

Abstract
Section:
Translational Cancer mechanisms and therapy.
Purpose:
TNF-related apoptosis inducing ligand (TRAIL) expression by immune cells contributes to anti-tumor immunity. A naturally occurring splice variant of TRAIL, called TRAILshort, antagonizes TRAIL-dependent cell killing. It is unknown whether tumor cells express TRAILshort and if it impacts anti-tumor immunity.
Experimental Design:
We used an unbiased informatics approach to identify TRAILshort expression in primary human cancers, and validated those results with immunohistochemistry (IHC) and in situ hybridization (ISH). TRAILshort specific monoclonal antibodies were used to determine the effect of TRAILshort on tumor cell sensitivity to TRAIL, and to immune effector cell dependent killing of autologous primary tumors.
Results:
As many as 40% of primary human tumors express TRAILshort by both RNAseq and IHC analysis. By ISH, TRAILshort expression is present in tumor cells and not bystander cells. TRAILshort inhibition enhances cancer cell lines sensitivity to TRAIL dependent killing both in vitro and in immunodeficient xenograft mouse models. Immune effector cells isolated from patients with B cell malignancies killed more autologous tumor cells in the presence compared to the absence of TRAILshort antibody (P<0.05).
Conclusion:
These results identify TRAILshort in primary human malignancies, and suggest that TRAILshort blockade can augment the effector function of autologous immune effector cells.
Keywords: TRAIL, apoptosis, Immunotherapy, oncology
Graphical Abstract
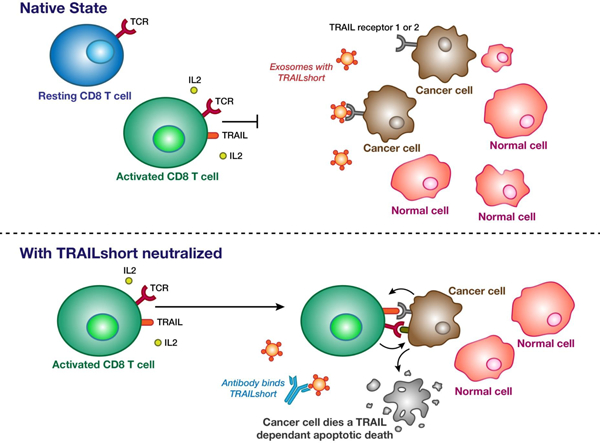
Following activation, immune effector cells express TRAIL, but are unable to kill TRAIL-receptor 1 or 2 expressing tumor cells, due to the presence of TRAILshort. When TRAILshort is neutralized by a specific antibody, immune effector cell killing of tumor cells is enhanced.
INTRODUCTION
The human immune system is capable of identifying and eliminating tumor cells, and it is increasingly recognized that effective cancer therapy requires the presence of an effective immune response. New therapies which target T cell inhibitory pathways have revolutionized cancer therapy, and yet only a minority of cancer patients derives benefit from immunotherapy. Consequently, there remains a need to identify new targets for intervention, which further enhance the ability of immune effector cells to kill tumor targets.
TNF-related apoptosis inducing ligand (TRAIL) is a proapoptotic death-inducing ligand that can cause the death of tumor cells, or virally infected cells in vitro while sparing untransformed or uninfected cells[1]. TRAIL is expressed by activated T cells and NK cells [2], arguing for a role of TRAIL in the innate and adaptive immune response. Knockout studies support that contention, since TRAIL −/− mice have normal lymphoid and myeloid cell development [3], yet develop spontaneous lymphoid and stromal tumors after >500 days [4]. Similarly in response to the genotoxic carcinogen methylcholanthrene, TRAIL−/− mice have enhanced rates of fibrosarcoma development in comparison to WT parental mice [5]. Notwithstanding that the regulation of TRAIL in mice is less complex than in humans (mice have 1 TRAIL receptor, whereas humans have 4), the loss of TRAIL-Receptor in mice has similar effects. Deletion of the TRAIL receptor in mice enhances tumorigenesis in a lymphoma-prone Eμ-myc mouse, and increases diethylnitrosamine-induced (DEN-induced) hepatocarcinogenesis [6]. Similarly, tumor growth and progression (metastasis) are accelerated in the TRAIL−/− [4] and TRAILR−/− mice [6]. Finally, neutralizing antibodies against TRAIL, which prevent it from interacting with the TRAIL receptor, cause increased oncogenesis in mice treated with methylcholanthrene; and in mice implanted with tumors, antibody neutralization of TRAIL increase rates of tumor growth and metastasis [7, 8]. Thus, neutralization of the TRAIL/TRAIL receptor axis favors tumor development.
TRAIL is a transmembrane protein that forms homo-trimers and this trimerization is critical for TRAIL’s pro-apoptotic effect [9]. TRAIL can bind to four membrane bound receptors, TRAIL-Receptor R1, R2, R3 and R4, and to one soluble receptor, osteoprotegerin. Apoptotic signaling is mediated by the two membrane-bound TRAIL receptors, TRAIL-R1 and TRAIL-R2, which signal through intracellular death domains (DD), to recruit intracellular FAS-associated death domain protein (FADD) leading to activation of initiator caspases 8 and 10, through proximity-induced autocatalysis. Caspase 8/10 activation leads to cleavage and activation of downstream effector caspases, including caspase 3, which induces cleavage of a wide array of cellular proteins and caspase-activated nucleases, ultimately resulting in the phenotypic changes of apoptosis [10].
In our previous studies on the effects of HIV infection on regulation of the TRAIL:TRAIL receptor axis, we discovered that cells from HIV infected patients make a novel splice variant of TRAIL, that is present in plasma and tissue culture supernatants, which antagonizes the pro-apoptotic effects of TRAIL [11]. This protein, named TRAILshort, is a 101-amino acid polypeptide that shares the first 90 amino acids with full-length TRAIL, but has a distinct 11-amino acid C terminus. Because TRAILshort is produced as a consequence of a splicing event that excises exons 3 and 4 from the normal 5 exon protein, TRAILshort is missing cysteine 230, and exists as a monomer, and lacks apoptosis-inducing activity[11]. Moreover, TRAILshort binds preferentially to TRAIL-R1 and -R2, and inhibits the pro-apoptotic activity of full-length TRAIL, thereby acting as a dominant negative ligand [12]. Interestingly TRAILshort is found in extracellular vesicles, which are elaborated from TRAILshort expressing cells, and these vesicles can confer TRAIL resistance upon otherwise TRAIL sensitive bystander, neighboring cells [12]. Finally, we have observed that interfering with TRAILshort function using inhibitory antibodies alters T cell dynamics following acute HIV infection in vitro [13, 14].
We now know that TRAILshort is induced by type I interferon signaling and Toll-like receptor (TLR) activation, suggesting that other diseases may be associated with elevated TRAILshort expression. In the current report we sought to identify other disease states in which TRAILshort is expressed.
MATERIALS AND METHODS:
Cell Culture
Uninfected primary peripheral blood mononuclear cells (PMBC) were isolated from donor apheresis cones using density gradient centrifugation. The following cell lines were also used: Jurkat (ATCC), HBL-1 (Ansell Lab, Mayo Clinic), OCI-LY3 (DSMZ), JeKo-1 (ATCC), RPMI-8226 (ATCC), BCWM.1 (Ansell Lab, Mayo Clinic), Ovcar8 (NCI-DTP), Hovtax2 (John Copland Lab, Mayo Clinic), Cov362 (ECACC), Caov3 (ATCC), Ovcar5 (NCI-DTP) PEO1 (ECACC) TYK-nu (JCRB), L3.6 (Daniel Billadeau Lab, Mayo Clinic), BxPC-3 (ATCC;), HepG2 (ATCC), HLE (JCRB), EM-Meso (Tobias Peikert Lab, Mayo Clinic); MDA-MB231 (ATCC), and Sk-Mel-28 (ATCC). TRAIL knockout Jurkat T cells were previously described [13].
Animal Studies
Mouse protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Mayo Clinic and animal procedures followed IACUC guidelines.
(Gene Expression Omnibus) Analysis
All publicly available RNA-Seq data from NCBI’s Sequencing Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra) were downloaded and processed with an internal gene expression quantification pipeline. In brief, FASTQ files were run through the pre-processing package fastp (https://github.com/OpenGene/fastp) to obtain high quality reads. Gene and isoform expression levels were estimated using salmon (https://salmon.readthedocs.io/en/latest/salmon.html). Each sample from the SRA was further annotated with the nferX Natural Language Processing (NLP) software (proprietary & unpublished) for disease phenotypes based on available sample descriptions in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). Significance was determined with the Fisher’s Exact Test, where the first group was presence in the top 5% expressing samples for a transcript (based on Transcripts per Million (TPM)) and the second was presence in the bottom 25% expressing samples. The final output was an enrichment score (reported as -log(p-val)) for each disease phenotype (Table S1).
TCGA (The Cancer Genome Atlas) Analysis
Isoform-level gene expression data from TCGA (http://gdac.broadinstitute.org) was used to estimate transcript abundance. The non-normalized isoform counts and transcript abundance estimates were generated by the TCGA Research Network: https://cancergenome.nih.gov/. Isoform expression values from Broad GDAC were named in the general format: (SUBTYPE.rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_isoforms__data.data.txt). The “scaled estimate” value found in these files was transformed to transcripts per million (TPM) by multiplication with 1e6. We then extracted the expression values for TRAILshort (‘uc003fie.2’) and visualized this across multiple tissues. R code and data for this plot has been deposited in github ((https://github.com/egarciarivera/rcode/tree/master/trail_short_tcga). Cutoffs were set as follows, with 3 TPM representing baseline level of expression for any gene,[15] 10 TPM being the ~90th percentile (high), and 20 TPM being the ~95th percentile (very high) of TRAILshort expression.
