi has minimal activity, despite the presence of BRCA1/2 mutations and presumed HR-deficiency. Moreover, the majority of tumors are initially responsive, but then develop resistance (17–24). Often, cancers that are HR-deficient go on to acquire molecular adaptations that transition them to an HR-proficient state (25,26). The paradigm that BRCA1 mutations are equal to HR deficiency may be over simplistic, and BRCA1 mutation-containing cancers likely adapt strategies to maintain some level of HR. In this review, we focus on the relationships between BRCA1 mutations, HR and cancer. We explore the concept that HR is not an all-or-nothing occurrence but a spectrum, and that where a tumor stands on this spectrum may have therapeutic relevance.
BRCA1 domains govern function
The human BRCA1 gene comprises over 81 kilobases of DNA on chromosome 17. The largest transcript encodes an 1863 amino acid (aa) protein with an approximate mass of 220 kDa, referred to as BRCA1 full-length or p220. Within the full-length peptide, there are conserved RING, coiled-coil (CC), and BRCT domains, as well as a central unstructured region encoded by exon 11 that accounts for over half of the total protein (Figure 1A) (27,28). The molecular activity of BRCA1 within HR stems from protein interactions and the formation of distinct multi-protein complexes. BRCA1-BARD1 interact via their respective N-terminal RING domains. The BRCA1 BRCT repeats engender a phosphopeptide binding region that facilitates protein interactions, including CtIP, ABRAXAS, and BACH1. In contrast, the BRCA1 CC domain is only known to form a single direct protein interaction with the CC domain of PALB2 (27). Although the function of exon 11 is not clear, due to its large size, mutations in this region account for nearly 30% of pathogenic BRCA1 mutations (29).
Figure 1.
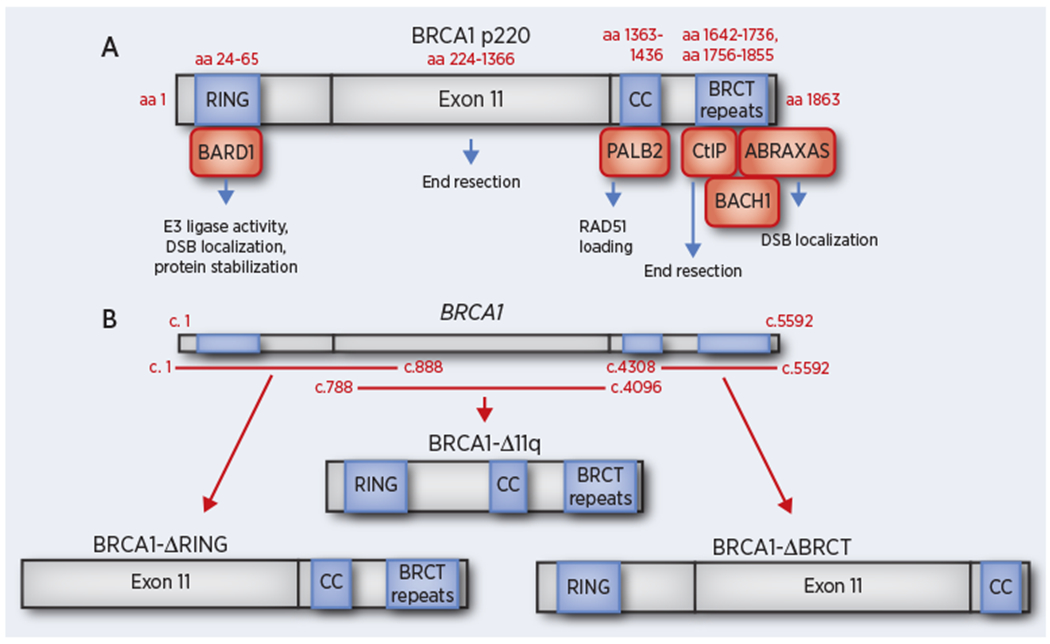
Schematic representation of BRCA1 full-length and hypomorphic proteins that have been detected in cancers. (A) The full-length (p220) BRCA1 protein is depicted along with known functional domains and corresponding protein interactions. Amino acid (aa) residues corresponding to predicted domain start and end sites are indicated as well as potential functional activities of each domain. (B) BRCA1 cDNA is shown (above) with corresponding base pairs indicated. Redlines indicate regions where frameshift mutations may be present but remain capable of expressing either the BRCA1-ΔRING, BRCA1-Δ11q, or BRCA1-ΔBRCT proteins.
BRCA1-BARD1 and HR
BRCA1 has several well-defined functions that contribute to the overall proficiency of HR. The BRCA1-BARD1 heterodimer is an E3 ubiquitin ligase, but the importance of this activity in HR is unclear (30–34). BRCA1-BARD1 E3 ligase substrates have been reported to include histone H2A and CtIP (33,34). The BRCA1I26A mutation is not found in cancer patients but disrupts E3 ligase activity, and there are conflicting reports regarding the degree to which this mutation is deleterious, with multiple studies showing little or no impact on HR or DNA damage sensitivity (31,35–37). In contrast, the physical association of BRCA1-BARD1 is crucial for the stability of each protein. The BRCA1C61G missense mutation is found in cancer patients and prevents BARD1 heterodimerization. Consequently, there is decreased expression of both proteins, loss of HR and DNA damage sensitivity (38–43). BRCA1-BARD1 have also been shown to directly interact with, and enhance the recombinase activity of, RAD51 (44).
