

Abstract
Alterations in immune-related pathways are common hallmarks of cancer. A comprehensive understanding of how cancer mutations rewire immune signaling networks and functional output across cancer types is instrumental to realize the full potential of immunotherapy. Here we systematically interrogated somatic mutations involved in immune signaling that alter immune responses in cancer patients. To do so, we developed a Network-based Integrative model to Prioritize Potential immune respondER genes (NIPPER). Identified mutations were enriched in essential protein domains and genes identified by NIPPER were associated with responsiveness to multiple immunotherapy modalities. These genes were used to devise an interactome network propagation framework integrated with drug-associated gene signatures to identify potential immunomodulatory drug candidates. Together, our systems-level analysis results help interpret the heterogeneous immune responses among patients and serve as a resource for future functional studies and targeted therapeutics.
Introduction
Immunotherapy has been considered as a promising strategy for treatment of various types of cancer. Although these immunotherapy methods have made remarkable success, only a minority of patients are observed to respond to the treatment with cytotoxic T lymphocyte antigen-4 (CTLA-4) or programmed death receptor-1 (PD-1) blockade (1). There is a clinical need to identify predictive biomarkers and to understand the potential mechanisms of immunotherapy resistance.
With the development of high-throughput sequencing technology, recent evidence has pointed to a number of predictive biomarkers, such as tumor mutational burden (TMB) (2), neoantigen burden (3), PD-L1 or PD-L2 mRNA expression (4), epigenetic markers and immune cell infiltration profiles (5). With these predictive biomarkers in hand, machine learning-based methods are being built to predict the immunotherapy response. However, it is challenging to integrate different biomarkers. Moreover, increasing studies are beginning to reveal the underlying mechanisms of immunotherapy resistance. The expression of immune-related genes (6), insufficient immune cell infiltration (7), oncogenic pathway activity (8) as well as metabolism dysregulation (9) have all been related to therapy resistance. Nonetheless, additional insights are still needed for a comprehensive understanding of the mechanisms of immunotherapy resistance.
In addition, genes do not function in isolation but rather interact extensively with each other, and therefore any genetic abnormality is not restricted to the gene product that it encodes (10). The emerging tools of network- or pathway-based medicine offer a valuable platform to explore the predictive biomarkers as well as to understand the dysregulation pattern of cancer-related pathways. Previous studies by The Cancer Genome Atlas (TCGA) have systematically analyzed the alteration landscape of cancer-related signaling pathways (11). However, we still lack the knowledge about the alteration pattern of immune-related pathways across cancer type.
To further refine both the genomic and transcriptomic derived events associated with immune infiltration and response to immunotherapy, we first charted the detailed perturbation landscape of immune-related pathways. A network-based immunogenomics model with scoring systems was designed to identify modulation in signaling networks associated with immunogenicity. Furthermore, using a network propagation framework integrated with drug associated gene signatures we discovered potential immunomodulatory drug candidates. These results can help elucidate the functionally relevant mechanisms of immune-related pathway alterations and might inform potential immunotherapy treatment options.
Materials and Methods
Human Immunome
Human immune-related genes were obtained from the ImmPort project (http://www.immport.org) (12). In total, 1811 genes in 17 immune-related pathways were obtained. In addition, we also obtained genes that are identified to be essential to immune response from recent literature (13,14). These genes were defined as essential immunotherapy genes and grouped into the 18th pathway. In total, 2,273 immune-related genes were obtained.
TCGA Patient Cohort
The results in our analysis are based upon datasets generated by TCGA Research Network (http://cancergenome.nih.gov/). We analyzed 33 different TCGA projects, each project represents a specific cancer type, including KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; KICH, kidney chromophobe; LGG, brain lower grade glioma; GBM, glioblastoma multiforme; BRCA, breast cancer; LUSC, lung squamous cell carcinoma; LUAD, lung adenocarcinoma; READ, rectum adenocarcinoma; COAD, colon adenocarcinoma; UCS, uterine carcinosarcoma; UCEC, uterine corpus endometrial carcinoma; OV, ovarian serous cystadenocarcinoma; HNSC, head and neck squamous carcinoma; THCA, thyroid carcinoma; PRAD, prostate adenocarcinoma; STAD, stomach adenocarcinoma; SKCM, skin cutaneous melanoma; BLCA, bladder urothelial carcinoma; LIHC, liver hepatocellular carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; ACC, adrenocortical carcinoma; PCPG, pheochromocytoma and paraganglioma; SARC, sarcoma; LAML, acute myeloid leukemia; PAAD, pancreatic adenocarcinoma; ESCA, esophageal carcinoma; TGCT, testicular germ cell tumors; THYM, thymoma; MESO, mesothelioma; UVM, uveal melanoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; CHOL, cholangiocarcinoma.