Immunohistochemical Staining
Five micron sections of formalin-fixed paraffin-embedded (FFPE) tissues were retrieved for 20 minutes using Epitope retrieval 2 (EDTA; Leica), and incubated in protein block (Dako) for 5 minutes. The TRAILshort primary antibody (clone 2.2 at 1 µg/µl,) or appropriate isotype control antibody were diluted to 1:800 in background reducing diluent (Dako) and incubated for 15 minutes. Slides were counterstained with hematoxylin, washed, and dehydrated in ethanol prior to permanent coverslipping in xylene-based medium.
RNA in situ Hybridization
Deparaffinized FFPE slides were pretreated with RNAScope Hydrogen Peroxidase (Advanced Cell Diagnostics [ACD]), then 1X RNAScope Target Retrieval Reagents (ACD) followed by RNAScope Protease III (ACD) at 40°C. Slides were hybridized with a positive control probe BA-Hs-PPIB-3zz (ACD), a negative control probe BA-DapB-3zz (ACD), and a target probe BA-Hs-TNFSF10-tv2-E2E3 (ACD) for 2 hours at 40°C inside a HybEZ oven. Thereafter, amplification was performed per manufacturer’s instructions. Signal detection was performed using a mix of BaseScope Red A and Red B (ACD) at a ratio of 60:1 at room temperature for 10 minutes. Slides were then rinsed in tap water, counterstained, and mounted.
IncuCyte Cell Killing Assay
Adherent cells were seeded in a flat-bottom 96-well plate at a density of 104cells/well. For suspension cells, 96-well plates were pre-coated with 50 µL of poly-L-ornithine for 1 hour at RT then washed twice. Assays were performed as described previously [12] using the IncuCyte platform (Essen BioScience) and IncuCyte caspase 3/7 apoptosis detection reagent that was added immediately after cells were plated, but before the indicated treatments. Cells were monitored every 2 hours for up to 60 hours by anti-active caspase 3 staining in triplicate. The pan-caspase inhibitor Q-VD was used where indicated (Sigma Aldrich).
Flow Cytometry and PCR
Cells were collected and washed twice, blocked in 5% fetal bovine serum for 30 min on ice, and stained using a CF555-labeled TRAILshort (Clone 2.2) antibody, a phycoerythrin (PE)-labeled CD253 TRAIL antibody (Abcam), and a fluorescein isothiocyanate (FITC)-labeled death receptor 5 (DR5) antibody (ThermoFisher) for one hour on ice. TUNEL staining was performed according to manufacturer’s protocol (Roche). TMRE staining was performed as previously described.[11]. Flow cytometry was performed using a Becton Dickinson LSR Fortessa X20 and FACS Diva Software. The primers and conditions for real-time PCR assessment of TRAILshort have been described previously [11].
Antibody Humanization
Variable heavy (VH) and light (VL) regions of the TRAILshort murine antibody were sequenced and compared using BLASTp (NCBI) to identify homology with known human VH and VL antibody sequences. The four most homologous candidates to the murine sequences were identified taking into account framework homology, maintenance of key framework residues, and canonical loop structure (based on a combined IMGT/Kabat CDR labelling approach). The 4 humanized VH domains and 4 humanized VL domains were synthesized and cloned into the pD2610-v13 mammalian expression vector (Atum) in a combinatorial manner on an IgG1 Fc backbone. Resultant recombinant chimeric antibodies were purified and assessed by surface plasmon resonance (Figure S1) using a Biacore 2000 instrument for kinetic interaction analyses (Biaffin). This identified clone HC2LC3, with an equilibrium dissociation constant (KD) of 3.8 × 10–12 M, (Figure S3) that also effectively synergized with sk-TRAIL to kill Jurkat T cells in vitro (Figure S4). The HC2LC3 clone was selected for further testing.
TRAILshort antibody in vivo Safety Assessment
An in vivo safety assessment of anti-TRAILshort antibody was performed by injecting 10 C57-BL/6 mice (5 male, 5 female) with 10 mg/kg anti-TRAILshort antibody (clone 2.2) and observing mice twice daily for 7 days.
NSG Mouse Xenograft Model
A single cell clone of Jurkat cells stably expressing firefly luciferase (Jurkat-Luc; obtained from Dr. Douglas K. Graham, Emory University, GA [16]), was used for mouse experiments, and five million cells injected via tail vein injection into NSG mice as previously described [16]. HBL-1cells harvested during log phase growth were injected subcutaneously into the right dorsal region NSG mice as previously described [17]. In vivo tumor burden for Jurkat-Luc cells was determined twice weekly by measuring the whole-body luciferase activity with an IVIS200 imaging system or, in the case of HBL-1 cells, by blinded measurement using calipers. After ~20 days (range 20–30 days) when tumors had become established, mice were randomly assigned to treatment groups and were monitored daily.
Quantification and statistical analysis
For the time-dependent cell killing experiments, the area under the curve was compared using the two-sample t-test. For mouse xenograft experiments, the log-rank test was used to assess differences in overall survival between treatment groups. Changes in tumor volume (or luminescence) over time were compared between treatment groups using linear regression, with an endpoint reflecting the last day when all mice were alive. Statistical analyses were performed using R (version 3.5.1.) [18]. Statistical significance was defined as p < 0.05, and all tests were two-sided.
Data and software availability
Study Approval
All the human-derived samples were obtained in compliance with IRB-approved protocols (Mayo IRB #s 118–01 and 1827–00).
RESULTS
TRAILshort transcripts are associated with human disease.
To determine the potential clinical significance of TRAILshort expression we used unbiased isoform level gene expression data from 253,200 samples across 6,600 studies in Gene Expression Omnibus (GEO), and identified TRAILshort transcripts in 117,500 samples, and significant enrichment (>10) of TRAILshort expression in patients with active infectious diseases consistent with our earlier publications [11–14] (Table S1), and identified a novel association of TRAILshort enrichment with human malignancy, as 15/25 disease states in which enrichment was observed occurred in patients with malignant diagnoses.
TRAILshort transcripts are highly prevalent in human cancers
The Cancer Genome Atlas (TCGA) database [19] identified that ~40% of primary human malignancies express TRAILshort transcripts. Specifically TRAILshort expression of >3 TPM, (which represents the baseline level of expression for any gene[15]) was observed in 40% of cases, while 10% of malignancies express TRAILshort at high levels (>10 TPM), and ~5% of malignancies had very high levels of TRAILshort (>20 TPM) (Figure 1A). TRAILshort transcripts were significantly co-associated with 66 different transcripts (P<0.05, Figure 1B) and 63/66 (95%) of those genes are type I interferon-regulated, suggesting a common pathway of induction. Interestingly HPV positive tumors expressed more TRAILshort transcripts that HPV-negative tumors (mean +/− SE of HPV-positive 17.6 +/− 1.9 TPM, vs HPV-negative 11.9 +/− 0.6 TPM, P=0.006, Figure 1C) consistent with HPV activation of TLR7 [20, 21], and the known ability of TLR7 activation to induce TRAILshort expression [12].
Figure 1. TRAILshort is prevalent within human cancer tissues.
