

SUMMARY
Three-dimensional (3D) visualization of vitrified cells can uncover structures of subcellular complexes without chemical fixation or staining. Here, we present a pipeline integrating three imaging modalities to visualize the same specimen at cryogenic temperature at different scales: cryo-fluorescence confocal microscopy, volume cryo-focused ion beam scanning electron microscopy, and transmission cryo-electron tomography. Our proof-of-concept benchmark revealed the 3D distribution of organelles and subcellular structures in whole heat-shocked yeast cells, including the ultrastructure of protein inclusions that recruit fluorescently-labelled chaperone Hsp104. Since our workflow efficiently integrates imaging at three different scales and can be applied to other types of cells, it could be used for large-scale phenotypic studies of frozen-hydrated specimens in a variety of healthy and diseased conditions with and without treatments.
Keywords: Airyscan microscopy, cryo correlative-light and electron microscopy (cryoCLEM), volume cryo-focused ion bean scanning electron microscopy (cryoFIB-SEM), cryo-electron tomography (cryoET), Hsp104 chaperone, protein aggregation, amyloid, protein misfolding, saccharomyces cerevisiae (yeast), cellular tomography
Graphical Abstract
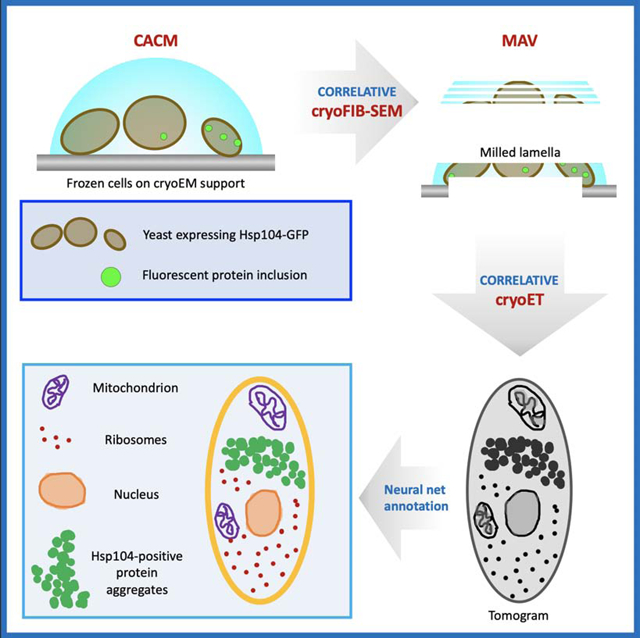
eTOC blurb
Wu, Mitchell & Galaz-Montoya et al. integrated three advanced cryogenic imaging modalities combining fluorescence, focused ion beam and electron microscopy to visualize protein aggregates and subcellular components in vitrified yeast cells under heat shock. Their pipeline can be applied to other types of cells in normal and diseased states.
INTRODUCTION
Visualizing cells in 3D is a powerful approach to study structures and interactions of organelles and macromolecular complexes. Among 3D imaging techniques for cell biology, cryogenic electron tomography (cryoET) is emerging as a leading approach for in situ structure determination (Beck and Baumeister, 2016). However, several limitations preclude its wider application, including the thickness of some samples and difficulties in locating and identifying features of interest within them. To visualize molecular details in thicker samples by cryoET, such as regions in eukaryotic cells away from the thin cell periphery, cryogenic focused ion beam scanning electron microscopy (cryoFIB-SEM) has been used to generate thin lamellae from vitrified cells (Marko et al., 2007, Hayles et al., 2007, Rigort et al., 2012, Strunk et al., 2012) (i.e., thin layers through the bulky cell) with a process called “ion beam milling”, enabling many exciting biological observations inside the cell (Hagen et al., 2015). However, technical challenges remain in ensuring that the milled lamellae contain the features of interest. Correlative light and electron microscopy (CLEM) can overcome this challenge by fluorescently labelling targets (Timmermans and Otto, 2015, Hampton et al., 2017), thereby guiding cryoFIB-SEM milling (Schertel et al., 2013, Gorelick et al., 2019) and the selection of optimal imaging areas for cryoET experiments, as well as aiding the interpretation of observed features. However, even the smallest eukaryotic cells are typically several microns thick and the desirable lamella thickness range is ~100–500 nm; thus, targeting nanoscale features along the z direction remains extremely challenging. Here, as a proof-of-principle, we present a pipeline to address this challenge by using high-resolution CryoAiryscan Confocal Microscopy (CACM) to determine the z position of fluorescent targets within cells that were vitrified on electron microscopy (EM) grids, followed by cryoFIB-SEM “mill and view” (cryoMAV) imaging, which provides a 3D view of whole cells with resolvable organelles as they are being milled to produce a lamella containing the target of interest, and ending with visualization of molecular details of regions of interest by cryoET.
RESULTS
Our 3D multi-scale correlative imaging pipeline integrates three different microscopy platforms with different resolution ranges and contrast mechanisms, operated at cryogenic temperature (Figure 1A,B, Supplementary Figure 1, & Supplementary Movie 1). To test our workflow, we applied all steps to the same vitrified Saccharomyces cerevisiae cells (yeast, ~5–10 μm in diameter) in which the chromosomal gene hsp104 coding for chaperone heat shock protein Hsp104 bears an in-frame C-terminal green fluorescent protein (GFP) tag (hereafter Hsp104-GFP). Integration of such tag does not affect Hsp104 functions nor cell viability (Specht et al., 2011, Malinovska et al., 2012, Sontag et al., 2017). Upon heat stress, Hsp104-GFP is recruited to inclusions that appear as bright puncta in fluorescence microscopy images (Sanchez de Jimenez et al., 1995).
Figure 1. “Mill and view” by cryoFIB-SEM of cryogenically-preserved yeast cells reveals the cellular context proximal to protein aggregates recruiting fluorescently-tagged HSP104.
