

Abstract
We investigated the role of PRMT5 in MPN pathogenesis and aimed to elucidate key PRMT5 targets contributing to MPN maintenance.
PRMT5 is overexpressed in primary MPN cells and PRMT5 inhibition potently reduced MPN cell proliferation ex vivo. PRMT5 inhibition was efficacious at reversing elevated hematocrit, leukocytosis and splenomegaly in a model of JAK2V617F+ polycythemia vera (PV) and leukocyte and platelet counts, hepatosplenomegaly and fibrosis in the MPLW515L model of myelofibrosis (MF). Dual targeting of JAK and PRMT5 was superior to JAK or PRMT5 inhibitor monotherapy, further decreasing elevated counts and extramedullary hematopoiesis in vivo.
PRMT5 inhibition reduced expression of E2F targets and altered the methylation status of E2F1 leading to attenuated DNA damage repair, cell cycle arrest and increased apoptosis.
Our data link PRMT5 to E2F1 regulatory function and MPN cell survival and provide a strong mechanistic rationale for clinical trials of PRMT5 inhibitors in MPN.
Keywords: PRMT5, MPN, JAK2V617F
Introduction
Protein arginine methylation is a key post-translational protein modification which is catalyzed by the family of protein arginine methyl transferases (PRMTs)(1). These enzymes transfer methyl groups, provided by the cofactor S-adenosyl-methionine (SAM), to arginine residues on histones and on non-histone proteins(2). PRMT activity can result either in asymmetric dimethyl arginine (ADMA) or symmetric dimethyl arginine (SDMA). PRMTs are thus classified as either class I (ADMA) or class II (SDMA) methyltransferases(2–4). By methylating arginine residues on histones, PRMTs act as epigenetic writers whose interplay with epigenetic readers(5) and erasers leads to modification of the histone code that contributes to the regulation of transcription, DNA replication and DNA repair. Arginine methylation of non-histone proteins has also been implicated in cell signaling, ribosome biogenesis, RNA transport and splicing(6,7).
PRMT5 is the primary type II PRMT responsible for symmetric dimethylation of arginine residues and has a known role in normal development and an emerging role in different malignant contexts. PRMT5 knockout (KO) mice are not viable(8) and conditional PRMT5-KO mice show alterations in hematopoiesis(9) suggesting that PRMT5 activity has a key role in development and hematopoiesis. Dysregulation of PRMT5 occurs in a variety of malignancies and is associated with increased cancer cell proliferation and dismal prognosis(7,10–12). The relationship between methylthioadenosine phosphorylase (MTAP) allelic status/function and PRMT5 activity has suggested PRMT5 as a potential therapeutic target in different malignancies(13,14), and recent data linking PRMT5 function to the spliceosome(15,16) has further highlighted the potential of PRMT5 inhibition as a cancer therapeutic strategy. As such, therapeutic agents targeting PRMT5 have entered early clinical trials (NCT02783300, NCT03854227, NCT03573310, NCT03886831, NCT04089449).
Myeloproliferative Neoplasms (MPN) are clonal myeloid malignancies initiated by somatic mutations in hematopoietic stem cells (HSC)(17). The most common BCR-ABL negative MPN are polycythemia vera (PV), essential thrombocythemia (ET) and myelofibrosis (MF). These diseases are characterized by overproduction of terminally differentiated blood cells along with an increased bleeding and thrombosis risk, and increased risk of transformation to bone marrow failure and/or acute leukemia. Genetic profiling has shown that the most common mutations in MPN result in dysregulated JAK-STAT signaling, most commonly JAK2V617F, across all three diseases(18–21). Based on these discoveries, the JAK1/2 inhibitor ruxolitinib was developed for MPN therapy and approved for the treatment of MF in 2011 and PV in 2014 and a selective JAK2 inhibitor fedratinib has recently been approved for treatment of MF. Although ruxolitinib therapy results in a substantial reduction in constitutive symptoms and spleen size (22–25), JAK inhibitor therapy with ruxolitinib or other agents in clinical development does not reduce mutant allele burden or provide disease modification. This underscores the need to explore different therapeutic targets in MPN either as monotherapy or in combination with JAK kinase inhibitors.
In addition to mutations in the JAK-STAT pathway, genomic studies have identified recurrent disease alleles in epigenetic modifiers in myeloid malignancies, including MPN (26–28). These data underscore the relevance of epigenetic alterations to MPN pathogenesis and suggest a potential role for epigenetic therapies in MPN. Previous studies suggested a functional link between constitutive JAK2 activity, PRMT5 phosphorylation, and MPN pathogenesis(29), however the therapeutic potential of PRMT5 inhibition in MPN has not been studied. We investigated the role of PRMT5 in MPN pathogenesis through the use of C220, a potent and selective, small molecule PRMT5 inhibitor and aimed to elucidate key PRMT5 targets contributing to the maintenance of MPN cells.
Results
PRMT5 is overexpressed in MPN and its inhibition by C220 is anti-proliferative in vitro and in patient-derived specimens
To assess the requirement for PRMT5 in cells harboring a JAK2V617F mutation, we performed in vitro proliferation assays with different concentrations of C220 – a potent and highly selective small molecule PRMT5 inhibitor (Figures S1A, S1B, S1C). C220 treatment caused a dose-dependent inhibition of proliferation in Ba/F3-EpoR cells expressing JAK2V617F, with a 2.5-fold lower IC50 compared to cells expressing JAK2WT or empty vector (Figure 1A). C220 dose-dependently reduced the proliferation of the JAK2V617F mutant cell lines SET2, UKE1 and HEL in vitro. (Figure 1B). We previously developed models of JAK inhibitor “persistent” MPN cells which survive in the setting of prolonged exposure to ruxolitinib and other JAK kinase inhibitors (30,31). To address if ruxolitinib persistent cells (Figure S2A) could be targeted by PRMT5 inhibition, we performed proliferation assays in ruxolitinib persistent SET2 cells compared to naïve SET2 cells and found similar sensitivity to C220 (Figure 1C). C220 exposure for 6 days did not alter phosphorylation of JAK2, STAT5, AKT or ERK in SET2 cells at doses which reduced SDMA, indicative of target inhibition (Figure 1D). These data suggest a distinct mechanism by which PRMT5 inhibition impacts the survival of MPN cells. Consistent with these data, combined PRMT5 inhibition with JAK1/2 inhibition showed additive, but not synergistic effects as quantified by a ZIP synergy score of 5.3 for the JAK2V617F Ba/F3 and 3.7 for the SET2 cells (Figures 1E, 1F, S2B, S2C). No additive effects were observed with the combination in JAK2WT Ba/F3 cells (Figures S2D, S2E).
Figure 1: Inhibition of PRMT5 by C220 shows efficacy in vitro and ex vivo.
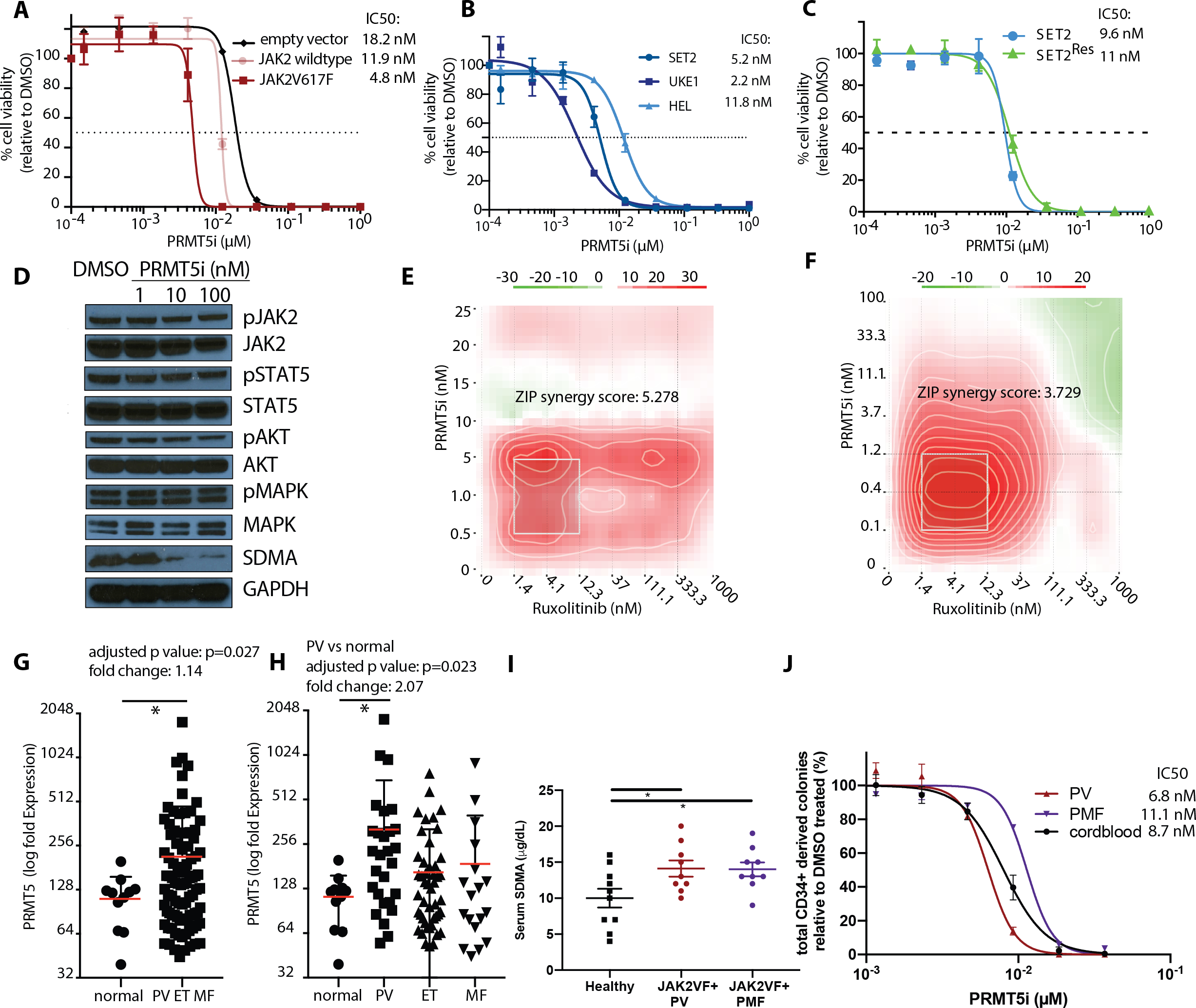
(A) Proliferation with increasing concentration of C220 (μM) relative to DMSO is depicted for Ba/F3 cells transduced with Jak2V617F, Jak2-wt or empty vector. IC50 values for C220 are indicated on the right. Data are indicated as mean ± SEM.
(B) Proliferation with increasing concentration of C220 (μM) relative to DMSO is depicted for SET2, UKE1 and HEL cells harboring either heterozygous (SET2) or homozygous (UKE1, HEL) Jak2V617F mutations, respectively. IC50 values for C220 are indicated on the right. Data of indicated as mean ± SEM..
(C)Proliferation with increasing concentration of C220 (μM) relative to DMSO is depicted for SET2 and Ruxolitinib-resistant SET2 cells (SET2Res). IC50 values for C220 are indicated on the right. Data of indicated as mean ± SEM..