Another critical function of BARD1 is to localize BRCA1 to sites of DNA damage. The BARD1-BRCT domain binds to poly-ADP ribose (PAR) and targets the heterodimer to double stranded DNA breaks (DSBs) early on in the response to DNA damage (45). However, at later time points, the BARD1-BRCT domain interacts with Lysine 9-dimethylated histone H3 (H3K9me2), which is mediated primarily by HP1γ, and is required for focal accumulation (46). Moreover, a screen identified the BARD1-ANK domain as efficiently binding H4 not methylated at lysine 20 (H4K20me0). BARD1ΔAnk failed to be recruited to chromatin and resulted in cellular sensitivity to PARPi (37). The BRCA1-BRCT domain also contributes to recruitment to DSBs through an ABRAXAS-RAP80 complex (47–49). The interplay between these various mechanisms of BRCA1-BARD1 recruitment to chromatin and DSB sites is unclear.
BRCA1 and DNA end resection
The degradation of double stranded (ds)DNA to single stranded (ss)DNA overhangs by nucleases is termed DNA end resection and is required to initiate HR. Perhaps the most recognized function of BRCA1 in HR is in counteracting the 53BP1-RIF1-shieldin complex (50,51). A plethora of studies have emerged over recent years that have identified proteins that comprise the 53BP1-RIF1-shieldin-complex, which functions to inhibit DNA end resection. However, the precise biological mechanism by which BRCA1 counters this complex and activates resection is less clear. BRCA1 may physically displace 53BP1 (31,52), or recruit phosphatases that dephosphorylate 53BP1 and result in loss of RIF1 binding (53). Additionally, the exon 11 region and BRCT domain of BRCA1 have independently been shown to prevent RIF1 accumulation at DSBs (54,55). Moreover, the BRCA1-BRCT domain promotes CtIP localization to DSB foci, which in turn stimulates MRE11 nuclease activity. Key studies have shown that the BRCA1-CtIP interaction, although not crucial for end resection to occur, increases the efficiency and speed of resection (56–59).
BRCA1-PALB2 promotes RAD51 loading
Seminal studies published in 2009 uncovered a principal function of BRCA1 within HR. Here, BRCA1 was shown to directly bind to PALB2 via each proteins respective CC domains, resulting in the formation of a macro complex consisting of BRCA1-PALB2-BRCA2-RAD51 (60–62). The latter promotes the loading of RAD51 onto resected ssDNA, generating RAD51 filaments that are primed for strand invasion. Cell lines engineered to express PALB2 or BRCA1 mutations that disrupt the CC domains are defective for RAD51 foci formation (60–62). However, there remains debate regarding whether this is a critical interaction and function of BRCA1, or something that can be readily bypassed. Specifically, because 53BP1 knockout (KO) can rescue HR via the restoration of DNA end resection in BRCA1 mutant cells, the significance of the BRCA1-PALB2 interaction is contentious. Recent studies have addressed this question, demonstrating that BRCA1 hypomorphs that retain the CC domain are required for efficient 53BP1 KO-mediated restoration of RAD51 foci (55,63,64). Other studies have shown that cell lines expected to be BRCA1 null had efficient RAD51 foci in the setting of 53BP1 deficiency (65–67). Therefore, in the context of 53BP1 KO, BRCA1-independent PALB2-BRCA2-RAD51 loading appears to be highly variable, and potentially cell line-dependent. Moreover, BRCA1-independent PALB2 loading has been demonstrated to occur via mechanisms that depend on ATR and RNF168 activity (68–71). In summary, the BRCA1 protein has a multi-faceted and complex role within HR, with prominent actions in initiating DNA end resection and promoting RAD51 loading.
BRCA1 and replication fork protection
Proteins that are involved in HR are often also required to protect DNA fibers from nuclease mediated degradation in response to DNA replication fork stalling agents such as hydroxyurea (HU). The fork protection (FP) function of BRCA1 and BRCA2 appears to be distinct from their HR activities, with several separation-of-function mutations documented (72–74). For example, Brca1 CC domain mutations are highly disruptive to HR, but do not impact FP (55). Similarly, in mouse cells, Brca1-ΔRING was defective for HR but proficient for FP (70). In contrast, the Brca1-Δ11 protein is defective for both FP and HR (55,70,75). Some studies have shown FP disruptive mutations have minor effects on cell viability, others indicate more substantial effects on viability and response to therapy (74–77). Interestingly, proteins that are defective for fork protection, such as Brca1-ΔRING and Brca1-Δ11, retain the ability to provide residual HR, and can play a role in PARPi resistance upon selection pressure (29,78), although the precise interplay between partial HR and FP has not been addressed.
A requirement for HR in genome replication
HR is required to restart replication forks that pause after encountering spontaneously arising breaks and problematic DNA sequences or structures. While yeast and bacteria can replicate their genomes in the absence of essential HR genes such as RecA/RAD51 and RAD52, the same is not true for vertebrate cells (5,79–82). BRCA1, BRCA2 and RAD51 genetic KO all result in early embryonic lethality in mice (6–12). BRCA2 deficiency as well as mutations that specifically disrupt the HR activity of the protein were shown to trigger replication stress that is transmitted to the next cell cycle through DNA under replication. The latter caused chromosome segregation and the presence of 53BP1 nuclear bodies in G1 phase human breast epithelial cells, followed by senescence or apoptosis-associated loss of viability (76). Studies using DT40 chicken cells showed that genetic inactivation of RAD51 caused cells to accumulate in G2/M before dying. Chromosome analyses presented multiple breaks spontaneously occurring in proliferating cells (83). Surprisingly, DT40 cells with BRCA1 KO cell can proliferate, although at a slower speed, possibly due to RAD51 overexpression (84). In contrast, the human haploid HAP1 cell line is highly dependent on BRCA1 for viability, and a KO-knockin (KI) approach was able to identify potential pathogenic variants based on their ability to rescue cell proliferation (85).
