

Abstract
This study aimed to understand the functional expression of KATP channel subunits in distinct lymphatic cell types, and assess the consequences of altered KATP channel activity on lymphatic pump function. KATP channel subunits Kir6.1 and SUR2B were expressed in mouse lymphatic muscle by PCR, but only Kir6.1 was expressed in lymphatic endothelium. Spontaneous contractions of popliteal lymphatics from WT (C57BL/6J) mice, assessed by pressure myography, were very sensitive to inhibition by the SUR2-specific KATP channel activator pinacidil, which hyperpolarized both mouse and human lymphatic smooth muscle (LSM). In vessels from mice with deletion of Kir6.1 (Kir6.1−/−) or SUR2 (SUR2[STOP]) subunits, contractile parameters were not significantly different from those of WT vessels, suggesting that basal KATP channel activity in LSM is not an essential component of the lymphatic pacemaker, nor exerts a strong influence over contractile strength. However, these vessels were >100-fold less sensitive than WT vessels to pinacidil. Smooth muscle-specific expression of a Kir6.1 gain-of-function (GoF) subunit resulted in severely impaired lymphatic contractions and hyperpolarized LSM. Membrane potential and contractile activity was partially restored by the KATP channel inhibitor, glibenclamide. In contrast, lymphatic endothelium-specific expression of Kir6.1 GoF subunits had negligible effects on lymphatic contraction frequency or amplitude. Our results demonstrate a high sensitivity of lymphatic contractility to KATP channel activators through activation of Kir6.1/SUR2-dependent channels in LSM. In addition, they offer an explanation for the lymphedema observed in patients with Cantú Syndrome, a disorder caused by gain-of-function mutations in genes encoding Kir6.1/SUR2.
Keywords: Cantú Syndrome, lymphedema, lymphatic dysfunction, electrophysiology, pressure myography
Introduction
The lymph pump system is an essential component of interstitial fluid balance and tissue homeostasis, combining extrinsic (passive) and intrinsic (active) forces to move lymph from the peripheral tissues to the central venous system against a hydrostatic pressure gradient. Contrary to the classical view that passive forces predominate, it is now clear that effective lymph propulsion requires competent intraluminal valves, robust contractions of lymphatic muscle cells and synchronized contraction waves (Scallan et al., 2016; Castorena-Gonzalez et al., 2018b). These contractions are driven by spontaneous action potentials (APs), similar in shape to sino-atrial node APs and involving a similar palette of underlying voltage-dependent ion channels (Cotton et al., 1997; McCloskey et al., 1999; Imtiaz et al., 2007; Imtiaz et al., 2010; Telinius et al., 2014b; Zawieja et al., 2018b; Zawieja et al., 2019), including Na+ and Ca2+ channels (Hollywood et al., 1997a, b; Telinius et al., 2015). Prior studies have also indicated the presence of ATP-sensitive (KATP) currents in lymphatic tissue, and a marked sensitivity of contraction to pharmacological activators of these channels (Mizuno et al., 1999; Mathias & von der Weid, 2013; Telinius et al., 2014b). KATP channels are composed of heterooctameric complexes of pore-forming (Kir6) and sulphonylurea receptor (SUR) subunits. Two Kir6 genes, KCNJ8 (Kir6.1) and KCNJ11 (Kir6.2), and two SUR genes, ABCC8 (SUR1) and ABCC9 (SUR2), encode mammalian KATP channels (Nichols, 2006), with alternative splicing of ABCC9 giving rise to at least two protein variants, referred to as SUR2A and SUR2B (Chutkow et al., 1996; Foster & Coetzee, 2016). Expression of Kir6.1 and SUR2B subunits has been detected in rat mesenteric lymphatics (by RT-PCR of whole vessel samples) and these KATP channel subunits were suggested to mediate the inhibitory effects of nitric oxide on spontaneous contractions (von der Weid et al., 2012; Mathias & von der Weid, 2013). Telinius et al. confirmed that pinacidil inhibited spontaneous contractions of isometric strips of human thoracic duct smooth muscle and that response was reversed by glibenclamide, implying the presence of functional KATP channels (Telinius et al., 2014a).
Activated by intracellular ADP and inhibited by intracellular ATP, KATP channels can act to couple metabolic state to electrical activity (Ashcroft, 1988; Nichols, 2006), but whether these channels can normally suppress lymphatic pacemaking activity and/or can be activated under hypoxic or ischemic conditions as in other tissues (Foster & Coetzee, 2016) remains unknown. Recent studies have shown that Cantu Syndrome (CS), a complex multi-organ disorder, is caused by gain-of-function (GoF) mutations in KCNJ8 (Kir6.1) or ABCC9 (SUR2) (Harakalova et al., 2012; van Bon et al., 2012; Brownstein et al., 2013; Cooper et al., 2014). CS patients are characterized by a constellation of symptoms which typically includes congenital hypertrichosis and dysmorphic facial features (Cantu et al., 1982; Grange et al., 2019). 80% of CS patients exhibit cardiovascular abnormalities, including hypotension, cardiomegaly, patent ductus arteriosus and pulmonary hypertension (Grange et al., 1993; Nichols et al., 2013; Levin et al., 2016). 50% of CS patients have symptomatic patent ductus arteriosus, compared to 0.05-0.2% in the general population, that requires surgical ligation or, if untreated, eventually leads to pulmonary hypertension (Nichols et al., 2013; Cooper et al., 2014). KATP GoF in these patients causes reduced excitability and reduced contractility in arterial smooth muscle, which in turn leads to a chronic reduction in systemic blood pressure (Li et al., 2013), eliciting reflex enhancement of cardiac output, with consequent ventricular hypertrophy and high-output cardiac failure (Levin et al., 2016; Huang et al., 2018). One prominent feature of CS that remains unexplained is >50% incidence of lymphedema (Garcia-Cruz et al., 2011; Grange et al., 2019). Lymphedema presents as subcutaneous fluid, and later fat, accumulation in the extremities that is typically bilateral, but can be unilateral or variable (Szuba & Rockson, 1998; Rockson, 2006). As with other forms of primary and secondary lymphedema, the only current treatment options for lymphedema in CS patients are palliative measures such as compression bandages and manual lymph drainage/massage. Understanding the mechanisms underlying lymphedema in CS patients could enable pharmacologic intervention to treat the cause(s) rather than the symptom(s).
We hypothesize that hyperactive Kir6.1/SUR2-dependent KATP channels, if present and functional in lymphatic vessel smooth muscle, would interfere with pacemaking and action potential (AP) generation by those cells. This would in turn inhibit the spontaneous, propulsive lymphatic contractions that are required to transport lymph against adverse hydrostatic pressure gradients in lymphatic networks of dependent extremities (Scallan et al., 2016), potentially resulting in lymphedema. In this study we examined the expression of KATP subunit isoforms in specific lymphatic cell types and, as an initial test of the above hypothesis, measured the electrical properties and contractile function of lymphatic smooth muscle in mice that selectively express a GoF KATP channel mutation in lymphatic cells.
Methods
Ethical Approval.
All procedures were reviewed and approved by the animal care committees at the University of Missouri or Washington University, St. Louis, and complied with the standards stated in the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health, revised 2011). Human mesenteric lymphatic vessels were isolated from bariatric surgery samples obtained after written, informed consent was secured from participating subjects at the University of Missouri. The use of human samples was approved by the University of Missouri Office of research Institutional Research Board (Project #1203035 HS) and conformed to the principles of the Declaration of Helsinki.
Mice.
Smooth-muscle specific Kir6.1 GoF mice were described previously (Li et al., 2013). Briefly, point mutations were engineered in two c-terminal residues of Kir6.1, Gly343Asp, Gln53Arg, hereafter referred to as Kir6.1[GD-QR], to render the channel insensitive to ATP, and this was engineered into a transgenic construct (Tong et al., 2006) under tamoxifen-inducible Cre-recombinase control. Mice carrying this transgene were crossed to SMMHC-Cre(Myh11Cre)ERT2 mice (JAX #01979; males only, as the transgene is carried on the y-chromosome) to generate double-transgenic SMMHC-CreERT2;Kir6.1[GD-QR mice with an inducible GoF Kir6.1 transgene expressed specifically in smooth muscle. To test the consequences of expressing the transgene selectively in lymphatic endothelium, Kir6.1[GD-QR] mice were crossed to Prox1CreERT2 mice (a gift from Guillermo Oliver, Feinberg Institute, Northwestern University). Transgenic mice were identified by PCR on mouse-tail DNA using green fluorescent protein–specific oligonucleotide primers (Li et al., 2013). For genotyping, genomic DNA was extracted from tail clips using the HotSHOT method. Genotypes were determined by PCR with 2x PCR Super Master Polymerase Mix (Catalog # B46019, Bimake, Houston, TX) according to the provider’s instructions. For analysis of gene expression specifically in lymphatic muscle cells, wild-type (WT) male mice (15-35 g) on the C57BL/6J background were crossed to ROSA26mT/mG reporter mice (JAX, #007676) and induced with tamoxifen (TMX). Mice were induced by five consecutive i.p. injections of 100 mg tamoxifen (10mg/ml in safflower oil). Kir6.1−/− mice (Miki et al., 2002) were a gift from Susumu Seino (Kobe University). Generation of SUR2-STOP mice was described previously (Ran et al., 2013). Briefly, an indel mutation in ABCC9 (c.3446_3450delACTTCinsGA) resulting in introduction of a premature stop codon (p.Y1148Stop) was generated using CRISPR/Cas9-mediated genome engineering (Smeland et al., 2019). Heterozygous SUR2wt/Stop mice were intercrossed to generate homozygous SUR2Stop/Stop (referred to as SUR2[STOP]) and wild-type littermates, which were used for experiments.
Human Lymphatic Tissues.
A discarded section of the intestinal wall, with attached fat, was collected from patients undergoing roux-en-y gastric bypass surgery and placed in cold physiological saline solution for subsequent experimentation. The samples were maintained in BSA-containing Krebs buffer at 4°C prior to and during dissection. Lymphatic collectors were located within the mesenteric adipose tissue adjacent to the jejunal wall, dissected out, cleared of fat and cannulated as described previously (Castorena-Gonzalez et al., 2018b).
Vessel Isolation, Pressure Myograph, and Data Acquisition.
Mice were anesthetized by intraperitoneal injection of pentobarbital-sodium (Nembutal; 60 mg/kg) and placed in the prone position on a heated tissue dissection/isolation pad. The superficial saphenous vein was exposed by a proximal-to-distal incision along the calf area beginning at the ankle and the popliteal afferent lymphatic vessels on each side of the vein were isolated as previously described (Scallan et al., 2016). Each vessel was then pinned with short segments of 40 μm stainless steel wire onto the Sylgard-coated surface of a dissection chamber filled with BSA-containing Krebs buffer at room temperature. Once secured, the majority of the surrounding adipose and connective tissues were removed by microdissection. An isolated vessel was then transferred to a 3-mL observation chamber, cannulated, pressurized to 3 cmH2O using two glass micropipettes (50-60 μm outside diameter) and transferred to the stage of a Zeiss inverted microscope. Lymphatic segments used in these studies contained 1 valve. To facilitate accurate diameter tracking, each cannulated vessel segment was then cleared of remaining connective and adipose tissue. Polyethylene tubing was attached to the back of each glass micropipette and connected to a computerized pressure controller (Scallan et al., 2013). To minimize diameter-tracking artifacts associated with longitudinal bowing at higher intraluminal pressures, input and output pressures were briefly set to 10 cmH2O at the beginning of every experiment, and the vessel segment was stretched axially to remove any longitudinal slack.