Molecular and Clinical Datasets across Cancer Types
Somatic mutations were obtained from the publicly available TCGA MAF file (“MC3”) which covers 10,224 patients (15). This dataset was directly downloaded from Synapse under the number of syn7214402. Six calling methods were applied and numbers of filters were applied. All these mutations were subjected to ANNOVAR (16) and we obtained the conservation and MetaSVM score for each mutation (17).
RNA-seq data were obtained from the TCGA project via the R-package “TCGAbiolinks”, which was specifically developed for integrative analysis with GDC data. We downloaded the Fragments Per Kilobase of transcript per Million mapped reads (FPKM)-based gene expression for 33 types of cancer. In addition, we also obtained the raw read counts for each gene in these samples. The clinical information for patients of 33 cancer types was downloaded from TCGA project via the R-package “TCGAbiolinks”, including the survival status, stages, grades, survival time.
Moreover, we obtained the gene expression of two cohorts treated with immunotherapy. The first cohorts (GSE35640) included metastatic melanoma patients treated with MAGE-A3 immunotherapeutic (18). Clinical benefit included objective responders (complete and partial) according to RECIST 1.0 (19). Only 22 responders and 34 non-responders were used in our analysis. Another cohort (GSE78220) included melanoma biopsies treated with anti-PD-1 therapy (20). We analyzed gene expression of 5 complete responders and 13 progressive disease samples.
Functional assay of mutations in cell lines
All cell lines used in the study were obtained from ATCC, and were authenticated by short tandem repeat (STR) profiling and tested for the absence of mycoplasma. Cells were passaged twice from collection or thawing for use in experiments. In vitro cell viability assays for mutations were adapted from one of our recent studies (21). Two cell lines BaF3 and MCF10A were used and mutation functional calls, including activating, inactivating, inhibitory, non-inhibitory and neutral were determined by comparing with the corresponding wild-type counterparts. We used the consensus calls that were consistent in both cell lines.
Tumor Purity Estimation and Immune Cell Deconvolution
ESTIMATE (Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data) was used to estimate the immune score, stromal score and tumor purity of each patients across 33 cancer types (22). The FPKM-based gene expression profiles were used as input of this method. In addition, TIMER (https://cistrome.shinyapps.io/timer/) was used to estimate the abundances of member cell types in a mixed cell population, using gene expression data (23).
Pathway Mutation Burden Score
Pathway Mutation Burden (PMB) score was defined to evaluate whether the immune pathways were likely to mutate in a specific cancer type. This score considers the proportion of mutated genes as well as the coverage of patients in each cancer type. For an immune pathway i in cancer type j, the score is defined as following:
Where is the number of mutated immune genes in pathway i; is the total number of genes in pathway i; is the number of patients of cancer j that with gene mutations in pathway i; and is the total number of patients sequenced in cancer j.
Immune Response-related Scoring System
Here, we considered three immune response-related scores estimated from gene expression. The immune score that represented the infiltration of immune cells in tumor tissue was assessed based on ESTIMATE (22). The MHC score was formulated from the gene expression levels of the “core” MHC-I set (including HLA-A, HLA-B, HLA-C, TAP1, TAP2, NLRC5, PSMB9, PSMB8 and B2M) (24). The FPKMs of genes were first log-transformed and then median-centered. The mean expression levels of these core genes was defined as the MHC score. The cytolytic activity (CYT score) was based on transcript levels of two key cytolytic effectors, granzyme A (GZMA) and perforin (PRF1), found in previous studies (25).