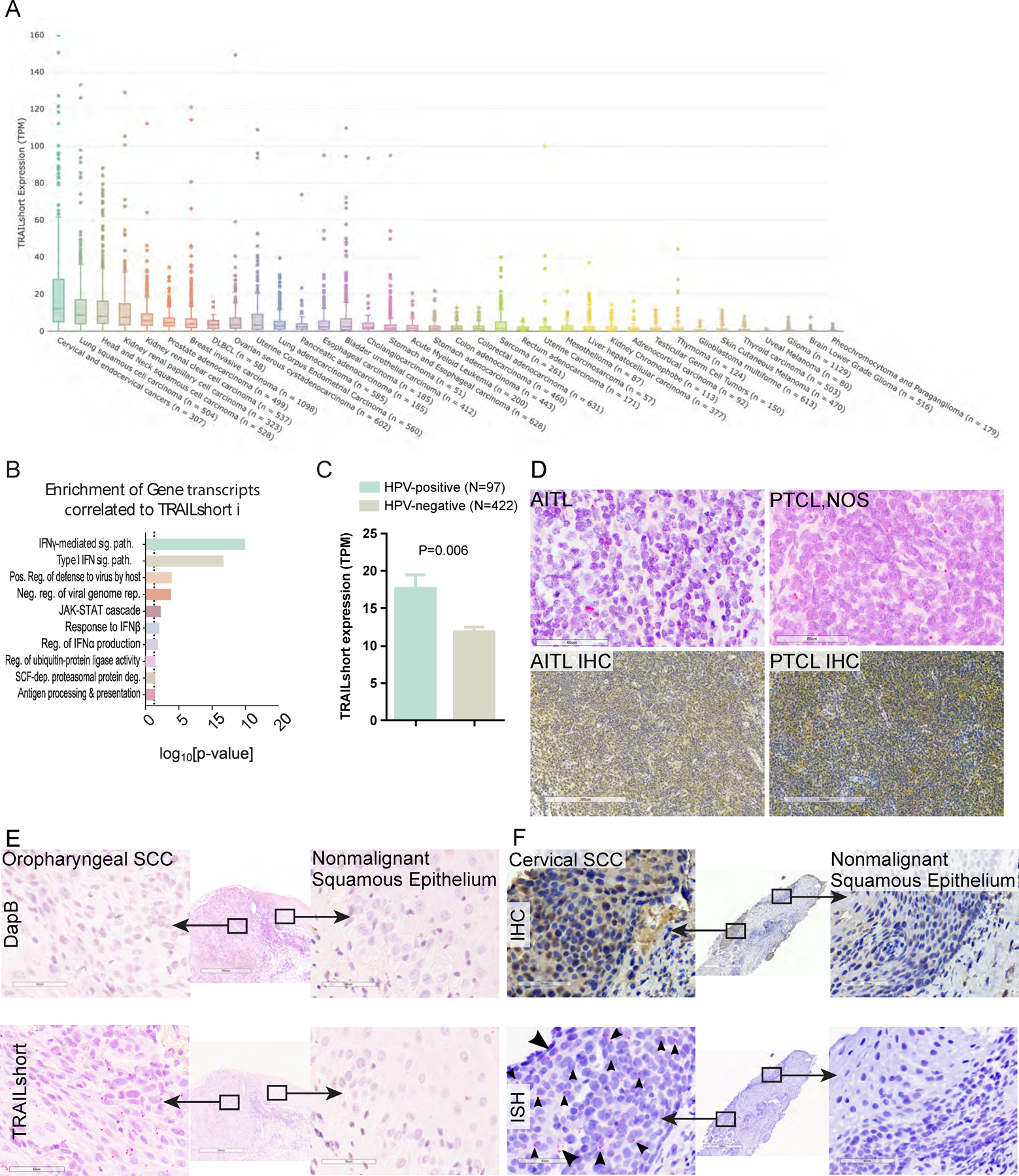
(A) Tissues within the TCGA dataset were queried for the presence of TRAILshort transcripts, and data are expressed as transcripts per million (TPM) according to tissue type. (B) Pearson’s correlation coefficients (PCC) identified 66 gene transcripts co-associated with expression of TRAILshort transcripts, and the functional clustering is shown. (C) TRAILshort transcripts were compared between HPV-positive (N=97) and HPV-negative (N=422) tumors in the TCGA dataset; depicted are mean +/− standard error; P=0.0061 by two-tailed Mann-Whitney test. (D-F) The indicated tissues were stained by immunohistochemistry (IHC) for TRAILshort protein expression (brown staining), or by in situ hybridization (ISH) for TRAILshort mRNA expression (red dots indicated with black arrowheads). (D) Angioimmunoblastic T-cell lymphoma (AITL) and peripheral T-cell lymphoma not otherwise specified (PTCL NOS) stain positive for TRAILshort mRNA ( red dots with black arrowheads). (top panels, RNA ISH, scale bar = 60 µm) as well as by IHC (bottom panels, scale bar = 300 µm). (E) Oropharyngeal squamous cell carcinoma (SCC) tissue was stained by ISH using a negative control probe, DapB top panels, and no signal is detected, yet TRAILshort mRNA is detected by ISH in the malignant tissue indicated by the red dots (highlighted by black arrowheads), but not in the non-malignant adjacent tissue Scale bar = 60 µm for all panels. (F) TRAILshort protein detected by IHC (top panels) or TRAILshort mRNA detected by ISH (bottom panels) in malignant (left panels) or non-malignant (right panels) cervical squamous epithelium.
TRAILshort protein and message are detected in human cancer biopsies
We assessed primary human cancer tissues for TRAILshort protein by immunohistochemistry (IHC), using a mouse monoclonal anti-TRAILshort antibody which we have previously validated [11], and by TRAILshort message specific in situ hybridization (ISH). The anti-TRAILshort antibody was developed and validated to recognize the unique C-terminal 11 amino acids encoded by TRAILshort [13]. The TRAILshort ISH probes target the novel splice junction not present in full length TRAIL message. Staining was optimized using the TRAILshort negative HeLa cell line (transfected with control vector), or Hela cells transfected with TRAILshort (Figure S1A, and S1B). Once optimized, TRAILshort message and protein were evaluated in multiple primary cancer specimens (Figure S1 C–E). TRAILshort message was confined to tumor cells, while TRAILshort protein was more diffuse, consistent with TRAILshort being contained with extracellular vesicles, and present within the microenvironment [11, 12].
Multiple tumor types identified by informatics analyses to be TRAILshort positive, stained positive for TRAILshort mRNA by ISH, while some tissues (eg. urothelial carcinoma) were negative for TRAILshort mRNA (Figure S1E). We also assessed lymphomas for example angioimmunoblastic T-cell lymphoma (AITL) and peripheral T-cell lymphoma not otherwise specified (PTCL NOS), had detectable TRAILshort expression by both ISH and IHC (Figure 1D, top and bottom panels respectively). In total ISH was carried out for 82 human cancer tissue sections. Thirty-one (37.8%) of these tissues were positive for TRAILshort by ISH, and positive staining was detected most commonly in squamous cell carcinomas (from cervical, tonsillar, and/or oropharyngeal sources), lymphomas, and pancreatic adenocarcinomas (Table S2). Within single tissue sections that contained both malignant tissue (eg. oropharyngeal squamous cell carcinoma) and nonmalignant tissue (non-neoplastic squamous epithelium) TRAILshort ISH detected TRAILshort in neoplastic cells but not the non-neoplastic squamous epithelium (Figure 1E). Similarly, TRAILshort protein and mRNA were detected in malignant squamous epithelium from the cervix (Figure 1F left panels), but not in nonmalignant squamous epithelium from the same tissue section (Figure 1F right panels).
TRAILshort expression protects human cancer cell lines from the death inducing effects of TRAIL agonists.
TRAILshort protects HIV infected T cells from death induced by TRAIL[13]. Using the same antibody we used to neutralize TRAILshort, we now tested the effect of that antibody on TRAIL induced killing of TRAILshort expressing cancer cell lines. In initial experiments, a TRAIL agonist (super killer (sk)-TRAIL) induced minimal death of the TRAILshort expressing cell line HBL-1, and that killing was significantly augmented by the addition of a TRAILshort neutralizing antibody (Figure 2A); by contrast the TRAILshort antibody had no impact on TRAIL induced killing of the TRAILshort negative RPMI8226 cell line. Across the cell lines tested, 6 / 23 cell lines (26%) tested showed a significant increase in cell killing imparted by the TRAILshort antibody (Figures 2C–G), while other tumors (Figure 2D) and non-cancerous peripheral blood mononuclear cells (PBMCs, Figure 2I) were unaffected by anti-TRAILshort antibody, suggesting that TRAILshort antibody might selectively impact TRAIL sensitivity of a subset of malignant cells. Transcriptional profiles of five responsive cell lines and nine non-responsive cell lines, were compared (Figure 2H), revealing 27 differentially expressed transcripts, with significant enrichment (P<0.0001) of Type I interferon induced genes.
Figure 2. Neutralization of TRAILshort enhances TRAIL induced cell killing.
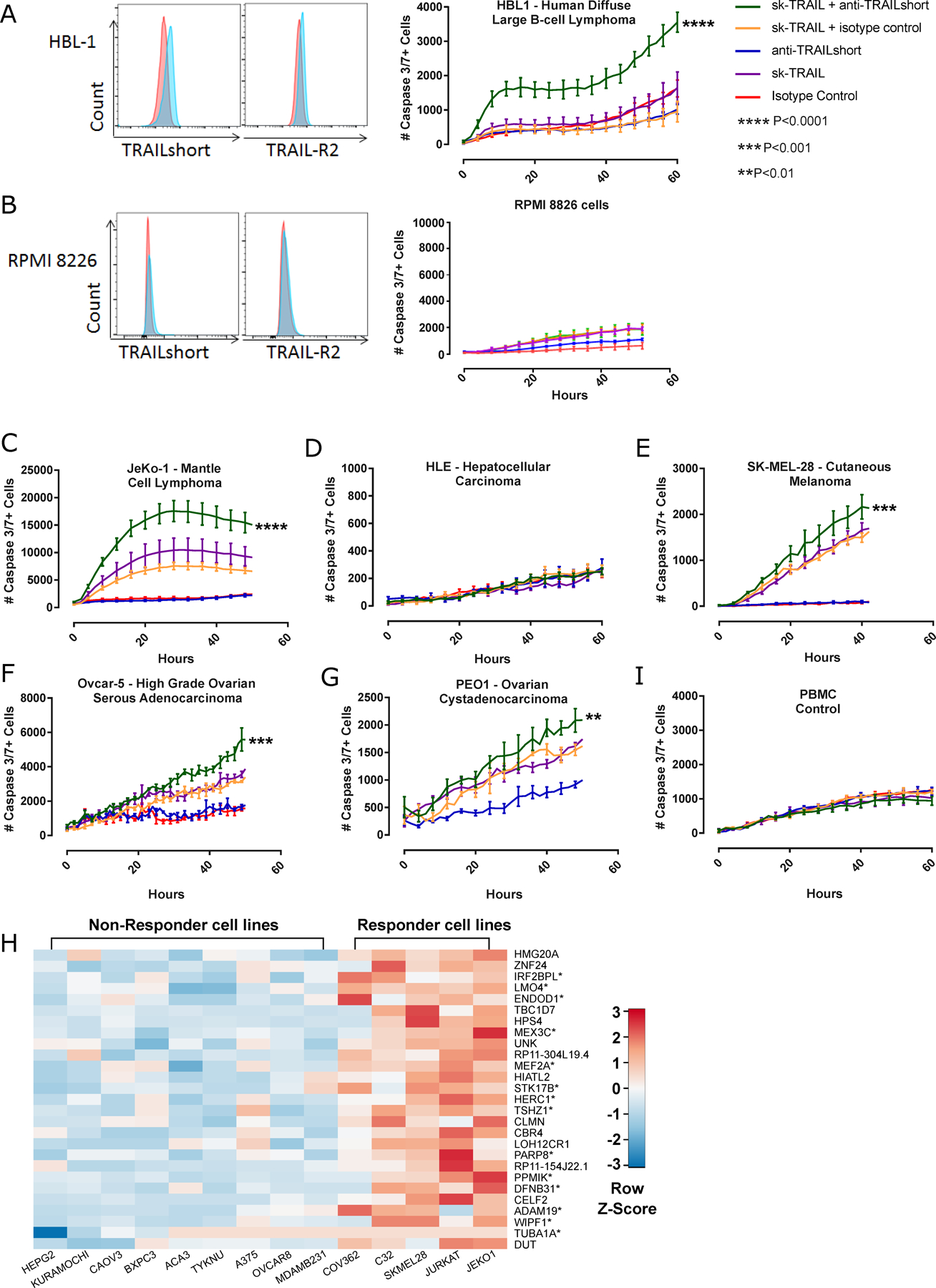
Human cancer cell lines were assessed for TRAILshort and TRAIL receptor 2 expression, and in parallel tested for cell killing in response to skTRAIL, with or without TRAILshort antibody. TRAILshort and TRAIL receptor 2 expression is shown for (A) HBL1 cells or (B) RPMI 8226 cells (Red histogram= isotype control staining; blue =TRAILshort or TRAILR2), and these cells were tested for cell killing in response to recombinant TRAIL (skTRAIL) alone, anti TRAILshort antibody alone, or the combination. Cell killing curves performed as in A and B, shown for (C) JeKo-1, (D) HLE, (E) SK-MEL-28, (F) Ovcar-5, and (G) PEO1, (I) primary peripheral blood mononuclear cells (PBMCs). (H) The transcriptional profile of anti-TRAILshort-responsive cell lines was compared to that of anti-TRAILshort-non-responsive cell lines to generate a heat map, which identified differentially expressed genes. Interferon response genes are indicated by an asterisk.