Once cannulated, a lymphatic vessel was allowed to equilibrate at 37°C with inflow and outflow pressures set to 3 cmH2O. Constant exchange of Krebs buffer was maintained using a peristaltic pump at a rate of 0.5 mL/min. After temperature stabilized, popliteal lymphatic vessels typically began to exhibit spontaneous contractions within 10-15 min that stabilized in a consistent pattern within ~30 minutes. Human mesenteric vessels were brought to 37°C over the course of 1 hour and required another 1-2 hrs in Krebs solution to develop spontaneous contractions. Custom LabVIEW programs (National Instruments; Austin, TX) acquired real-time analog data and digital video through an A-D interface (USB-6216, National Instruments) and detected the inner diameter of the vessel (Davis et al., 2006). Videos of the contractile activity of lymphatic vessels were recorded for further analyses under bright-field illumination at 30 fps using a Basler A641fm firewire camera.
Assessment of ex vivo Contractile Function and Pressure Responses.
The contractile parameters of each vessel were characterized at different levels of intraluminal pressure spanning the physiological range from 0.5 to 10 cmH2O (in successive steps as follows: 3, 2, 1, 0.5, 3, 5, 8, and 10 cmH2O). Spontaneous contractions were recorded at each pressure in typical intervals of 2 minutes, although up to 5 min was sometimes allowed at the lowest pressure, if needed, to obtain multiple contractions. Basal contractile function was assessed at 3 cmH2O but popliteal lymphatic vessels exhibited their strongest contractions at pressures between 1-2 cmH2O. During the pressure response protocol, both the input and output pressures were maintained at equal levels so that there was no imposed pressure gradient for forward flow. At the end of every experiment, all vessels were equilibrated by perfusion with calcium-free Krebs buffer containing 3 mM EGTA for 30 minutes, and passive diameters were obtained at each level of intraluminal pressure.
Ex vivo Contractile Function Parameters.
Once an experiment was completed, internal diameter traces (from a single representative region of interest) and/or 30-fps bright-field videos of spontaneous contractions were analyzed using custom-written LabVIEW programs to detect end diastolic diameter (EDD), end systolic diameter (ESD), and contraction frequency (FREQ, computed on a contraction-by-contraction basis and averaged over a 2-5 min period). These data were used to calculate several commonly reported parameters that characterize the contractile function of lymphatic vessels:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
where DMAX represents the maximum passive diameter (obtained after incubation with calcium-free Krebs solution) at a given level of intraluminal pressure. When frequency was zero, a value for amplitude was omitted. Each of these contractile parameters represent the average of all the recorded contractions during the 2-5 min time periods corresponding to the different intraluminal pressures.
In vivo Imaging of Popliteal Lymphatic Contractile Function.
The mouse hindlimb was treated with depilating cream and washed with PBS to remove hair. After removal of the superficial layer of the skin, lymphatic collectors were visualized via injection of 10 to 20 μl (s.c.) of DyLight 594–conjugated tomato lectin (Vector Laboratories) into the footpad. Images of popliteal lymphatics were collected using a customized Leica SP8 2-photon microscope equipped with a ×25/0.95 NA water-dipping objective. A Mai Tai HP DeepSee Laser (Spectra- Physics) tuned to 900 nm provided the excitation light. Fluorescence emission was separated by dichroic mirrors at 458, 495, and 560 nm (Semrock) and directed to external detectors. 2-Photon excitation produced a second harmonic signal from collagen. Vessel diameters were measured using Imaris (Bitplane) and Matlab (Mathworks). After the 594 nm channel was isolated in Imaris, a Matlab script was used to break the vessel image into 20 sections, and then the width was measured randomly within each section. The average value of all 20 measurements gave the average vessel diameter at each point in time. Graphs were generated from the average thickness at each point over the 1000 frames collected at 14 fps.
FACS analysis.
Inguinal-axillary lymphatic vessels from tamoxifen-treated SMMHC-CreERT2;Rosa26mTmG mice were dissected as described previously (Zawieja et al., 2018b). Cleaned vessel segments were transferred to a 1-ml tube of low-Ca2+ PSS containing (in mM): 137 NaCl, 5.0 KCl, 0.1CaCl2, 1.0 MgCl2, 10 HEPES, 10 Glucose, and 1 mg/ml BSA at room temperature for 10 min. The solution was decanted and replaced with a similar solution containing 26 U/ml papain (Sigma, St. Louis, MO) and 1 mg/ml dithioerythritol. The vessels were incubated for 30 min at 37°C with occasional agitation, then transferred to a new tube containing low-Ca2+ PSS containing 25 mg/ml collagenase H (FALGPA U/ml, Sigma), 0.7 mg/ml collagenase F (Sigma), 20 mg/ml trypsin inhibitor (Sigma), 1mg/ml elastase (Worthington), and incubated for 6 min at 37°C. The resultant dispersed cells from digested vessels were sedimented by centrifugation (300g, 4 min), resuspended in 0.6 ml PSS containing 1mM Ca solution, and filtered through a nylon filter with 35-μm mesh size to obtain single cell suspension. Smooth muscle cells expressing eGFP were sorted by fluorescence‐activated cell sorting (FACS) with a Beckman-Coulter MoFlo XDP instrument using an excitation laser (488 nm) and emission filter (530/40 nm), with 70 μm nozzle at a sheath pressure of 45 psi and sort rate of 100 events per second. Sorting was performed at the Cell and Immunobiology Core facility at the University of Missouri.
Isolation of Popliteal Lymphatic Endothelial Cell Tubes and Smooth Muscle.
Popliteal lymphatic endothelial cell tubes (LECTs) were prepared as recently described (Behringer et al., 2017) by incubation in low Ca2+-PSS for 20 min at 34°C. Low Ca2+-PSS contained 0.62 mg/ml papain (≥ 6 units), 1.5 mg/ml collagenase (≥ 15 units), 1.0 mg/ml dithioerythritol, 0.1% bovine serum albumin (USB Corp.; Cleveland, OH; USA) and 0.1 mmol/L CaCl2. To separate muscle cells from the endothelial layer, a vessel segment was gently triturated using aspiration and ejection from a micropipette during visual inspection at 200X. Collection pipettes were prepared from borosilicate glass capillary tubes [1.0 mm outer diameter (OD)/ 0.58 mm ID; World Precision Instruments (WPI), Sarasota, FL; USA], pulled (P-97; Sutter Instruments; Novato, CA; USA) and heat-polished at one end (tip internal diameter: ~50–70 μm). The resulting LEC tube or LMCs were then rinsed with PSS lacking enzymes at room temperature and spun at 1000 rpm for 5 min before RNA extraction.
End-Point RT-PCR.
Total RNA was extracted from lymphatic vessels or sorted cells using the Arcturus PicoPure RNA isolation kit (Thermo Fisher Scientific, Waltham, MA) with on-column DNase I treatment (Qiagen, Valencia, CA) according to the manufacturer’s instructions. RNA was eluted with 20 μL nuclease-free water. Purified RNA was then used for cDNA synthesis via reverse transcription using random hexamer and oligod(T) primers, and SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, Waltham, MA). PCR was performed in a reaction mixture containing first-strand cDNA as the template, 2 mM MgCl2, 0.25 μM primers, 0.2 mM deoxynucleotide triphosphates; and GoTaq® Flexi DNA polymerase (Promega, Madison, WI). The PCR program comprised an initial denaturation step at 95°C for four minutes; followed by 35 repetitions of the following cycle: denaturation (94°C, 30s), annealing (58°C, 30s) and extension (72°C, 30s). This was followed by a final elongation step for 5 min at 72°C. PCR amplification products were separated on a 2% agarose gel by electrophoresis, stained with SYBR-Safe (Thermo Fisher Scientific, Waltham, MA), and visualized by UV transillumination. All primers were designed to amplify intron-spanning DNA regions; primer sequences are listed in Table 1.
Table 1.
Primer sequences used in this study.*
| Gene | Strand | Sequence | Amplicon (bp) |
|---|---|---|---|
| Kcnj8 | s | GAG GAC GCG TGT GGA GGA AA | 301 |
| as | TGG TGA AGA TGA CCA GCG TG | ||
| Kcnj11 | s | ACA TGC AGG TGG TGC GCA AG | 430 |
| as | AGG GCA TCC AGC AGA CTG CG | ||
| Abcc9 | s | GGA GGC TTA GAT GCC ACT GT | 465 (2A) |
| as | TTT CCG GGG TGT CGT ATT CC | 289 (2B) | |
| Acta2 | s | GTG AAG AGG AAG ACA GCA CAG | 146 |
| as | GCC CAT TCC AAC CAT TAC TCC | ||
| Cdh5 | s | TCA ACG CAT CTG TGC CAG AGA T | 116 |
| as | CAC GAT TTG GTA CAA GAC AGT G |
Solutions and Chemicals.
Krebs buffer contained: 146.9 mM NaCl, 4.7 mM KCl, 2 mM CaCl2·2H2O, 1.2 mM MgSO4, 1.2 mM NaH2PO4·H2O, 3 mM NaHCO3, 1.5 mM Na-HEPES, and 5 mM D-glucose (pH = 7.4). An identical buffer was prepared with the addition of 0.5% bovine serum albumin (“Krebs-BSA”). During cannulation Krebs-BSA buffer was present both luminally and abluminally; however, during the experiment the abluminal solution was constantly exchanged with plain Krebs. Ca2+-free Krebs was Krebs with 3 mM EGTA replacing CaCl2·2H2O. All chemicals were obtained from Sigma-Aldrich (St. Louis, MO), with the exception of BSA (US Biochemicals; Cleveland, OH), MgSO4 and Na-HEPES (ThermoFisher Scientific; Pittsburgh, PA). Pinacidil and glibenclamide (Sigma-Aldrich) were each prepared in DMSO as stock solutions and then further diluted in Krebs solution to reach the final concentrations stated.
Membrane Potential Recording.
Sharp electrode impalements into lymphatic smooth muscle cells were made on pressure myograph preparations using intracellular glass micro-electrodes (250-350MΩ) filled with a 1M KCl solution. Addition of 2 μM wortmannin (Tocris), to blunt vessel movement associated with contractions, facilitated the intracellular impalements. Membrane potential was sampled at 1-5 KHz using NPI SEC-05x and/or AxoClamp2A amplifiers and digitized through an A-D interface (USB-6216, National Instruments).
Statistical Tests.
The number n refers to the number of vessels included per group. Statistical differences between the various functional parameters were assessed via 1) unpaired t-test (parametric) with Welch’s correction when performing a single comparison between 2 groups (Figs. 4,9); 2) 1-way ANOVAs followed by Dunnett’s post-hoc tests for pinacidil concentration-response curves when comparing individual values to control values (Fig. 2); 3) mixed model ANOVAs (allowing for missing values) with Tukey’s multiple comparisons tests when comparing pinacidil responses between WT, Kir6.1−/− and SUR2[STOP] (Fig. 2); 4) 2-way ANOVAs with Tukey’s multiple comparison tests to compare contraction parameters between WT, Kir6.1−/− and SUR2[STOP] (Fig. 3); or 5) mixed model ANOVAs (allowing for missing values) with Sidak’s multiple comparisons tests when comparing data from 2 groups with multiple treatments (i.e. control vs glibenclamide, and their interaction with intraluminal pressure, in tamoxifen induced and non-induced mice) (Figs. 7,8). Statistical analyses were performed using Prism8 (Graphpad). Data are plotted as mean±SD with significance set at p<0.05. See Statistical Summary Document for detailed results.
Fig. 4.