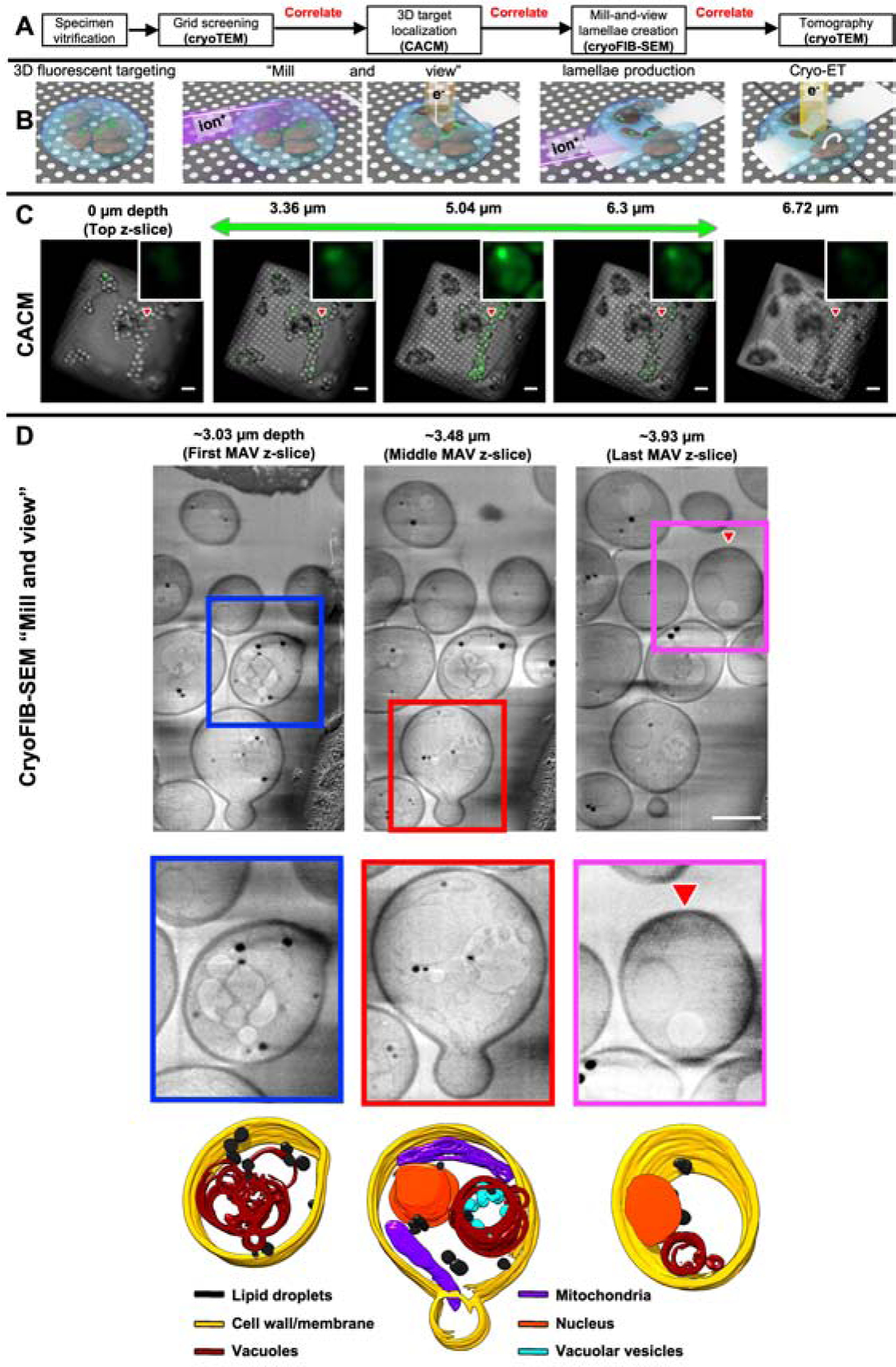
(A) Experimental workflow including multiple correlative imaging steps under cryogenic conditions via different imaging platforms. (B) Cartoon representation from left to right: vitrified yeast cells on an EM grid containing a tagged target; MAV-mode imaging by cryoFIB-SEM; lamella generation; cryoET imaging. (C) Representative CACM images showing slices through heat-shocked yeast cells expressing Hsp104-GFP on an EM grid, recorded at different z heights (scale bar=10 μm). The green signals come from our tagged target and the red triangle points to the location of a bright fluorescent punctum, indicative of a large inclusion containing Hsp104-GFP, highlighted in the zoomed-in insets. (D) Representative MAV images at different z heights (starting at ~3.36 μm depth from the cell surface, as guided by the CACM images in (C)). The colored boxes highlight individual yeast cells showcasing different contextual subcellular features, annotated in the bottom row, as labelled (scale bar=5 μm), and the red triangle points to densities at the location of the same punctum singled out in (C).
We first imaged our vitrified specimens using CACM. To determine the relative z-height of each region of interest (ROI) containing fluorescent puncta inside the yeast cells, we counted the number of z-slices from the top of the stack to the center of each fluorescent punctum in the CACM z-stacks. The punctum shown in Figure 1C became visible at ~2.94 μm depth from the top surface of the cell and persisted through an additional ~3.36 μm. We used the correlative software ZEN Connect (Zeiss) to overlay a CACM image of the ROI with a corresponding cryoFIB-SEM image and readily located the target punctum containing Hsp104-GFP in 3D within the achievable z-resolution of the optical microscope (Supplementary movie 2). We used this CACM data as a guide to mill away a large portion from the top of the cell (~3 μm) to approach the ROI indicated by the fluorescent inclusion. Then, we used the “serial milling block-face imaging” (Schertel et al., 2013, Vidavsky et al., 2016) mode of the cryoFIB-SEM to mill 31 finer slices (~30 nm thickness for each slice or a total of ~930 nm) and collected an SEM image of the freshly-exposed surface in each slice, a process hereafter referred to as “mill and view” (MAV) for frozen, hydrated specimens in a near-native state without exogenous heavy-metal stains. This imaging mode provided rich contextual structural information of our vitrified specimen above the region ultimately prepared as a lamella, thereby generating a 3D atlas of multiple whole yeast cells at intermediate resolution as we approached the target (Figure 1D). Our MAV images revealed abundant lipid droplets, vacuoles with vesicle-like features, mitochondria, cell nuclei, and daughter cells in the process of budding. Importantly, we minimized the electron beam dose to reduce radiation damage during milling to optimally preserve the structural integrity of the specimen for subsequent cryoET experiments. The time to carry out this entire process will depend on the experience of the investigators, the type of specimen and targets being visualized, and laboratory environmental conditions and experimental parameters. Supplementary Table S1 summarizes the average clock time and the rate of completion for each step to allow the execution of the next step in this workflow for the yeast specimens used in our present study. After extensive optimization of each step and experience, the time required for each experiment can be reduced with improved completion rates.