(D) Western Blot assessment of JAK2, STAT5, AKT and MAPK phosphorylation and signaling in SET2 cells treated for 6 days with increasing concentrations of C220 (nM).
(E) Dose-response matrix from proliferation synergy assay for the combination of PRMT5 inhibition by C220 and Jak1/2 inhibition by ruxolitinib in Jak2V617F transduced Ba/F3 cells is visualized as a heatmap..
(F) Dose-response matrix from proliferation synergy assay for the combination of PRMT5 inhibition by C220 and JAK1/2 inhibition by ruxolitinib in SET2 cells is visualized as a heatmap..
(G) PRMT5 expression profiling in peripheral blood granulocytes from 93 MPN patients is compared to 11 healthy controls. Analysis of publicly available data (32). (*p<0.05).
(H) PRMT5 expression profiling of in peripheral blood granulocytes from 28 PV, 47 ET, 18 MF is compared to 11 healthy controls. Analysis of publicly available data (32). (*p<0.05)..
(I) Assessment of serum SDMA levels in patients with JAK2V617F mutated PV (n=9) and JAK2V617F mutated PMF (n=9) compared to healthy controls (n=10) (*p<0.05 each, respectively)..
(J) Proliferation with increasing concentration of C220 (μM) relative to DMSO is depicted for CD34+ cells from JAK2V617F mutated PV and PMF patients compared to wild-type CD34+ cells derived from cord blood (n=3). IC50 values for C220 are indicated on the right. Data are indicated as mean ± SEM.
To further address the relevance of PRMT5 function to MPN pathogenesis we assessed PRMT5 expression in human peripheral blood granulocytes using previously published RNA expression profiling data(32). 93 patients with MPN displayed a median PRMT5 expression that was significantly higher compared to 11 age matched normal subjects (p=0.027; Figure 1G). PRMT5 expression was significantly higher in 28 PV patients compared to healthy controls (p=0.023) and showed a trend towards higher expression in 18 MF and 47 ET patients (p=0.597, p=0.302, respectively, Figure 1H).
The presence of a JAK2V617F mutation was associated with significantly higher PRMT5 expression in MPN (p=0.027) and specifically in JAK2V617F mutated PV (p=0.0064) patient samples, and there was a trend towards higher PRMT5 expression in JAK2V617F mutated MF patients compared to MF patients with other mutations (Figures S3A, S3B).
Patients harboring homozygous JAK2V617F mutations (defined by Taqman value of >50) showed significant higher PRMT5 expression compared to normal controls (MPN: p<0.001, PV: p<0.0001, MF: p=0.048), and compared to patients with heterozygous JAK2V617F (MPN: p=0.0015, PV: p=0.0046)/CALR (MPN: p<0.0001)/MPL (MPN: p=0.037) mutations or who were triple negative with respect to JAK2, MPL, or CALR mutations (MPN: p=0.011) (Figures S3C, S3D).
Furthermore, we observed a significant positive correlation between PRMT5 expression in granulocytes or SDMA serum levels and JAK2V617F expression measured by Taqman (p<0.0001 and p=0.012, respectively, Figures S3E, S3F).
SDMA serum levels were significantly increased in 9 JAK2V617F PV patients and 9 JAK2V617F PMF patients as compared to 10 healthy controls (p=0.031 and p=0.028, respectively) (Figure 1I). Additionally, human CD34+ cells from PV/PMF patients demonstrated sensitivity to C220 in proliferation assays ex vivo (Figure 1J) at concentrations of C220 that inhibited SDMA (Figures S3G, S3H). These data further support PRMT5 as a therapeutic target in MPN. Wild-type CD34+ cells also showed similar sensitivity in these assays, such that the impact of PRMT5 inhibition was assessed in MPN cells and in normal hematopoietic cells in subsequent in vivo studies.
PRMT5 inhibition by C220 shows significant efficacy in vivo in the Jak2V617F model of PV
We next assessed the efficacy of C220 in the Jak2V617F-driven murine MPN model of PV in vivo. Bone marrow of Jak2V617F CD45.2 Mx1-Cre conditional knock-in mice(33) was transplanted in a 50:50 ratio with CD45.1 Jak2 wild-type bone marrow into lethally irradiated recipients. C220 administered at a dose of 12.5 mg/kg orally qd 5 days on, 2 days off for 4 weeks demonstrated potent target inhibition (Figure 2A), as evidenced by 63% and 59% reduction of SDMA protein levels in the bone marrow and spleen, respectively, compared to vehicle treated control mice (Figure 2B). Monotherapy with C220 was well tolerated and did not reduce overall mouse body weights or spleen and liver weights as compared to sex- and age-matched wildtype (WT) transplant recipient controls (Figures S4A, S4B, S4C). C220 did not attenuate Jak2/Stat5 signaling in vivo (Figure 2C). PV mice treated with C220 displayed significantly lower hematocrits (mean 63% vs. 76 %, p<0.05), white blood cell counts (WBC) (mean 13.5 G/l vs. 20.5 G/l, p<0.01), reticulocytes (mean 276 G/l vs. 1612 G/l, p<0.001) and splenomegaly (mean weight 95 mg vs. 254 mg, p<0.001) compared to mice treated with vehicle (Figures 2D, 2E, 2F, 2G, 2H). C220 therapy reduced the proportion of CD105+CD150- progenitors (p<0.0001), committed erythroid progenitors (Pre-CFU-E, p=0.0004), Pre-Meg-E (p=0.0157) as well as megakaryocyte–erythroid progenitors (MEP, p=0.0139), Ter119medCD71high pro-erythroblasts (p<0.0001), Ter119highCD71high basophilic erythroblasts (p<0.0001) and CD11b+Gr1- cells (p= 0.0023) relative to vehicle. Although treatment with C220 did not reduce mutant allele chimerism in the peripheral blood (Figure S5A), C220 significantly reduced JAK2V617F+(CD45.2+)/CD45.1 chimerism in Lin-Sca1-cKit+ stem/progenitors (p=0.0214, p=0.0003) and MEP (p=0.0148, p<0.0001), in BM and spleen, respectively, as well as Pre-CFU-E (p=0.0132), Pre-Meg-E (p=0.0477), pre-granulocyte macrophage progenitors (Pre-GM, p=0.0421) in the BM and orthochromatophilic erythroblasts in the spleen (p=0.0238) (Figures S5B, S5C, S5D, S5E, S5F, S5G). Additionally, there was a trend to lower chimerism in CD105+CD150- progenitors, pro-erythroblasts and chromatophilic erythroblasts as well as in CD11b+Gr1+ populations (Figures S5H, S5I, S5J, S5K, S5L, S5M, S5N).
Figure 2: PRMT5 inhibition by C220 in the conditional Jak2V617F knock-in mouse model of PV.
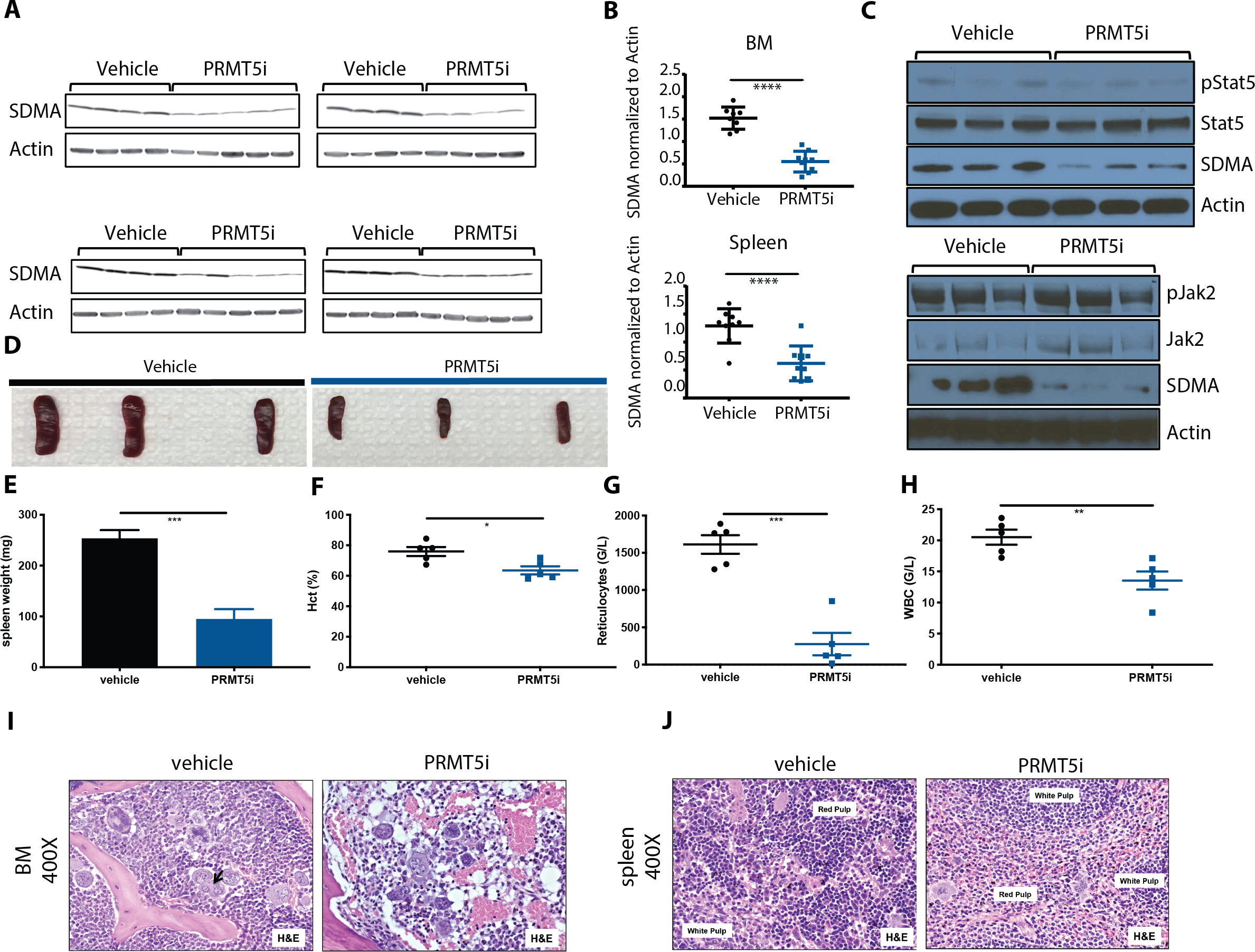
(A) SDMA expression in total BM and spleen is assessed by Western blot after 4 weeks of treatment with 12.5 mg/kg C220 in the conditional knock-in model of PV.
(A) SDMA expression in total BM and spleen normalized to actin. Data are represented as mean +/− SEM (****p<0.0001 versus vehicle group)
(B) Western Blot assessment of Jak2 and Stat5 phosphorylation and signaling in total BM after 4 weeks of treatment with C220 versus vehicle in the conditional knock-in model of PV.
(C) Spleen size at 4 weeks of treatment with C220 at 12.5 mg/kg or vehicle.
(D) Spleen weights at 4 weeks of treatment with C220 at 12.5 mg/kg or vehicle. Data are represented as mean +/− SEM (***p<0.001 versus vehicle group)
(E) Hematocrit at 4 weeks of treatment with C220 at 12.5 mg/kg or vehicle. Data are represented as mean +/− SEM (*p<0.05 versus vehicle group)
(F) Reticulocyte counts at 4 weeks of treatment with C220 at 12.5 mg/kg or vehicle. Data are represented as mean +/− SEM (***p<0.001 versus vehicle group)
(G) WBC at 4 weeks of treatment with C220 at 12.5 mg/kg or vehicle. Data are represented as mean +/− SEM (**p<0.01 versus vehicle group)
(H) Representative images of bone marrow histology (H&E) are shown in vehicle versus C220 treated mice. Magnification 400X.