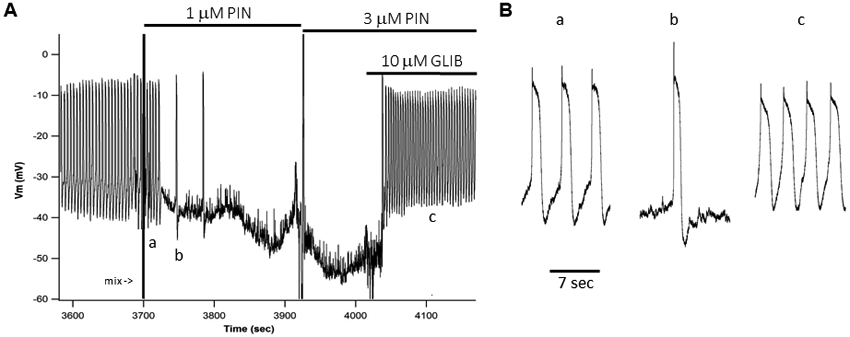
A) Membrane potential (Vm) recording made with an intracellular electrode (150 MΩ) showing spontaneous APs firing in a lymphatic muscle cell of a WT mouse popliteal lymphatic vessel (pressurized to 3 cmH2O). Expanded time scale (B) reveals a “resting” potential between −35 and −40 mV and APs with peak ~−5 mV and plateau at −8 mV lasting ~1.5 sec. Each AP corresponds to a single contraction. The application of 1 μM PIN (A) first slows the firing rate from ~13/min to 2/min before Vm hyperpolarizes to ~−45 mV and APs stop. A higher dose of 3 μM PIN further hyperpolarizes the cell to ~−55 mV. Subsequent application of 10 μM GLIB depolarizes the cell to between −32 and −36 mV, triggering APs at a higher rate than during the control period. The recording was made in the presence of 2 μM wortmannin to blunt contractions that otherwise would dislodge the microelectrode.
Fig. 9.

A) In vivo images of popliteal lymphatic collectors from control mice (left) and TMX-treated SMMHC-CreERT2;Kir6.1[GD-QR] mice taken at the minimum diameter of the lymphatic contraction cycle. Red= lectin; Green = GFP, Blue = 2nd harmonic (collagen); calibration bar = 80 μm. An intraluminal valve (with GFP+ leaflets) is visible in the latter. Below each image is an example of the diameter change from the vessel midpoint over a 1.2 min period, measured from the lectin channel; it shows that cyclical diameter changes in the control vessel and absence of diameter change in the TMX-treated SMMHC-CreERT2;Kir6.1[GD-QR] vessel. B) Summary of amplitude and frequency of diameter fluctuations in control (n=9) and GOF (SMMHC-CreERT2;Kir6.1[GD-QR]) (n=7) vessels. * = p< 0.0002; # = p<0.0007.
Fig. 2.
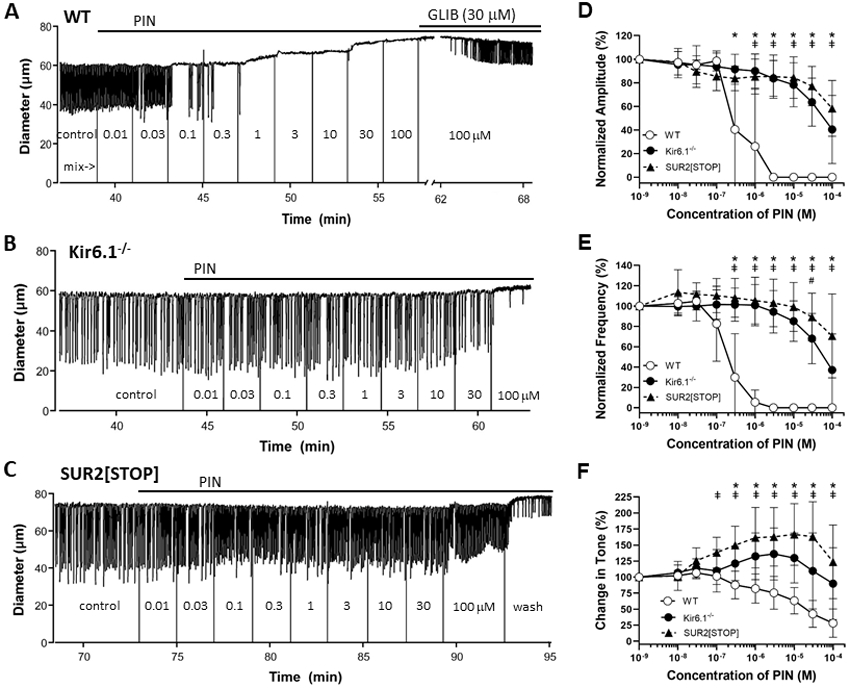
Recordings of spontaneous contractions from popliteal lymphatic vessels, pressurized to 3 cmH2O, from A) WT, B) Kir6.1−/− and C) SUR2[STOP] mice, before and after addition of pinacidil (PIN) to the bath. Solid vertical lines from zero (from blanked video) indicate addition of PIN and subsequent mixing of the bath by pipetting. PIN silenced contractions and dilated the WT vessel starting at a concentration of 100 nM, but had little effect on the Kir6.1−/− and SUR2[STOP] vessels at concentrations <100 μM. Glibenclamide (GLIB, 30 μM) partially reversed the inhibition by 100 μM PIN in the WT vessel. Analysis of the effect of PIN on normalized D) contraction amplitude, E) contraction frequency and F) tone, with values referenced to the control period, for vessels from the 3 strains, showing >100-fold right-shift in the IC50 values of the concentration-response curves for Kir6.1−/− and SUR2[STOP] vessels compared to WT. IC50 values are listed in Table 2. Means ± SD, n=9 for WT vessels, n=9 Kir6.1−/− and n=9 SUR2[STOP]. * p<0.05 for WT vs SUR2[STOP]; ǂ for WT vs Kir6.1−/− (no significant values); # for Kir6.1−/− vs SUR2[STOP] (no significant values). Significant differences for comparisons to control values are listed in the Statistical Summary Document.
Fig. 3.
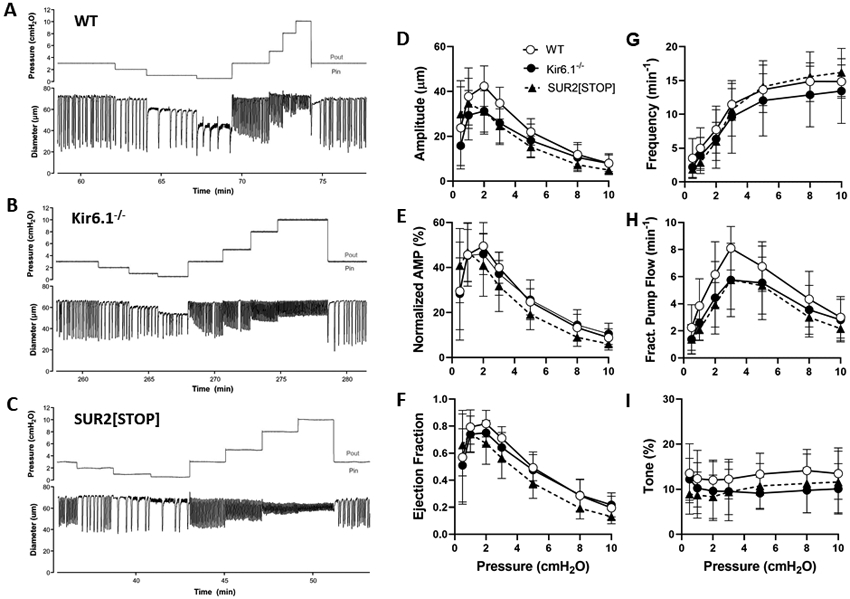
Contractile parameters of popliteal lymphatic vessels from WT, Kir6.1−/− and SUR2[STOP] mice, studied at seven intraluminal pressures from 0.5 to 10 cmH2O. There were no significant differences between any of the groups at any equivalent pressures. Means ± SD. n=16 for WT, n=19 for Kir6.1−/−, n=14 for SUR2[STOP].
Fig. 7.
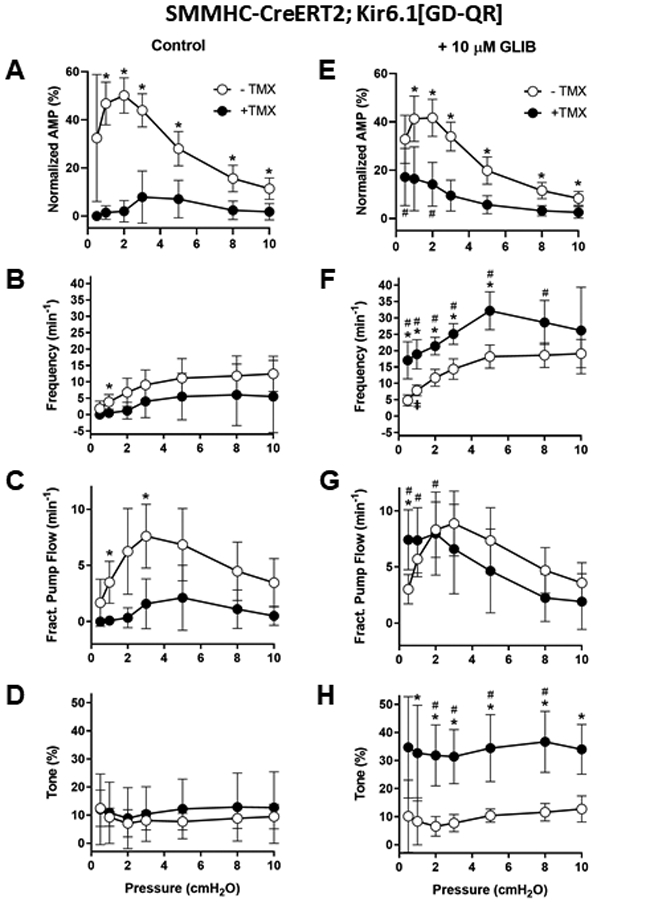
A-D) Summary of the contractile properties of popliteal lymphatics from induced and non-induced SMMHC-CreERT2;Kir6.1[GD-QR] mice. Expression of the GOF channel selectively in LMCs resulted in profound blunting of contraction amplitude at almost all pressures, ~no contractions at pressures ≤2 cmH2O and contraction frequencies ~1/2 of normal at other pressures. Calculated fractional pump flows in GOF vessels were 60-90% lower than in non-induced vessels at most pressures. Surprisingly, there were no significant differences in tone between the two groups. E-H) The application of 10 μM GLIB: reduced amplitude slightly at all pressures in vessels from non-induced mice (compare open symbols in A and E), but improved amplitude at all pressures (compare solid points in A and E); substantially increased frequency at all pressures in both induced and non-induced vessels; almost normalized fractional pump flow in the induced vessels; increased tone in the induced vessels by ~3-fold at all pressures. Means ± SD, n= 8 induced; n = 7 not-induced; * p < 0.05 between induced and non-induced; ǂ p < 0.05 between non-induced with vs without GLIB; # p < 0.05 between induced with vs without GLIB.
Fig. 8.