BRCA1 and TP53 mutations
Given the importance of HR for DNA replication, BRCA1 defective cells and mice are inviable. Thus, cells that undergo transformation require epi/genetic events that permit for loss of BRCA1 to become advantageous and subject to positive selection. The most well-defined mechanism is the presence of TP53 mutations, which are invariably detected in BRCA1 mutant cancers (86,87). Loss of the p53-p21 signaling axis prevents DNA damage from triggering cell death (12,88). The ability of p53 and p21 KO to rescue viability in the presence of Brca1 mutations has been illustrated using mouse genetics. Nonetheless, the efficiency of rescue is variable, and dependent on the specific Brca1 mutation. For example, p53 KO rescues the embryonic viability of mice that are homozygous for hypomorphic Brca1 alleles (89–91). In contrast, p53 KO had little effect, beyond an extra day of embryonic life, on homozygous Brca1 null allele embryos (12,88,92). The latter suggests that TP53 mutations alone may be insufficient for some cancers to maintain viability; and that BRCA1 hypomorphic protein products, in conjunction with TP53 mutations, sustain viability. Significantly, while TP53 mutations abrogate DNA damage checkpoints, they do not explain how BRCA1 mutant cancers replicate their genome in the absence of HR.
BRCA1 hypomorphic proteins
The transformation process is thought to initiate when epithelial cells acquire TP53 mutations, followed by loss of heterozygosity (LOH) at the BRCA1 locus (93). When the wild-type copy of BRCA1 is lost, the mutation-containing allele remains behind, which is unable to support HR and consequently lacks tumor suppressor activity. BRCA1 mutant cancers have extensive genomic instability; thus, a reasonable prediction might be that the remaining mutation-containing locus might also eventually be lost in a portion of cells. However, BRCA1 mutations are frequently detectable in established cancers, with few cases in which the locus is entirely absent (94–98). Similarly, established BRCA1 mutant cancer cell lines invariably retain the mutation-containing allele, regardless of passage number or culture conditions (29,99). Thus, in the absence of wild-type BRCA1, mutant alleles may be subject to positive selection. Indeed, several copies of the mutation-containing allele are often observed in tumors (discussed in more detail below).
BRCA1 missense mutations produce full-length proteins that can have hypomorphic activity. For example, mice with Brca1C61G mutant mammary cancers rapidly developed PARPi and cisplatin resistance due to increased expression of the Brca1C61G protein (100). Moreover, mice containing some Brca1 hypomorphic mutations can survive embryogenesis. Brca1CC/CC mice were born with Fanconi anemia-like defects, and Brca1 CC in-frame mutant proteins were capable of displacing RIF1-shieldin, thereby promoting DNA end resection, but were defective for RAD51 loading (55). In contrast, frameshift mutations that block the production of proteins are unable to contribute to HR. Nevertheless, there are several examples of BRCA1 frameshift mutations that are predicted to be unable to generate protein, but in fact produce unanticipated BRCA1 isoforms.
BRCA1 mutations that arise in the germline of patients can be broadly grouped by mutation location (Figure 1B). BRCA1 5’ located mutations, including 185delAG, can generate proteins that lack the RING, but contain all other domains, through initiating translation from start sites that are downstream of the mutation-induced stop codon (78,101). Meanwhile, BRCA1 exon 11 frameshift mutations remain capable of expressing the BRCA1-Δ11q alternative splice isoform, which is without exon 11, but contains all conserved functional domains (29,102). BRCA1-Δ11q can provide residual growth in the presence of PARPi or cisplatin, but remains defective relative to the full-length protein. BRCA1 BRCT domain mutations are highly destabilizing and result in proteasomal degradation (103,104). However, gene re-arrangements can generate proteins that lack the entire BRCT domain, including the destabilizing mutation (105).
Proteins with folding issues may also be stabilized by HSP90 (106). BRCA1- ΔRING, -ΔBRCT, and -Δ11q proteins can be readily detected under PARPi selection pressure and are capable of partially supporting RAD51 loading due to retention of the CC domain (63) (Figure 1B). However, lower levels of BRCA1 mutant protein expression are sometimes detected in the absence of PARPi selection pressure and likely support residual HR activity.
Typical methods used to quantify HR activity include the DR-GFP reporter, RAD51 foci formation, and PARPi colony assays. However, while these methods are effective at uncovering HR defects, they may lack the sensitivity required to detect residual HR activity. Our laboratory has adopted a modified colony assay that is capable of uncovering low levels of HR activity afforded by BRCA1 mutant proteins. The MDA-MB-436 breast cancer cell line has undetectable BRCA1 protein expression and is used to generate stable BRCA1 cDNA addbacks with a lenti-virus expression system. We seed cells at increasing densities, and incubate with a low concentration of PARPi that prevents the negative control cDNA-expressing cells from forming colonies, but has no effect on cells that express the BRCA1 wild-type positive control. Hypomorphic proteins enable colonies to form when cells are seeded at higher densities, whereas negative control cells do not form any colonies. Using this approach, the ability of BRCA1 mutant proteins to provide residual HR can be visualized and quantified.
BRCA1 locus modifications
The paradigm for BRCA mutant cancer development is centered on the “two-hit” hypothesis, where one allele is lost, and the remaining has a mutation that results in loss-of-function (107). However, recent insights suggest further complexity. In an analysis of patients with pathogenic germline mutations, depending on the cohort, 16-27% of BRCA1/2 tumors analyzed fit the classic hypothesis where one allele was lost and the mutant allele remained. However, 23-42% lost the wild-type allele but had 2 copies of the mutant allele, and 16-36% had 3 copies of the mutant allele. Another 4-36% of tumors retained both the wild-type and mutant gene copy (98).
Retention of the wild-type allele has obvious implications for cancer development and therapeutic outcomes. Ovarian cancers that did not demonstrate LOH were associated with worse survival (98), likely from poorer response to platinum or PARPi therapies. Cancers that do not demonstrate LOH have additional driver mutations, such as KRAS or EGFR, and are better characterized by tumor lineage as opposed to BRCA mutation status (108). Possible explanations for retention of wild-type BRCA1 could be that HR re-wiring adaptations are not in place to support survival in the event of LOH. Alternatively, cancer promoting alterations occur prior to BRCA1 LOH and make the latter unnecessary.