Classification of Patients with Different Immune Responses
To investigate whether the immune-related scores could be used to classify the patients with different immune responses to immunotherapy, we obtained gene expression profiles of patients with metastatic melanoma treated with MAGE‑A3 immunotherapy (25) or anti-PD1 (20). The three types of immune response-related scores were calculated for each patient. Next, we used the Receiver Operator Characteristic (ROC) Curve Analysis and precision-recall curve to evaluate the effects of these scores. This process was performed by using ‘ROCR’ package in R program. Additional analysis was performed using patients from the TIDE server (26).
Identification of Protein Regions Associated with Immune Response
The domainXplorer algorithm was used to identify the protein regions that are associated with immune scores (27). The protein functional regions were defined as protein domains in Pfam or intrinsically disordered regions identified by Foldindex. In addition, potential domains identified by AIDA (28) were also included in our analysis. Here, we only focused on immune genes-related functional regions.
In brief, domainXplorer used three statistical tests to evaluate whether mutations in a functional protein region were correlated with the immune response-related scores (27). The first linear model was defined as follow:
Where “ImS” is the immune score of each patient, “T” is the tissue of origin of each patient (here is the cancer type), and “D” indicates whether the functional region is mutated or not. In the next step, Wilcox rank sum test was used to compare the immune response-related scores in patients with mutations in the functional region being analyzed against those with mutations in other regions of the same immune gene (Test-2) or those without mutations in the immune gene at all (Test-3). Protein regions with p1<0.05, p2<0.05 and p3<0.05 were identified as to be associated with immune response. In addition, we also identified the protein regions that were correlated with the proportion of immune cell infiltration based on the same method.
Differential Expression Analysis
To identify differentially expressed genes in each cancer type, we downloaded raw RNA-seq counts for all tumor samples, compared to normal tissues. We then used Limma-Voom (29,30) to identify the genes and considered the tumor purity as a factor. Genes with p-values less than 0.05 and four-fold changes in expression were identified as differentially expressed genes in each cancer type.
To evaluate the similarity of cancer types based on the differentially expressed immune genes, we calculated the Jaccard index for each pair of cancer
Where and are the differentially expressed immune genes in cancer type i and j. The directions of differentially expressed genes were not considered in our analysis.
Gene Set Enrichment Analysis
Gene Set Enrichment Analysis was used to identify the enriched immune pathways by differentially expressed genes in each cancer type (31). First, genes were ranked by negative log10 of the differential expression analysis-derived FDR multiplied by the sign of the logFC (log fold change). The “weighted” enrichment statistics were calculated for enrichment or depletion of each immune pathway in specific cancer type.
Prediction of Neoantigens and Functional Screen of Mutations
The somatic mutations were collected from TCGA project and we obtained the HLA alleles for tumor samples from TCIA (https://tcia.at/home) (32). Next, NetMHCpan 4.0 was employed for neoantigen prediction based on the information on mutations and HLA alleles (33). All the wild-type and mutated peptides with 8–11 amino acids were extracted and predicted for the binding affinity. Rank of the predicted affinity was compared to a set of random natural peptides. Rank threshold for strong binding peptides was 0.5% and for weak binding peptides was 2.0%. A cutoff of 0.5% was used to define mutations giving rise to neoantigens. The predicted binding affinity was in nanomolar units.
Network-based Prioritization of Immunome-related Genes and Drug Discovery
Human protein-protein network was integrated into our analysis to prioritize immune dysregulated genes. First, we extracted the subnetwork formed by immune-related genes from an integrated functional network (34). The main component of this subnetwork consists 6,060 interactions among 1,202 immune-related genes. Next, the immune genes were ranked based on the correlation between gene expression and immune scores, MHC scores and CYT scores. Top 50 genes with higher positive- or negative-correlation with immune, MHC and CYT scores were selected as seed genes. In addition, the genes which mutations are associated with immune-related scores were also integrated as seed genes. To simulate the propagation of the genetic alterations through the network, we employed the random walk with restart (35). When an immune gene in the network is ranked in top 50, a value “1” is initially assigned to the gene. Then the genetic alterations are propagated along the neighbors such that the genetic alteration probability was calculated for all genes in the network according to the following equation:
Where is the initial binary probability vector, A denotes a degree-normalized adjacency matrix of the immune subnetwork, and determines the diffusion degree of the genetic influence through the network. We used the optimal value () in our analysis (36). After numbers of iteration, we obtained the final probability for each gene in the network. Genes with greater score than average probability were identified as immune dysregulated genes for each cancer type.