Mechanism of action of TRAILshort neutralization
To further understand the mechanism by which TRAILshort antagonism alters cell fate, we focused on Jurkat T cells, which we have previously observed to further upregulate TRAILshort in response to HIV infection [12, 13]. Because TRAIL receptor signaling can lead to apoptosis in susceptible cells or cell proliferation in resistant cells [22], we investigated the effects of TRAILshort antagonism on apoptosis and cell proliferation. First we demonstrated that Jurkat T cells are partially susceptible to TRAIL-induced apoptosis, which is inhibited by the pan-caspase inhibitor QVD (Figure 3A). As TRAILshort is a splice variant of TRAIL, genetic deletion of TRAIL necessarily deletes TRAILshort. Taking advantage of this, we next compared cell death induced by TRAIL in wild type or CRISPR Cas/9 deleted TRAIL (and thus TRAILshort) Jurkat T cells. When wild type (WT) or TRAIL knockout (KO) Jurkat T cells were treated with skTRAIL, TRAIL-induced apoptosis (measured by loss of mitochondrial outer membrane potential (Figure 3B) or DNA end-nick labeling (Figure 3C) was markedly increased in the TRAIL deficient Jurkats compared to the WT Jurkats, consistent with TRAILshort antagonizing the proapoptotic effects of TRAIL. We then confirmed surface TRAILshort expression in uninfected wild type Jurkat T cells (Figure 3D).
Figure 3. Neutralization of TRAILshort does not alter cell proliferation nor activation, but enhances apoptotic cell death.
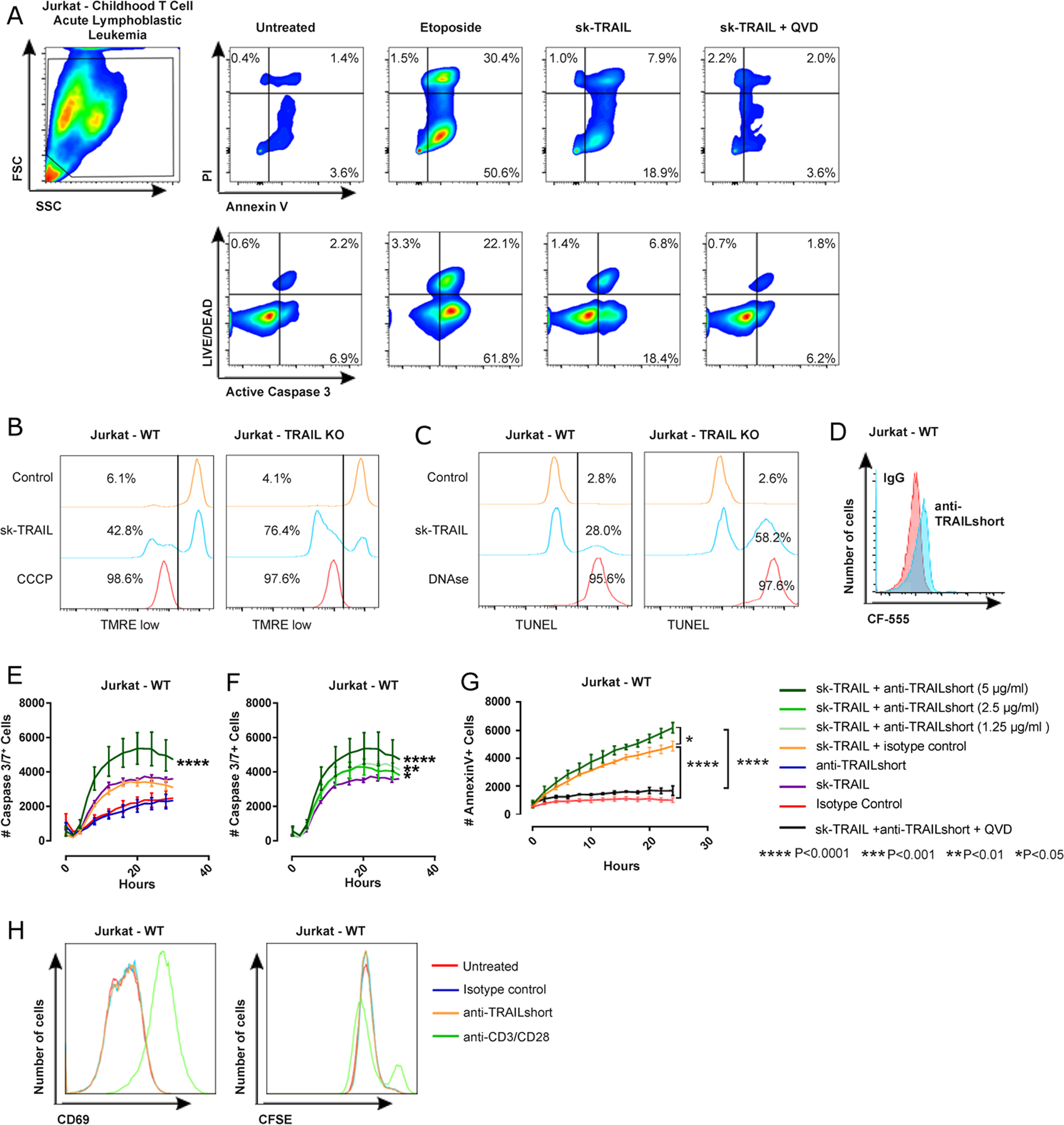
(A) Jurkat T cells were treated with control, sk-TRAIL, or sk-TRAIL plus the pan-caspase inhibitor QVD for 24 hours and assessed for apoptosis by Annexin V/PI staining and active caspase 3/ LIVE/DEAD staining by flow cytometry. Etoposide was used as a positive control. (B) Wild type (WT) Jurkat T cells or Jurkat T cells genetically deficient in TRAIL by CRISPR (TRAIL KO) were treated with skTRAIL or vehicle control for 12 hours and mitochondrial depolarization assessed by tetramethylrhodamine, ethyl ester, perchlorate (TMRE) staining and flow cytometry. Carbonyl cyanide m-chlorophenylhydrazone (CCCP) was used as a positive control. Percentage of TMRE low cells is enumerated. (C) WT and TRAIL KO Jurkat T cells were treated with skTRAIL or control for 12 hours and DNA fragmentation assessed by TUNEL staining and flow cytometry. DNAse treatment was used as positive control. The percentage of TUNEL positive cells is enumerated. (D) Jurkat T cells were assessed for surface expression of TRAILshort by flow cytometry. (E – F) Ten thousand Jurkat T cells were treated with either an isotype control antibody (5µg/ml), recombinant pre-aggregated sk-TRAIL (1 ng/ml, unless unresponsive in which case 10 ng/ml was used), mouse anti-TRAILshort antibody (5 µg/ml, unless otherwise noted), or a combination of isotype control and either sk-TRAIL or mouse anti-TRAILshort antibody at low, medium, or high concentration (1.25, 2.5 or 5 µg/ml), plus sk-TRAIL as indicated. The number of caspase 3/7 positive cells was monitored every 2 hours. All experiments were performed in triplicate. Data are represented as mean ± SE. Significance shown in A and B compares sk-TRAIL + anti-TRAILshort versus sk-TRAIL alone. (G) Jurkat cells were treated with sk-TRAIL plus anti-TRAILshort antibody in the presence of or absence of the pan-caspase inhibitor Q-VD to test the caspase-dependence of apoptosis. (H) WT Jurkat cells were treated with nothing, isotype control, anti-TRAILshort antibody or anti-CD3/CD28 microbeads and assessed for surface expression of CD69 (at 3 days) and CFSE dilution (at 5 days).