(I) Representative images of splenic architecture (H&E) are shown in vehicle versus C220 treated mice. Magnification 400X.
C220 therapy reduced cellularity and normalized the proportion of myeloid lineage cells in C220 treated mice compared to vehicle treated mice (Figure 2I). The splenic architecture was also restored in the setting of C220 therapy, with decreased extramedullary hematopoiesis consistent with reduced myeloproliferation (Figure 2J). C220 significantly reduced serum cytokines in the Jak2V617F model, consistent with inhibition of the systemic inflammation characteristic of MPN(34).
Dual PRMT5 and JAK1/2 inhibition is superior to monotherapy in Jak2V617F mutant disease in vivo
We next assessed the efficacy of combined C220 and ruxolitinib in vivo. Combination therapy was well tolerated and did not reduce mouse body weights (Figure S6A). Importantly, combination C220/ruxolitinib therapy showed further reduction in hematopoietic parameters in the Jak2V617F model, including hematocrit (mean 37.7% vs. 50.2% or 78.9%, p=0.0089 and p=0.0009) and WBC (mean 2.2 G/l vs. 4.7 G/l or 7.6 G/l, p=0.0015 and p<0.0001) (Figures 3A, 3B) compared to single agent C220 and ruxolitinib. Mutant allele chimerism in the peripheral blood was not affected by combination therapy, but we observed normalization of reticulocyte numbers, hemoglobin levels, and circulating CD11b+/Gr1- cells with combination therapy (p<0.0001 each versus vehicle, Figures 3C, S6B, S6C, S6D). Furthermore, dual targeting of JAK1/2 and PRMT5 was superior to monotherapy with either agent with respect to reducing splenomegaly (mean weight 39 mg vs. 76 mg or 134 mg, p=0.0004 and p<0.0001), BM hypercelluarity (mean femur counts 4.3 vs. 20.0 vs. 27.2 × 106 total lysed cells, both p<0.0001) and hepatomegaly (mean 1076 mg vs. 1369 mg or 1372 mg, p=0.0006 and p=0.0031) (Figures 3D, 3E, 3F). Spleen weights of mice receiving combination therapy were lower than WT transplant recipient controls (39 mg versus 77 mg, p<0.0001). Liver weights in combination treated mice were not different compared to WT transplant recipient controls (1076 mg versus 1128 mg, p=n.s.) (Figures S4B, S4C).
Figure 3: Dual PRMT5 and JAK1/2 inhibition is superior to C220 or ruxolitinib monotherapy in the conditional Jak2V617F knock-in model of PV.
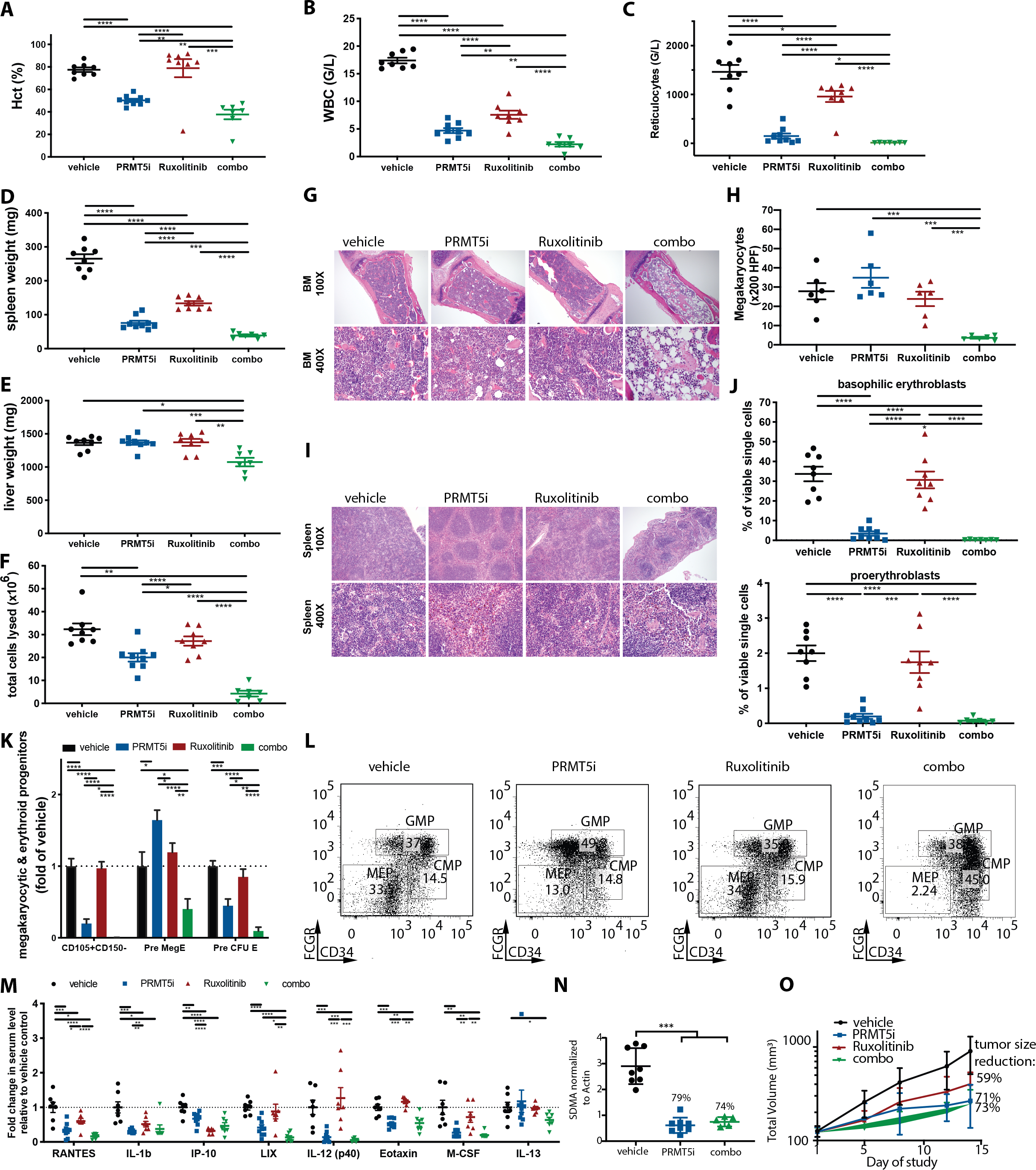
(A) Hematocrit at 4 weeks of treatment with C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM (****p<0.0001, ***p<0.001, **p<0.01).
(B) WBC at 4 weeks of treatment with C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM (****p<0.0001, **p<0.01).
(C) Reticulocyte counts at 4 weeks of treatment with C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM (*p<0.05).
(D) Spleen weights at 4 weeks of treatment with C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM (****p<0.0001, ***p<0.001).
(E) Liver weights at 4 weeks of treatment with C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM (***p<0.001, **p<0.01, *p<0.05).
(F) Total lysed cells in the bone marrow of 1 femur at 4 weeks of treatment with C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM (****p<0.0001, **p<0.01, *p<0.05).
(G)Representative images of bone marrow histology (H&E) are shown in C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle treated mice. Magnification 100X and 400X.
(H) Absolute number of megakaryocytes per 200 HPF in the bone marrow histology is shown for C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle treated mice. Data are represented as mean +/− SEM (***p<0.001).
(I) Representative images of splenic architecture (H&E) are shown in C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle treated mice. Magnification 100X and 400X.
(J)The Ter119medCD71high proerythroblast and Ter119highCD71high basophilic erythroblast populations in C220 (12.5 mg/kg), ruxolitinib (60 mg/kg), combination of both or vehicle treated animals are illustrated as means +/− SEM (****p<0.0001, ***p<0.001).
(K) Lin-cKithighCD41-FcgR-CD150-CD105+, Lin-cKithighCD41-FcgR-CD150+CD105+ committed erythroid progenitors (Pre-CFU-E) and Lin-cKithighCD41-FcgR-CD150+CD105- bipotential megakaryocyte-erythroid progenitors (Pre-Meg-E) are assessed after 4 weeks of treatment with C220 at 12.5 mg/kg, ruxolitinib at 60 mg/kg or combination of both versus vehicle treated mice. Data are illustrated as means +/− SEM (****p<0.0001, ***p<0.001**p<0.01, *p<0.05).
(L) The impact of C220, ruxolitinib, combination and vehicle treatment on Lin-Sca1-Kit+FcgR-CD34- megakaryocytic-erythroid progenitors (MEP) in the BM in the conditional Jak2V617F knock-in model of PV is assessed. Representative flow plots are depicted (C220 versus vehicle p<0.05, C220 versus ruxolitinib p<0.01, combo versus vehicle p<0.0001, combo versus ruxolitinib p<0.0001, combo versus C220 p<0.001).
(M) Serum cytokine levels measured after 4 weeks of treatment with C220 at 12.5 mg/kg, ruxolitinib at 60 mg/kg or combination of both versus vehicle treated mice are illustrated as individual points indicating fold change relative to vehicle. Only cytokines significantly attenuated by combo versus vehicle group are shown. Data are illustrated as means +/− SEM (****p<0.0001, ***p<0.001**p<0.01, *p<0.05).
(N) SDMA expression in s.c. tumors of SET2 cell derived xenografts after oral treatment with C220 at 15 mg/kg, ruxolitinib at 60 mg/kg or combination of both versus vehicle is assessed after 14 days of treatment (***p<0.001 versus vehicle group).
(O) Tumor volume of s.c. tumors in SET2 cell derived xenografts after oral treatment with C220 at 15 mg/kg, ruxolitinib at 60 mg/kg or combination of both versus vehicle treated mice is depicted.
Ruxolitinib and C220 monotherapy did not reduce the aberrant expansion of megakaryocytes seen in this model, whereas combination treatment significantly reduced megakaryocytes compared to vehicle treatment (counted by x200 HPF, p<0.001) (Figures 3G, 3H). Combination therapy was able to abrogate the aberrant myeloid infiltrate in spleens of Jak2V617F mice and restore splenic architecture, which was only partially achieved with C220 or ruxolitinib monotherapy (Figure 3I).