Next, we integrated drug associated gene signatures to identify the drug candidates for each cancer type. We first obtained the expression signature genes from The Connectivity Map (also known as CMAP) (37). For each drug, there are two types of signature genes, one is the up-regulated genes and the other one is the down-regulated genes. We used hypergeometric test to evaluate whether the drug-perturbed gene signatures were enriched in cancer-specific immune gene sets. For each cancer type, we obtained the p-value and then all the p-values across 33 cancer types were combined to a combined p-value.
Where w is the weight for the individual p-values, and z is the z-score of p-values. Here, we set the same weights for all individual p-values. The drugs with adjusted p-values less than 0.01 were identified. We obtained the drugs that had up-regulated/down-regulated gene signatures enriched for genes positively/negatively correlated with immune scores.
Survival Analysis
The patients were divided into two groups based on the median expression of specific immune gene. Log-rank test was used to evaluate the survival difference between two groups. Genes with p-values less than 0.05 were considered as significant.
Results
Genome and Transcriptome-based Immune Response Prediction
Tumor mutational burden (TMB), a measurement of the overall number of genetic alterations observed in a cancer sample, has been suggested as a potentially prognostic marker for immunotherapy (2). In addition, several gene signatures have also been proposed to predict the immune response of patients (Figure 1A). We first evaluated whether if three distinct proposed immune gene expression signatures were associated with immunotherapy response: 1) immune score, which is a measure of total immune infiltrate into the tumor; 2) MHC score, which is a measure of antigen presentation required for tumor cell recognition by T cells and subsequent T cell-mediated killing; and 3) cytolytic activity, which is a measure of cytolytic enzymes used by immune cells to kill tumor cells. Recombinant MAGE-A3 protein combined with immunostimulant AS02B or AS15, has been administered to patients with early metastatic melanoma (18). Based on the gene expression data in response to treatment with MAGE‑A3 immunotherapy, we found that the patients that responded to immunotherapy had significantly higher immune scores (p=0.003), MHC scores (p=1.7e-4) and cytolytic activity (p=0.007, Figure 1B). Moreover, we found that these three immune-related scores indeed distinguished the responder from non-responder patients with area under the curve of the receiver operator characteristic (AUROC) from 0.71 to 0.79 (Figure 1C). To further validate this, we analyzed another gene expression dataset for patients with melanoma treated with anti-PD-1 immune checkpoint blockade (20). We also found that patients which were responsive to the PD-1 immunotherapy exhibited higher immune-related scores (Figure S1A). The AUROCs for classification ranged from 0.52 to 0.85, and the precision-recall curves showed clear separation of responders versus non-responders (Figure S1B-D), which was similar to the results obtained from gene signatures in previous studies (38). These results suggest that the immune-related scores can potentially reflect the immune milieu of tumors in patients and help identify specific factors that may modulate these environments.
Figure-1. Immune response prediction signatures in cancer.

(A) Literature curated immune response prediction signatures. (B) Boxplots show the distribution of Immune score, MHC score and CYT score in immune responder vs non-responder patients. (C) Receiver operating characteristics (ROC) analysis of immune score, MHC score and CYT score from each prediction. The AUCs and 95% confidence levels are labeled. (D) The distribution of immune score, MHC score and CYT score across 33 cancer types in TCGA cohort. Cancers are ranked by their median scores. (E) Scatter plots show the correlation between immune score, MHC score and CYT score across cancer types. ***p<0.001.