Cancer cell lines and PDX tumors often harbor several copies of the BRCA1 mutation-containing allele (105). Extra copies permit for transcription from multiple alleles and may enhance mutant protein production. Moreover, the presence of a second allele has been shown to facilitate Alu-mediated rearrangements in the BRCA1 locus that enables the production of hypomorphic proteins (105). In summary, the presence and retention of single or multiple BRCA1 mutant alleles enables truncated protein isoforms to be expressed, which support residual HR and the viability of cancers.
RNF168-53BP1 and basal viability
While PARPi treatments induce high levels of expression of BRCA1 hypomorphs, during cancer initiation and prior to chemotherapy selection pressure, truncated BRCA1 protein expression may be low or absent, and alternative re-wiring mechanisms are required to support HR. Deletion of 53BP1 can restore resection and HR in Brca1 mutant mice and cancers (50,51). In the setting of Brca1 null alleles, 53bp1 KO was insufficient to provide robust HR and PARPi resistance, or genome stability and protection from tumor development. Nonetheless, 53bp1 KO did restore the ability of Brca1 null embryonic cells to divide and produced pups at close to the expected Mendelian rate (63,64). Thus, in addition to therapy resistance, modulation of the 53BP1 pathway may be pertinent to BRCA1 mutant tumor initiation (Figure 2).
Figure 2.
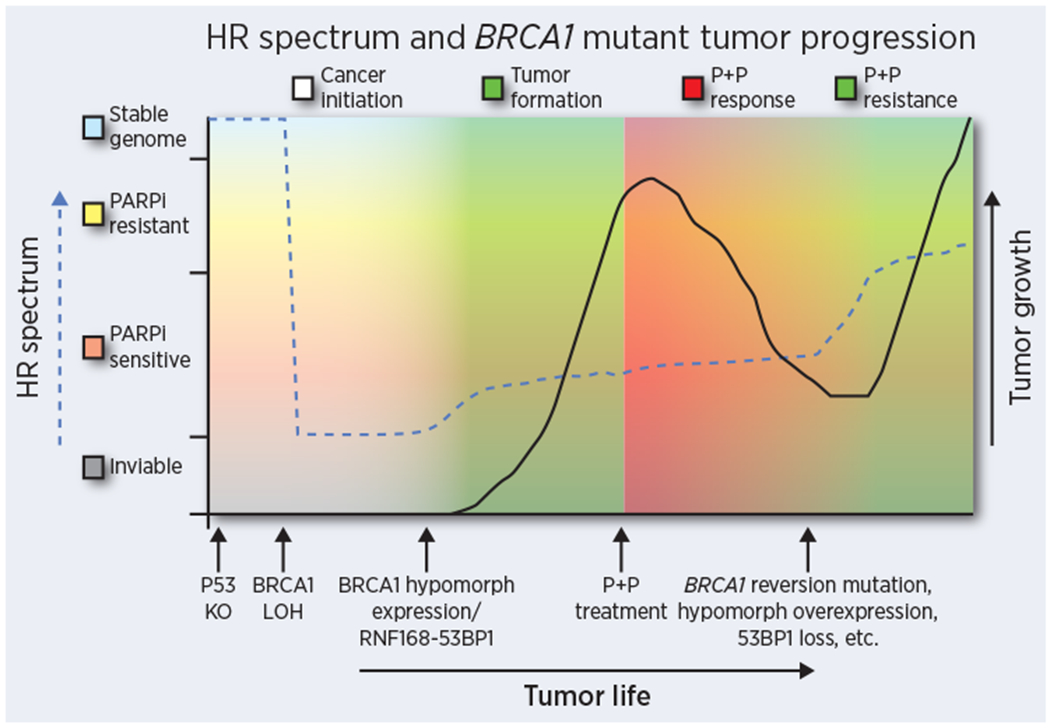
The HR spectrum over the life course of a BRCA1 mutant cancer. Partial HR activity is likely required for tumor initiation and growth, but may also play a role in therapy resistance, and could account for some patients demonstrating poor responses in the treatment naïve setting. Dashed line indicates HR activity and solid line indicates cancer growth over time. Arrows along the x axis indicate significant events over the course of cancer development and progression. Shading colors correspond to cancer development, cancer growth, PARPi and Platinum therapy (P+P) responsiveness and resistant growth.
Genetic disruption of proteins that are either up or downstream of 53BP1 can also restore end resection and HR. The RIF1-shieldin complex acts downstream of 53BP1, and KO of components have been shown to restore HR and promote PARPi resistance in a manner similar to 53BP1 (109). TRIP13 amplification also contributes to loss of shieldin complex activity (110). The RNF8-RNF168 pathway functions upstream and is required to recruit 53BP1 to DSB sites. In response to DNA damage, RNF8 binds to phosphorylated MDC1 and promotes the recruitment of RNF168, both are E3 ubiquitin ligases that conjugate Lys 63-linked ubiquitin chains onto histone H2A. Additionally, RNF168 monoubiquitinates H2A at K13/15, which serves as a recruitment module for 53BP1 (111). Thus, depletion of RNF168 and RNF8 have both been shown to decrease 53BP1 recruitment and consequently promote DNA end resection in the absence of BRCA1 (70,112,113). Moreover, the TIRR protein interferes with 53BP1 recruitment and can influence HR activity (114).