Flow cytometric analysis showed that combination therapy significantly reduced erythroid progenitors such as Ter119medCD71high pro-erythroblasts (BM/spleen: 0.07%/0.12% versus 0.20%/0.68% [p=0.172/p=0.0003] versus 1.74%/2.24% [p=0.0002/p<0.0001] versus 2.00%/5.77% [p<0.0001/<0.0001]) and Ter119highCD71high basophilic erythroblasts (BM/spleen: 0.3%/0.2% versus 3.5%/9.2% [p=0.0258/p=0.0388] versus 30.7%/49.9% [p<0.0001/p<0.0001] versus 33.7%/20.9% [p<0.0001/p<0.0001]) compared to C220, ruxolitinib, or vehicle treatment, both in the BM (Figure 3J) and spleen (Figures S6E, S6F, S6G). Furthermore, combination treatment was superior to single agent C220 or ruxolitinib treatment in reducing BM erythroid and megakaryocytic progenitors including CD105+CD150- progenitors (0.1% vs. 9.0% vs. 43.8% of FcGR negative cells, p=0.0146 and p<0.0001), Pre-MegE (2.4% vs 9.7% vs 7.0% of FcGR negative cells, p<0.0001 and p=0.0011) and Pre-CFU-E (0.7% vs. 3.1% vs. 5.9% of FcGR negative cells, p=0.0095 and p<0.0001) (Figure 3K). MEP cells were significantly reduced by dual PRMT5/JAK1/2 targeting compared to either monotherapy or vehicle in the bone marrow (6.0% of BM Lin−Sca1−cKit+ vs. 21.1% vs. 31.8% vs. 30.0%, p=0.0002, p<0.0001 and p<0.0001) (Figure 3L and S6H) and spleen (37.4% of spleen Lin−Sca1−cKit+ vs. 54.4% vs. 89.4%, vs. 91.6%, p=0.11, p<0.0001 and p<0.0001) (Figure S6I). Combination treatment, but neither single agent treatment, also significantly decreased the portion of Lin−Sca1+cKit+ CD48− CD150− multipotent myeloid progenitors (MPP) in the bone marrow (p<0.0001) and Lin−Sca1−cKit+ myeloid progenitors (MP) in the spleen (p=0.0014) (Figures S6J, S6K). A significant decrease of CD41+ HSC in the bone marrow and the spleen was achieved by combination treatment (p=0.0002, p<0.0001) (Figures S6L, S6M) but not with single agent treatment. C220 monotherapy and combination treatment both significantly reduced serum cytokines (specifically Eotaxin [p<0.0001, p=0.0031], M-CSF [p=0.0023, p=0.0031], IL-12p40 [p=0.0003, p=0.0004], LIX [p<0.0001, p<0.0001], RANTES [p=0.0003, p<0.0001], IP-10 [p=0.0049, p=0.0008], IL-1b [p=0.0002, p=0.0075]), respectively, compared to vehicle control (Figures 3M, S6N), consistent with inhibition of the systemic inflammation characteristic of MPN. Of note, levels of Eotaxin, IL-12 (p40), LIX and M-CSF were not reduced by ruxolitinib treatment alone but were attenuated by both C220 monotherapy and combination treatment (Figures 3M, S6N). In addition, PRMT5 inhibitor monotherapy also significantly lowered KC (p=0.0068), MCP-1 (p=0.0397), MIP-1a (p=0.0074) and MIP-2 (p=0.0312) serum levels; levels of these cytokines were not altered by ruxolitinib monotherapy nor combination treatment (Figure S6N). Taken together, this suggests that distinct inflammatory cytokines are regulated downstream of JAK2 and PRMT5 in MPN cells.
We also investigated the efficacy of combination therapy in an in vivo SET2 xenograft model. Adequate suppression of PRMT5 was achieved by C220 and combination treatment as demonstrated by potent reduction in SDMA levels with in vivo therapy (Figure 3N); SDMA levels remained unaffected by monotherapy ruxolitinib treatment (Figure S6O). Consistent with the data in our murine model, we observed significant efficacy with C220 monotherapy (71% tumor reduction) and combination therapy (73% tumor reduction) with 14 days of treatment which was greater than that observed with JAK inhibition (59%) (p=0.0006, p=0.0039, 0.0040 compared to vehicle treated mice, respectively; p=0.0545 and p=0.0464 compared to ruxolitinib treated mice, respectively) (Figure 3O).
Dual PRMT5 and JAK1/2 inhibition shows increased efficacy in the MPLW515L model
We next investigated the efficacy of PRMT5 inhibition and of combined PRMT5/JAK inhibition in a MPLW515L bone marrow transplant (BMT) model of myelofibrosis. C220 therapy at 15 mg/kg oral, once daily (5 days on/2 days off) was sufficient to potently suppress SDMA levels in vivo (100% suppression compared to vehicle, p<0.005) (Figures S7A, S7B). C220 monotherapy significantly reduced splenomegaly (mean: 151 mg versus 423 mg, p=0.0042), hepatomegaly (mean:1156 mg versus 1 663 mg, p=0.0025), WBCs (mean: 12.8 G/l versus 170.6 G/l, p=0.0225) and platelet counts (mean: 233 G/l versus 1868 G/l, p=0.049) (Figures S7C, S7D, S7E, S7F) compared to vehicle treated mice.
C220 treated mice showed a significant reduction of peripheral blood mutant allele burden (27.4% versus 83.6% of GFP+ cells, p=0.0003), and significantly lower neutrophils (1.4% versus 49%, p=0.0028) and monocytes (7.7% versus 23.5%, p=0.0374) (Figures S7G, S7H, S7I) compared to vehicle treated mice. In the BM, there was a trend towards fewer Lin- cells and MEPs (7.6% versus 12.3%) in the PRMT5 inhibitor treated mice (Figures S7J, S7K).
We next investigated the efficacy of combined JAK/PRMT5 inhibition. Ruxolitinib in combination with the higher dose of C220 (15 mg/kg, qd 5/2), which showed efficacy in this more aggressive model, resulted in significant pancytopenia. Therefore, combination studies were done with ruxolitinib at 60 mg/kg po bid and C220 at 10 mg/kg po qd (5/2). Combined JAK/PRMT5 inhibition induced more significant suppression of splenomegaly (mean weight 137 mg versus 459 mg, p=0.006), hepatomegaly (mean weight 1114 mg versus 1651 mg, p=0.019), leukocytosis (mean WBC 6.1 G/l versus 149.3 G/l, p=0.044) and hematocrits (mean 31% versus 51%, p=0.0296) and resulted in a trend towards lower platelet counts (mean platelet count 804 G/l versus 3225 G/l, p=0.1502) compared to vehicle treated mice (Figures 4A, 4B, 4C, 4D, S8A, S8B). Combination treatment reduced reticulocytes significantly more compared to ruxolitinib treatment (p=0.023) (Figure S8C).
Figure 4: Dual PRMT5 and JAK1/2 inhibition shows increased efficacy in the MPLW515L model.
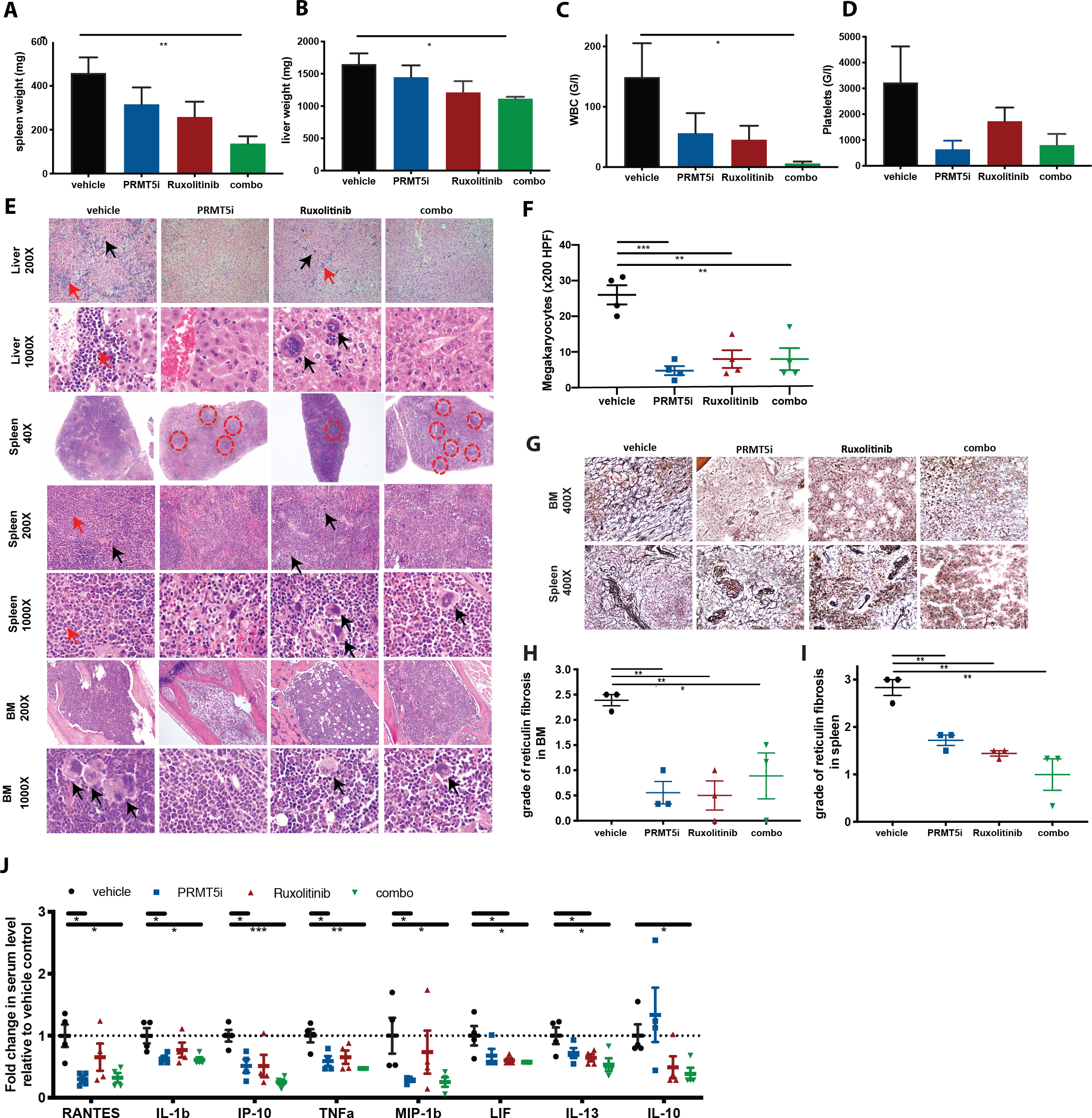
(A) Spleen weights at 3 weeks of treatment with C220 at 10 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM (**p<0.01).
(B) Liver weights at 3 weeks of treatment with C220 at 10 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM (*p<0.05).
(C) WBC at 3 weeks of treatment with C220 at 10 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM (*p<0.05).
(D) Platelet counts at 3 weeks of treatment with C220 at 10 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle. Data are represented as mean +/− SEM.
(E) Representative images of liver, spleen and bone marrow histology (H&E) are shown after 3 weeks of treatment in C220 at 10 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle treated mice. Magnification 1000X, 200X and 40X. Black arrows point to megakaryocytes. Red arrows point to myeloid infiltrate. Red circles surround follicles in spleen.
(F) Absolute number of megakaryocytes per 200 HPF in the bone marrow histology is shown for C220 at 12.5 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle treated mice. Data are represented as mean +/− SEM (***p<0.001, **p<0.01).
(G) Representative images of bone marrow and spleen fibrosis (reticulin stain) are shown after 3 weeks of treatment in C220 at 10 mg/kg, ruxolitinib 60 mg/kg or combination of both versus vehicle treated mice. Magnification 400X.
(H) Histologic grade of reticulin fibrosis in the bone marrow in the 4 treatment arms is illustrated. For each sample, degree of fibrosis was evaluated in 6 HPF (0=no fibrosis, 1=focal mild fibrosis, 2=marked, but non-diffuse fibrosis, 3=diffuse fibrosis). Each data point reflects the mean value of fibrosis for each mouse (3 mice per group). Data are illustrated as means +/− SEM (**p<0.01, *p<0.05).