We next explored the generalizability of these scoring systems in predicting patient immune responses across cancer types. We first calculated the three immune-related scores (immune score, MHC score and cytolytic activity) for all tumor patients from the TCGA project (Figure 1D). We found that the distributions of the immune-related scores were diverse across 33 cancer types. For example, tumors from patients with kidney renal clear cell carcinoma (KIRC) exhibited higher immune-related scores, and immune checkpoint blockade has shown promising results for front-line treatment of KIRC in multiple phase III clinical trials (39). Moreover, although the three immune-related scores appeared to be correlated with each other, the Pearson Correlation Coefficients (PCC) ranged from 0.51–0.79 (Figure 1E). This observation implied that these scores might be complementary with each other, therefore integrating these scores would facilitate the identification of novel candidate gene targets for immunotherapy.
Immune Mutational Burden Analysis Identifies Critical Pathways and Genes
It has been suggested that the mutations in immune pathways could impact tumor-immune interactions (40). It is unclear, however, how the immune mutational burden (IMB) is associated with patient outcome. Moreover, it seems that the mutation burdens across pathways are varied (11). Therefore it remains elusive if and how mutations in immune-related pathways contribute to higher mutation burden. To systematically investigate the global functional genomic landscape of immune-related genes, we manually curated 2,273 immune-related genes from literature (12,13). These genes were further classified into 18 immune-related pathways (Figure 2A). Next, we analyzed whole exomes from 100 melanoma patients treated with CTLA-4 blockade (41). We first calculated the number of mutations (tumor mutation burden, TMB) and the number of mutations located in immune-related genes (defined as immune mutation burden, IMB). We further compared TBM and IMB between responders and non-responders. The mutation burden and immune burden of responders were significantly higher than those of non-responders (Figure S2A, p<0.05). In addition, mutation burden and immune burden could predict immune response with similar power (Figure S2B, AUCs=0.68). Furthermore, we calculated the IMB for each patient in TCGA cohort. We found that the cancer types that are likely to respond well to immunotherapy had higher proportion of immune-related gene mutations, such as skin cutaneous melanoma (SKCM) and kidney cancer (Figure S2C). These results suggest that the immune burden is a useful indicator of immune response; target sequencing of immune-related genes would help predict the immune response of patients.
Figure-2. Critical mutated immunological pathways and genes in cancer.
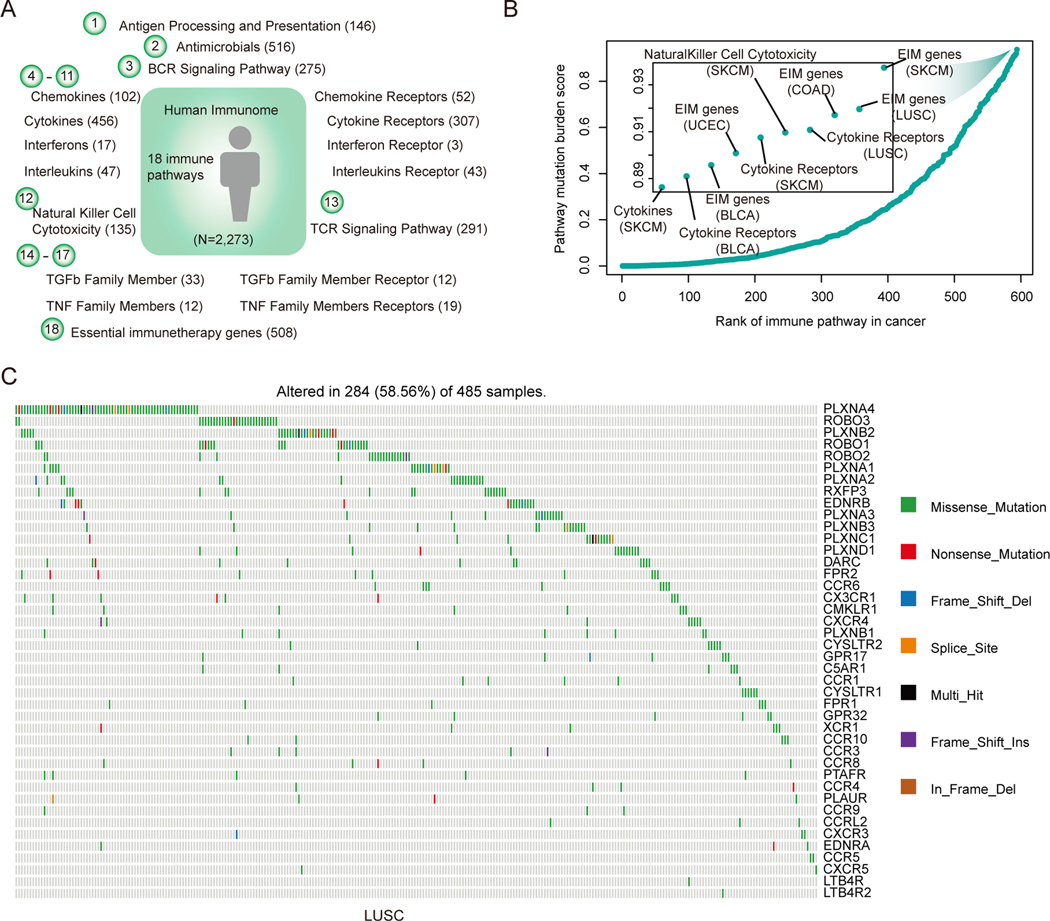