The correlation between CACM (Figure 2A) and cryoFIB-SEM MAV (Figure 2B) data in 3D using Zen Connect software (Zeiss) (Figure 2C) allowed to identify areas on the lamella (Figure 2D,E) with fluorescent puncta from which to collect cryoET tilt series. We also collected tilt series from areas with diffuse or no fluorescence as internal controls. We generated ~290–500 nm-thick lamellae to include as much of the 3D context proximal to the inclusions as permitted by the penetration power of the 300 keV transmission electron microscope. The tomograms from regions containing the bright puncta (i.e. Hsp104-GFP-positive inclusions) exhibited large and dense pleomorphic structures (Figure 2F), likely consisting of misfolded proteins. Interestingly, unlike some prior observations by traditional electron microscopy (Zhou et al., 2014), these inclusions did not appear as a continuous or completely amorphous mass but rather as a collection of pleomorphic compact granules with distinct boundaries. These granules were similar in size, and appeared to cluster in one specific region of the cell (Kaganovich et al., 2008), giving rise to inclusions that, with lower resolution microscopy approaches, may appear to be continuous structures.
Figure 2. CryoET of cryoFIB-SEM lamellae containing yeast cells with intracellular protein aggregates recruiting HSP104-GFP reveals that the unfolded proteins are compacted into globular densities.

(A). Z-directional, maximum intensity projection of CACM z-stack images of yeast cells expressing Hsp104-GFP. (B) The last cryoFIB-SEM MAV slice, right before creating the lamella. (C) Superposition between (A) and (B) to correlate fluorescent signals with areas seen by cryoFIB-SEM to select optimal regions on the lamella that both contain the target and are amenable for subsequent cryoET experiments. (D) Top and (E) slanted views of the final milled lamella (white arrow highlights an estimated thickness of ~260–300 nm). (F) Slice (~3 nm thick) through a representative cryoET tomogram collected from a region with a bright fluorescent punctum and corresponding annotation of features (bottom row) in different colors (green:inclusions containing Hsp104-GFP; yellow:a multivesicular body made of lipid membranes; pink:ribosomes. Scale bars: A-C, 5 μm; D,E 2.5 μm; F, 100 nm).
The fluorescence intensities seen by CACM (Figure 2A) correlated with the abundance and size of pleomorphic Hsp104-GFP-containing clusters in cryoET tomograms (Figure 2F, Figure 3, & Supplementary movie 2). Notably, tomograms from regions with diffuse GFP fluorescence also contained clusters made of pockets with a similar size and shape (Figure 3A,B) as those observed in the large inclusion (Figure 2F), but they were more sparsely distributed through the cytoplasm. On the other hand, tomograms from regions with no fluorescence (Figure 3C) or from control cells without heat shock lacked these clusters altogether. In addition to providing insight into the morphology of Hsp104-GFP clusters, the tomograms revealed ribosomes, multi-vesicular bodies (MVBs), mitochondria, vesicles budding into vacuoles, nuclei with nuclear pores, among other subcellular features, some of which we annotated semi-automatically using neural networks in EMAN2 (Chen et al., 2017) (bottom row in Figure 2 & 3, Supplementary movie 2).
Figure 3. CryoET of cryoFIB-SEM lamellae containing yeast cells with intracellular protein aggregates recruiting HSP104-GFP reveals different amounts of globular densities corresponding to compacted protein aggregates from cellular areas with different levels of fluorescence.

(A,B) Slices through representative cryoET tomograms collected from regions with diffuse fluorescence, and (C) an area with virtually no fluorescence, and corresponding annotation of features (bottom row) in different colors (green:inclusions containing Hsp104-GFP; yellow:cell wall and membranes, including a multivesicular body in (A) and (C) and nuclear membrane in (B); pink:ribosomes). The red arrows highlight holes in the nuclear membrane, corresponding to nuclear pores. Scale bars: 100 nm.
DISCUSSION
The goal of visualizing entire cellular atlases for cultured cells and tissues across resolutions is propelling the rapid development of cryoFIB technologies (Schaffer et al., 2019) and correlative approaches with multiple imaging techniques in structural biology. These advances are accelerating our ability to visualize intact cellular and subcellular structures in their native context to better understand their inter-relationships and the structural mechanisms underlying their functions. The formation of the Hsp104-GFP-positive clusters we observed here or similar structures may underlie the reduction in cytotoxic effects from protein misfolding by chaperones (Shahmoradian et al., 2013, Escusa-Toret et al., 2013, Darrow et al., 2015) and may be central to the reversibility of protein aggregation in heat-shocked yeast when cells are returned to normal temperatures (Wallace et al., 2015). Indeed, Hsp104 is known to solubilize some protein aggregates (Sweeny and Shorter, 2016) and to modulate prion propagation (Chernoff et al., 1995). More research is needed to determine whether the formation of these granules or their clustering is mediated through phase separation (Peskett et al., 2018). Furthermore, the workflow we present could also be applied to visualize the structural organization of prion aggregates with heterogenous compositions in yeast (Bagriantsev et al., 2008) at nanometer resolution and their spatial distributions with respect to other subcellular components to understand possible differences underlying the mechanisms of toxic variants (Bradley and Liebman, 2003).