(I) Histologic grade of reticulin fibrosis in the spleen in the 4 treatment arms is illustrated. For each sample, degree of fibrosis was evaluated in 6 HPF (0=no fibrosis, 1=focal mild fibrosis, 2=marked, but non-diffuse fibrosis, 3=diffuse fibrosis). Each data point reflects the mean value of fibrosis for each mouse (3 mice per group). Data are illustrated as means +/− SEM (**p<0.01).
(J) Serum cytokine levels measured after 3 weeks of treatment with C220 at 10 mg/kg, ruxolitinib at 60 mg/kg or combination of both versus vehicle treated mice are illustrated as individual points indicating fold change relative to vehicle. Only cytokines significantly attenuated by the combo versus vehicle group are shown. Data are illustrated as means +/− SEM (***p<0.001**p<0.01, *p<0.05).
In terms of toxicity, spleen weights of mice receiving combination therapy were still larger compared to MIGR1 (MSCV-IRES-GFP empty vector) transplant recipient controls (137 mg versus 69 mg, p=0.0079). Liver weights in combination treated mice were comparable to MIGR1 transplant recipient controls (1114 mg versus 1148 mg, p=n.s.) (Figures S8D, S8E).
C220 monotherapy, and combination treatment, but not single agent ruxolitinib treatment, significantly reduced the proportion of CD11b+Gr1- cells in peripheral blood (p=0.0101 and p=0.0181) and BM (p=0.0219 and p=0.0240) (Figure S8F). Histopathologic analysis showed that the myeloid infiltrates in the liver seen in vehicle treated mice were decreased by single agent PRMT5 or JAK kinase inhibition and more markedly reduced with combination therapy (Figure 4E). C220 monotherapy, and to a greater extent combination therapy, significantly decreased splenic extramedullary hematopoiesis and partially restored splenic architecture (Figure 4E). BM hypercellularity was significantly decreased by C220 and combination therapy compared to ruxolitinib treated mice (p=0.006 and p=0.012) (Figures 4E, S8G). The marked increase in megakaryocytes seen in this model was significantly reversed by PRMT5 inhibition, JAK inhibition or combination therapy (p=0.0004, p=0.003, p=0.004, respectively) (Figure 4F). Reticulin fibrosis in bone marrow and spleen was increased in vehicle treated mice but significantly reduced in all 3 treatment cohorts (Figures 4G, 4H, 4I). C220 monotherapy and combination therapy significantly lowered levels of circulating IL-1b (p=0.0312 and p=0.0257), IP-10 (p=0.0143 and p=0.0004), MIP-1b (p=0.0482 and p=0.0466), RANTES (p=0.0102 and p=0.0139) and TNFalpha (p=0.0216 and p=0.0024), which were not attenuated by ruxolitinib monotherapy (Figures 4J, S8H). Combination treated mice displayed reduced circulating levels of LIF (p=0.0323) and IL-13 (p=0.0324), both of which were also reduced by ruxolitinib (p=0.0474 and p=0.0497, respectively). Of note, IL-10 levels were only significantly reduced by dual PRMT5/JAK inhibition (p=0.0257), but not by treatment with either agent alone.
PRMT5 inhibition alters the post-translational methylation of E2F1, leading to altered expression of E2F1 downstream targets, including genes involved in cell cycle and DNA damage repair
To further understand the mechanism by which C220 leads to therapeutic efficacy in MPN, we performed RNA-seq in sorted Jak2V617F+ MEP (Figure S9A) from the Jak2V617F model and from SET2 xenografts after vehicle or C220 in vivo therapy. In both models, principal component analysis (PCA) of RNA-sequencing data separated C220 treated from control groups (Figures S9B, S9C). The transcriptome of C220 and vehicle treated SET2 xenografts or Jak2V617F+MEP revealed a total of 4219 and 2862 genes which were differentially expressed, respectively, of which 2223 and 874 were significantly down-regulated (Figures 5A, 5B, S9D). Gene set enrichment analysis (GSEA) revealed a significant enrichment for p53 pathway activation signatures and reduced expression of gene expression signatures associated with E2F targets (including DNA repair, G2M checkpoint) and interferon-α and interferon-γ response (Figures 5C, 5D, 5E, 5F) with PRMT5 inhibition.
Figure 5: In vivo PRMT5 inhibition reduces expression of gene sets associated with Interferon signaling and E2F targets - that are upregulated in CD34+ JAK2V617F positive MPN patients.
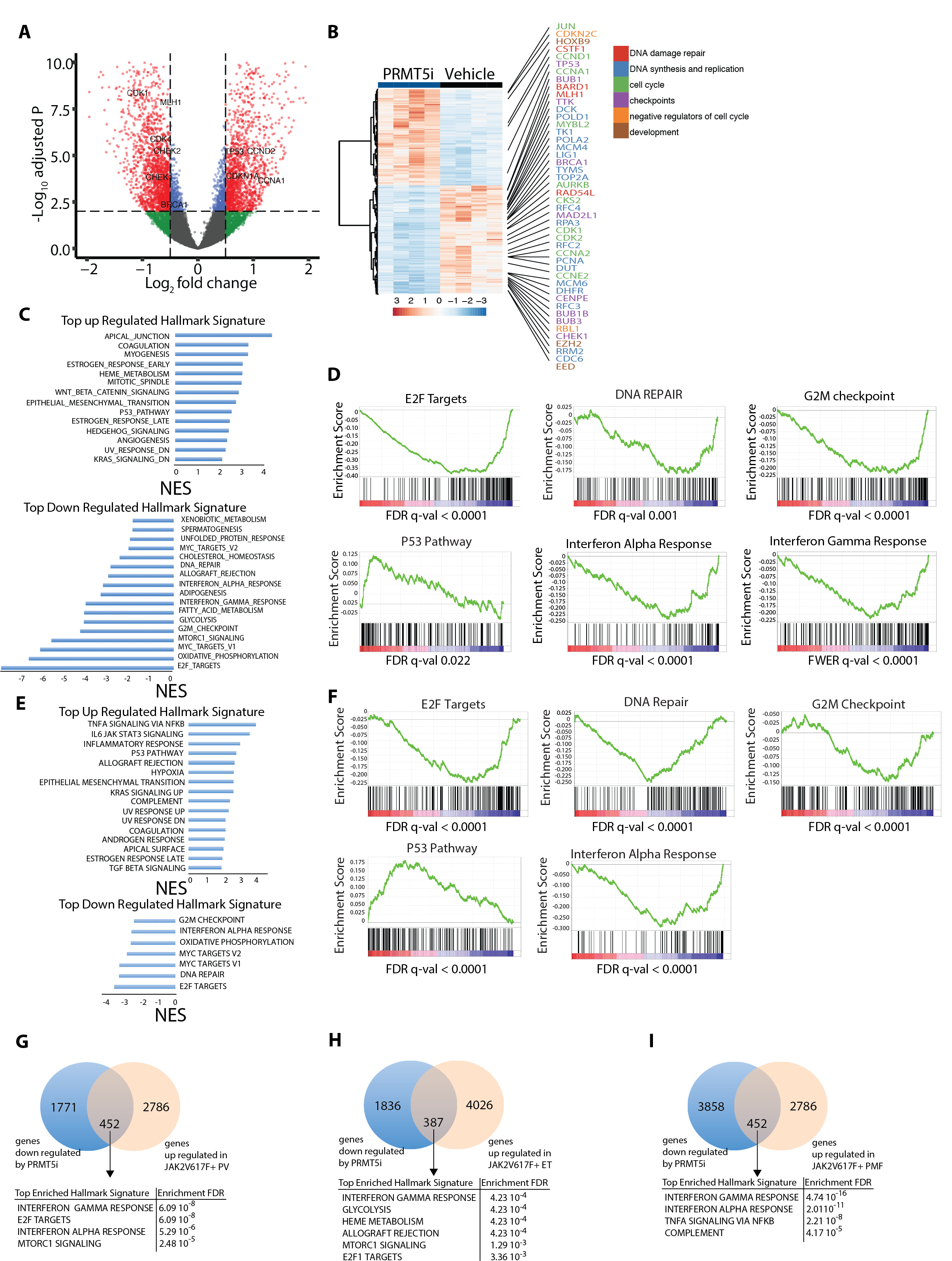
(A) Volcano plot representing differences in gene expression detected by RNA sequencing of tumors derived from SET2 xenografts in mice that have been treated with C220 or vehicle for 2 weeks. The significant events an inclusion level >0.5 log fold change and an FDR corrected p-value <0.0001 are shown in red. Among these, examples of genes related to cell cycle and DNA damage repair have been labeled. Genes with a log fold change <0.5 and a p-value <0.0001 are labeled in blue. Genes with a log fold change >0.5 and a p-value >0.0001 are labeled in green. Genes with a log fold change <0.5 and a p-value >0.0001 are labeled in black.
(B) Heatmap showing the expression of differentially expressed genes detected by RNA sequencing of tumors derived from SET2 xenografts in mice that have been treated with C220 or vehicle for 2 weeks. Representative E2F target genes involved in DNA damage repair (red), DNA synthesis and replication (blue), cell cycle (green), checkpoints (purple), cell cycle (orange) and development (brown) are depicted.
(C) HALLMARK gene set enrichment analysis of genes affected by PRMT5 inhibition in SET2 xenografts treated with C220 or vehicle for 2 weeks. NES: normalized enrichment score.
(D) The E2F targets DNA repair, G2M checkpoint, p53 pathway, interferon alpha and interferon gamma response gene expression signatures were tested for enrichment by GSEA in SET2 xenografts in mice that have been treated with C220, or vehicle for 2 weeks.
(E) HALLMARK gene set enrichment analysis of genes affected by PRMT5 inhibition in Jak2V617F- positive MEP derived from the Jak2V617F conditional BMT model after treatment with C220 or vehicle for 2 weeks. NES: normalized enrichment score.
(F) The E2F targets such as DNA repair, G2M checkpoint, p53 pathway, interferon alpha and interferon gamma response gene expression signatures are tested for enrichment by GSEA in Jak2V617F- positive MEP derived from the Jak2V617F conditional BMT model after treatment with C220 or vehicle for 2 weeks.
(G) Venn diagram showing the overlap between upregulated genes in CD34+ JAK2V617F-positive PV (36) and downregulated genes by PRMT5i in SET2 xenografts. The cutoffs were fold change >0.05 with a false discovery rate (FDR) adjusted p-value of <0.01.
(H) Venn diagram showing the overlap between upregulated genes in CD34+ JAK2V617F-positive ET (36) and downregulated genes by PRMT5i in SET2 xenografts. The cutoffs were fold change >0.05 with a false discovery rate (FDR) adjusted p-value of <0.01.
(J) Venn diagram showing the overlap between upregulated genes in CD34+ JAK2V617F-positive PMF (35) and downregulated genes by PRMT5i in SET2 xenografts. The cutoffs were fold change >0.05 with a false discovery rate (FDR) adjusted p-value of <0.01.
GSEA of ATAC-seq data revealed a reduced accessibility of gene sets associated with E2F targets, DNA damage repair and interferon-α and interferon-γ response with C220 (Figure S9E), Genes with significant reduced accessibility and gene expression during C220 therapy included genes involved in immune system processes (including CCL3 and CCL4), metabolism and DNA binding (Figures S9F, S9G). Motif enrichment on peaks with lower accessibility in ATAC seq and concurrently reduced gene expression revealed and enrichment of Myc and NF-E2 targets (Figure S9H).