(A) Human immune pathways and number of genes in each pathway. In total, 2,273 immune-related genes in 18 pathways are analyzed. (B) The PMB score for each immune-related pathway in cancer. Each dot represents a pathway in a specific cancer type. The immune score is calculated as the product of the proportion of mutated genes in the pathway and the percentage of mutated samples. Pathways are ranked by the PMB score. (C) The mutations of genes in cytokine receptor pathway in LUSC. Genes were ranked by the mutation frequency.
Next, we defined a Pathway Mutation Burden (PMB) score to evaluate to what extent each immune-related pathway is perturbed by genomic alterations in tumor. This score is calculated based on the mutation frequency and the proportion of mutated genes in each pathway. We found that the essential genes identified by CRISPR-Cas9 KO screens (13) had higher PMB scores across cancer types (Figure 2B). Moreover, we observed another pathway-cytokine receptors (CRs) with higher PMB scores in LUSC, SKCM and DLBC (Figure 2B). We next focused on the CR mutations in LUSC (Figure 2C). Consistent with previous studies, the mutations in CR pathway exhibited a mutual exclusive pattern (42). Moreover, the mutations in CR pathway had higher functional impact scores than other mutations evaluated by metaSVM (17) (Figure S2D). The top-1 gene (PLXNA4) ranked by mutation frequency was mutated in approximately 19% of lung cancer patients. PLXNA4 was shown previously to promote tumor progression and tumor angiogenesis by activating VEGF and FGF signaling (43). We further found that the expression of PLXNA4 was correlated with the patients’ survival in LUSC (Figure S2E, p=0.017). These results suggest that the mutations in cytokine receptors pathway might play critical roles in cancer and the proposed PMB index could help prioritizing key mutations in cancer pathways.
Widespread Transcriptional Alterations of Human Immunome across Cancer Types
Next, we interrogated if and to what extent the human immunome is altered in cancer at the gene expression level. We identified differentially expressed genes across 18 cancer types with more than five normal samples. As tumor purity may affect the identification of differentially expressed genes (44), we first estimated the tumor purity of each sample (Figure S3A) and identified the differentially expressed genes using tumor purity as a co-variable in the regression model. We found that the immune-related genes were significantly perturbed in cancer when compared to other coding genes (Figure 3A and Figure S3B, p-values<0.05 for all cancer types, Fisher’s exact test). The Odd Ratios (ORs) of immune genes vs non-immune genes were higher than 1.0 in all cancer types. These results indicate that these genes could play a role in promoting tumor growth.
Figure-3. Widespread transcriptome alterations of human immunome in cancer.