TRAIL induced apoptosis of Jurkat T cells was augmented in the presence of TRAILshort antibody (Figure 3E), an effect that was dose dependent (Figure 3F) and inhibited by the pan caspase inhibitor QVD (Figure 3G). These data argue that the antibody effect impacts cell death signaling. Altogether, these data indicate that TRAILshort protects Jurkat T cells from TRAIL-induced killing, and that cell death occurring in the setting of anti-TRAILshort antibody plus recombinant TRAIL is apoptotic in nature.
Finally we assessed the impact of anti-TRAILshort antibody alone on proliferation, and found that treatment of Jurkat T cells with anti-TRAILshort antibody does not lead to activation nor proliferation (Figure 3H) compared to isotype control antibody.
Anti-TRAILshort antibody exerts antitumor effect in human leukemia/lymphoma xenografts
We next assessed putative anti-tumor effects of TRAILshort antibodies in vivo. An in vivo safety assessment of anti-TRAILshort antibody in C57-BL/6 mice demonstrated no morbidity, mortality, abnormalities in hematology or chemistry, or pathology at necropsy (Supplemental Document S1). For these proof-of-concept studies, we did not measure or optimize pharmacokinetics.
Next, non-obese diabetic, severe combined-immunodeficient, common γ-chain deficient (NSG) mice were implanted with luciferease expressing Jurkat T leukemia cells, and the tumor was allowed to become established for ~20 days [16]. Starting on day 20 after tumor injection, mice received 10 mg/kg of either anti-TRAILshort antibody or isotype control; the treatment was repeated weekly. Treatment with anti-TRAILshort antibody alone resulted in both decreased tumor burden through day 31 (the last day all mice remained alive, Figure 4A, P=0.04) and prolonged survival over the full period of observation (Figure 4B, P<0.0001) compared to isotype control antibody.
Figure 4. Anti-TRAILshort antibody alone or in combination with a TRAIL agonist has antitumor effects in Jurkat T cell and HBL-1 xenograft models.
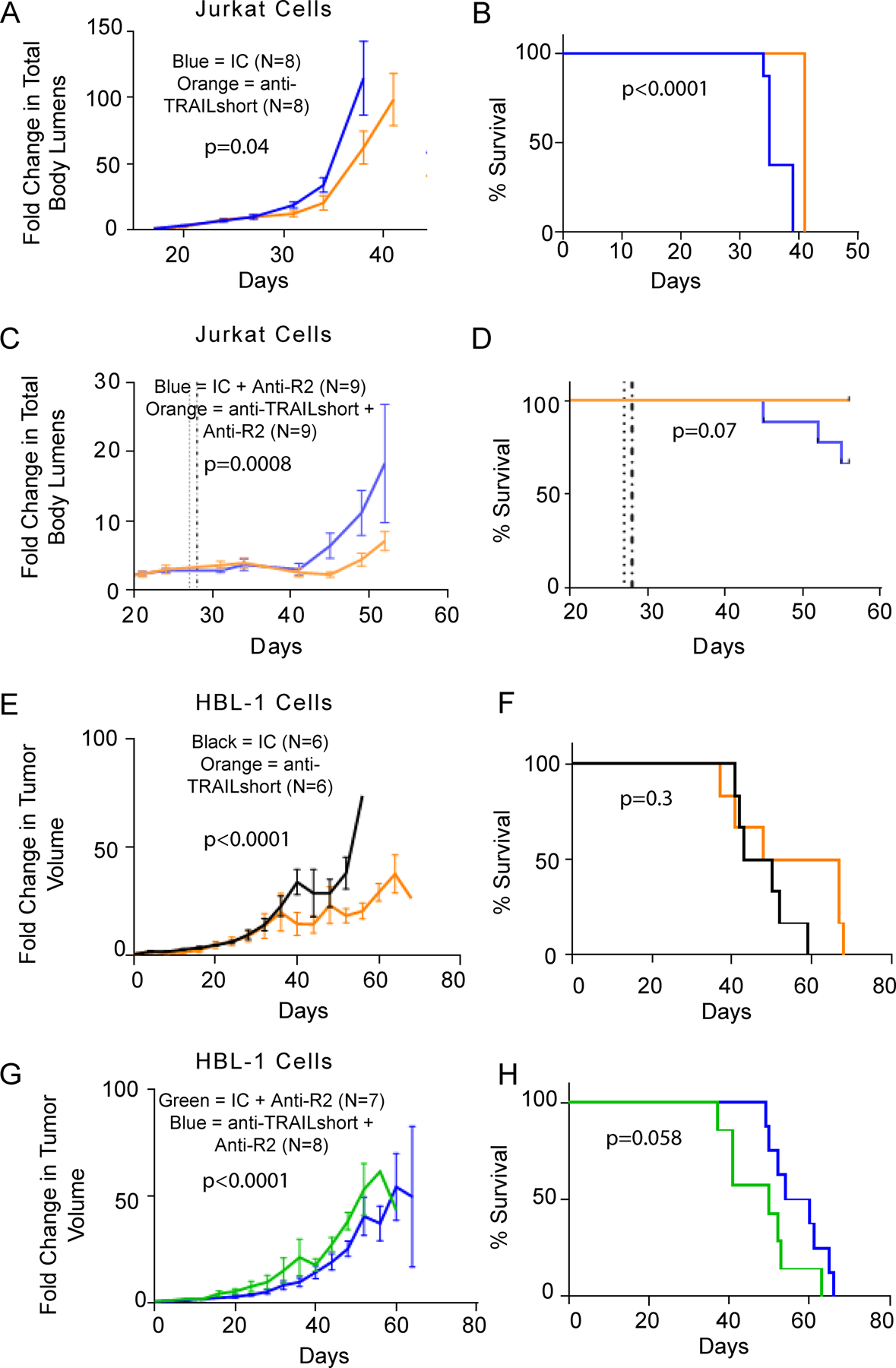
(A-B) Non-obese diabetic, severe combined-immunodeficient, common γ-chain deficient (NSG) mice were implanted with Jurkat human T leukemia cells expressing luciferase by IV injection, and tumors were allowed to become established. Mice were treated weekly with mouse anti-TRAILshort antibody (clone 2.2) alone or isotype control antibody. Mice were imaged twice weekly and followed for (A) total body luminescence over time and (B) survival. (C-D) Mice injected with luciferase expressing Jurkat T cells were administered a single injection of mouse anti-TRAILshort antibody (clone 2.2) followed 24 hours later by a single injection of anti-TRAIL-R2 antibody, and followed for (C) mouse whole body luminescence over time (P=0.0008) and (D) Mouse survival (P=0.07). (E-H) NSG mice were implanted with HBL-1 cells by IV injection, and tumors were allowed to become established. These mice were treated weekly with anti-TRAIL short antibody or isotype control alone (E-F), or followed the next day by anti-TRAILR2 antibody (G-H), and tumors measured and followed for (E and G) tumor size and (F and H) survival. Results shown are mean ± SD.
Next, we combined TRAILshort antibody with a TRAIL agonist, in this case anti-TRAIL R2 antibody [23]. NSG mice engrafted with luciferase expressing Jurkat cells (as in Figure 4A) were treated with a single dose of either anti-TRAILshort antibody (clone 2.2) or isotype control, followed 24 hours later with anti-TRAIL-R2 [24]. Mice receiving the anti-TRAILshort antibody followed by the TRAIL agonist had significantly decreased tumor burden compared to isotype antibody plus TRAIL agonist-treated mice (Figure S2, representative images at day 45, Figure 4C, P=0.0008). Furthermore, while 3/9 mice treated with isotype control antibody succumbed to the tumor, all anti-TRAILshort antibody treated mice survived until the end of observation on day 56 (the predefined end of the experiment, Figure 4D, P=0.07).
In a second, orthogonal, validated in vivo xenograft model [17], NSG mice were injected subcutaneously with HBL-1 cells, and subsequently treated with anti-TRAILshort antibody or isotype control alone (Figure 4E–F) or in combination with a TRAIL receptor agonist antibody (anti-TRAIL-R2, Figure 4G–H). Mice treated with anti-TRAILshort antibody alone had significant reductions in tumor volumes compared to isotype control treatments through day 37 (the last day all mice remained alive, Figure 4E) with apparent but non-significant effect on survival (Figure 4F). When antiTRAIL-R2 antibody was added to anti-TRAILshort antibodies in this experimental system, mice receiving anti-TRAILshort antibody plus anti-TRAIL-R2 also had significantly reduced tumor volumes (P<0.0001, Figure 4G) but also had improvement in survival compared to mice treated with isotype control plus anti-TRAIL-R2 (P=0.058, Figure 4H).
These xenograft data indicate that anti-TRAIL short antibody has modest anti-tumor effects both alone and in combination with a TRAIL agonist, suggesting in vivo relevance of inhibiting TRAILshort with therapeutic antibodies. Optimizing of pharmacokinetics and/ or combining this therapy with tumor specific T cells might further enhance this modest therapeutic effect, and further improve survival rates in vivo.
Effect of TRAILshort antibody on fresh tumor cells isolated from patients undergoing splenectomy for malignant diagnoses.
We next obtained cells freshly harvested from patients undergoing splenectomy for suspected malignancy. These splenocyte preparations are known to contain mixtures of tumor cells (most of them of B cell lineage) and immune effector cells including T cells. For these experiments we used a fully humanized anti TRAILshort antibody clone HC2LC3, with an equilibrium dissociation constant (KD) of 3.8 × 10−12 M, (Figure S3 and S4).