We next integrated genes that were significantly downregulated by PRMT5 inhibition with genes that were upregulated in gene expression data derived from CD34+ JAK2V617-mutated PV, ET or primary MF (PMF) patients(35,36). There was an overlap of 452, 387 and 452 genes for PV, ET and PMF respectively which were upregulated in primary MPN cells and significantly attenuated with PRMT5 inhibition in SET2 xenografts (Figures 5G, 5H, 5I). GSEA of overlapping genes showed a highly significant enrichment for interferon response in all 3 MPN subtypes and for E2F targets in PV and ET. In concordance with these results, the ranked difference of normalized enrichment scores between SET2 xenografts and CD34+ JAK2V617F+ PV, ET or PMF indicated IFN response and inflammatory response as well as MTORC1 signaling as pathways that were up-regulated in these MPN and down-regulated by C220 (Figures S9I, S9J, S9K).
E2F targets were one of the two major target gene sets down-regulated upon treatment with C220; this included E2F targets involved in a variety of processes including DNA damage repair (e.g. RAD54L), checkpoint control (e.g. BRCA1), and cell cycle entry (e.g. CDK 1,2)(37). We focused on elucidating 1) if PRMT5 was needed for the survival of SET2 cells and 2) the mechanism by which PRMT5 inhibition affects E2F function and downstream target gene expression. We performed a CRISPR knock-out competition assay in SET2 cells stably transduced with Cas9, and first showed reduced fitness of PRMT5 knockout cells validating our pharmacologic data with C220 (Figures 6A, S10A). Likewise, knockout of E2F1, E2F2 and E2F3 decreased proliferation in SET2 cells (Figure 6A, S10A, S10B, S10C and S10D, Tables S1 and S2).”
Figure 6: PRMT5 inhibition alters the methylation status of E2F1, leading to altered expression of E2F1 downstream targets, including genes involved in cell cycle and DNA damage repair.
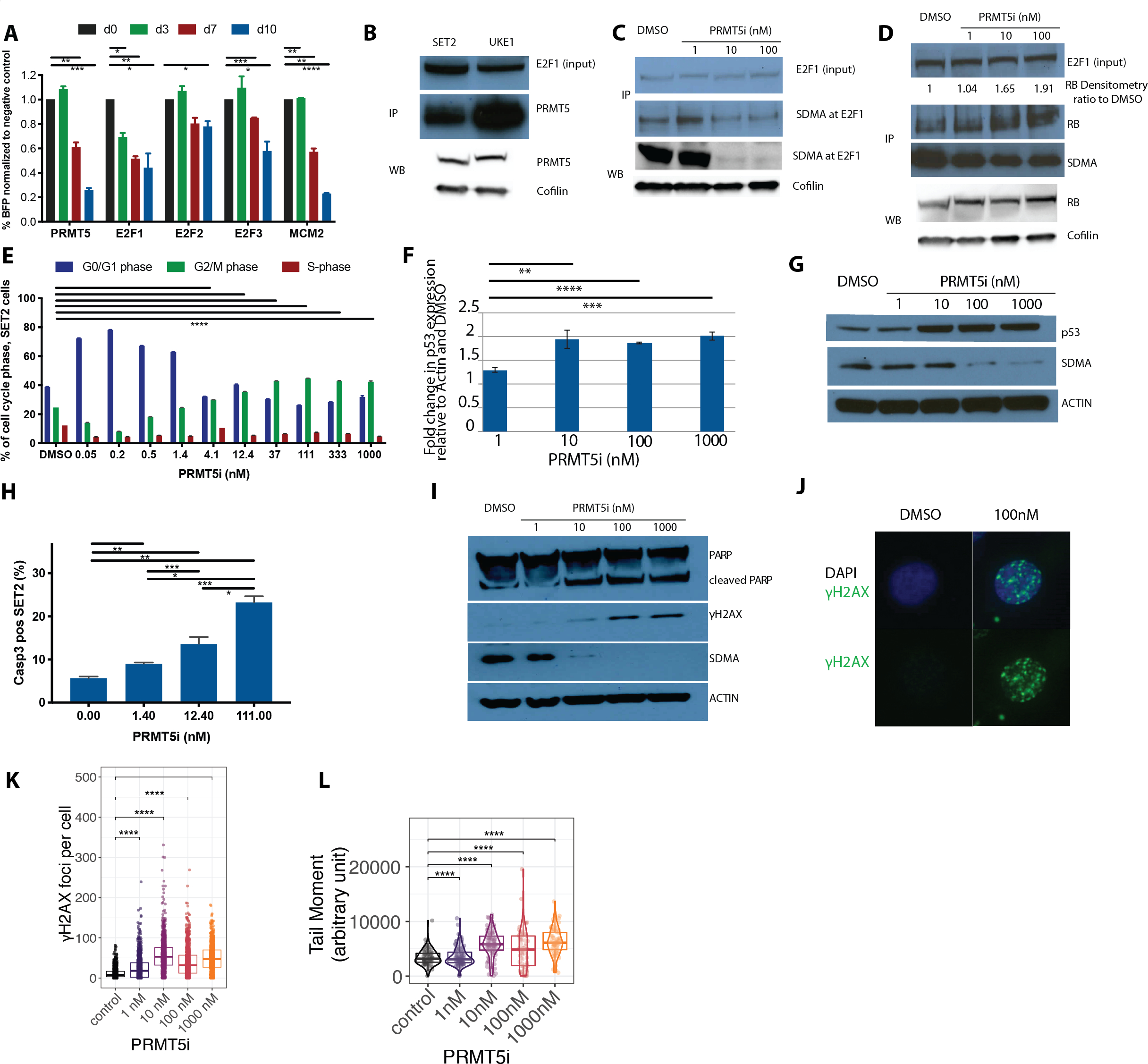
(A) Competition assays to assess the requirement of PRMT5, E2F1, E2F2 and E2F3 for proliferation in SET2 cells. Stably expressing Cas9 SET2 cells were transduced with sgRNA against PRMT5, E2F1, E2F2, or E2F3. Percentage of BFP-positive cells (sgRNA-expressing) was monitored over time. MCM2 is used as a positive control.
(B) Western blot assessment of PRMT5 expression in total protein lysate and after immunoprecipitation of E2F1 in SET2 and UKE cells is illustrated1.
(C) Western blot assessment of expression of symmetrically dimethylated E2F1 (SDMA at E2F1) in total protein lysate and after immunoprecipitation of E2F1 in SET2 cells treated with different doses of inhibitor or DMSO control for 6 days is illustrated1.
(D) Western blot assessment of RB protein expression in total protein lysate and after immunoprecipitation of E2F1 in SET2 cells treated with different doses of inhibitor or DMSO control for 6 days is illustrated1.
(E) Cell cycle proportion is determined by flow cytometry of SET2 cells treated with different doses of inhibitor or DMSO control for 6 days using the FITC BRDU Flow Kit (BD). Data are indicated as mean ± SEM (****p<0.0001, for comparison of % of cells in G2/M phase)
(F) Relative mRNA expression of p53 derived from SET2 cells treated with increasing doses of C220 or DMSO control for 6 days. Results are presented as the ratio of the C220 treated value to the DMSO treated value, normalized to actin. The experiment was performed in triplicate. Data are indicated as mean ± SEM (**p<0.01,***p<0.001,****p<0.0001).
(G) Expression of p53 protein and SDMA is assessed in SET2 cells treated with different doses of inhibitor or DMSO control for 6 days.
(H) Induction of apoptosis in SET2 cells treated with increasing doses of C220 or DMSO control for 6 days is measured as proportion of activated caspase-3 positive cells in flow cytometry and plotted as bar graphs. Data are indicated as mean ± SEM (*p<0.05,**p<0.01,***p<0.001).
(I) Western Blot assessment of PARP, cleaved PARP, γH2AX and SDMA in SET2 cells treated with increasing concentrations of C220 (nM) or DMSO control for 6 days followed by irradiation with 10Gy, 6 hours prior to lysate preparation.
(J) Representative picture of γH2AX foci detected by immunofluorescence staining in SET2 cells treated with increasing doses of C220 or DMSO control for 6 days, followed by irradiation with 10Gy, 6 hours prior to immunofluorescence staining.
(K) Quantification of the number of γH2AX foci in SET2 cells treated with increasing doses of C220 or DMSO control for 6 days, followed by irradiation with 10Gy, 6 hours prior to immunofluorescence staining (****p<0.0001).
(L) Neutral comet assay performed in SET2 cells treated with increasing doses of C220 or DMSO control for 6 days, followed by irradiation with 10Gy, 6 hours prior to Comet assay.Violin Blot and Box Plot displaying the median and interquartile range (box). The Tail Moment is calculated by the formula:
Tail Moment = (Tail Intensity/(Head Intensity+Tail Intensity))* Tail width*100.
(****p<0.0001).
1Note: For WB and IP analyses, all images that appear in blue scale were acquired with conventional film and scanned, all images that appear in gray and white scale were electronically acquired.
We performed immunoprecipitation studies with E2F1 followed by western blot analysis of PRMT5 in SET2 and UKE1 cells and observed that PRMT5 co-immunoprecipitated with E2F1 (Figure 6B, Table S3). We also immunoprecipitated PRMT5 and were able to detect E2F1 and E2F2 but to a much lesser extent E2F3 (Figure S10E). To test the hypothesis that PRMT5 affects E2F targets by modulating E2F1 and E2F2 methylation status, SET2 cells were treated with different C220 concentrations, followed by immunoprecipitation for E2F1 or E2F2 and Western blot for SDMA in both, the total lysate and the co-immunoprecipitates (Figures 6C, S10F). C220 exposure resulted in attenuated symmetric dimethylation of E2F1, but not of E2F2 (Figures 6C, S10F). This suggests that there may be increased interaction between demethylated E2F1 and known co-factors, including the retinoblastoma (Rb) tumor suppressor protein. C220 exposure did not affect total Rb levels, but increased association between E2F1 and Rb (Figure 6D). These data suggest that PRMT5 inhibition may attenuate E2F target gene activation through differential activation of transcriptional activators and repressors based on E2F methylation status (Figure S10G).