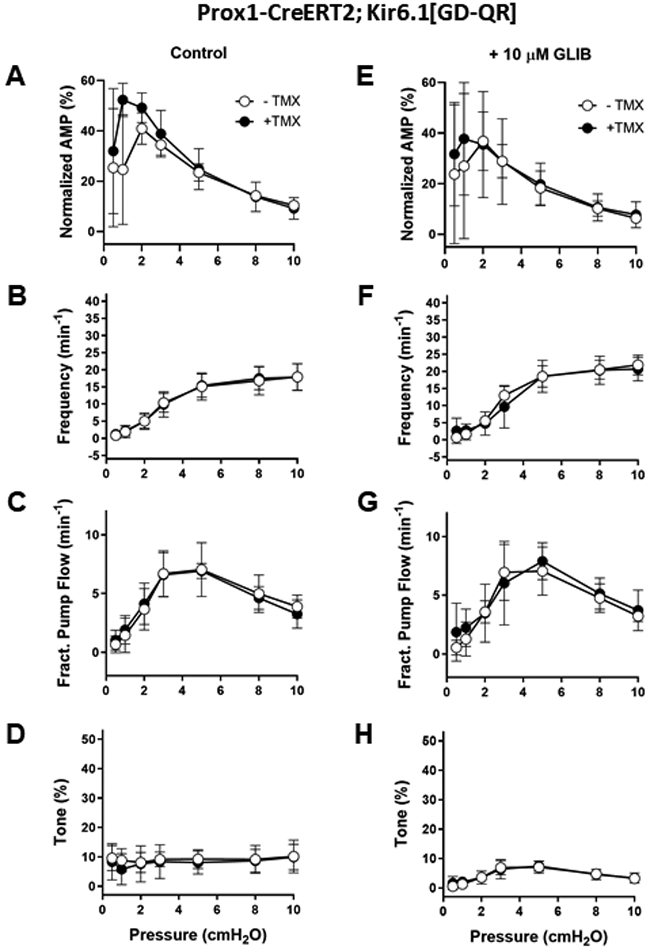
A-D) Summary of the contractile properties of popliteal lymphatics from induced and non-induced Prox1-Cre;Kir6.1[GD-QR] mice. Expression of the GOF channel was without significant effect on any of the contractile parameters at any pressure. E-H) The application of 10 μM GLIB reduced amplitude slightly at all pressures in vessels from both non-induced and induced mice, led to only modest increased frequencies at many pressures in both induced and non-induced vessels (none significant), and did not significantly alter fractional pump flow or tone (with a single exception) in either group. Means ± SD; n = 6 induced; n = 6 not-induced; * p < 0.05 between induced and non-induced (no significant values); ǂ p < 0.05 between non-induced with vs without GLIB (no significant values); # p < 0.05 between induced with vs without GLIB (no significant values). Axes are scaled the same as in Fig. 7 to facilitate comparisons between figures.
Results
Kir6.1 and SUR2B underlie lymphatic smooth muscle KATP
Kir6.1 and SUR2B were previously shown to be expressed in whole lymphatic vessels from guinea pig mesentery (Mathias & von der Weid, 2013), but that analysis did not differentiate between expression in endothelium and smooth muscle. Because whole lymphatics contain additional cell types, including fibroblasts, mast cells, macrophages, dendritic cells and nerve endings, we developed methods to specifically purify mouse lymphatic smooth muscle cells (LMCs) and lymphatic endothelial cells (LECs), using two different approaches. First, mouse popliteal afferent lymphatic vessels were isolated from the popliteal fossa of anesthetized mice, as previously described (Scallan et al., 2016). After excision the vessels were cleaned of adventitial fat and connective tissue and isolated LEC tubes were obtained by partial digestion and trituration (Behringer et al., 2017). A typical LEC tube, containing a single valve, is shown in Fig. 1A. End-point PCR identified message for CDH5 and absence of α-SMA confirmed that the sample was nearly pure endothelium. Message was also identified for Kir6.1 but not Kir6.2 or, SUR2. In contrast, analysis of the sheath of LMCs remaining after expulsion of the LEC tube (Fig. 1B) revealed message for Kir6.1 and SUR2 (but not Kir6.2 or SUR1, not shown). The band for SUR2 represents SUR2B rather than SUR2A, based on the amplicon size (see Table 1). The sample showed strong expression of α-SMA but also weak expression of CDH5, suggesting it contained mostly LMCs but with minor contamination from LECs or another cell type.
Fig. 1.
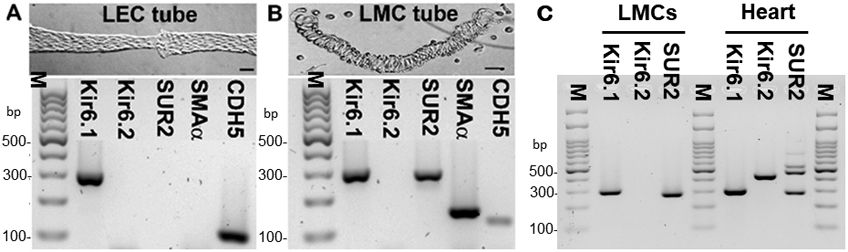
A) An image of an isolated LEC tube dissociated from a mouse popliteal lymphatic vessel (top), and PCR results (bottom) showing message for Kir6.1 and CDH5 (VE-cadherin) but not Kir6.2, SUR2 or SMA α-actin; B) An image of LMCs remaining after expulsion of an LEC tube (top), and PCR results (bottom) showing message for Kir6.1, SUR2 and SMA α-actin, with some contamination from CDH5. C) PCR results from LMCs purified by FACS using 4 pooled, inguinal-axillary lymphatic vessels from a tamoxifen-treated SMMHC-CreERT2;Rosa26mTmG mouse, showing message for Kir6.1 and SUR2, but not Kir6.2; heart was used as a positive control. Kir6.1 (Kcnj8) = 301 bp; Kir6.2 (Kcnj11) = 430 bp; SUR2A (Abcc9) = 465 bp; SUR2B (Abcc9) = 289 bp; SMA = 146 bp; CDH5 = 116 bp. PCR results are representative of 3 experiments. Calibration bars = 50 μm.
As a second approach, we selectively expressed a GFP reporter in smooth muscle by generating SMMHC-CreERT2;Rosa26mTmG mice, which allowed the use of FACS analysis to obtain a purified lymphatic muscle cell population. For this approach we used inguinal-axillary lymphatics from the flank of TMX-induced SMMHC-CreERT2;Rosa26mTmG mice, providing 2-4 lymphatic vessels ~120 μm in diameter and 1-2 cm in length, per animal. The vessels were cleaned of fat, connective tissue and any small blood capillaries, then pooled and enzymatically digested prior to FACS analysis. After selection of a narrow gating window for the GFP channel (i.e. only recombined, GFP+ cells), we collected and extracted RNA from 500 to 2,000 cells. Analysis of this sample revealed message for Kir6.1 and SUR2B (Fig.1C). A sample of cardiac tissue (cardiac muscle and associated blood vessels) was used as a positive control and predictably showed message for Kir6.1, Kir6.2, SUR2A, and SUR2B (Fig.1C)(Morrissey et al., 2005).
Having demonstrated that mouse lymphatic muscle expresses message for SUR2B and Kir6.1 (but not Kir6.2) channels, we used Kir6.1−/− and SUR2[STOP] mice to test the responses of lymphatic vessels lacking Kir6.1 or SUR2 subunits, respectively, to the KATP agonist, pinacidil. Kir6.1−/− knock-out mice were previously shown to completely lack KATP channels in vascular smooth muscle, resulting in a hypertensive, Prinzmetal angina-like phenotype (Miki et al., 2002; Li et al., 2013). SUR2[STOP] mice completely lack KATP channels in cardiac muscle and arterial smooth muscle, but the lymphatic consequences of loss of Kir6.1 or SUR2 subunits are unknown. Representative recordings of the effects of pinacidil on the spontaneous contractions of popliteal lymphatics from WT, Kir6.1−/− and SUR[STOP] mice are shown in panels A, B and C, respectively, of Fig. 2. Pinacidil essentially stopped contraction of WT vessels at concentrations as low as 100 nM, while higher concentrations additionally resulted in tonic dilation (Fig. 2A). These vessels normally only develop 5-10% tone (Scallan & Davis, 2013; Zawieja et al., 2018b), and hence pinacidil-induced dilations were modest compared to those in most arteries. Weak spontaneous contractions reappeared when GLIB (30 μM) was added with pinacidil (100 μM) still present. In contrast, the contraction frequency and amplitude of the Kir6.1−/− vessel changed little until much higher pinacidil concentrations (>10-30 μM) were reached (Fig. 2B). SUR2[STOP] vessels were similarly insensitive to pinacidil (Fig. 2C). Summary data for WT, Kir6.1−/− and SUR2[STOP] vessels, shown in panels D, E and F of Fig. 2, reveal dramatic loss of sensitivity, with IC50 values (listed in Table 2) for pinacidil inhibition of contraction amplitude and frequency (from Hill equation fits) right-shifted >100 fold for Kir6.1−/− and SUR2[STOP] vessels, compared to WT vessels. Tone was progressively lost with increasing pinacidil concentrations in WT vessels but, in contrast, pinacidil led to slight increases in tone in Kir6.1−/− and SUR2[STOP] vessels (Fig. 2F). In the latter two groups the tone changes were biphasic and could not be fit to the Hill equation for calculation of IC50 values. The vehicle (DMSO) had no significant effects at any of the concentrations used, as determined in a separate set of protocols (not shown). The impaired responses of Kir6.1−/− and SUR2[STOP] lymphatic vessels to pinacidil are consistent with Kir6.1 and SUR2 subunits being essential for normal expression of KATP channels in those vessels.
Table 2.
IC50 values determined from fits of pinacidil concentration-response relationships (using Hill equation) for popliteal lymphatics from WT, Kir6.1−/− and SUR2[STOP] mice.
| Parameter | WT | Kir6.1−/− | SUR2[STOP] |
|---|---|---|---|
| Normalized AMP IC50 (M) | 3.4 ·10−7 | 1.4 ·10−4 | 7.0 ·10−4 |
| Normalized FREQ IC50 (M) | 1.3 ·10−7 | 8.3 ·10−5 | 4.1 ·10−4 |
| Change in Tone IC50 (M) | n/a | n/a | n/a |
All parameters were normalized to those averaged over the 2-min period prior to administration of the first pinacidil dose. M = molar. n/a = could not be fit to Hill equation.
At a baseline pressure of 3 cmH2O, Kir6.1−/− and SUR2[STOP] mice exhibited apparently normal spontaneous lymphatic contractions (Fig. 2B,C). We then tested whether any of the major contractile parameters (amplitude, frequency, ejection fraction, fractional pump flow, or even tone, were significantly different from those of WT vessels at 7 different pressure levels between 0.5 and 10 cmH2O, spanning the presumed physiologic working range of these vessels in the mouse. Example diameter recordings from each of the three strains in response to pressure steps are shown in Fig. 3A-C. Analyses of the average contractile parameters from 16 WT vessels, 19 Kir6.1−/− vessels and 14 SUR2[STOP] vessels are shown in Fig. 3D-I: there were no significant differences between any of the parameters between groups at any pressure. This finding suggests little or no KATP activity under basal conditions in WT vessels. However, the exquisite sensitivity to pinacidil suggests that, once activated, KATP channels can profoundly inhibit spontaneous contractions.