A recent study (Schaffer et al., 2019) applied a cryo-correlative approach to generate lamella from a very thick bulk specimen, namely intact nematode cells with fluorescently-tagged centrosomes, cryogenically fixed with high-pressure freezing (HPF). The frozen specimens were cryo-planed to remove bulk frozen material, rendering them amenable to imaging by cryo-spinning disk confocal microscopy, which in turn allowed to position the target of interest within ~10 μm of the frozen surface, a requirement when performing a “cryo-lift-out”. Cryofluorescence images were overlaid with corresponding SEM block-face images by 2D correlation to determine the lateral position of the fluorescent target beneath the specimen surface and cryoET-usable lamellae were captured perpendicular to the optical imaging axis. While this challenging technique is an exciting development, it is not yet widely available and would provide no advantage if the intention of the experimentalist were to extract thicker volumes and apply cryoMAV during lamellae production to investigate the cellular context proximal to the labelled target(s). Additionally, image alignments of 3D CACM, cryoMAV, and/or cryoET datasets for correlation are more straightforward as all 3D datasets can be acquired in a nearly-parallel orientation. In our view, the two methods are complementary and, in principle the lift-out approach using HPF bulk samples is fully compatible with cryoMAV (Schertel et al., 2013, Vidavsky et al., 2016). Ultimately, HPF is appropriate when dealing with large bulk specimens, whereas plunge-freezing is suitable for sparsely-distributed, individual cells, which in principle can be as small as bacterial or synthetic cell systems.
While the examples above focused on demonstrating our guided workflow for Hsp104-GFP-positive inclusions at multiple scales (Supplementary Movie 2), many additional subcellular components were captured in our 3D “mill and view” cryoFIB-SEM (cryoMAV) and cryoET data such as vacuoles, lipid droplets, nuclei, ribosomes, mitochondria, and MVBs, among others (Supplementary Movie 3; the 3D CACM volume for the fluorescent target can be seen between seconds 2 and 3 of this movie, before the cryoFIB-SEM overlay), offering ample opportunity for observation-based discoveries. Indeed, we demonstrate that volume cryoMAV can directly visualize multiple vitrified cells at the same time at subcellular resolution, which should allow for large-scale, individual whole-cell imaging and quantification studies of cellular phenotypes under different conditions, as shown for bulk specimens (Schertel et al., 2013, Vidavsky et al., 2016, Khalifa et al., 2016), and recently for freeze-substituted, resin-embedded cells of various types, including neurons (Hoffman et al., 2020). In our work, knowing when to stop milling was facilitated by the guidance provided from the CACM data. In other potential applications, a priori knowledge about the target’s localization may serve to guide milling with a precision higher than that afforded by CACM; for example, if the target is known to be proximal to prominent subcellular features that are readily recognizable in cryoMAV images, these may serve as landmarks that can inform on where to stop milling. Indeed, mapping of fluorescent targets by 3D CACM and intermediate resolution and contrast cryoMAV images of proximal organelle reference points may allow to triangulate the location of structures smaller than allowed by CACM resolution limits alone. Further research is necessary to test such a hypothesis.
While our instrument selection at each step was influenced by availability, each of these instruments can potentially be substituted by other commercial or home-built instruments as long as the selected optics and detection systems are adequate for cryo work and the particular biological question and feature of interest. For instance, one could use a different light microscope for the CACM component of our pipeline, including cryo correlative single molecule super-resolution light and electron microscopy, which was recently demonstrated to be capable of identifying individual molecules within tens of nanometers in small bacterial cells without the use of cryoFIB-SEM milling (Dahlberg et al., 2020). Additionally, one must keep in mind that the required lateral resolution and optical section thickness and sensitivity of various optical imaging platforms will influence detectability and precision when targeting features of interest in lamellae.
Here, we have applied our proposed workflow to multiple grids from different cultures of yeast cells, demonstrating the pipeline’s reproducibility. Furthermore, while most institutions may not have all the instruments and expertise for the described experiments, our study demonstrates the feasibility of transferring the fragile milled lamellae across facilities thousands of miles apart (see Methods). These integrated, multi-scale imaging techniques could be applied to a wide variety of cell types to probe cellular and subcellular structures under normal, pathological, and treatment conditions.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wah Chiu, wahc@stanford.edu
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Representative tomogram containing an Hsp104-GFP-positive aggregate densities: EMD-21953. Raw CACM, cryoFIB-SEM, and cryoET data necessary to replicate our analyses can be made available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Engineering of yeast expressing GFP-tagged Hsp104 and cell culture
Endogenously-tagged Hsp104-GFP (S65T) yeast from the Yeast GFP Clone Collection (Huh et al., 2003) were grown at room temperature (28 °C) in YPD medium to an OD600 of 0.4. The culture was then incubated at 37 °C for 60 min before vitrification on cryoEM grids for the heat-shocked batch and at room temperature (28 °C) for control cells.
METHOD DETAILS
Sample preparation, vitrification, and pre-screening
Yeast cells were vitrified on Quantifoil 200 mesh gold TEM grids with R2/2 holey carbon film by plunge freezing using a Leica EMGP (5s back blotting) and clipped into ThermoFisher FIB AutoGrid rings modified for cryoFIB-SEM milling (referred to as “cryo-FIB AutoGrids”; Thermo Fisher part number 1205101). The clipped grids containing Hsp104-GFP yeast were pre-screened using a Talos Arctica transmission electron microscope (ThermoFisher) operated at 200 kV to evaluate grid integrity and ice thickness, and to identify suitable cells for subsequent experiments. Such screening can be easily done by using the grid montaging function of SerialEM software (Mastronarde, 2005). After a full-grid montage is assembled by SerialEM for each grid, the microscopist can rapidly determine which grids are suitable for subsequent cryoFIB-SEM milling. A “suitable grid”, as characterized in a transmission electron microscope, typically shows apparent cells along with areas where the holes in the carbon support film are visible, suggesting that the cells are sparsely distributed on the grid and that the ice is not too thick.