In the setting of BRCA1 and 53BP1 deficiency, RNF8 and RNF168 ubiquitin activity contributes to BRCA1-independent RAD51 loading, likely through a direct interaction between RNF168 and PALB2 (69–71,113). Therefore, in BRCA1 and 53BP1 deficient cells, RNF168 may play a key role in regulating HR. In BRCA1 null cancers, RNF168 protein expression is finely regulated. Here, low levels of RNF168 protein expression were detected, and consequently reduced 53BP1 recruitment activated resection and provided residual HR. However, RNF168 remained essential and was required for RAD51 loading in the absence of BRCA1. Indeed, a further decrease in RNF168 expression abolished HR and was detrimental to cellular viability. Of note, BRCA1 mutant cells that expressed high levels of hypomorphic proteins retained the ability to promote residual HR without modulation of RNF168 expression, and were not dependent on reduced RNF168 for viability (113). Hence, residual HR may be supported by a variety of means, which are potentially determined by whether mutant alleles are capable of expressing high enough levels of hypomorphic proteins.
RAD18 is another H2A-ubiquitin binding protein and is recruited to DSBs in an RNF168-dependent manner. In studies where RNF168 was ectopically overexpressed in BRCA1 null cancers, DNA end resection was blocked via 53BP1 recruitment. Moreover, DNA repair events at stalled replication forks were redirected toward a break-induced replication (BIR)-like mechanism through a RAD18-SLF1 signaling axis (115). Significantly, mutational signatures associated with tandem duplications arise from BIR-like events can be readily viewed in BRCA1 mutant cancer genomes (116,117). Thus, RNF168 is a master regulator of multiple aspects of DNA repair, particularly in the setting of BRCA1-deficiency.
BRCA1 mutations and therapeutics
Tumor HR status is a critical determinant of PARPi and platinum therapy response. In the standing paradigm, the BRCA1 wild-type genotype confers therapy resistance, and mutations sensitivity (13,14). In the clinical setting, BRCA1/2 mutant cancers demonstrate overall better progression free survival (PFS) than wild-type cancers from PARPi (17–24). However, PARPi’s have been particularly successful in ovarian cancers, where efficacy is observed in both BRCA wild-type and mutant cancers, and there is a wide spectrum of responses (118). BRCA wild-type ovarian cancers are thought to have defects in other HR genes that account for sensitivity, including PALB2, RAD51D, and multiple FANC genes (119,120).
Whether a particular group of BRCA mutations confer worse or better response to PARPi or impact the duration of effectiveness is an important question, but one that has not yet been fully resolved (121). Analyses of cohorts of ovarian cancer patients showed that BRCA1 exon 11 mutation carriers may have worse overall response to platinum and overall survival (OS) compared to other mutation types (29,122). This may be due to expression of the BRCA1-Δ11q splice variant that can promote therapy resistance. However, there are many examples of patients with similar mutations, including those in BRCA1 exon 11, which have a complete, partial, as well as no response to PARPi. Thus, it does not appear that specific BRCA1 mutations can be used to predict therapy efficacy or the duration of PARPi response. A range of factors likely determine response and resistance, much of which, in the clinical setting, are unknown. The collection of paired PARPi pre- and post-resistant tumor samples from patients is currently limited and future efforts along this front will be crucial.
Insights into PARPi resistance mechanisms have largely come from studies using BRCA1 mutant human cancer cell lines and mouse models, and are too numerous to review here (26). It should be noted they include non-HR related events; however, we will focus on mechanisms that restore HR. With respect to the role of BRCA1 mutations, we and others have observed increased expression of a range of BRCA1 truncated proteins in multiple human cancer cell lines (29,78,100,101,105,106,123). Importantly, RNAi or CRISPR/cas9 targeting of truncated BRCA1 proteins reversed PARPi resistance. However, ectopic overexpression of truncated BRCA1 proteins only provided partial or low levels of HR and PARPi resistance. Although BRCA1 isoforms were not as efficient as wild-type, they did enable significantly more colonies to grow in the presence of PARPi compared to cells that were null for BRCA1 expression (29,78,105). Thus, BRCA1 hypomorphs likely promote resistance in combination with additional events that together provide robust PARPi resistance. In several instances, we have observed reductions in 53BP1 protein levels in conjunction with BRCA1 hypomorph expression in cells that acquired PARPi resistance (63,106). In another example, BRCA1 hypomorphic protein expression was critical for PARP1 mutation-induced PARPi resistance (124).
BRCA1 mutant proteins might not be expressed at high enough levels or may have insufficient HR activity to induce resistance. Here, secondary mutations that restore the reading frame and expression of the full-length protein can promote resistance (26). Full-length BRCA1 proteins are more efficient at promoting PARPi resistance, and a preference for reversion mutations might be expected. However, reversion mutations are only detected in a portion of PARPi resistant cancers (125), and determinants of the acquisition of reversion mutations versus other mechanisms of resistance are unclear. Conceivably, reversion mutations may be more readily generated than alternative translation, alternative splicing, and protein folding re-wiring events that are required for BRCA1 hypomorph protein expression. Of note, specific mutations do not appear to determine whether reversions occur or hypomorphs are expressed, as there are examples of both mechanisms in RING, exon 11 and BRCT mutation-containing cancers. Moreover, intra-tumoral heterogeneity could play a role in PARPi resistance. Additional studies of patient tumors are required to determine whether multiple mechanisms are engaged within the same tumor.
Implications and future directions
Despite progress, there is still much unknown, and hereditary BRCA mutations continue to significantly contribute to breast and ovarian cancer mortality. The process of HR is fundamental to the study of BRCA cancer. HR re-wiring mechanisms are emerging, but there are likely many additional means by which cells adapt to survive in the absence of wild-type BRCA1. Further complexities, such as the relationship between replication fork protection and HR will be important to decipher, particularly in determining their relative impact on cell viability and therapeutic response. Accumulating DNA damage also triggers luminal to basal/mesenchymal trans-differentiation events that permit for cells to tolerate BRCA1 loss (126). An understanding of these events and pathways could reveal additional therapeutic vulnerabilities.