Lymphatic SM KATP can regulate electrical activity and contractility
Intracellular recordings of membrane potential (Vm) were made using sharp electrodes in LMCs of pressurized popliteal lymphatics (Fig. 4). Because large-amplitude contractions almost always dislodged the recording pipette, the vessels were treated with 2 μM wortmannin to inhibit myosin light chain kinase. This dose was chosen to suppress contractions adequately for recordings, but still leave residual contractions so we could ensure the vessel remained spontaneously active. At this dose there is no significant effect of wortmannin on resting Vm, or lymphatic action potential (AP) shape (Zawieja et al., 2018c), although action potential (AP) firing rate typically increased slightly. APs activated spontaneously following diastolic depolarization from −40 to −35 mV, with a rapid upstroke and peak at ~−8 mV, and a plateau at −10 mV that lasted 1-1.5 s followed by repolarization to −40 mV. Addition of 1 μM pinacidil to the bath, followed by careful mixing so as not to dislodge the recording pipette, resulted in a profound slowing of pacemaker depolarization and AP frequency, and then compete cessation of APs (Fig. 4A). The maximum diastolic potential hyperpolarized slightly from −40 mV to ~−45 mV. APs that fired immediately after pinacidil application were typically larger in amplitude than normal (Fig. 4B), consistent with hyperpolarization-driven relief of steady-state voltage-gated Ca2+ channel inactivation. Subsequent addition of 3 μM pinacidil produced further hyperpolarization to ~−53 mV, with continued absence of APs. Addition of 10 μM glibenclamide, with pinacidil still present, produced an immediate depolarization and restoration of spontaneous AP activity. These results are consistent with activation of Kir6.1/SUR2B channels in LMCs by pinacidil and reversal by the KATP channel inhibitor glibenclamide.
To test whether functional KATP currents are also present in human lymphatic smooth muscle, we measured Vm in LMCs of pressurized human mesenteric lymphatics obtained from subjects undergoing bariatric surgery. A typical recording is shown in Fig. 5A. Resting Vm was slightly more negative (−45 mV) in human than in mouse. The human LMC AP had a higher upstroke velocity and a notch between the spike and the plateau, with a plateau around −20 mV (Fig. 5B,d). As in mouse, 1 μM pinacidil caused only a very subtle hyperpolarization but completely silenced the firing of spontaneous APs; 3 μM pinacidil produced ~15 mV hyperpolarization in this vessel. Glibenclamide (10 μM) rapidly reversed the effect of pinacidil, restoring the spontaneous firing of APs. Also, similar to mouse, there was a slight potentiation in the amplitude of the AP spike immediately following the application of glibenclamide (Fig. 5B). The changes in Vm in response to pinacidil and glibenclamide are summarized in Fig. 5C; on average 2 and 3 μM pinacidil produced ~4 and 12 mV hyperpolarizations, which were reversed by glibenclamide.
Fig. 5.
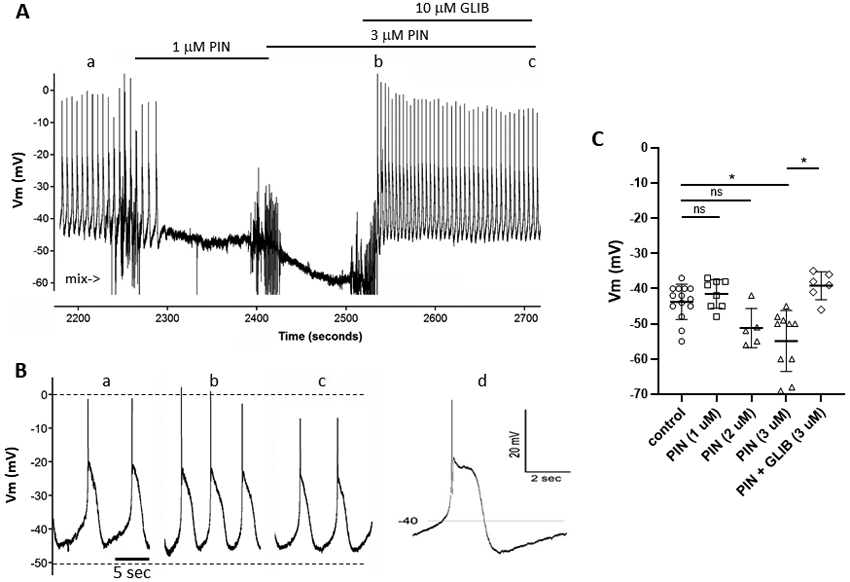
A) Membrane potential (Vm) recording from a lymphatic muscle cell of a human mesenteric lymphatic vessel (pressurized to 3 cmH2O). Spontaneous APs fire at a constant rate until 1 μM PIN is applied, after which the firing rate slows and Vm hyperpolarizes from −45 to ~−47 mV; the latter is sufficient to silence APs. 3 μM PIN further hyperpolarizes the cell to ~−60 mV. Subsequent application of 10 μM GLIB depolarizes the cell to −45 mV and APs resume firing at a higher rate. B) Expanded time scale showing APs at times a, b, c; d shows an even more expanded scale, revealing a notch between the spike and plateau. Recordings were made in the presence of 2 μM wortmannin. C) Summary of resting Vm measurements before and after PIN addition showing progressive hyperpolarization with higher PIN concentration and reversal by GLIB. Error bars are ±SD. Control (n=14); 1 μM PIN (n=8); 2 μM PIN (n=5); 3 μM PIN (n=10); 3 μM PIN+3 μM GLIB (n=6).
Lymphatic contractility is highly sensitive to SM KATP GoF
As mentioned in the Introduction, CS patients suffer from a high incidence of unexplained lymphedema, which we hypothesize may result from reduced lymphatic contractility as a consequence of increased LMC KATP channel activity. To examine this, we used pressure myography and measured contractility of popliteal lymphatics from mice with GoF KATP channels in lymphatic cells as a result of transgenic expression of ATP-insensitive Kir6.1[G343D,Q53R] mutant subunits (Li et al., 2013). Smooth muscle-specific expression of the Kir6.1[GD-QR] GoF transgene was previously shown to cause a significant hypotensive phenotype as a result of enhanced K+ conductance and reduced blood vessel contractility in arterial smooth muscle (Li et al., 2013), but the effect on lymphatic function is unknown. The first noticeable difference between lymphatic vessels from tamoxifen-induced SMMHC-CreERT2;Kir6.1[GD-QR] mice compared to un-induced or WT mice was the length of time required to develop spontaneous contractions during the equilibration period. Once cannulated, pressurized to 3 cmH2O, and equilibrated at 37°C, popliteal lymphatics from non-induced SMMHC-CreERT2;Kir6.1[GD-QR] mice developed ~10% spontaneous tone and exhibited large-amplitude, spontaneous contractions within ~15 min; the contraction pattern typically stabilized at a steady rate and amplitude after an additional 15 min (Fig 6A). This is similar to what we reported previously for C57BL/6J popliteal lymphatics (Zawieja et al., 2018b). In contrast, half the popliteal vessels from induced SMMHC-CreERT2;Kir6.1[GD-QR] mice did not develop any spontaneous contractions even after 50 min (Fig. 6B), whereas the other half developed only weak, irregular contractions (not shown).
Fig. 6.
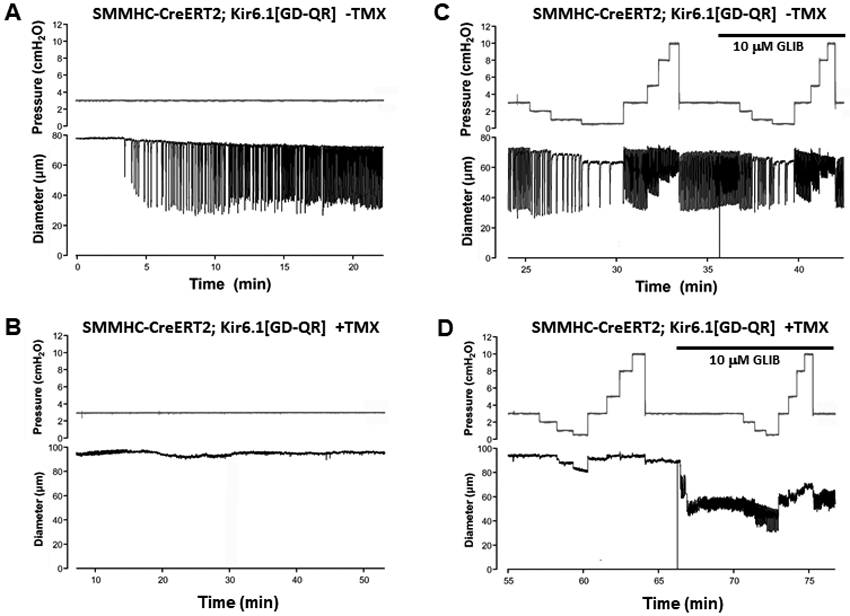
A) Diameter recording of a popliteal lymphatic from a non-induced SMMHC-CreERT2;Kir6.1[GD-QR] mouse showing typical development of spontaneous contractions within a few minutes after equilibration at 37°C. B) In contrast, a popliteal lymphatic from a SMMHC-CreERT2;Kir6.1[GD-QR] mouse treated with TMX (2 weeks prior) does not develop spontaneous contractions even after 50 min. C) The vessel from the non-induced mouse shows a typical response to imposed pressure steps: an increase in contraction frequency and slight reduction in amplitude as pressure is elevated from 0.5 to 10 cmH2O. GLIB (10 μM GLIB) elevates basal frequency and slight reduces amplitude. D) The vessel from the induced mouse does not show contractions at any pressure, but develops weak, high-frequency contractions in the presence of GLIB.
After the 30-60 min equilibration period, vessels were subjected to different intraluminal pressures between 0.5 and 10 cmH2O. Lowering pressure from 3 to 2, 1, and 0.5 cmH2O in a non-induced SMMHC-CreERT2;Kir6.1[GD-QR] vessel led to the typical response described previously for C57Bl/6J vessels (Scallan & Davis, 2013; Zawieja et al., 2018b): a progressive reduction in frequency, and a biphasic amplitude response with a maximum at 1-2 cmH2O. Elevating pressure progressively from 0.5 to 10 cmH2O led to progressive increases in frequency and reductions in amplitude (Fig. 6C). Application of glibenclamide (10 μM) produced an increase in spontaneous contraction frequency and slight reduction in amplitude at all pressures (Fig. 6C). This effect is evident by comparing the frequency at 1 cmH2O before and after addition of glibenclamide (5.4 vs 10.5 contractions/min, respectively). In marked contrast, most vessels from induced SMMHC-CreERT2;Kir6.1[GD-QR] mice exhibited a notable absence of spontaneous contractions even at the highest pressures (Fig. 6D); other vessels that did not show contractions at low pressures developed a few weak, irregular contractions at pressures > 5 cmH2O (not shown). Application of glibenclamide to induced SMMHC-CreERT2;Kir6.1[GD-QR] vessels evoked spontaneous contractions, but contraction amplitude was typically much lower than in WT or control vessels and contraction frequency was higher. Once contractions developed, they were modulated by pressure as in control vessels, but with reduced amplitude and elevated frequency at each pressure (Fig. 6D).
Summary data for the contractile properties of popliteal lymphatics from induced and non-induced SMMHC-CreERT2;Kir6.1[GD-QR] vessels are shown in Fig. 7A-D. Contraction parameters (normalized amplitude, frequency, FPF, tone) were calculated as described in Methods. These analyses reveal severe contractile dysfunction of popliteal lymphatic vessels from SMMHC-CreERT2;Kir6.1[GD-QR] mice, due in large part to dramatically reduced contraction amplitude and in part to lowered contraction frequency. The FPF curve is the most comprehensive index of overall pumping action (approximating the flow rate) and indicates that pumping was compromised by ~70% at intermediate pressures and by ~95% at the lowest three pressures in induced SMMHC-CreERT2;Kir6.1[GD-QR] vessels (Fig. 7C).