To begin, freshly harvested splenocytes (containing malignant B cells as well as immune effector cells) were treated immediately with anti-TRAILshort antibody (HC2LC3), or isotype control antibody, alone or in combination with the TRAIL agonist skTRAIL, and cell death analyzed. A total of 26 samples were tested (Table S3), and 21 of those ultimately had a malignant diagnosis, while 5 cases were non-malignant immune thrombocytopenia purpura (ITP) or reactive follicular hyperplasia.
Of the 21 cases with a malignant diagnosis, 9 (43%) - mostly B cell lymphomas - were killed in response to anti TRAILshort antibody in combination with sk-TRAIL (representative examples in Figure 5A). Reassuringly, none of the cells harvested from patients with non-malignant diagnoses died in response to TRAILshort antibody alone or in combination with skTRAIL (Table S3) similar to what we previously observed with healthy PBMC (Figure 2I). Thirteen of the malignant tumors were also tested by ISH for TRAILshort expression and 6 were positive (Table S3, representative example in Figure 5C–D), and of those tumors shown to be TRAILshort positive by ISH, 5/6 (83%) died in response to TRAILshort antibody in combination with skTRAIL.
Figure 5. Humanized anti-TRAILshort antibodies have antitumor activity in primary human tumor samples ex vivo.
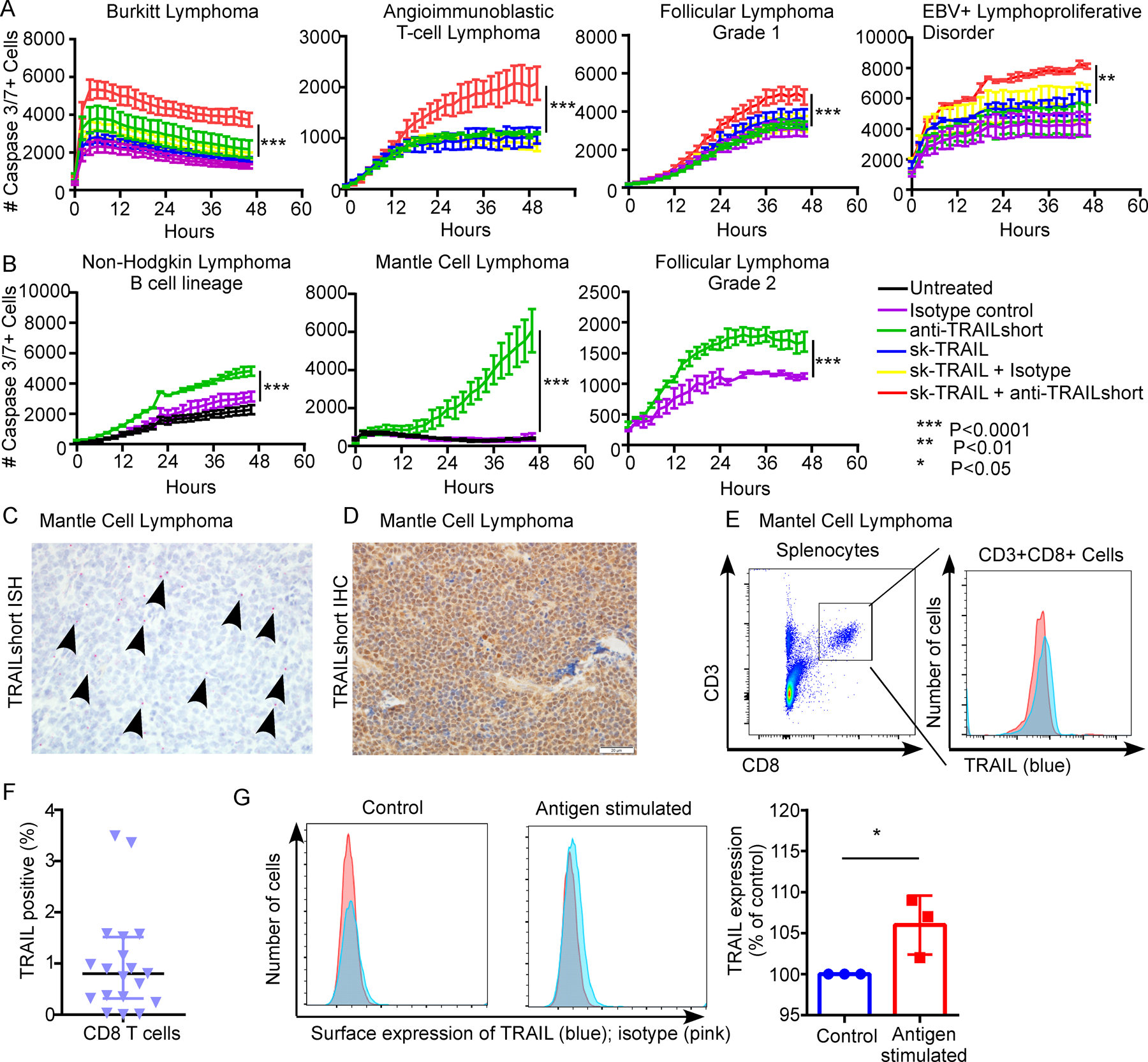
Ten thousand splenocytes harvested from human patients with suspected malignancies were treated within 4 hours of harvest with isotype control antibody (5µg/ml), recombinant pre-aggregated sk-TRAIL (1 ng/ml), humanized anti-TRAILshort antibody (5 µg/ml), the combination of isotype control antibody and sk-TRAIL, or humanized anti-TRAILshort antibody at low medium or high dose (1.25, 2.5 or 5 µg/ml) plus sk-TRAIL as indicated. (A) Representative patient samples in which splenocyte death was augmented by the addition of anti-TRAILshort antibody to the TRAIL agonist skTRAIL. (B) Representative patient samples in which splenocyte death was induced with anti-TRAILshort antibody treatment alone. (C) Splenic tissue from the patient with Mantle cell lymphoma from B, was stained for TRAILshort expression by ISH (red dots and arrows), and (D) IHC ( brown staining). (E) CD8 T cells from the splenocyte suspension were analyzed by flow cytometry using CD3/CD8 and TRAIL antibodies; TRAIL expression in CD3+CD8+ cells is shown. (F) Surface TRAIL expression was examined in peripheral CD3+CD8+ T cells from healthy donors by flow cytometry, in unstimulated conditions or (G) stimulated with tetanus toxoid (recall antigen) ex vivo for 24 hours, histogram shows pooled results from 3 donors.
In a subset of patient samples above, splenocytes were killed by TRAILshort antibody alone (Figure 5B), suggesting the presence of an endogenous source of TRAIL. Tumor infiltrating T cells have been reported to express high levels of TRAIL [25–27], and T cell activation by phorbol esters or mitogen further enhances TRAIL expression [28]. We therefore assessed purified CD8 positive cells from a mantle cell lymphoma case and observed TRAIL expression by CD8 positive cells (Figure 5E). Moreover freshly isolated CD3+CD8+ T cells from healthy donors, expressed low levels of surface TRAIL (Figure 5F), and consistent with the original descriptions of TRAIL expression by T cells, we observed that T cell stimulation by recall antigen (in this case tetanus toxoid) further enhances T cell expression of TRAIL (P<0.05, Figure 5G).
Given that some tumor cells express TRAILshort, that TRAILshort blocks TRAIL mediated cell death, and that both antigen stimulated CD8 T Cells and tumor infiltrating CD8 positive cells express TRAIL, we tested the hypothesis that the ability of tumor infiltrating immune effector cells to kill autologous tumors might be antagonized by TRAILshort, and enhanced by neutralization of TRAILshort (Figure 6). Cells from splenocyte suspensions were separated into CD8 positive cells and the remaining cells pooled, stained with a lipophilic red die and considered as tumor target cells. Autologous activated CD8 positive cells, and red target cells were mixed at varying effector: target cell ratios (according to the number of available cells, range 1:1 to 5:1) and cell death assessed in the red tumor target cells (Figure 6A). The number of tumor cells (red) that became caspase 3/7 positive (green) was minimal when tumor cells were incubated alone. The addition of CD8 positive cells alone to the tumor target cells did not significantly alter tumor cell killing. However, activated CD8 positive cells plus TRAILshort antibody, but not control antibody, resulted in significantly increased CD8 positive cell killing of target tumor cells (P<0.05, Figure 6 B–D), in 3 (23%) of 13 patient samples tested. Altogether, these data indicate that anti-TRAILshort antibody can augment the ability of autologous effector cells to kill tumor targets.
Figure 6. Humanized anti-TRAILshort antibodies augment antitumor activity of immune effector cells ex vivo.