ZEISS cryoAiryscan confocal microscopy screening and subcellular fluorescence target localization
For each EM grid containing vitrified samples, the entire grid was imaged on a Zeiss LSM800 cryoAiryscan microscope equipped with the Linkam CMS196M cryo-stage (maintained at −196 °C) using the 5x EC Plan-Neofluar objective lens (NA 0.16) to acquire low magnification overview images and assess grid quality, sample flatness, and gross ice contamination. Samples were simultaneously imaged in transmitted and fluorescence modes. This allowed us to evaluate the density and distribution of the sample and ensure that the side of the grid containing the cells were cell-side up (facing the objective lens) while providing an overview image for later correlation and re-localization. A coarse z-stack in the reflected-light channel at low magnification (5x) allowed recognition of the “notch”, a directional machined cutout on the FIB AutoGrid used as a landmark to orient the grid and identify targets of interest in optimal areas for subsequent cryoFIB-SEM experiments (Schaffer et al., 2015). After assessing grid and sample conditions, we restricted imaging to the central region of the grid (most accessible with the gallium ion beam) at intermediate magnification in transmitted and reflected light, as well as Airyscan fluorescence to localize the targets of interest. Once we found suitable targets, we recorded their positions as ROIs in the Zen Blue (Zeiss imaging software). Next, the 100x LD EC Epiplan-Neofluar objective lens (NA 0.75) with a 4.1 mm working distance was used to collect 3D confocal z-stacks through the specimen using the Airyscan detector. The whole process took ~30 min for each grid, including the time for collecting screening images of the entire grid at 5X and confocal optical slices for 4 ROIs close to the central area of the grid.
This Airyscan capability provided high signal-to-noise, high-resolution images using long working-distance air immersion optics under cryo conditions (Huff, 2015), which facilitated the 3D localization of the targets during correlation across imaging techniques. A coordinate-based overview map with images overlaid as target locations was created using the ZEN Connect module of Zen Blue (Zeiss) and the data was transferred to the cryoFIB-SEM for correlation and target localization. Following Airyscan imaging, the cryo-stage was dismounted from the instrument, and the samples were cryogenically transferred under cryogenic conditions out of the cryo-stage in preparation for cryoFIB-SEM. All experiments up to this point were carried out at Stanford University and Stanford Linear Accelerator Center (SLAC), National Accelerator Laboratory (Stanford-SLAC).
Cryo focused ion beam and scanning electron microscopy (cryoFIB-SEM)
The clipped FIB AutoGrids with screened frozen samples were then shipped in a vapor cryogenic shipper (CX100, 3.7 L, Worthington Industries) from Stanford-SLAC to the Peterson Nanotechnology Materials Facility at the Massachusetts Institute of Technology (M.I.T.) via FedEx one-day shipping to proceed with cryoFIB-SEM experiments. Prior to loading of a sample, the cryo-stage and anti-contaminator (located within the FIB-SEM sample chamber) were pre-cooled to −150°C and −180°C, respectively, and allowed to equilibrate for 20–30 mins. The specimen grids were then mounted into the FIB-SEM cryo-shuttle (pre-tilted autogrid holder with cryo-shield) under liquid nitrogen in the Leica EM Vacuum Cryo Manipulation (VCM) system. This shuttle was then loaded into the Vacuum Cryo Transfer (VCT) system (Leica EM VCT500) for cryogenic transfer under vacuum into the chamber of the Zeiss Crossbeam 540 FIB-SEM. The samples were inserted through an airlock on a transfer rod to minimize contamination and maintain high vacuum in the FIB-SEM chamber.
Following transfer, the grids were coated with a ~2–3 μm-thick layer of platinum using the gas injection system (GIS). This helps to reduce charging of and damage to beam-sensitive samples during imaging and milling. For coating, the Pt-GIS was set to 25 °C and the stage was moved to −2 mm from the FIB/SEM coincidence point, before coating for ~1–2 min. After coating, the grids were screened using the SEM and the FIB. The SEM was used to screen the grids at 538X (0.21 μm/pixel), 2.0 KV, 3.2 pA beam current using the SESI detector to collect secondary electrons and the FIB at ~273X (409 nm/pixel), 30 kV, 50 pA beam current using the InLens secondary electron detector. The fluorescence maps from the cryoAiryscan were loaded into the ZEN Connect software (Zeiss) and aligned to these SEM and FIB images, allowing for navigation and target selection. The InLens detector allowed for higher-resolution and higher-contrast imaging of unstained samples (Griffin, 2011). Since the samples are very sensitive to gallium ion beam damage, we only used the FIB to image for beam alignment.
Coarse targeting in z with CACM correlation
The ability to recognize ROIs in all three imaging modalities is helpful for fast and accurate correlation. Features that can be localized in z are particularly valuable, because they can guide where to stop milling. In our case, the pleomorphic clusters correlating with puncta were visible in MAV and low-mag EM images. Nyquist sampling was used to set the z-interval between every slice in CACM to be ~0.42 μm, which is typically 2–3x finer than the microscope’s optical resolution in z. As we imaged pre-selected ROIs in z-stacks, we used the z-interval and determined the number of z-slices from the top surface of yeast cells before arriving at bright puncta corresponding to Hsp104-GFP signals. Though the cryoFIB-SEM milling slices were much thinner than the z-resolution of CACM, the latter technique provided an informed estimate for how much material to remove during coarse milling. Note that the milling angle was not accounted for during the coarse targeting in z.
The overview images taken using the SEM and FIB in combination with the correlated cryoAiryscan data were used to target ROIs using Zen Connect software. The stage was moved to the center of the ROI and tilted to ~15–17° to allow for a milling angle of ~6–8° relative to the grid. Coarse milling was carried out at a pixel size of 39.19 nm, 30 kV, and 700 pA. For monitoring the milling, the SEM was used with a pixel size of 20–50 nm while the voltage and beam currents were 2 kV and 25 pA, respectively. The coarse milling step to remove ~3 μm from the top surface of the yeast cell took ~10–15 min.
Refinement of z-targeting using cryoFIB-SEM “mill and view”
We used cryo “mill and view” (cryoMAV) to further refine the lamella milling boundaries in z, and to obtain contextual 3D information about the regions of the sample that are removed during lamella generation. In general, cryoMAV uses a FIB to remove a thin layer of sample, as thin as ~3 nm (Wei et al., 2012), and images the milled surface with SEM before removing the next thin layer. In our workflow, this procedure can prevent both insufficient and over-milling so long as 1) the puncta are not ablated by coarse milling (this becomes increasingly challenging the smaller the target, given the large step size in CACM stacks), 2) the targets have a depth larger than the cryoMAV slicing interval, and 3) densities corresponding to the targets are recognizable in the cryoFIB-SEM images. CryoMAV records general contextual information of the material removed, allowing us to evaluate cellular features and their positions with respect to ROIs. This imaging technique may find useful applications in large-scale, intermediate-resolution phenotype analyses of sparsely-distributed, and relatively small vitrified cells in normal, diseased, and treatment conditions, as shown previously for bulk tissue samples and organisms (Vidavsky et al., 2016, Khalifa et al., 2016).