During the development of BRCA1 mutant cancer, HR deficiency is advantageous, but in established tumors is an Achilles heel, providing a therapeutic window for targeting cancer cells over non-transformed cells. Consequently, HR-deficiency is often transient, and determined by therapy selection pressure (Figure 2). Retaining a state of HR-deficiency would prolong PARPi sensitivity and patient survival. While efforts are underway to develop therapeutics that target HR defective cells, the selective targeting of cancers that have acquired HR-proficiency may provide a complementary approach to PARPi. Moreover, loss of HR activity confers dependencies on alternative DNA repair pathways. Microhomology-mediated end joining (MMEJ) has been shown to be one such pathway (127,128), and DNA polymerase theta inhibitors are under development (129,130). Continued research into the basic biology of DNA repair pathways is of critical importance for the discovery of new and effective therapeutic targets. Finally, routine collection of tumor material from patients who have progressed on PARPi therapy will be of enormous benefit in understanding resistance and designing new treatments.
Acknowledgments
This work was supported by US National Institutes of Health (NIH) Grants R01CA214799. J.J.K. was supported by an American Cancer Society - Tri State CEOs Against Cancer Postdoctoral Fellowship, PF-19-097–01–DMC, Ovarian Cancer Research Alliance and Phil and Judy Messing grant 597484, and T32 CA009035.
Footnotes
Conflicts disclosure
The authors declare no conflicts of interest.
References
- 1.Petrucelli N, Daly MB, Feldman GL. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genetics in medicine : official journal of the American College of Medical Genetics 2010;12:245–59 [DOI] [PubMed] [Google Scholar]
- 2.Chen CC, Feng W, Lim PX, Kass EM, Jasin M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu Rev Cancer Biol 2018;2:313–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell 1999;4:511–8 [DOI] [PubMed] [Google Scholar]
- 4.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997;88:265–75 [DOI] [PubMed] [Google Scholar]
- 5.Michel B, Flores MJ, Viguera E, Grompone G, Seigneur M, Bidnenko V. Rescue of arrested replication forks by homologous recombination. Proc Natl Acad Sci U S A 2001;98:8181–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, et al. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci U S A 1996;93:6236–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol 1996;16:7133–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gowen LC, Johnson BL, Latour AM, Sulik KK, Koller BH. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat Genet 1996;12:191–4 [DOI] [PubMed] [Google Scholar]
- 9.Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 1996;85:1009–23 [DOI] [PubMed] [Google Scholar]
- 10.Liu CY, Flesken-Nikitin A, Li S, Zeng Y, Lee WH. Inactivation of the mouse Brca1 gene leads to failure in the morphogenesis of the egg cylinder in early postimplantation development. Genes Dev 1996;10:1835–43 [DOI] [PubMed] [Google Scholar]
- 11.Suzuki A, de la Pompa JL, Hakem R, Elia A, Yoshida R, Mo R, et al. Brca2 is required for embryonic cellular proliferation in the mouse. Genes Dev 1997;11:1242–52 [DOI] [PubMed] [Google Scholar]
- 12.Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A. Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev 1997;11:1226–41 [DOI] [PubMed] [Google Scholar]
- 13.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913–7 [DOI] [PubMed] [Google Scholar]
- 14.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21 [DOI] [PubMed] [Google Scholar]
- 15.Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res 2001;61:4842–50 [PubMed] [Google Scholar]
- 16.Quinn JE, Kennedy RD, Mullan PB, Gilmore PM, Carty M, Johnston PG, et al. BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res 2003;63:6221–8 [PubMed] [Google Scholar]
- 17.Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol 2014;25:32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol 2016;17:1579–89 [DOI] [PubMed] [Google Scholar]
- 19.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 2014;15:852–61 [DOI] [PubMed] [Google Scholar]
- 20.Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1274–84 [DOI] [PubMed] [Google Scholar]
- 21.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010;376:235–44 [DOI] [PubMed] [Google Scholar]
- 22.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009;361:123–34 [DOI] [PubMed] [Google Scholar]
- 23.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol 2010;28:2512–9 [DOI] [PubMed] [Google Scholar]
- 24.Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med 2017;377:523–33 [DOI] [PubMed] [Google Scholar]
- 25.Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annual review of medicine 2015;66:455–70 [DOI] [PubMed] [Google Scholar]
- 26.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med 2013;19:1381–8 [DOI] [PubMed] [Google Scholar]
- 27.Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol 2010;11:138–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 2012;12:68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res 2016;76:2778–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mallery DL, Vandenberg CJ, Hiom K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J 2002;21:6755–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, Daza-Martin M, et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol 2016;23:647–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thakar A, Parvin J, Zlatanova J. BRCA1/BARD1 E3 ubiquitin ligase can modify histones H2A and H2B in the nucleosome particle. Journal of biomolecular structure & dynamics 2010;27:399–406 [DOI] [PubMed] [Google Scholar]
- 33.Kalb R, Mallery DL, Larkin C, Huang JT, Hiom K. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell reports 2014;8:999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev 2006;20:1721–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reid LJ, Shakya R, Modi AP, Lokshin M, Cheng JT, Jasin M, et al. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc Natl Acad Sci U S A 2008;105:20876–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 2011;334:525–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura K, Saredi G, Becker JR, Foster BM, Nguyen NV, Beyer TE, et al. H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat Cell Biol 2019;21:311–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joukov V, Chen J, Fox EA, Green JB, Livingston DM. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc Natl Acad Sci U S A 2001;98:12078–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev 2006;20:34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fabbro M, Rodriguez JA, Baer R, Henderson BR. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J Biol Chem 2002;277:21315–24 [DOI] [PubMed] [Google Scholar]