The application of glibenclamide (10 μM) to vessels from induced SMMHC-CreERT2;Kir6.1[GD-QR] mice led to substantial increases in amplitude at low pressures, and ~6x increase in frequency and ~3x increase in active tone at most pressures (compare solid symbols in Fig 7A and E, in Fig 7D and H, respectively). After glibenclamide, FPF was nearly normalized in vessels from induced SMMHC-CreERT2;Kir6.1[GD-QR] mice, due to enhancement of contraction amplitude at low pressures and of contraction frequency at all pressures. In contrast, the effects of glibenclamide on vessels from non-induced mice were slight decreases in amplitude and increases in frequency and FPF at all pressures. Curiously, tone was not significantly different between control and induced SMMHC-CreERT2;Kir6.1[GD-QR] vessels (Fig 7D) at any pressure, but glibenclamide (10 μM) significantly elevated tone only in vessels from induced SMMHC-CreERT2;Kir6.1[GD-QR] mice. These results indicate that glibenclamide can substantially rescue the impairment of contractile function in SMMHC-CreERT2;Kir6.1[GD-QR] GoF vessels.
We also tested the consequences of expressing the Kir6.1[GD-QR] GoF transgene selectively in lymphatic endothelium using a tamoxifen-inducible Prox1Cre. Popliteal lymphatics from induced Prox1-CreERT2;Kir6.1[GD-QR] mice had nearly identical frequency, FPF and tone as non-induced control vessels (Fig. 8A-D). Similar to the results shown in Fig. 6, and consistent with those in Fig. 3, glibenclamide led to slight increases in frequency and slight reductions in amplitude at almost all pressures for vessels in each group, suggesting that expression of the Kir6.1[GD-QR] GoF transgene in LECs had little effect on contractile function.
In vivo imaging confirms contractile dysfunction in GOF popliteal lymphatics
We used a multiphoton microscope to image vessel activity in vivo. This allowed assessment of the contractility of intact lymphatic collectors in their natural environment, surrounded by fat and connective tissue, and presumably subject to normal in vivo pressures and flows, at least to the degree that these are not artificially altered by injection of upstream tracers required for imaging (Zawieja et al., 2018a). RFP-tagged lectin was injected into the dorsal foot, and the superficial layer of skin was removed to expose the popliteal lymphatics adjacent to the saphenous vein. The vessel lumen was thus visible in the red channel. Unrecombined cells expressing GFP (Li et al., 2013), including LECs and valve leaflets, were visible in the green channel (see Fig. 9A). The second harmonic provided an image of the collagen surrounding the vessel. In control mice spontaneous diameter fluctuations were consistently present in 9 of 9 popliteal lymphatics, as reported in other publications using other tracers (Castorena-Gonzalez et al., 2018b). The amplitudes of the contractions were variable from vessel to vessel and animal to animal but the frequency was consistently around 5 per minute. Based on the frequency-pressure relationships of ex vivo popliteal vessels shown in Fig. 7B, this would suggest an in vivo intraluminal pressure around 3 cmH2O. In contrast, only 1 of 7 vessels in SMMHC-Cre;Kir6.1[GD-QR] mice exhibited diameter fluctuations of a magnitude that were consistent with contractile activity. Analyses of the amplitude and frequency of the contractile activity of popliteal lymphatics in vivo are summarized in Fig. 9B. These findings indicate a severe decrease of in vivo lymphatic pump function as a result of LSM KATP GoF, consistent with the ex vivo findings in Fig. 7.
KATP activation hyperpolarizes lymphatic SM
Electrophysiological studies of intact lymphatic vessels from induced SMMHC-CreERT2;Kir6.1[GD-QR] mice revealed that the membrane potentials of smooth muscle cells in popliteal lymphatics were more hyperpolarized than those of control vessels under basal conditions. A typical Vm recording from a WT vessel (Fig 10A) shows the characteristic APs described earlier and their corresponding contractions (amplitude was blunted by wortmannin treatment but frequency was increased). In contrast, a recording from an induced SMMHC-CreERT2;Kir6.1[GD-QR] vessel shows a sharp negative deflection associated with successful impalement and a single AP (likely due to current leak around the micropipette tip while the membrane was resealing) followed by a slow hyperpolarization, settling at ~−50 mV before the pipette dislodged (Fig. 10B). Summary data from 9 recordings in WT vessels and 11 recordings in SMMHC-CreERT2;Kir6.1[GD-QR] vessels show that LMCs had maximum diastolic potential Vm values between −20 to −56 mV (Fig. 10C). None of the cells in SMMHC-CreERT2;Kir6.1[GD-QR] vessels exhibited APs, apart from the cell in Fig. 10B, which may have been induced as an artifact during impalement. For WT cells that were firing APs, the “resting” Vm was taken as the peak after-hyperpolarization (open triangles in Fig. 10A). On average LMCs from induced SMMHC-CreERT2;Kir6.1[GD-QR] vessels were 11 mV more hyperpolarized than cells in WT vessels (Fig. 10C).
Fig. 10.

A) Vm recording from a lymphatic muscle cell of a WT popliteal lymphatic (pressurized to 3 cmH2O), showing spontaneous action potentials preceding blunted contractions (see below). Open triangle denotes maximum negative Vm level during the cycle and closed triangle denotes the threshold prior to action potential firing. B) Vm recording in a lymphatic muscle cell of a popliteal lymphatic from an induced SMMHC-CreERT2;Kir6.1[GD-QR] mouse showing a single AP activated by electrode penetration after which the membrane gradually hyperpolarizes to ~−50 mV before the electrode dislodged. Open circle indicates the maximum negative Vm. C) Analysis of threshold Vm and maximum negative Vm for recordings from 9 WT and 11 SMMHC-CreERT2;Kir6.1[GD-QR] lymphatic muscle cells. No APs were recorded in the latter after Vm stabilized. On average Vm was −11 mV more hyperpolarized in SMMHC-CreERT2;Kir6.1[GD-QR] lymphatic muscle cells. All recordings were made in the presence of 2 μM wortmannin. Means ± SD.
Discussion
Kir6.1/SUR2B-dependent KATP channels control LSM but not LEC function
Given the paucity of data regarding KATP channels and lymphatic function, this study aimed to understand the functional expression of KATP channel subunits in distinct lymphatic cell types, and assess the consequences of altered KATP channel activity on lymphatic pump function. We first assessed expression of KATP channel subunits in mouse lymphatic muscle by PCR detection. Kir6.1 and SUR2B isoforms were present in purified lymphatic smooth muscle cell samples, but only Kir6.1 was identified in lymphatic endothelial cell preparations (Fig. 1). Spontaneous contractions of WT popliteal lymphatic vessels were very sensitive to inhibition by the SUR2-specific KATP channel activator pinacidil. In contrast, vessels from Kir6.1−/− and SUR2[STOP] mice were completely insensitive to low doses of pinacidil (Fig. 2). Pinacidil had little effect on the contraction frequency of either Kir6.1−/− or SUR2[STOP] lymphatic vessels until the concentration exceeded 1 μM (IC50 =8.3·10−5 M), but caused progressive blunting of contraction amplitude at concentrations from 10 nM to 10 μM [concentrations of KATP activators often used in vascular and lymphatic studies (Mathias & von der Weid, 2013; Telinius et al., 2014b)], suggesting an off-target effect. Interestingly, WT popliteal lymphatic contractions were much more sensitive to pinacidil (IC50 = 3.4·10−7 for amplitude, 1.4·10−7 for frequency) than has been reported previously for arteries from various vascular beds, e.g. IC50 from 1·10−6 to 10·10−6 (Aziz et al., 2015; Foster & Coetzee, 2016). This suggests that lymphatic smooth muscle may be a particularly sensitive type of smooth muscle to the activity of KATP channels, potentially explaining the high incidence of lymphedema in Cantu syndrome, a condition resulting from GoF mutations in Kir6.1 or SUR2 subunits (see below).
The observation that Kir6.1−/− and SUR2[STOP] lymphatic vessels have spontaneous contractions with essentially normal frequency, amplitude and tone (Fig. 3) suggests that basal KATP channel activity in lymphatic smooth muscle is not an essential component of the pacemaker, or exerts a strong influence over the generation of lymphatic tone or contractile strength. However, acute application of the KATP channel inhibitor, glibenclamide (10 μM) to non-induced Kir6.1 GoF vessels (Figs. 6-7) led to consistent increases in contraction frequency and decreases in contraction amplitude, implying at least a minimal basal KATP channel activity that contributes a slight, tonic hyperpolarizing influence. (The effects on tone were less consistent: glibenclamide increased tone (at some or all pressures) in 3 of the 4 groups (Fig. 6D, Fig.7D).) It is unclear how to reconcile these observations, although the glibenclamide concentrations used to test acute responses could have off-target effects (Lee & Lee, 2005), and/or some sort of compensation may occur in the global knock-out mouse models (El-Brolosy & Stainier, 2017).
Our results are consistent with a previous finding that KATP channel activators such as pinacidil or cromakalim can completely inhibit spontaneous lymphatic contractions (Mathias & von der Weid, 2013). In the present study the expression of Kir6.1 GoF subunits in mouse lymphatic smooth muscle (LSM) silenced contractions in some vessels but produced only weak, lower-frequency contractions in other vessels (Figs. 6-7). In contrast, expression of Kir6.1 GoF subunits in Prox1Cre; Kir6.1[GD-QR] lymphatic endothelial cells (LEC) was essentially without effect on frequency or amplitude of contraction (Fig. 8). This confirms that it is indeed within the LSM that KATP channels are active, and suggest the possibility that, lacking the SUR2B subunit (Fig. 1), Kir6.1 subunits in LEC do not generate functional KATP channels. The lack of effect of Kir6.1[GD-QR] transgene expression in lymphatic endothelium (Fig. 8) is notable in this regard. Not only were contraction strength and frequency unimpaired in Prox1Cre; Kir6.1[GD-QR] vessels, contractions at one (low) pressure were significantly enhanced compared to control vessels. The explanation for this effect is unclear but it is possible that overexpression of the channel in LECs leads to increased NO production, which at low levels can lower contraction frequency but enhance amplitude (Gasheva et al., 2006). Whether this effect might be blocked by L-NAME or whether the actions of endothelium-dependent dilators are compromised in Kir6.1−/− vessels remain to be determined. These findings add supporting evidence for another unique feature of lymphatic vessels. Endothelial cell hyperpolarization in arterial smooth muscle is readily conducted across myoendothelial gap junctions (MEGJs) to the smooth muscle layer; thus, activation of KATP channels in arterial endothelial cells would be expected to result in smooth muscle cell hyperpolarization and dilation (Aziz et al., 2017). The observation that the expression of GoF KATP channels in LECs (although possibly without the SUR2 subunit, Fig. 1) does not substantially impair lymphatic contractions therefore is consistent with weak or non-existent electrical coupling between LECs and LMCs as indicated by recent studies (Castorena-Gonzalez et al., 2018b; Hald et al., 2018); this may be a feature of the lymphatic vessel wall that is required for the initiation and conduction of APs along the smooth muscle layer, without shunting of current into the endothelium.