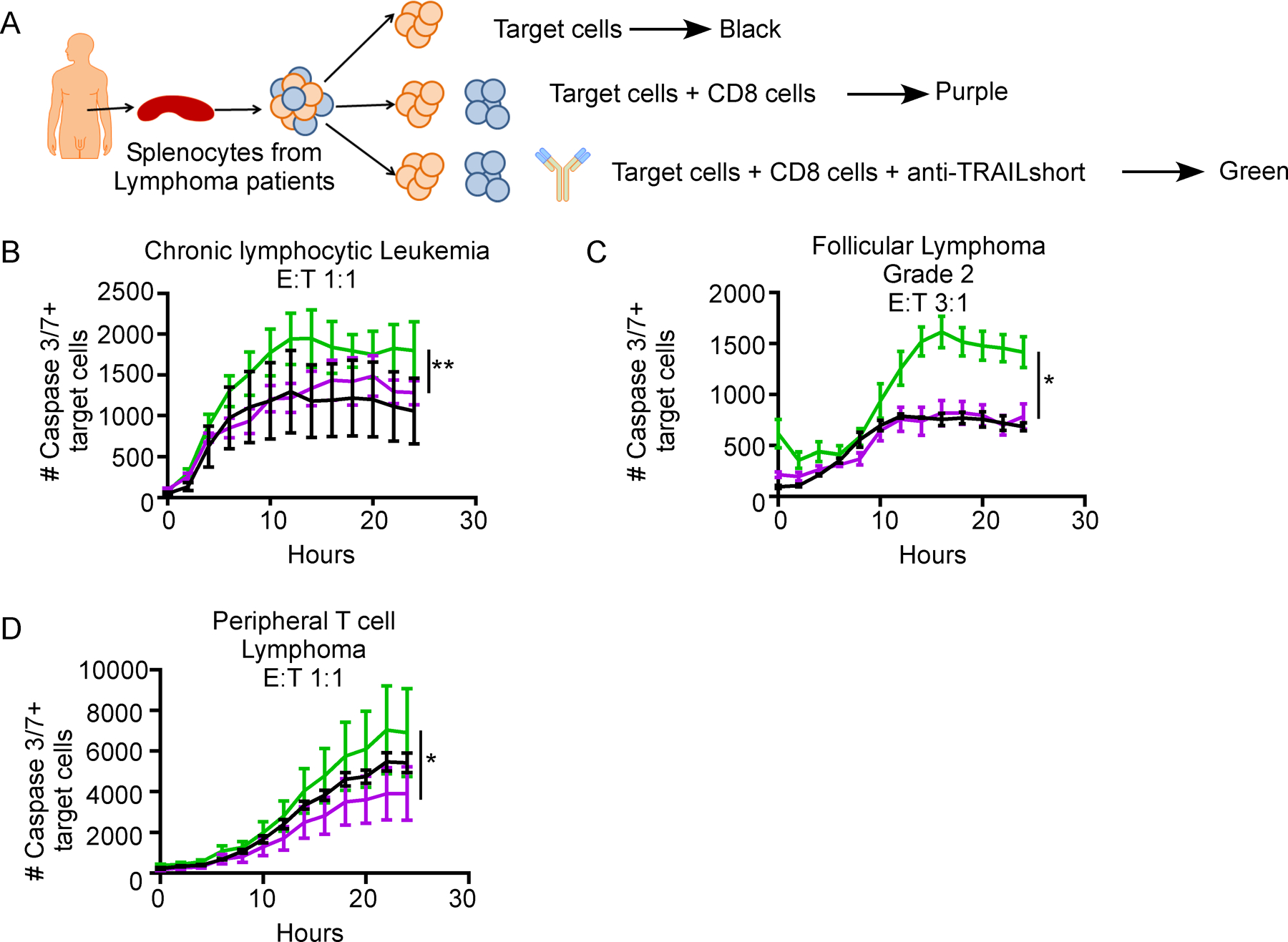
(A) CD8 positive cells were positively selected from bulk patient splenocytes, and co-cultured with autologous target cells (bulk splenocytes after CD8 cell removal stained with cell tracker red) at the indicated effector to target ratios, in the presence or absence of anti-TRAILshort antibody, and cell death was measured over time in the red target cell population. (B-D) Representative data of the number of Caspase 3/7 positive target cells in co-culture from three patient samples with response to anti-TRAILshort antibody.
DISCUSSION:
TRAIL is upregulated on activated T cells including tumor infiltrating T cells [2], and their interaction with TRAIL receptor 1 and/or 2 expressing tumor cells should logically result in the killing of the tumor cells. Paradoxically that does not occur, leading multiple groups to speculate and test the hypothesis that supplying more, or more powerful, TRAIL agonists might result in effective tumor killing – to limited success. Here we show that TRAILshort is highly expressed in a subset of primary human cancers, and we provide proof of concept that inhibiting the production of TRAILshort genetically or inhibiting the function of TRAILshort using first generation antibodies, enhances autologous immune effector cell killing of tumors.
Since the concept of immune surveillance against malignant and/or infected cells was first proposed, it has become an overarching principle of pathophysiology. As new immune effector mechanisms are described, new pathways of immune escape are invariably unmasked. In such cases Darwinian pressures favor the survival of cancerous cell clones which escape immune surveillance pathways. It therefore follows that immune cell killing of cancer cells through TRAIL would be antagonized in cancer clones in which TRAILshort is expressed. Moreover it suggests that this mechanism of immune escape would eventually be shared across a diverse collection of cancerous cell types and virally infected cells.
Alterations in the TRAIL:TRAIL receptor axis are known to impact outcomes of both infectious diseases, as well as cancer. For example, TRAIL receptor deficiency in mice increases influenza virus replication and morbidity [29], increases rates of lymphomagenesis, and enhances spread of certain malignancies [3–6, 30]. TRAIL axis inactivation in humans may have similar implications, as null mutations in TRAIL receptor genes have been linked to primary human cancers [31, 32]. Consistent with that model, TRAILshort antagonism of TRAIL mediated immune surveillance results has the following pathophysiologic correlates: elevated TRAILshort is associated with higher viral loads [11] and low levels of TRAILshort is associated with Elite control of HIV infection [14], and as we demonstrate herein using preclinical models, that TRAILshort impairs immune control of cancer.
Alternative splicing enhances the diversity of proteins expressed by a single gene often resulting in protein variants with disparate or opposing functions [33, 34]. Splice variants of genes that impact apoptosis, cell cycle and repair, cell-cell and cell-matrix interactions, have been associated with neoplasia and metastasis [35, 36]. For example, CD44 is normally involved in cell adhesion, migration, and cell-matrix interactions; however, CD44 splice variant 6 independently imparts metastatic potential to an otherwise non-metastatic pancreatic adenocarcinoma cell line [37, 38], and this effect can be inhibited by monoclonal antibodies targeting CD44 [39]. The observation of that TRAILshort impairs T cell control of tumors furthers the importance of alternate splicing in immune surveillance [40].
There is also ample precedent for soluble isoforms of membrane bound proteins altering biology. B cell Maturation antigen (BCMA) is a cell surface protein expressed universally on myeloma cells, and is required for optimum survival of plasma cells [41]. BCMA serves as a receptor for the cytokines BAFF and APRIL, which promote B cell growth, maturation and survival. Soluble BCMA exists in plasma of patients with multiple myeloma, where it binds circulating BAFF, thus preventing BAFF mediated normal B cell development and plasma cell survival [42]. Similarly soluble PD-1 (sPD-1) occurs due to alternative splicing [43], and sPD1 isoforms enhance T cell proliferation in vitro. In contrast, whereas membrane associated PDL1 binding to the PD1 receptor reduces T cell proliferation and inhibits apoptosis of regulatory T cells, soluble PDL1 (sPDL1) results from matrix metalloprotease mediated cleavage [44], and exposure of T cells to sPDL-1 causes their death by apoptosis [45]. Furthermore, high levels of sPDL-1 in sera are associated with impaired survival of patients with myeloma, lymphoma [46] and melanoma [47].
While no tumors that are TRAILshort negative are impacted by TRAILshort antibody, it is noteworthy that not all cell types that express TRAILshort become sensitized to TRAIL mediated killing following the addition of TRAILshort antibodies. This situation has parallels to the emerging observation that PDL1 positive tumors are not always sensitive to treatment with PDL1 inhibitors [48, 49]. From a biologic perspective it is likely that other factors which impact TRAIL signaling should modify the biologic response to TRAILshort inhibition, for example as the relative expression of death inducing versus decoy TRAIL receptors, levels of anti-apoptotic Bcl2 family members, and expression of different cFLIP isoforms, or IAP family members. Future work will explore these hypotheses at the molecular level.
Polymorphisms in TRAIL have been associated with an increased risk of lymphoma, suggesting a role for TRAIL in B-cell lymphomagenesis [50]. Consequently, the TRAIL pathway has been targeted in B-cell malignancies for example using TRAIL agonists in patients with non-Hodgkin lymphoma and multiple myeloma, as well as other malignancies, with uniformly disappointing results. Our results may shed light on a reason for these failures – failure to eliminate TRAIL receptor bearing tumors is not due to TRAIL deficiency (as often T cells from these patients have sufficient TRAIL expression) but rather there is an excess of TRAILshort which antagonizes the effect of endogenous TRAIL.
Our results establish proof of concept that both in vitro and in vivo, TRAILshort expressed by human cancers antagonizes the TRAIL:TRAIL receptor axis, that TRAILshort is a functional dominant negative TRAIL receptor ligand that inhibits the pro-apoptotic effects of full-length TRAIL, and finally that immune escape afforded by TRAILshort can be partially reversed by TRAILshort specific antibody.
Supplementary Material
Statement of Translational Relevance.
This work shows that primary human tumors express TRAILshort, which antagonizes tumor cell killing by immune effector cells, yet this restriction can be overcome by inhibiting TRAILshort with TRAILshort specific antibodies.
ACKNOWLEDGEMENTS
Some results presented here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. We would like to thank Eric C. Polley (Department of Biomedical Statistics and Informatics) for his assistance with data analysis and critical manuscript review.
Financial Support: This work was supported by grants AI110173 and AI120698 from NIAID, by P50CA97274 and P50CA102701 from the NCI.
Footnotes
Conflict of Interest:
One or more of the investigators associated with this project and Mayo Clinic have a Financial Conflict of Interest in technology used in the research and that the investigator(s) and Mayo Clinic may stand to gain financially from the successful outcome of the research and this research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and is being conducted in compliance with Mayo Clinic Conflict of Interest policies.