E2F gene regulatory networks play a major role in regulating checkpoint (e.g. BUB1b, BUB3) and cell cycle entry (e.g. CDK1,2 CCNA2, CCNE2). Modulation of E2F gene expression by PRMT5 inhibition caused a G2/M cycle arrest in SET2 (44.8%, 35.5%, 30.0% and 24.7% of cells in G2/M phase in 111nM, 12.4 nM, 4.1 nM and DMSO treated SET2 cells, respectively, p<0.0001 for each vs. DMSO control) (Figure 6E) and UKE1 cells (16.4%, 6.7%, 3.3% and 1.0% of cells in G2/M phase in 111nM, 12.4 nM, 4.1 nM and DMSO treated UKE1 cells, respectively, p<0.0001 for each vs. DMSO control) (Figure S10H). We observed increased apoptosis as demonstrated by upregulation of p53 (100 nM vs. DMSO, p=0.0013) (Figures 6F, 6G, Table S4) and a significant increase in activated caspase 3 expression (111 nM vs DMSO control: p=0.0006 in SET2, p<0.0001 in UKE1 cells) (Figures 6H, S10I). Moreover, a subset of E2F target genes have known roles in DNA damage repair (e.g. RAD54L, MLH1, BRCA1) (Figure 5B). C220 treated SET2 cells showed a significant increase in radiation induced double strand breaks as assessed by γH2AX Western Blot (Figure 6I), γH2AX immunofluorescence staining (100 nM vs DMSO control: γH2AX foci/cell p<0.0001) (Figures 6J, 6K) and a neutral comet assay (100 nM vs DMSO control: arbitrary units tail moment p=0.019) (Figure 6L). In addition, repair of single strand breaks was impaired in the setting of PRMT5 inhibition, with increased PARP expression/cleavage and increased activated caspase 3 expression in C220 treated cells compared to vehicle (Figure 6I and 6H). These findings prompted us to explore combination therapy with C220 plus inhibitors targeting DNA damage repair pathways, including ATM (AZD0156) and PARP (Olaparib). Indeed, combined PRMT5 and ATM inhibition, as well as combined PRMT5 and PARP inhibition, showed additive effects in SET2 cells (ZIP synergy scores of 2.8 and 2.8, respectively) (Figures S10J, S10K) consistent with increased efficacy of the combination in vitro.
Discussion
The discovery of somatic JAKV617F mutations resulting in constitutively active JAK2/STAT5 signaling underscored activated JAK-STAT signaling as a disease underlying mechanism in PV, ET and PMF. These findings led to the clinical development of JAK kinase inhibitors and subsequent approval of ruxolitinib for PV and MF and fedratinib for MF. Despite these important genetic and therapeutic insights, current JAK1/2 inhibitors can achieve significant clinical benefits without altering the natural history of the disease, suggesting a need for new therapeutic approaches that show efficacy alone and in combination with JAK inhibition in MPN.
Dysregulated PRMT5 expression occurs in a variety of malignancies. Moreover, previous work has suggested that PRMT5 may be a specific therapeutic target in MTAP deleted tumors and in malignancies with spliceosome component mutations. As such, PRMT5 inhibition is currently being tested as a novel promising therapy(12) in a variety of solid cancers, lymphoma and MDS in clinical trials (JNJ-64619178, PF-06939999, GSK3326595, PRT543, PRT811) (www.clinicaltrials.gov). The role of PRMT5 in the pathogenesis and therapy of MPN has been unclear. Liu et al. showed that JAK2V617 phosphorylates PRMT5 impairing its methylation activity, suggesting that certain aspects of PRMT5 function might be attenuated in MPN cells(29). However, it is not clear if PRMT5 phosphorylation completely attenuates PRMT5 catalytic activity, or if PRMT5 phosphorylation is altered in the setting of preclinically/clinically achievable JAK inhibitor doses. We found that PRMT5 is significantly overexpressed in JAK2V617F+ PV, and that the transcriptional effects of PRMT5 inhibition in MPN cells had significant overlap with the set of genes which are altered in expression in MPN cells. These data suggest that there is an aberrant gene expression signature mediated by PRMT5 in MPN cells, and we demonstrate a potential therapeutic role for PRMT5 inhibition in MPN. PRMT5 inhibition, alone and in combination with ruxolitinib, showed significant efficacy in Jak2V617F and MPLW515L MPN in vivo models, with reduced myeloproliferation, pathologic responses, and attenuated inflammatory cytokine secretion. Despite the significant, potent reduction in many hallmark disease features of MPN, the smaller spleens seen with combination JAK/PRMT5 inhibitor therapy (Figure S4B) suggests that the impact of dual JAK/PRMT5 inhibitor therapy on normal hematopoiesis will need to be studied in clinical studies.
Chronic inflammation is a hallmark of MPN(38). The increase of several inflammatory serum cytokines has a crucial role in MPN disease maintenance and progression. Therefore, targeting inflammation at early disease stages is a therapeutic pillar to prevent myelofibrosis. PRMT5 inhibition potently reduced the serum levels of multiple cytokines, including TNFalpha – a cytokine that has been shown to be essential for clonal expansion in a JAK2V617F mouse model(39).
Levels of circulating IL-8, IL-2R, IL-12 and IL-15 have been shown to be independently prognostic in patients with primary myelofibrosis and associated with impaired outcome(40). IL-8 levels in particular are an independent prognostic factor that correlates inversely with leukemia-free survival. In the Jak2V617F model, therapy with C220 or C220/ruxolitinib combination therapy significantly lowered cytokine levels of the murine IL-8 analogs LIX and MIP1a. Moreover, the combination of C220 and ruxolitinib was superior to either agent as monotherapy - with respect to reducing elevated circulating levels of LIX. This was also evident in the MPLW515L model in which we observed a trend of lower levels of IL-8 analogs LIX and MIP1a (Figure S8H). The epigenetic mechanisms that lead to aberrant cytokine secretion, including IL-8 analogs in preclinical models and IL-8 in human MPN cells, require further investigation; our data suggest a key role for PRMT5 in this aberrant inflammatory circuit.
Most importantly, although C220 and ruxolitinib therapy induced coordinate changes in gene expression, such as those involved in the interferon response (Figures S11A, S11B, Table S5), C220 exposure down-regulated more than 1808 genes that are not down-regulated by ruxolitinib (Figures S11A, S11C, S11D, Table S6). These data suggest that PRMT5 inhibition induces therapeutic efficacy through an orthogonal mechanism not shared with JAK kinase inhibition. We observed increased expression of E2F target genes and of interferon pathway members in MPN cells which was potently suppressed with PRMT5 inhibitor therapy in vivo. In addition, integrated RNA-seq and ATAC-seq analyses uncovered that C220 potently reduced expression and accessibility of targets of the transcription factor NF-E2 which is overexpressed in the majority of MPN patients and causes an MPN phenotype with a high rate of leukemic transformation in mice when overexpressed in mice(41). The role of interferon signaling and other inflammatory mediators in the bone marrow microenvironment and molecular mechanisms are postulated to drive progression of bone marrow fibrosis in myelofibrosis although the exact pathophysiology is not well understood. Our data suggest that modulation of interferon signaling by PRMT5 inhibition may have therapeutic importance in MPN cells and in a broader suite of malignant contexts.
Kim et al. suggest a link between Rb/E2F and TPO signaling via JAK2 in hematopoietic stem cell homeostasis, however, the role of E2F and E2F target gene expression in MPN pathogenesis has not been studied(42). Our data illuminate a potential biologic and therapeutic role for this pathway in MPN. Arginine methylation of E2F1 has been discovered in solid cancer cell lines but never previously studied in hematologic malignancies(43). We provide evidence that expression of PRMT5, E2F1, E2F2 and E2F3 is needed for proliferation and survival of JAK2-mutant MPN cells. Consistent with the potent suppression of E2F target genes in vivo, we found that E2F1 and E2F2 physically interact with PRMT5 in JAK2-mutant MPN cells. PRMT5 inhibition reduced dimethylated E2F1 leading to a higher affinity between E2F1 and the tumor suppressor Rb protein and resultant reduced expression of E2F1 downstream targets. Amongst those targets were proteins involved in DNA damage repair, and we observed increased DNA damage in C220 treated MPN cells and cooperativity between PRMT5 inhibition and either ATM or PARP inhibition in MPN cells. Previous work by Mullally(44) and colleagues and by Skorski et al.(45). have implicated aberrant DNA damage responses in MPN pathogenesis, and our data suggest PRMT5 as a therapeutically tractable mediator of these responses with pathogenetic relevance. Further mechanistic studies of the interaction between PRMT5 and E2F1 in the context of MPN will be of importance to delineate the link between aberrant JAK-STAT signaling, MPN pathogenesis, and PRMT5/E2F1 mediated gene regulation.
Taken together, our data provide evidence that PRMT5 is a therapeutic target in MPN, alone and in combination with JAK inhibition. These data demonstrate a novel link between PRMT5, E2F1 gene regulatory function, and the survival of MPN cells and provide a strong mechanism-based rationale for therapeutic studies of PRMT5 inhibitors in MPN. Based on these studies, PRT543, a novel and selective PRMT5 inhibitor, is currently being evaluated in a Phase I clinical trial, including in MPNs (NCT03886831).
Methods
Inhibitors
C220 is a derivative of a published PRMT5 inhibitor (46) and was synthesized by Wuxi Apptec. For in vitro assays, it was stored as a 10 mM solution in DMSO. For in vivo assays, C220 was dissolved in a 0.5% methylcellulose, 0.1% Tween-20 solution. C220 was administered as oral gavage once daily for 5 days a week, for a total of 4–6 weeks. AZD0156 and Olaparib were purchased from MedChemExpress and stored as 10 mM DMSO solution.
Cell lines
Ba/F3 stably expressing the EPOR were transduced with viral supernatants containing MSCV- Jak2V617-IRES-GFP, MSCV-Jak2WT-IRES-GFP, MSCV-IRES-GFP empty vector, flow sorted for GFP and maintained in RPMI1640/10% fetal calf serum (FCS) supplemented with 10 ng/ml IL-3 in the EPOR/Jak2WT and EPOR/EV cells. HEL and SET2 cells were grown in RPMI1640 medium containing 10% or 20% FCS, respectively. UKE1 cells were cultured in RPMI1640 supplemented with 10% horse serum (StemCell Techn.) and 50 μM hydrocortisone (Sigma-Aldrich). SET2 cells transduced with Cas9 were selected for in the presence of Blasticidin at a concentration of 10 μg/ml. Ruxolitinib persistent SET2 cells were generated by incubating SET2 cells at increasing doses of ruxolitinib over 10 weeks. SET2 cells were authenticated by and obtained from DSMZ <6 months before use. All cell lines underwent mycoplasma testing (Lonza, MycoAlert™ Mycoplasma Detection Kit, #LT07–318) before use.
In vitro proliferation assays
For proliferation assays, 10 000 cells/200 μl medium were plated in triplicate and supplemented with increasing doses of inhibitor using 9-point, 3-fold dilutions with a top concentration of 1000 nM for C220. After 3 and 6 days, cells were split 1:4 and supplemented with fresh medium and inhibitor. Proliferation was assessed at 3, 6 and 10 days using the CellTiter-Glo luminescent cell viability assay (Promega) and normalized proliferation in media with an equivalent volume of DMSO. IC50 was determined using Graph Pad Prism 8.0.
Methocult experiments in patients
Mononuclear cells were isolated by Ficoll from peripheral blood of untreated JAK2V617F mutated PV and PMF de-identified patients. CD34+ cells were enriched for by AUTOMACS using the CD34 MicroBead Kit human (Miltenyi Biotec, Cat#130-046-702). CD34+ cells were plated at a density of 2500 cells in cytokine supplemented methylcellulose medium (H4435; STEMCELL Technologies) with increasing concentrations of C220 or DMSO control. Colonies were scored at day 10. All experiments were performed in triplicate using 5 different patient samples (3 PV, 2 PMF).
SET2 cell derived mouse xenografts (CDX)
SET2 CDX studies were conducted in 8 weeks old female SCID Beige mice at Charles River (CR) Discovery Sciences (Morrisville, NC). CR Discovery Services specifically complies with the recommendations of the Guide for Care and Use of Laboratory Animals with respect to restraint, husbandry, surgical procedures, feed and fluid regulation, and veterinary care. The animal care and use program at CR Discovery Services is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC), which assures compliance with accepted standards for the care and use of laboratory animals.