Pathophysiological consequences of LSM Kir6.1/SUR2B-dependent KATP channel overactivity
Overactive LSM KATP channels are predicted to reduce contractility and hence lymphatic pump function, by reducing excitability. The pathological relevance of the GoF KATP channel is confirmed by the in vivo imaging studies shown in Fig. 9; 6 out of 7 popliteal vessels in mice expressing the Kir6.1[GD-QR] transgene exhibited no spontaneous contractile activity, compared to 9 out of 9 vessels with varying degrees of contractile activity in control mice. Vm measurements in LMCs from control and Kir6.1 GoF of popliteal lymphatics showed that Kir6.1 GoF vessels were on average 11 mV more hyperpolarized than control vessels. Based on our such recordings, only a few mV of hyperpolarization appear to be required to substantially slow spontaneous AP firing; 11 mV hyperpolarization is comparable to the effect produced by 3 μM pinacidil (Fig. 4) and likely would completely silence most vessels. LMCs in which the Kir6.1[GD-QR] transgene is variably expressed, due to incomplete Cre recombination, would be protected from full hyperpolarization and might still be able to initiate pacemaking in a vessel in which other LMCs are hyperpolarized.
Our results show that glibenclamide can partially rescue impaired contractile function in SMMHC-CreERT2;Kir6.1[GD-QR] GoF vessels. However, that rescue is only partial, as glibenclamide treatment (at a dose of 10 μM) results in low-amplitude, high-frequency contractions which may not translate to effective lymph propulsion under an adverse pressure gradient (Castorena-Gonzalez et al., 2018a). The reasons for incomplete rescue by glibenclamide are unclear, but may be related to excessive depolarization caused by 10 μM glibenclamide (Fig. 5C). Vm recordings in lymphatic muscle cells indicate that maximal KATP inhibition by glibenclamide reduces the amplitude and duration of the basal AP (Figs. 4-5) which may result from glibenclamide depolarization leading to incomplete recovery of L-type Ca2+ channels from inactivation. Conversely, low level activation of KATP channels by low doses of pinacidil increases the amplitude of the AP in lymphatic muscle (Figs. 4-5), possibly by partial relief of voltage-induced L-type Ca2+ channel inactivation (Cota & Stefani, 1989). Glibenclamide has also been shown to have more direct inhibitory effects on L-type Ca2+ channels at some doses (Lee & Lee, 2005). It is thus conceivable that this off-target action reduces the restoration of contraction amplitude by glibenclamide, and perhaps lower glibenclamide doses might reverse the effect of the Kir6.1[GD-QR] transgene without compromising the amplitude of lymphatic spontaneous contractions. Since many patients with chronic lymphedema from other causes exhibit large and dilated lymphatic vessels that may indicate a loss of the LSM contractile phenotype and altered transcriptional program, whether or not lymphatic contractile dysfunction in Cantu syndrome can be restored, and lymphedema resolved, simply by KATP channel inhibition, such as with glibenclamide, after the onset of gross lymphedema is an important question.
In contrast to the classical view that lymphatic transport is predominately driven by extrinsic factors (Alitalo et al., 2005), it is now clear that intrinsic contractility of lymphatic smooth muscle is the dominant mechanism whereby fluid is transferred through the lymphatic system in humans (Engeset et al., 1977; Olszewski & Engeset, 1980). Whether impaired lymphatic contractile activity alone leads to lymphedema remains unclear because the preponderance of human primary lymphedemas is linked to genetic disruption of lymphatic endothelial cell proliferation or valvulogenesis (Mortimer & Rockson, 2014). Potentially revealing in this regard, Cantú Syndrome patients suffer from a high incidence of clinical lymphedema (Garcia-Cruz et al., 2011; Grange et al., 2019), and an obvious potential explanation from the present study is that this indeed results from failure of lymphatic pumping due to KATP channel GoF-induced inexcitability. However, both onset and extent of lymphedema are variable in Cantú patients (Grange et al., 2019), which may be related to the degree of channel overactivity, or may depend on secondary factors such as immunological activation and fibrosis. Mice experience very low imposed hydrostatic pressures as a result of gravitational forces, and peripheral lymphedema is seldom observed in mice unless there is a profound structural defect in the lymphatic system (Rutkowski et al., 2010). Even mice that are unable to recruit mural cells (LMCs and pericytes) to collecting lymphatic vessels, but which otherwise retain a normal lymphatic network with apparently functional valves, fail to develop lymphedema (Wang et al., 2017). Despite a 70-95% reduction in estimated lymph flow in vessels from SMMHC-CreERT2;Kir6.1[GD-QR] mice (Fig. 7C), we did not observe development of peripheral lymphedema in those animals, even several months after TMX induction. Lymphedema typically only develops in dependent extremities of humans (including Cantú patients), and a recent study suggests that subtle lymphatic defects may not be obvious in mice unless they are subjected to a gravitational load (Castorena-Gonzalez et al., 2018a). Thus, insight to the pathological evolution of lymphedema in Cantu patients may prove difficult to obtain in mice.
Conclusions
We present evidence that lymphatic vessels are sensitive to Kir6.1- and SUR2-dependent KATP channel activation in LSM, but not LEC. Although KATP channel activity is not significant under basal conditions, vessels are very sensitive to pharmacological activation. We show that LSM expression of gain-of-function KATP channels, as are found in Cantú syndrome, causes dramatic impairment of lymphatic contractility under both ex vivo and in vivo conditions. Finally, we provide evidence that the KATP channel inhibitor, glibenclamide, can partially reverse the effects of KATP channel gain-of-function, validating a potential therapeutic approach to restoration of lymphatic contractile activity in patients with Cantú syndrome-associated lymphedema.
Supplementary Material
Key Points.
Spontaneous contractions are essential for normal lymph transport and these contractions are exquisitely sensitive to the KATP channel activator, pinacidil. KATP channel Kir6.1 and SUR2B subunits are expressed in mouse lymphatic smooth muscle (LSM) and form functional KATP channels as verified by electrophysiological techniques.
Global deletion of Kir6.1 or SUR2 subunits results in severely impaired lymphatic contractile responses to pinacidil.
Smooth muscle-specific expression of Kir6.1 gain-of-function mutant (GoF) subunits results in profound lymphatic contractile dysfunction and LSM hyperpolarization that is partially rescued by the KATP inhibitor, glibenclamide. In contrast, lymphatic endothelial-specific expression of Kir6.1 GoF has essentially no effect on lymphatic contractile function.
The high sensitivity of LSM to KATP channel GoF offers an explanation for the lymphedema observed in patients with Cantú Syndrome, a disorder caused by gain-of-function mutations in genes encoding Kir6.1 or SUR2, and suggests that glibenclamide may be an appropriate therapeutic agent.
Translational Perspective.
Cantu Syndrome is a complex multi-organ disorder caused by gain-of-function mutations in KCNJ8 (Kir6.1) or ABCC9 (SUR2). Cantu patients are characterized by a constellation of symptoms that includes >50% incidence of lymphedema. We hypothesized that hyperactive Kir6.1/SUR2-dependent KATP channels, simulating the mutant channels expressed in patients with Cantu syndrome, would interfere with lymphatic pacemaking and action potential generation in lymphatic smooth muscle. Indeed, smooth-muscle specific expression of a gain-of-function Kir6.1 channel in mice results in profound lymphatic contractile dysfunction, assessed both ex vivo and in vivo, that can be partially rescued by the KATP channel inhibitor, glibenclamide. Our results offer an explanation for the lymphedema observed in patients with Cantú Syndrome, and suggest the use of glibenclamide as a potential therapeutic agent to rescue lymphatic contractile dysfunction in these patients.
Acknowledgments
Funding
This research was supported by National Institutes of Health grants R01 HL‐122608 and HL‐122578 to M.J.D, and by R35 HL-140024 to CGN. S.D.Z. was supported in part by NIH grant K99 HL-143198 and J.A.C-G was supported in part by NIH grant K99 HL-141143.
Abbreviations
- CS
Cantú Syndrome
- GoF
gain-of-function
- ATP
adenosine trisphosphate
- KATP
ATP-sensitive K+ channel
- Kir
inward rectifying K+ channel
- SUR
sulphonylurea receptor
- AMP
contraction amplitude
- FREQ
contraction frequency
- FPF
fractional pump flow
- RFP
red fluorescence protein
- GFP
green fluorescence protein
- LMC
lymphatic muscle cell
- LEC
lymphatic endothelial cell
- LSM
, lymphatic smooth muscle
- WT
wild-type
- DMSO
dimethyl sulfoxide
- EF
ejection fraction
- AP
action potential
- Vm
membrane potential
- FACS
fluorescence-activated cell sorting
- SMA
smooth muscle α-actin
Footnotes
Competing Interests
The authors have no competing interests to declare or conflicts of interest to disclose. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Alitalo K, Tammela T & Petrova TV. (2005). Lymphangiogenesis in development and human disease. Nature 438, 946–953. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM. (1988). Adenosine 5’-triphosphate-sensitive potassium channels. Annu Rev Neurosci 11, 97–118. [DOI] [PubMed] [Google Scholar]
- Aziz Q, Li Y, Anderson N, Ojake L, Tsisanova E & Tinker A. (2017). Molecular and functional characterization of the endothelial ATP-sensitive potassium channel. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz Q, Li Y & Tinker A. (2015). ATP-sensitive potassium channels and vascular function. Channels (Austin) 9, 3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ, Scallan JP, Jafarnejad M, Castorena-Gonzalez JA, Zawieja SD, Moore JE Jr., , Davis MJ & Segal SS. (2017). Calcium and electrical dynamics in lymphatic endothelium. J Physiol 595, 7347–7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein CA, Towne MC, Luquette LJ, Harris DJ, Marinakis NS, Meinecke P, Kutsche K, Campeau PM, Yu TW, Margulies DM, Agrawal PB & Beggs AH. (2013). Mutation of KCNJ8 in a patient with Cantu syndrome with unique vascular abnormalities - support for the role of K(ATP) channels in this condition. Eur J Med Genet 56, 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu JM, Garcia-Cruz D, Sanchez-Corona J, Hernandez A & Nazara Z. (1982). A distinct osteochondrodysplasia with hypertrichosis- Individualization of a probable autosomal recessive entity. Hum Genet 60, 36–41. [DOI] [PubMed] [Google Scholar]
- Castorena-Gonzalez JA, Zawieja SD, Li M, Srinivasan RS, Simon AM, de Wit C, de la Torre R, Martinez-Lemus LA, Hennig GW & Davis MJ. (2018a). Mechanisms of Connexin-Related Lymphedema. Circ Res 123, 964–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castorena-Gonzalez JA, Zawieja SD, Li M, Srinivasan S, Simon AM, Hennig G, de la Torre R, Martinez-Lemus LA & Davis MJ. (2018b). Mechanisms of connexin-related lymphedema: roles for Cx37, Cx43, Cx47 and Cx45 in the entrainment of spontaneous lymphatic contractions. Circulation Research (in revision). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chutkow WA, Simon MC, Le Beau MM & Burant CF. (1996). Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATP channels. Diabetes 45, 1439–1445. [DOI] [PubMed] [Google Scholar]