- 41.McCarthy EE, Celebi JT, Baer R, Ludwig T. Loss of Bard1, the heterodimeric partner of the Brca1 tumor suppressor, results in early embryonic lethality and chromosomal instability. Mol Cell Biol 2003;23:5056–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem 2001;276:14537–40 [DOI] [PubMed] [Google Scholar]
- 43.Ruffner H, Joazeiro CA, Hemmati D, Hunter T, Verma IM. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci U S A 2001;98:5134–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao W, Steinfeld JB, Liang F, Chen X, Maranon DG, Jian Ma C, et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017;550:360–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li M, Yu X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013;23:693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu W, Nishikawa H, Fukuda T, Vittal V, Asano M, Miyoshi Y, et al. Interaction of BARD1 and HP1 Is Required for BRCA1 Retention at Sites of DNA Damage. Cancer Res 2015;75:1311–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007;316:1194–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007;316:1198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007;316:1202–5 [DOI] [PubMed] [Google Scholar]
- 50.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 2010;17:688–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010;141:243–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. Journal of cell science 2012;125:3529–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Isono M, Niimi A, Oike T, Hagiwara Y, Sato H, Sekine R, et al. BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell reports 2017;18:520–32 [DOI] [PubMed] [Google Scholar]
- 54.Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 2013;49:872–83 [DOI] [PubMed] [Google Scholar]
- 55.Nacson J, Di Marcantonio D, Wang Y, Bernhardy AJ, Clausen E, Hua X, et al. BRCA1 Mutational Complementation Induces Synthetic Viability. Mol Cell 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 2009;284:9558–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polato F, Callen E, Wong N, Faryabi R, Bunting S, Chen HT, et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J Exp Med 2014;211:1027–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reczek CR, Szabolcs M, Stark JM, Ludwig T, Baer R. The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. The Journal of cell biology 2013;201:693–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cruz-Garcia A, Lopez-Saavedra A, Huertas P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell reports 2014;9:451–9 [DOI] [PubMed] [Google Scholar]
- 60.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A 2009;106:7155–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Molecular cancer research : MCR 2009;7:1110–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol 2009;19:524–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell reports 2018;24:3513–27 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen J, Li P, Song L, Bai L, Huen MSY, Liu Y, et al. 53BP1 loss rescues embryonic lethality but not genomic instability of BRCA1 total knockout mice. Cell Death Differ 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Belotserkovskaya R, Raga Gil E, Lawrence N, Butler R, Clifford G, Wilson MD, et al. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nature communications 2020;11:819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 2018;20:954–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 2017;31:318–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luijsterburg MS, Typas D, Caron MC, Wiegant WW, van den Heuvel D, Boonen RA, et al. A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zong D, Adam S, Wang Y, Sasanuma H, Callen E, Murga M, et al. BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol Cell 2019;73:1267–81 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakada S, Yonamine RM, Matsuo K. RNF8 regulates assembly of RAD51 at DNA double-strand breaks in the absence of BRCA1 and 53BP1. Cancer Res 2012;72:4974–83 [DOI] [PubMed] [Google Scholar]
- 72.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011;145:529–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012;22:106–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daza-Martin M, Starowicz K, Jamshad M, Tye S, Ronson GE, MacKay HL, et al. Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature 2019;571:521–7 [DOI] [PubMed] [Google Scholar]
- 75.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016;535:382–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feng W, Jasin M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nature communications 2017;8:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ding X, Ray Chaudhuri A, Callen E, Pang Y, Biswas K, Klarmann KD, et al. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nature communications 2016;7:12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI, et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J Clin Invest 2016;126:3145–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Michel B, Sinha AK, Leach DRF. Replication Fork Breakage and Restart in Escherichia coli. Microbiol Mol Biol Rev 2018;82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kolinjivadi AM, Sannino V, de Antoni A, Techer H, Baldi G, Costanzo V. Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett 2017;591:1083–100 [DOI] [PubMed] [Google Scholar]
- 81.Branzei D, Szakal B. Building up and breaking down: mechanisms controlling recombination during replication. Crit Rev Biochem Mol Biol 2017;52:381–94 [DOI] [PubMed] [Google Scholar]
- 82.Quinet A, Lemacon D, Vindigni A. Replication Fork Reversal: Players and Guardians. Mol Cell 2017;68:830–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, et al. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 1998;17:598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martin RW, Orelli BJ, Yamazoe M, Minn AJ, Takeda S, Bishop DK. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res 2007;67:9658–65 [DOI] [PubMed] [Google Scholar]
- 85.Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018;562:217–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chappuis PO, Nethercot V, Foulkes WD. Clinico-pathological characteristics of BRCA1- and BRCA2-related breast cancer. Semin Surg Oncol 2000;18:287–95 [DOI] [PubMed] [Google Scholar]
- 87.Tseng SL, Yu IC, Yue CT, Chang SF, Chang TM, Wu CW, et al. Allelic loss at BRCA1, BRCA2, and adjacent loci in relation to TP53 abnormality in breast cancer. Genes, chromosomes & cancer 1997;20:377–82 [PubMed] [Google Scholar]
- 88.Hakem R, de la Pompa JL, Elia A, Potter J, Mak TW. Partial rescue of Brca1 (5-6) early embryonic lethality by p53 or p21 null mutation. Nat Genet 1997;16:298–302 [DOI] [PubMed] [Google Scholar]
- 89.Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA, et al. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet 2001;28:266–71 [DOI] [PubMed] [Google Scholar]