The cryoMAV SEM imaging settings were 2.96 KX (pixel size=12.56 nm), 2.00 kV, 20 pA, using line integration for noise reduction and an inLens detector, with 30 kV and 50 pA for the focused ion beam and a z step size of 30 nm. The cryoMAV step generally took ~1–1.5 h. The cryoMAV images were aligned using linear stack alignment with SIFT (Fiji). Subsequently, the images were processed using de-striping, CLAHE, and non-local means filters (ORS Dragonfly) to enhance the contrast. Cellular features were manually segmented using Amira (Stalling et al., 2005) and visualized using UCSF Chimera (Pettersen et al., 2004).
Final lamella generation
After coarse milling and cryoMAV of the top surface of an ROI, we also milled away material from the bottom side to create the final lamella, which took ~15–20 min. We set the FIB to 30 kV and 700 pA for coarse milling to create ~2 μm-thick lamella, reduced the current to ~300 pA to thin the lamella to ~1 μm, and to ~100 pA to create the final ~290–500 nm thick lamellae. Finally, we “polished” both sides of the lamellae with a lower current (50 pA). On average, it took ~2–3 h to prepare a single lamella for our yeast specimens including cryoMAV. In comparison, standard lamella production takes ~1 h per lamella without cryoMAV, although recent methodological developments and automation offer the potential of reducing lamella production time to ~25 min per lamella (Zachs et al., 2020) and to run cryoFIB-SEM milling for lamella production overnight (Tacke et al., 2020).
Correlating cryoAiryscan and FIB-SEM images with cryoET views
The polished lamellae were obtained with correlative coordinates between the CACM and cryoFIB-SEM images that revealed the positions of aggregates containing Hsp104-GFP. CryoSEM images of polished lamellae show fewer details than cryoMAV images because they are taken with a lower electron dose to minimize electron damage, preserving as much information as possible for subsequent cryoET data collection; however, large aggregates containing Hsp104-GFP are identifiable in low-mag cryoEM screening images. The final lamellae were shipped in a Dry Shipper container (CXR100, 3.7 L, Worthington Industries) from the M.I.T. to the cryoET imaging facilities at Stanford-SLAC via FedEx one-day shipping to proceed with tilt series collection. The fragile samples appeared to be well-preserved through this transportation process, demonstrating that the multi-scale correlative imaging can be carried out with instrumentation in multiple facilities.
CryoET tilt series data collection, tomographic reconstruction, annotation, and subtomogram averaging
We collected tilt series of lamellae containing vitrified yeast cells with (n=17) and without (n=34) heat shock using a Titan Krios electron microscope (ThermoFisher) equipped with a BioQuantum Energy Filter (set to 20 eV) and operated at 300 kV in low-dose mode using SerialEM software (Mastronarde, 2005). At each tilt angle, we recorded “movies” with 5 frames each using a K2 camera (Gatan). The tilt series were collected bi-directionally from 25° to −60°, and then from 26° to +60° with a tilt step of 1°, target defocus of −8 μm and a cumulative dose of ~100 e/Å2. The images were motion-corrected with MotionCor2 software (Zheng et al., 2017). We used the IMOD software package (Kremer et al., 1996) for standard weighted-back projection tomographic reconstruction after patch-tracking alignment. Within each tilt series, unusable images (large drift, excessive ice contamination, etc.) were removed prior to tilt series alignment. The data were contrast transfer function (CTF) corrected with IMOD’s newly implemented 3D-CTF correction algorithm. Parallel reconstructions using a SIRT-like filter were computed for visualization purposes using heavily binned data (shrink factor=4) and post-reconstruction filters (lowpass, highpass, normalization, and thresholding at 3 standard deviations away from the mean). We carried out tomographic annotation of different features in the binned-by-4 tomograms using EMAN2’s semi-automated 2D neural network-based pipeline (Zheng et al., 2017), and performed manual clean-up of false positives in UCSF Chimera, which we also used to display all annotated densities and prepare the cryoET movies.
QUANTIFICATION AND STATISTICAL ANALYSIS
The development of this methodological pipeline requires no statistical analyses. Table S1 summarizes the average clock time and the rate of completion of each step to allow the execution of the next step in this workflow for the yeast specimens used. This was simply the number of trials that were completed without problems, over the total number of attempted trials.
Supplementary Material
Supplementary Movie 1. Conceptual depiction of multi-scale 3D cryo-correlative microscopy workflow, related to Figures 1A. Cartoon depiction of vitrified yeast cells containing a fluorescently labelled inclusion and imaged by “cryo mill and view” (cryoMAV) cryoFIB-SEM, followed by lamella production, and cryoET imaging.
Supplementary Movie 2. Experimental demonstration of multi-scale 3D cryo-correlative microscopy workflow, related to Figures 1, 2, & 3. This movie illustrates our entire multi-scale 3D cryo-correlative workflow with an experimental example of data from vitrified heat-shocked yeast cells expressing the endogenous protein chaperone Hsp104 bearing a C-terminal GFP fusion tag, imaged with CACM, followed by cryoFIB-SEM MAV and lamella generation, and ending with cryoET and annotation of features.