SET2 cells used for implantation were harvested during log phase growth and resuspended in phosphate buffered saline (PBS) containing 50% Matrigel™ (BD Biosciences). Each mouse was injected subcutaneously in the right flank with 1 × 107 tumor cells (0.1 mL cell suspension) while under isoflurane anesthesia to reduce ulcerations. Tumor growth was monitored as the average size approached the target range of 100 to 150 mm3. Twenty-one days later, designated as Day 1 of the study, mice were sorted according to tumor size and treated with C220 at 15 m/kg oral qd and ruxolitinib at 60 mg/kg oral bid as single agent or in combination for 2 weeks. Tumors were measured in two dimensions using calipers, and volume was calculated.
The MTV (n), the median tumor volume for the number of animals, n, on Day 14 was determined for each group. Percent tumor growth inhibition (%TGI) was defined as the difference between the MTV of the designated control group (Group 1) and the MTV of the drug-treated group, expressed as a percentage of the MTV of the control group: % × 100
PV and MF animal models
Bone marrow of 4 months old male Jak2V617F(33) CD45.2 primary mice was mixed in a 50:50 ratio with bone marrow of male CD45.1 C57BL/6 mice (obtained from Charles River) and transplanted into 6–8 weeks old lethally irradiated female recipients (obtained from JAX). After engraftment, mice were randomized by chimerism and blood counts into different treatment arms: vehicle, C220 (12.5 mg/kg, qd, 5/2 days), ruxolitinib (60 mg/kg bid), and combination of C220 (12.5 mg/kg, qd, 5/2 days) and ruxolitinib (60 mg/kg bid). Treatment was administered by oral gavage for 4–6 weeks.
In the conditional MPLW515L model of MF, male Balb/c mice received one dose of 5-fluoro uracil (0.150 mg/g mouse, i.p. injection). 5 days later, bone marrow was isolated, transduced with retroviral supernatant containing MSCV-hMPLW515L-IRES-GFP and injected into lethally irradiated female 6–8 weeks old Balb/c recipients (47). After engraftment, mice were randomized according to GFP and blood counts in to the different treatment arms: vehicle, C220 (10 mg/kg, qd, 5/2 days when used in combination trial; 15 mg/kg, qd, 5/2 days when used versus vehicle only), ruxolitinib (60 mg/kg bid, 7 days), and combination of C220 (10 mg/kg, qd, 5/2 days) plus ruxolitinib (60 mg/kg bid, 7 days).
The ruxolitinib dose of 60 mg/kg bid instead of 90 mg/kg bid in the MPLW515L model was chosen to allow for assessment of activity of the ruxolitinib-C220 combination treatment with tolerable BM toxicity with the combination. Mouse husbandry, care and all animal interventions were in strict compliance with the institutional animal care and use committee (IACUC) guidelines.
Please see data supplement for further details on methods.
Supplementary Material
Statement of Significance.
Expression of PRMT5 and E2F targets is increased in JAK2V617F+ MPN.
Pharmacological inhibition of PRMT5 alters the methylation status of E2F1 and shows efficacy in JAK2V617F/MPLW515L MPN models and primary samples. PRMT5 represents a potential novel therapeutic target for MPN, which is now being clinically evaluated.
Acknowledgement:
R.L.L. has received research support from Prelude Therapeutics.
W.X. has received research support from StemLine Therapeutics.
R.K.R has received research funding from Incyte, StemLine, Constellation.
F.P. has received research support from Prelude Therapeutics and the German research foundation PA 2541/1-1
Financial support:
R.L.L. has received research support from Prelude Therapeutics.
W.X. has received research support from StemLine Therapeutics.
R.K.R has received research funding from Incyte, StemLine, Constellation.
F.P. has received research support from Prelude Therapeutics and the German research foundation PA 2541/1-1
Footnotes
Conflict of interest statement:
R.L.L. is on the supervisory board of Qiagen and is a scientific advisor toLoxo(previous), Imago, C4 Therapeutics and Isoplexis, each of which include an equity interest.He has received research support from Prelude. He receives research support from and consulted for Celgene and Roche and has consulted for Lilly,Janssen, Astellas,MorphosysandNovartis. He has received honoraria from Roche, Lilly and Amgen for invited lectures and from Celgene and Gilead for grant reviews.
N.B., A.G., J.M., M.W., H.L., P.S. and K.V. are employees at Prelude Therapeutics.
W.X. has received research supports from StemLine Therapeutics.
A.D.V. received travel support from Mission Bio and is on the Editorial Advisory Board of Hematology News.
R.K.R. has consulted to Constellation, Incyte, Celgene, Promedior, CTI, Jazz Pharmaceuticals, Blueprint, Stemline, Galecto and has received Research funding from Incyte, Stemline, Constellation.
K.H. has been a consultant of Novo Holding A/S.
F.P. received funding from the German research foundation PA 2541/1–1, the Lauri Strauss Leukemia Foundation and the V Foundation (SU2C).
References
- 1.Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, et al. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci Signal. American Association for the Advancement of Science; 2016;9:rs9–rs9. [DOI] [PubMed] [Google Scholar]
- 2.Blanc RS, Richard S. Arginine Methylation: The Coming of Age. Mol Cell. 2017;65:8–24. [DOI] [PubMed] [Google Scholar]
- 3.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenblatt SM, Liu F, Nimer SD. Arginine methyltransferases in normal and malignant hematopoiesis. Exp Hematol. 2016;44:435–41. [DOI] [PubMed] [Google Scholar]
- 5.Gayatri S, Bedford MT. Readers of histone methylarginine marks. Biochim Biophys Acta. 2014;1839:702–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auclair Y, Richard S. The role of arginine methylation in the DNA damage response. DNA Repair (Amst). 2013;12:459–65. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. Nature Publishing Group; 2013;13:37–50. [DOI] [PubMed] [Google Scholar]
- 8.Tee W-W, Pardo M, Theunissen TW, Yu L, Choudhary JS, Hajkova P, et al. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. Cold Spring Harbor Lab; 2010;24:2772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu F, Cheng G, Hamard P-J, Greenblatt S, Wang L, Man N, et al. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J Clin Invest. American Society for Clinical Investigation; 2015;125:3532–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karkhanis V, Hu Y-J, Baiocchi RA, Imbalzano AN, Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem Sci. 2011;36:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. Springer Basel; 2015;72:2041–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao W, Chen X, Liu L, Shu Y, Zhang M, Zhong Y. Role of protein arginine methyltransferase 5 in human cancers. Biomed Pharmacother. 2019;114:108790. [DOI] [PubMed] [Google Scholar]
- 13.Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016;15:574–87. [DOI] [PubMed] [Google Scholar]
- 14.Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. American Association for the Advancement of Science; 2016;351:1214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radzisheuskaya A, Shliaha PV, Grinev V, Lorenzini E, Kovalchuk S, Shlyueva D, et al. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat Struct Mol Biol. Nature Publishing Group; 2019;26:999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fong JY, Pignata L, Goy P-A, Kawabata KC, Lee SC-W, Koh CM, et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. cancer cell. 2019;36:194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mead AJ, Mullally A. Myeloproliferative neoplasm stem cells. Blood. American Society of Hematology; 2017;129:1607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. [DOI] [PubMed] [Google Scholar]
- 19.James C, Ugo V, Le Couedic J-P, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. [DOI] [PubMed] [Google Scholar]
- 20.Levine RL, Loriaux M, Huntly BJP, Loh ML, Beran M, Stoffregen E, et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106:3377–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. [DOI] [PubMed] [Google Scholar]
- 22.Harrison CN, Vannucchi AM, Kiladjian J-J, Al-Ali HK, Gisslinger H, Knoops L, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. Nature Publishing Group; 2016;30:1701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrison C, Kiladjian J-J, Al-Ali H-K, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis N Engl J Med. Massachusetts Medical Society; 2012;366:787–98. [DOI] [PubMed] [Google Scholar]
- 24.Verstovsek S, Gotlib J, Mesa RA, Vannucchi AM, Kiladjian J-J, Cervantes F, et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol. BioMed Central; 2017;10:156–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delhommeau F, Dupont S, Valle Della V, James C, Trannoy S, Massé A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. [DOI] [PubMed] [Google Scholar]
- 27.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–6. [DOI] [PubMed] [Google Scholar]
- 28.Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788–800. [DOI] [PubMed] [Google Scholar]
- 29.Liu F, Zhao X, Perna F, Wang L, Koppikar P, Abdel-Wahab O, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. cancer cell. 2011;19:283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyer SC, Keller MD, Chiu S, Koppikar P, Guryanova OA, Rapaport F, et al. CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. cancer cell. 2015;28:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koppikar P, Bhagwat N, Kilpivaara O, Manshouri T, Adli M, Hricik T, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012;489:155–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel J-P, Mermel CH, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. American Society of Hematology; 2014;123:e123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. cancer cell. 2010;17:584–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleppe M, Kwak M, Koppikar P, Riester M, Keller M, Bastian L, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015;5:316–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calura E, Pizzini S, Bisognin A, Coppe A, Sales G, Gaffo E, et al. A data-driven network model of primary myelofibrosis: transcriptional and post-transcriptional alterations in CD34+ cells. Blood Cancer J. Nature Publishing Group; 2016;6:e439–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zini R, Guglielmelli P, Pietra D, Rumi E, Rossi C, Rontauroli S, et al. CALR mutational status identifies different disease subtypes of essential thrombocythemia showing distinct expression profiles. Blood Cancer J. Nature Publishing Group; 2017;7:638–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci. 2004;29:409–17. [DOI] [PubMed] [Google Scholar]
- 38.Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013;24:133–45. [DOI] [PubMed] [Google Scholar]
- 39.Fleischman AG, Aichberger KJ, Luty SB, Bumm TG, Petersen CL, Doratotaj S, et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood. 2011;118:6392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29:1356–63. [DOI] [PubMed] [Google Scholar]
- 41.Kaufmann KB, Gründer A, Hadlich T, Wehrle J, Gothwal M, Bogeska R, et al. A novel murine model of myeloproliferative disorders generated by overexpression of the transcription factor NF-E2. J Exp Med. 2012;209:35–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim E, Cheng Y, Bolton-Gillespie E, Cai X, Ma C, Tarangelo A, et al. Rb family proteins enforce the homeostasis of quiescent hematopoietic stem cells by repressing Socs3 expression. J Exp Med. 2017;214:1901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho E-C, Zheng S, Munro S, Liu G, Carr SM, Moehlenbrink J, et al. Arginine methylation controls growth regulation by E2F-1. EMBO J. 2012;31:1785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen E, Ahn JS, Sykes DB, Breyfogle LJ, Godfrey AL, Nangalia J, et al. RECQL5 Suppresses Oncogenic JAK2-Induced Replication Stress and Genomic Instability. Cell Rep. 2015;13:2345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nieborowska-Skorska M, Maifrede S, Dasgupta Y, Sullivan K, Flis S, Le BV, et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood. 2017;130:2848–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonday ZQ, Cortez GS, Grogan MJ, Antonysamy S, Weichert K, Bocchinfuso WP, et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med Chem Lett. American Chemical Society; 2018;9:612–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koppikar P, Abdel-Wahab O, Hedvat C, Marubayashi S, Patel J, Goel A, et al. Efficacy of the JAK2 inhibitor INCB16562 in a murine model of MPLW515L-induced thrombocytosis and myelofibrosis. Blood. American Society of Hematology; 2010;115:2919–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.