Disclosures unrelated to this work: SSK is inventor on patents in CART cell therapy that are licensed to Novartis (through an agreement between Mayo Clinic, University of Pennsylvania, and Novartis), Humanigen (through Mayo Clinic), and Mettaforge (through Mayo Clinic). SSK receives research funding from Kite, Gilead, Novartis, Juno, Celgene, Morphosys, Humanigen, Tolero, and Lentigen. ADB is a consultant and receives consulting fees or equity shares from Abbvie, Nference, and Xentalis. ADB is a founder and president of Splissen Therapeutics which has licensed patents related to TRAILshort.
REFERENCES
- 1.Walczak H, et al. , Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med, 1999. 5(2): p. 157–63. [DOI] [PubMed] [Google Scholar]
- 2.Mirandola P, et al. , Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood, 2004. 104(8): p. 2418–24. [DOI] [PubMed] [Google Scholar]
- 3.Sedger LM, et al. , Characterization of the in vivo function of TNF-alpha-related apoptosis-inducing ligand, TRAIL/Apo2L, using TRAIL/Apo2L gene-deficient mice. Eur J Immunol, 2002. 32(8): p. 2246–54. [DOI] [PubMed] [Google Scholar]
- 4.Zerafa N, et al. , Cutting edge: TRAIL deficiency accelerates hematological malignancies. J Immunol, 2005. 175(9): p. 5586–90. [DOI] [PubMed] [Google Scholar]
- 5.Cretney E, et al. , Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol, 2002. 168(3): p. 1356–61. [DOI] [PubMed] [Google Scholar]
- 6.Finnberg N, Klein-Szanto AJ, and El-Deiry WS, TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J Clin Invest, 2008. 118(1): p. 111–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeda K, et al. , Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med, 2001. 7(1): p. 94–100. [DOI] [PubMed] [Google Scholar]
- 8.Takeda K, et al. , Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med, 2002. 195(2): p. 161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bodmer JL, et al. , Cysteine 230 is essential for the structure and activity of the cytotoxic ligand TRAIL. J Biol Chem, 2000. 275(27): p. 20632–7. [DOI] [PubMed] [Google Scholar]
- 10.von Karstedt S, Montinaro A, and Walczak H, Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer, 2017. 17(6): p. 352–366. [DOI] [PubMed] [Google Scholar]
- 11.Schnepple DJ, et al. , Isolation of a TRAIL antagonist from the serum of HIV-infected patients. J Biol Chem, 2011. 286(41): p. 35742–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nie Z, et al. , Both HIV-Infected and Uninfected Cells Express TRAILshort, Which Confers TRAIL Resistance upon Bystander Cells within the Microenvironment. J Immunol, 2018. 200(3): p. 1110–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Natesampillai S, et al. , TRAILshort Protects against CD4 T Cell Death during Acute HIV Infection. J Immunol, 2019. 203(3): p. 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paim AC, et al. , HIV elite control is associated with reduced TRAILshort expression. AIDS, 2019. 33(11): p. 1757–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marinov GK, et al. , From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res, 2014. 24(3): p. 496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christoph S, et al. , Bioluminescence imaging of leukemia cell lines in vitro and in mouse xenografts: effects of monoclonal and polyclonal cell populations on intensity and kinetics of photon emission. J Hematol Oncol, 2013. 6: p. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fontan L, et al. , MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell, 2012. 22(6): p. 812–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Team RC, R: A language and environment for statistical computing. 2018, R Foundation for Statistical Computing: Vienna, Austria. [Google Scholar]
- 19.Cancer Genome Atlas Research, N., et al. , The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet, 2013. 45(10): p. 1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jouhi L, et al. , Toll-like receptor 5 and 7 expression may impact prognosis of HPV-positive oropharyngeal squamous cell carcinoma patients. Cancer Immunol Immunother, 2017. 66(12): p. 1619–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang X, Cheng Y, and Li C, The role of TLRs in cervical cancer with HPV infection: a review. Signal Transduct Target Ther, 2017. 2: p. 17055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehrhardt H, et al. , TRAIL induced survival and proliferation in cancer cells resistant towards TRAIL-induced apoptosis mediated by NF-kappaB. Oncogene, 2003. 22(25): p. 3842–52. [DOI] [PubMed] [Google Scholar]
- 23.Wiezorek J, Holland P, and Graves J, Death receptor agonists as a targeted therapy for cancer. Clin Cancer Res, 2010. 16(6): p. 1701–8. [DOI] [PubMed] [Google Scholar]
- 24.de Miguel D, et al. , Onto better TRAILs for cancer treatment. Cell Death Differ, 2016. 23(5): p. 733–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Most RG, et al. , Cyclophosphamide chemotherapy sensitizes tumor cells to TRAIL-dependent CD8 T cell-mediated immune attack resulting in suppression of tumor growth. PLoS One, 2009. 4(9): p. e6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorothee G, et al. , Tumor-infiltrating CD4+ T lymphocytes express APO2 ligand (APO2L)/TRAIL upon specific stimulation with autologous lung carcinoma cells: role of IFN-alpha on APO2L/TRAIL expression and -mediated cytotoxicity. J Immunol, 2002. 169(2): p. 809–17. [DOI] [PubMed] [Google Scholar]
- 27.Wiley SR, et al. , Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity, 1995. 3(6): p. 673–82. [DOI] [PubMed] [Google Scholar]
- 28.Jeremias I, et al. , TRAIL/Apo-2-ligand-induced apoptosis in human T cells. Eur J Immunol, 1998. 28(1): p. 143–52. [DOI] [PubMed] [Google Scholar]
- 29.Brincks EL, et al. , CD8 T cells utilize TRAIL to control influenza virus infection. J Immunol, 2008. 181(7): p. 4918–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson NS, et al. , Proapoptotic activation of death receptor 5 on tumor endothelial cells disrupts the vasculature and reduces tumor growth. Cancer Cell, 2012. 22(1): p. 80–90. [DOI] [PubMed] [Google Scholar]
- 31.Bin L, et al. , Tumor-derived mutations in the TRAIL receptor DR5 inhibit TRAIL signaling through the DR4 receptor by competing for ligand binding. J Biol Chem, 2007. 282(38): p. 28189–94. [DOI] [PubMed] [Google Scholar]
- 32.Shin MS, et al. , Mutations of tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1) and receptor 2 (TRAIL-R2) genes in metastatic breast cancers. Cancer Res, 2001. 61(13): p. 4942–6. [PubMed] [Google Scholar]
- 33.Lander ES, et al. , Initial sequencing and analysis of the human genome. Nature, 2001. 409(6822): p. 860–921. [DOI] [PubMed] [Google Scholar]
- 34.Modrek B and Lee C, A genomic view of alternative splicing. Nat Genet, 2002. 30(1): p. 13–9. [DOI] [PubMed] [Google Scholar]
- 35.Nissim-Rafinia M and Kerem B, Splicing regulation as a potential genetic modifier. Trends Genet, 2002. 18(3): p. 123–7. [DOI] [PubMed] [Google Scholar]
- 36.Philips AV and Cooper TA, RNA processing and human disease. Cellular and Molecular Life Sciences, 2000. 57(2): p. 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunthert U, et al. , A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell, 1991. 65(1): p. 13–24. [DOI] [PubMed] [Google Scholar]
- 38.Sneath RJ and Mangham DC, The normal structure and function of CD44 and its role in neoplasia. Mol Pathol, 1998. 51(4): p. 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo Y, et al. , Inhibition of human melanoma growth and metastasis in vivo by anti-CD44 monoclonal antibody. Cancer Res, 1994. 54(6): p. 1561–5. [PubMed] [Google Scholar]
- 40.Bejar R, Splicing Factor Mutations in Cancer. Adv Exp Med Biol, 2016. 907: p. 215–28. [DOI] [PubMed] [Google Scholar]
- 41.O’Connor BP, et al. , BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med, 2004. 199(1): p. 91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez E, et al. , Soluble B-Cell Maturation Antigen Mediates Tumor-Induced Immune Deficiency in Multiple Myeloma. Clin Cancer Res, 2016. 22(13): p. 3383–97. [DOI] [PubMed] [Google Scholar]
- 43.Nielsen C, et al. , Alternative splice variants of the human PD-1 gene. Cell Immunol, 2005. 235(2): p. 109–16. [DOI] [PubMed] [Google Scholar]
- 44.Chen Y, et al. , Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1+ cell lines. Cytokine, 2011. 56(2): p. 231–8. [DOI] [PubMed] [Google Scholar]
- 45.Frigola X, et al. , Soluble B7-H1: differences in production between dendritic cells and T cells. Immunol Lett, 2012. 142(1–2): p. 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossille D, et al. , High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: results from a French multicenter clinical trial. Leukemia, 2014. 28(12): p. 2367–75. [DOI] [PubMed] [Google Scholar]
- 47.Zhou J, et al. , Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol Res, 2017. 5(6): p. 480–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenkins RW, Barbie DA, and Flaherty KT, Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer, 2018. 118(1): p. 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Topalian SL, et al. , Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer, 2016. 16(5): p. 275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Travert M, et al. , CD40 ligand protects from TRAIL-induced apoptosis in follicular lymphomas through NF-kappaB activation and up-regulation of c-FLIP and Bcl-xL. J Immunol, 2008. 181(2): p. 1001–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Study Approval
All the human-derived samples were obtained in compliance with IRB-approved protocols (Mayo IRB #s 118–01 and 1827–00).