Supplementary Movie 3. Overlay of data from all steps in multi-scale 3D cryo-correlative workflow, Figures 1, 2, & 3. Superposition of 3D data and corresponding annotations derived from the visualization of frozen-hydrated heat-shocked yeast cells expressing Hsp104-GFP with three different imaging instruments and modalities. The data include a representative CACM volume stack (fluorescent green signals), cryoFIB-SEM MAV volume stack, and cryoET tomogram.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Tomogram in EMDB | This paper; @https://www.ebi.ac.uk/pdbe/emdb/ | EMD-21953 |
| Experimental Models: Organisms/Strains | ||
| Saccharomyces cerevisiae strain S65T | The Yeast GFP Clone Collection | S65T |
| Software and Algorithms | ||
| ZEN Connect module of Zen Blue | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html | N/A |
| SerialEM | https://bio3d.colorado.edu/SerialEM/ | N/A |
| IMOD | https://bio3d.colorado.edu/imod/ | N/A |
| Amira | https://www.thermofisher.com/us/en/home/industrial/electron-microscopy/electron-microscopy-instruments-workflow-solutions/3d-visualization-analysis-software/amira-life-sciences-biomedical.html | N/A |
| UCSF Chimera | https://www.cgl.ucsf.edu/chimera/ | N/A |
| EMAN2 | https://blake.bcm.edu/emanwiki/EMAN2 | N/A |
2–4 highlights.
3D imaging correlating 3 cryogenic microscopy techniques for cell structure studies
Same subcellular features in vitrified yeast visualized across resolution scales
Protein aggregates form compact pleomorphic globular structures in heat-shocked yeast
ACKNOWLEDGEMENTS
We thank Profs. Leslie Thompson and William Mobley for many helpful discussions, and Drs. Mei-Kuang Chen and Michael F. Schmid for their feedback on the manuscript, as well as Dr. David Mastronarde for extensive advice (and debugging) in using the latest 3D-CTF correction tools in IMOD.
Funding sources
This research is supported by grants from the U.S. National Institutes of Health (No. P01NS092525 to WC and JF, PO1AG054407 to JF, Nos. 5P41GM103832, R01AI148382 and S10OD021600 to WC) and the Department of Energy (No. BERFWP 100463 to WC), a postdoctoral fellowship from the Hereditary Disease Foundation, to GHW, and a postdoctoral fellowship from the U.S. National Institutes of Health (No. F32NS086253) to EMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
VG and JM are employed by Zeiss Research Microscopy Solutions. All other authors have no conflicts of interest to declare.
REFERENCES
- Bagriantsev SN, Gracheva EO, Richmond JE & Liebman SW (2008). Variant-specific [PSI+] infection is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition. Mol Biol Cell 19, 2433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M & Baumeister W (2016). Cryo-electron tomography: can it reveal the molecular sociology of cells in atomic detail? Trends in cell biol, 26, 825–837. [DOI] [PubMed] [Google Scholar]
- Bradley ME & Liebman SW (2003). Destabilizing interactions among [PSI(+)] and [PIN(+)] yeast prion variants. Genetics, 165, 1675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Dai W, Sun SY, Jonasch D, He CY, Schmid MF, Chiu W & Ludtke SJ (2017). Convolutional neural networks for automated annotation of cellular cryo-electron tomograms. Nat Methods, 14, 983–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG & Liebman SW (1995). Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science, 268, 880–4. [DOI] [PubMed] [Google Scholar]
- Dahlberg PD, Perez D, Su Z, Chiu W & Moerner WE (2020). Cryogenic correlative single-particle photoluminescence spectroscopy and electron tomography for investigation of nanomaterials. Angew Chem Int Ed Engl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow MC, Sergeeva OA, Isas JM, Galaz-Montoya JG, King JA, Langen R, Schmid MF & Chiu W (2015). Structural mechanisms of mutant huntingtin aggregation suppression by the synthetic chaperonin-like cct5 complex explained by cryoelectron tomography. J Biol Chem, 290, 17451–17461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escusa-Toret S, Vonk WI & Frydman J (2013). Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat Cell Biol, 15, 1231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick S, Buckley G, Gervinskas G, Johnson TK, Handley A, Caggiano MP, Whisstock JC, Pocock R & De Marco A (2019). PIE-scope, integrated cryo-correlative light and FIB/SEM microscopy. Elife, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin BJ (2011). A comparison of conventional Everhart-Thornley style and in-lens secondary electron detectors—a further variable in scanning electron microscopy. Scanning, 33, 162–173. [DOI] [PubMed] [Google Scholar]
- Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bauerlein FJ, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC & Grunewald K (2015). Structural basis of vesicle formation at the inner nuclear membrane. Cell, 163, 1692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton CM, Strauss JD, Ke Z, Dillard RS, Hammonds JE, Alonas E, Desai TM, Marin M, Storms RE, Leon F, Melikyan GB, Santangelo PJ, Spearman PW & Wright ER (2017). Correlated fluorescence microscopy and cryo-electron tomography of virus-infected or transfected mammalian cells. Nat Protoc, 12, 150–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayles MF, Stokes DJ, Phifer D & Findlay KC (2007). A technique for improved focused ion beam milling of cryo-prepared life science specimens. J microsc, 226, 263–269. [DOI] [PubMed] [Google Scholar]
- Hoffman DP, Shtengel G, Xu CS, Campbell KR, Freeman M, Wang L, Milkie DE, Pasolli HA, Iyer N, Bogovic JA, Stabley DR, Shirinifard A, Pang S, Peale D, Schaefer K, Pomp W, Chang CL, Lippincott-Schwartz J, Kirchhausen T, Solecki DJ, Betzig E & Hess HF (2020). Correlative three-dimensional super-resolution and block-face electron microscopy of whole vitreously frozen cells. Science, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J (2015). The Airyscan detector from ZEISS: confocal imaging with improved signal-to-noise ratio and super-resolution. Nat Methods, 12, i–ii. [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS & O’shea EK (2003). Global analysis of protein localization in budding yeast. Nature, 425, 686–91. [DOI] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R & Frydman J (2008). Misfolded proteins partition between two distinct quality control compartments. Nature, 454, 1088–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalifa GM, Kirchenbuechler D, Koifman N, Kleinerman O, Talmon Y, Elbaum M, Addadi L, Weiner S & Erez J (2016). Biomineralization pathways in a foraminifer revealed using a novel correlative cryo-fluorescence-SEM-EDS technique. J Struct Biol, 196, 155–163. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN & Mcintosh JR (1996). Computer visualization of three-dimensional image data using IMOD. J Struc Biol, 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Malinovska L, Kroschwald S, Munder MC, Richter D & Alberti S (2012). Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Mol Biol Cell, 23, 3041–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko M, Hsieh C, Schalek R, Frank J & Mannella C (2007). Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nat Methods, 4, 215–7. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol, 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Peskett TR, Rau F, O’driscoll J, Patani R, Lowe AR & Saibil HR (2018). A liquid to solid phase transition underlying pathological huntingtin exon1 aggregation. Mol Cell, 70, 588–601 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC & Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem, 25, 1605–12. [DOI] [PubMed] [Google Scholar]
- Rigort A, Bäuerlein FJ, Villa E, Eibauer M, Laugks T, Baumeister W & Plitzko JM (2012). Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proceedings of the National Academy of Sciences USA, 109, 4449–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez De Jimenez E, Medrano L & Martinez-Barajas E (1995). Rubisco activase, a possible new member of the molecular chaperone family. Biochem, 34, 2826–31. [DOI] [PubMed] [Google Scholar]
- Schaffer M, Engel BD, Laugks T, Mahamid J, Plitzko JM & Baumeister W (2015). Cryo-focused ion beam sample preparation for imaging vitreous cells by cryo-electron tomography. Bio Protoc, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer M, Pfeffer S, Mahamid J, Kleindiek S, Laugks T, Albert S, Engel BD, Rummel A, Smith AJ & Baumeister W (2019). A cryo-FIB lift-out technique enables molecular-resolution cryo-ET within native Caenorhabditis elegans tissue. Nat Methods, 16, 757–762. [DOI] [PubMed] [Google Scholar]
- Schertel A, Snaidero N, Han HM, Ruhwedel T, Laue M, Grabenbauer M & Mobius W (2013). Cryo FIB-SEM: volume imaging of cellular ultrastructure in native frozen specimens. J Struct Biol, 184, 355–60. [DOI] [PubMed] [Google Scholar]
- Shahmoradian SH, Galaz-Montoya JG, Schmid MF, Cong Y, Ma B, Spiess C, Frydman J, Ludtke SJ & Chiu W (2013). TRiCs tricks inhibit huntingtin aggregation. eLife, 2, e00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag EM, Samant RS & Frydman J (2017). Mechanisms and functions of spatial protein quality control. Annu Rev Biochem, 86, 97–122. [DOI] [PubMed] [Google Scholar]
- Specht S, Miller SB, Mogk A & Bukau B (2011). Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J Cell Biol, 195, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalling D, Westerhoff M & Hege H-C (2005). Amira: A highly interactive system for visual data analysis. The visualization handbook, 38, 749–67. [Google Scholar]
- Strunk KM, Wang K, Ke D, Gray JL & Zhang P (2012). Thinning of large mammalian cells for cryo-TEM characterization by cryo-FIB milling. J Microsc, 247, 220–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeny EA & Shorter J (2016). Mechanistic and structural insights into the prion-disaggregase activity of Hsp104. J Mol Biol, 428, 1870–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke S, Erdmann P, Wang Z, Klumpe S, Grange M, Kriz R, Dolník M, Mitchels J, Plitzko J & Raunser S (2020). A streamlined workflow for automated cryo focused ion beam milling. bioRxiv. [DOI] [PubMed] [Google Scholar]
- Timmermans FJ & Otto C (2015). Contributed review: Review of integrated correlative light and electron microscopy. Rev Sci Instrum, 86, 011501. [DOI] [PubMed] [Google Scholar]
- Vidavsky N, Akiva A, Kaplan-Ashiri I, Rechav K, Addadi L, Weiner S & Schertel A (2016). Cryo-FIB-SEM serial milling and block face imaging: Large volume structural analysis of biological tissues preserved close to their native state. J Struct Biol, 196, 487–495. [DOI] [PubMed] [Google Scholar]
- Wallace EW, Kear-Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM, Airoldi EM, Pan T, Budnik BA & Drummond DA (2015). Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell, 162, 1286–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D, Jacobs S, Modla S, Zhang S, Young CL, Cirino R, Caplan J & Czymmek K (2012). High-resolution three-dimensional reconstruction of a whole yeast cell using focused-ion beam scanning electron microscopy. Biotechniques, 53, 41–8. [DOI] [PubMed] [Google Scholar]
- Zachs T, Schertel A, Medeiros J, Weiss GL, Hugener J, Matos J & Pilhofer M (2020). Fully automated, sequential focused ion beam milling for cryo-electron tomography. Elife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y & Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods, 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Slaughter BD, Unruh JR, Guo F, Yu Z, Mickey K, Narkar A, Ross RT, Mcclain M & Li R (2014). Organelle-based aggregation and retention of damaged proteins in asymmetrically dividing cells. Cell, 159, 530–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1. Conceptual depiction of multi-scale 3D cryo-correlative microscopy workflow, related to Figures 1A. Cartoon depiction of vitrified yeast cells containing a fluorescently labelled inclusion and imaged by “cryo mill and view” (cryoMAV) cryoFIB-SEM, followed by lamella production, and cryoET imaging.
Supplementary Movie 2. Experimental demonstration of multi-scale 3D cryo-correlative microscopy workflow, related to Figures 1, 2, & 3. This movie illustrates our entire multi-scale 3D cryo-correlative workflow with an experimental example of data from vitrified heat-shocked yeast cells expressing the endogenous protein chaperone Hsp104 bearing a C-terminal GFP fusion tag, imaged with CACM, followed by cryoFIB-SEM MAV and lamella generation, and ending with cryoET and annotation of features.
Supplementary Movie 3. Overlay of data from all steps in multi-scale 3D cryo-correlative workflow, Figures 1, 2, & 3. Superposition of 3D data and corresponding annotations derived from the visualization of frozen-hydrated heat-shocked yeast cells expressing Hsp104-GFP with three different imaging instruments and modalities. The data include a representative CACM volume stack (fluorescent green signals), cryoFIB-SEM MAV volume stack, and cryoET tomogram.
Data Availability Statement
Representative tomogram containing an Hsp104-GFP-positive aggregate densities: EMD-21953. Raw CACM, cryoFIB-SEM, and cryoET data necessary to replicate our analyses can be made available upon request.