- Cooper PE, Reutter H, Woelfle J, Engels H, Grange DK, van Haaften G, van Bon BW, Hoischen A & Nichols CG. (2014). Cantu syndrome resulting from activating mutation in the KCNJ8 gene. Hum Mutat 35, 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota G & Stefani E. (1989). Voltage-dependent inactivation of slow calcium channels in intact twitch muscle fibers of the frog. Journal of General Physiology 94, 937–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton KD, Hollywood MA, McHale NG & Thorbury KD. (1997). Outward currents in smooth muscle cells isolated from sheep mesenteric lymphatics. Journal of Physiology 503, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Zawieja DC & Gashev AA. (2006). Automated measurement of diameter and contraction waves of cannulated lymphatic microvessels. Lymphat Res Biol 4, 3–10. [DOI] [PubMed] [Google Scholar]
- El-Brolosy MA & Stainier DYR. (2017). Genetic compensation: A phenomenon in search of mechanisms. PLoS genetics 13, e1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeset A, Olszewski W, Jaeger PM, Sokolowski J & Theodorsen L. (1977). Twenty-four hour variation in flow and composition of leg lymph in normal men. Acta Physiol Scandinavica 99, 140–148. [DOI] [PubMed] [Google Scholar]
- Foster MN & Coetzee WA. (2016). KATP Channels in the Cardiovascular System. Physiol Rev 96, 177–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cruz D, Mampel A, Echeverria MI, Vargas AL, Castaneda-Cisneros G, Davalos-Rodriguez N, Patino-Garcia B, Garcia-Cruz MO, Castaneda V, Cardona EG, Marin-Solis B, Cantu JM, Nunez-Reveles N, Moran-Moguel C, Thavanati PK, Ramirez-Garcia S & Sanchez-Corona J. (2011). Cantu syndrome and lymphoedema. Clinical dysmorphology 20, 32–37. [DOI] [PubMed] [Google Scholar]
- Gasheva OY, Zawieja DC & Gashev AA. (2006). Contraction-initiated NO-dependent lymphatic relaxation: a self-regulatory mechanism in rat thoracic duct. J Physiol 575, 821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange DK, Nichols CG & Singh GK. (1993). Cantu Syndrome and Related Disorders In GeneReviews((R)), ed. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K & Amemiya A. Seattle (WA). [PubMed] [Google Scholar]
- Grange DK, Roessler HI, McClenaghan C, Duran K, Shields K, Remedi MS, Knoers N, Lee JM, Kirk EP, Scurr I, Smithson SF, Singh GK, van Haelst MM, Nichols CG & van Haaften G. (2019). Cantu syndrome: Findings from 74 patients in the International Cantu Syndrome Registry. Am J Med Genet C Semin Med Genet 181, 658–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hald BO, Castorena-Gonzalez JA, Zawieja SD, Gui P & Davis MJ. (2018). Electrical Communication in Lymphangions. Biophys J 115, 936–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harakalova M, van Harssel JJ, Terhal PA, van Lieshout S, Duran K, Renkens I, Amor DJ, Wilson LC, Kirk EP, Turner CL, Shears D, Garcia-Minaur S, Lees MM, Ross A, Venselaar H, Vriend G, Takanari H, Rook MB, van der Heyden MA, Asselbergs FW, Breur HM, Swinkels ME, Scurr IJ, Smithson SF, Knoers NV, van der Smagt JJ, Nijman IJ, Kloosterman WP, van Haelst MM, van Haaften G & Cuppen E. (2012). Dominant missense mutations in ABCC9 cause Cantu syndrome. Nat Genet 44, 793–796. [DOI] [PubMed] [Google Scholar]
- Hollywood MA, Cotton KD, Thornbury KD & McHale NG. (1997a). Isolated sheep mesenteric lymphatic smooth muscle cells possess both T- and L-type calcium currents. Journal of Physiology 501, P109–P110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollywood MA, Cotton KD, Thornbury KD & Mchale NG. (1997b). Tetrodotoxin-sensitive sodium current in sheep lymphatic smooth muscle. Journal of Physiology 503, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, McClenaghan C, Harter TM, Hinman K, Halabi CM, Matkovich SJ, Zhang H, Brown GS, Mecham RP, England SK, Kovacs A, Remedi MS & Nichols CG. (2018). Cardiovascular consequences of KATP overactivity in Cantu syndrome. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imtiaz MS, von der Weid PY & van Helden DF. (2010). Synchronization of Ca2+ oscillations: a coupled oscillator-based mechanism in smooth muscle. Febs J 277, 278–285. [DOI] [PubMed] [Google Scholar]
- Imtiaz MS, Zhao J, Hosaka K, von der Weid PY, Crowe M & van Helden DF. (2007). Pacemaking through Ca2+ stores interacting as coupled oscillators via membrane depolarization. Biophys J 92, 3843–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY & Lee CO. (2005). Inhibition of Na+-K+ pump and L-type Ca2+ channel by glibenclamide in Guinea pig ventricular myocytes. J Pharmacol Exp Ther 312, 61–68. [DOI] [PubMed] [Google Scholar]
- Levin MD, Singh GK, Zhang HX, Uchida K, Kozel BA, Stein PK, Kovacs A, Westenbroek RE, Catterall WA, Grange DK & Nichols CG. (2016). KATP channel gain-of-function leads to increased myocardial L-type Ca2+ current and contractility in Cantu syndrome. Proc Natl Acad Sci U S A 113, 6773–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Knutsen RH, Zhang H, Osei-Owusu P, Moreno-Dominguez A, Harter TM, Uchida K, Remedi MS, Dietrich HH, Bernal-Mizrachi C, Blumer KJ, Mecham RP, Koster JC & Nichols CG. (2013). Hypotension due to Kir6.1 gain-of-function in vascular smooth muscle. J Am Heart Assoc 2, e000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias R & von der Weid PY. (2013). Involvement of the NO-cGMP-K(ATP) channel pathway in the mesenteric lymphatic pump dysfunction observed in the guinea pig model of TNBS-induced ileitis. Am J Physiol Gastrointest Liver Physiol 304, G623–634. [DOI] [PubMed] [Google Scholar]
- McCloskey KD, Toland HM, Hollywood MA, Thornbury KD & McHale NG. (1999). Hyperpolarization-activated inward current in isolated sheep mesenteric lymphatic smooth muscle. Journal of Physiology 521, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Suzuki M, Shibasaki T, Uemura H, Sato T, Yamaguchi K, Koseki H, Iwanaga T, Nakaya H & Seino S. (2002). Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med 8, 466–472. [DOI] [PubMed] [Google Scholar]
- Mizuno R, Ono N & Ohhashi T. (1999). Involvement of ATP-sensitive K(+) channels in spontaneous activity of isolated lymph microvessels in rats. Am J Physiol 277, H1453–1456. [DOI] [PubMed] [Google Scholar]
- Morrissey A, Rosner E, Lanning J, Parachuru L, Dhar Chowdhury P, Han S, Lopez G, Tong X, Yoshida H, Nakamura TY, Artman M, Giblin JP, Tinker A & Coetzee WA. (2005). Immunolocalization of KATP channel subunits in mouse and rat cardiac myocytes and the coronary vasculature. BMC Physiol 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer PS & Rockson SG. (2014). New developments in clinical aspects of lymphatic disease. J Clin Invest 124, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG. (2006). KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Singh GK & Grange DK. (2013). KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res 112, 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski WL & Engeset A. (1980). Intrinsic contractility of prenodal lymph vessels and lymph flow in human leg. American Journal of Physiology 239, H775–783. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA & Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockson SG. (2006). Lymphedema. Curr Treat Options Cardiovasc Med 8, 129–136. [DOI] [PubMed] [Google Scholar]
- Rutkowski JM, Markhus CE, Gyenge CC, Alitalo K, Wiig H & Swartz MA. (2010). Dermal collagen and lipid deposition correlate with tissue swelling and hydraulic conductivity in murine primary lymphedema. Am J Pathol 176, 1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scallan JP & Davis MJ. (2013). Genetic removal of basal nitric oxide enhances contractile activity in isolated murine collecting lymphatic vessels. J Physiol 591, 2139–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scallan JP, Wolpers JH & Davis MJ. (2013). Constriction of isolated collecting lymphatic vessels in response to acute increases in downstream pressure. J Physiol 591, 443–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scallan JP, Zawieja SD, Castorena-Gonzalez JA & Davis MJ. (2016). Lymphatic pumping: mechanics, mechanisms and malfunction. J Physiol 594, 5749–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeland MF, McClenaghan C, Roessler HI, Savelberg S, Hansen GAM, Hjellnes H, Arntzen KA, Muller KI, Dybesland AR, Harter T, Sala-Rabanal M, Emfinger CH, Huang Y, Singareddy SS, Gunn J, Wozniak DF, Kovacs A, Massink M, Tessadori F, Kamel SM, Bakkers J, Remedi MS, Van Ghelue M, Nichols CG & van Haaften G. (2019). ABCC9-related Intellectual disability Myopathy Syndrome is a KATP channelopathy with loss-of-function mutations in ABCC9. Nat Commun 10, 4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuba A & Rockson SG. (1998). Lymphedema: classification, diagnosis and therapy. Vasc Med 3, 145–156. [DOI] [PubMed] [Google Scholar]
- Telinius N, Kim S, Pilegaard H, Pahle E, Nielsen J, Hjortdal V, Aalkjaer C & Boedtkjer DB. (2014a). The contribution of K(+) channels to human thoracic duct contractility. Am J Physiol Heart Circ Physiol 307, H33–43. [DOI] [PubMed] [Google Scholar]
- Telinius N, Kim S, Pilegaard H, Pahle E, Nielsen J, Hjortdal VE, Aalkjaer C & Boedtkjer DM. (2014b). The contribution of potassium channels to human thoracic duct contractility. Am J Physiol Heart Circ Physiol. [DOI] [PubMed] [Google Scholar]
- Telinius N, Majgaard J, Kim S, Katballe N, Pahle E, Nielsen J, Hjortdal V, Aalkjaer C & Boedtkjer DB. (2015). Voltage-gated sodium channels contribute to action potentials and spontaneous contractility in isolated human lymphatic vessels. J Physiol 593, 3109–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Porter LM, Liu G, Dhar-Chowdhury P, Srivastava S, Pountney DJ, Yoshida H, Artman M, Fishman GI, Yu C, Iyer R, Morley GE, Gutstein DE & Coetzee WA. (2006). Consequences of cardiac myocyte-specific ablation of KATP channels in transgenic mice expressing dominant negative Kir6 subunits. Am J Physiol Heart Circ Physiol 291, H543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bon BW, Gilissen C, Grange DK, Hennekam RC, Kayserili H, Engels H, Reutter H, Ostergaard JR, Morava E, Tsiakas K, Isidor B, Le Merrer M, Eser M, Wieskamp N, de Vries P, Steehouwer M, Veltman JA, Robertson SP, Brunner HG, de Vries BB & Hoischen A. (2012). Cantu syndrome is caused by mutations in ABCC9. Am J Hum Genet 90, 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Weid PY, Rehal S, Dyrda P, Lee S, Mathias R, Rahman M, Roizes S & Imtiaz MS. (2012). Mechanisms of VIP-induced inhibition of the lymphatic vessel pump. J Physiol 590, 2677–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Jin Y, Mae MA, Zhang Y, Ortsater H, Betsholtz C, Makinen T & Jakobsson L. (2017). Smooth muscle cell recruitment to lymphatic vessels requires PDGFB and impacts vessel size but not identity. Development 144, 3590–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawieja SD, Castorena-Gonzalez JA, Dixon B & Davis MJ. (2018a). Experimental models used to assess lymphatic contractile function. Lymphat Res Biol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawieja SD, Castorena-Gonzalez JA, Scallan JP & Davis MJ. (2018b). Differences in L-type Ca(2+) channel activity partially underlie the regional dichotomy in pumping behavior by murine peripheral and visceral lymphatic vessels. Am J Physiol Heart Circ Physiol 314, H991–H1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawieja SD, Castorena-Gonzalez JA, To KHT, Gui P, Domeier TL & Davis MJ. (2018c). Electrical pacemaking in lymphatic vessels In Smooth Muscle Excitability, ed. Trebak M & Earley S. CRC Press. [Google Scholar]
- Zawieja SD, Castorena JA, Gui P, Li M, Bulley SA, Jaggar JH, Rock JR & Davis MJ. (2019). Ano1 mediates pressure-sensitive contraction frequency changes in mouse lymphatic collecting vessels. J Gen Physiol 151, 532–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.