- 90.Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev 2003;17:201–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cressman VL, Backlund DC, Avrutskaya AV, Leadon SA, Godfrey V, Koller BH. Growth retardation, DNA repair defects, and lack of spermatogenesis in BRCA1-deficient mice. Mol Cell Biol 2006;26:9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shen SX, Weaver Z, Xu X, Li C, Weinstein M, Chen L, et al. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene 1998;17:3115–24 [DOI] [PubMed] [Google Scholar]
- 93.Martins FC, De S, Almendro V, Gonen M, Park SY, Blum JL, et al. Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discov 2012;2:503–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A 2011;108:18032–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Friedman LS, Szabo CI, Ostermeyer EA, Dowd P, Butler L, Park T, et al. Novel inherited mutations and variable expressivity of BRCA1 alleles, including the founder mutation 185delAG in Ashkenazi Jewish families. American journal of human genetics 1995;57:1284–97 [PMC free article] [PubMed] [Google Scholar]
- 96.Takahashi H, Behbakht K, McGovern PE, Chiu HC, Couch FJ, Weber BL, et al. Mutation analysis of the BRCA1 gene in ovarian cancers. Cancer Res 1995;55:2998–3002 [PubMed] [Google Scholar]
- 97.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016;534:47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maxwell KN, Wubbenhorst B, Wenz BM, De Sloover D, Pluta J, Emery L, et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nature communications 2017;8:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Elstrodt F, Hollestelle A, Nagel JH, Gorin M, Wasielewski M, van den Ouweland A, et al. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res 2006;66:41–5 [DOI] [PubMed] [Google Scholar]
- 100.Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S, et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 2011;20:797–809 [DOI] [PubMed] [Google Scholar]
- 101.Drost R, Dhillon KK, van der Gulden H, van der Heijden I, Brandsma I, Cruz C, et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J Clin Invest 2016;126:2903–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hill SJ, Clark AP, Silver DP, Livingston DM. BRCA1 pathway function in basal-like breast cancer cells. Mol Cell Biol 2014;34:3828–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Williams RS, Chasman DI, Hau DD, Hui B, Lau AY, Glover JN. Detection of protein folding defects caused by BRCA1-BRCT truncation and missense mutations. J Biol Chem 2003;278:53007–16 [DOI] [PubMed] [Google Scholar]
- 104.Williams RS, Glover JN. Structural consequences of a cancer-causing BRCA1-BRCT missense mutation. J Biol Chem 2003;278:2630–5 [DOI] [PubMed] [Google Scholar]
- 105.Wang Y, Bernhardy AJ, Nacson J, Krais JJ, Tan YF, Nicolas E, et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nature communications 2019;10:5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A 2013;110:17041–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971;68:820–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019;571:576–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Setiaputra D, Durocher D. Shieldin - the protector of DNA ends. EMBO Rep 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clairmont CS, Sarangi P, Ponnienselvan K, Galli LD, Csete I, Moreau L, et al. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat Cell Biol 2020;22:87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Uckelmann M, Sixma TK. Histone ubiquitination in the DNA damage response. DNA Repair (Amst) 2017;56:92–101 [DOI] [PubMed] [Google Scholar]
- 112.Munoz MC, Laulier C, Gunn A, Cheng A, Robbiani DF, Nussenzweig A, et al. Ring Finger Nuclear Factor RNF168 Is Important for Defects in Homologous Recombination Caused by Loss of the Breast Cancer Susceptibility Factor BRCA1. J Biol Chem 2012;287:40618–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Krais JJ, Wang Y, Bernhardy AJ, Clausen E, Miller JA, Cai KQ, et al. RNF168-mediated ubiquitin signaling inhibits the viability of BRCA1 null cancers. Cancer Res 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Drane P, Brault ME, Cui G, Meghani K, Chaubey S, Detappe A, et al. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 2017;543:211–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Krais JJ, Johnson N. Ectopic RNF168 expression promotes break-induced replication-like DNA synthesis at stalled replication forks. Nucleic Acids Res 2020;48:4298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Menghi F, Barthel FP, Yadav V, Tang M, Ji B, Tang Z, et al. The Tandem Duplicator Phenotype Is a Prevalent Genome-Wide Cancer Configuration Driven by Distinct Gene Mutations. Cancer Cell 2018;34:197–210 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Willis NA, Frock RL, Menghi F, Duffey EE, Panday A, Camacho V, et al. Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature 2017;551:590–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Khalique S, Hook JM, Ledermann JA. Maintenance therapy in ovarian cancer. Curr Opin Oncol 2014;26:521–8 [DOI] [PubMed] [Google Scholar]
- 119.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med 2003;9:568–74 [DOI] [PubMed] [Google Scholar]
- 121.Hollis RL, Churchman M, Gourley C. Distinct implications of different BRCA mutations: efficacy of cytotoxic chemotherapy, PARP inhibition and clinical outcome in ovarian cancer. Onco Targets Ther 2017;10:2539–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dimitrova D, Ruscito I, Olek S, Richter R, Hellwag A, Turbachova I, et al. Germline mutations of BRCA1 gene exon 11 are not associated with platinum response neither with survival advantage in patients with primary ovarian cancer: understanding the clinical importance of one of the biggest human exons. A study of the Tumor Bank Ovarian Cancer (TOC) Consortium. Tumour Biol 2016;37:12329–37 [DOI] [PubMed] [Google Scholar]
- 123.Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, et al. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell 2009;35:327–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nature communications 2018;9:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov 2019;9:210–9 [DOI] [PubMed] [Google Scholar]
- 126.Wang H, Xiang D, Liu B, He A, Randle HJ, Zhang KX, et al. Inadequate DNA Damage Repair Promotes Mammary Transdifferentiation, Leading to BRCA1 Breast Cancer. Cell 2019;178:135–51 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015;518:258–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015;518:254–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Higgins GS, Boulton SJ. Beyond PARP-POLtheta as an anticancer target. Science 2018;359:1217–8 [DOI] [PubMed] [Google Scholar]
- 130.Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. Polymerase Theta Inhibition Kills Homologous Recombination Deficient Tumors. bioRxiv 2020:2020.05.23.111658 [Google Scholar]