(A) The proportion of differentially expressed immunogically relevant genes and all human genes. The odds ratios and 95% confidence levels of Fisher’s exact tests are shown in the right panel. All p-values are less than 0.05 for 18 cancer types with at least fiver normal samples. (B) Cluster of cancer types based on the similarity (Jaccard index) of differentially expressed immune genes. Cancer types of similar tissue origins are clustered together. (C) Heat map shows the p-values and enrichment scores for the differentially expressed genes enriched in immune-related pathways. Each row represents a cancer type and each column represents an immune-related pathway. The sizes of dots show the -log (p-values) and the colors of dots show enrichment scores. The bar plots below the heat map show the number of cancer types enriched for each pathway. (D) - (F) Gene Set Enrichment Analysis shows enrichment or depletion of cytokines, cytokines receptors and activated antigen processing and presentation in breast cancer. The horizontal bar in graded color from red to blue represents the rank-ordered, non-redundant list of all genes. The vertical black lines represent the projection of immune genes in each pathway onto the ranked gene list. (D) for antigen processing and presentation pathway, (E) for cytokines pathway and (F) for pathway cytokines receptors in breast cancer.
Cancer types with similar tissue origins often show similar transcriptome profiles (45). Next, we calculated the Jaccard index for each pair of cancer based on the similarity of differentially expressed immune-related genes (see details in Methods). We found that cancer lineages with similar tissue origin were clustered together (Figure 3B), such as lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), cholangiocarcinoma (CHOL) and liver hepatocellular carcinoma (LIHC), and three types of kidney cancer. These results indicate that these cancer types show immunome profiles within similar tumor microenvironment.
In addition, we ranked all genes based on their differential expression and used gene set enrichment analysis (GSEA) to explore which immune-related pathways were likely to show transcriptional perturbation. 16/18 immune-related pathways were significantly altered in at least one cancer type (Figure 3C). The antigen processing and presentation, cytokines and cytokines receptors pathways were perturbed in more than 16 cancer types (Table S1). Moreover, we found that these immune-related pathways showed consistent trend (enriched or depleted) across cancer types. For instance, while tumors have been observed to down-regulate antigen presentation for immune escape (46), we found antigen processing and presentation pathway was likely to be activated in breast invasive carcinoma and glioblastoma (BRCA and GBM, Figure 3C-D and Figure S3C). In addition, cytokines and cytokine receptors pathways were repressed in BRCA and GBM (Figure 3E-F and Figure S3D, FDR<0.001). Moreover, we performed expression analysis based on immunological gene signatures in MSigDB (47), which provides more specific immune pathways. In total, 4,872 immune-related gene signatures were analyzed. We found that approximately 67.3% of these immunological gene signatures were enriched in differentially expressed genes in at least one cancer type (Figure S4). Moreover, there were 27 gene signatures significantly enriched in >11 cancer types (Table S2). These results suggest that immunologically relevant genes are likely to be perturbed in cancer.
Critical Regions of Genes Associated with Immune Response
Growing evidence suggests that mutations in certain genes influence the immune response (27). Identification of these genes may provide valuable insights for improving immunotherapy. Thus, we explored ‘domainXplorer’ to identify critical regions of immune genes that are associated with immune response in cancer (27). Given the importance of the proposed immune-related scores in predicting immune response, we next determined whether cancer mutations in immunity candidate driver regions were significantly correlated with these scores (Table S3). Our analysis yielded a total of 209 protein regions in 145 immune-related genes that are likely associated with cancer immunity (Figure 4A). Specifically, mutations in seven genes (UBXN1, LTBP2, STAT1, NGFR, NRAS, MET and LTBP4) were significantly correlated with all of three types of immune-related scores. LTBP4, a member of the latent TGFβ binding protein gene family, had been linked to several human diseases (48). We found that patients with EGF_CA domain mutations exhibited significantly higher immune scores, MHC scores and cytolytic activity (Figure 4B, p<0.05). Moreover, we also identified mutations of STAT1 significantly correlated with these scores, which was consistent with previous observation that STAT1 played an important role in the innate immune response (49). These results collectively expand the catalog of potential cancer immune drivers and highlight the importance of taking into account the protein structural context to identify patients that are likely to clinically benefit from immunotherapy.
Figure-4. Critical protein-regions related to immune response in cancer.

(A) The distribution of p-values for testing the association of protein regions and immune response scores. The bottom Venn plot shows the overlap of protein-regions that are related with immune score, MHC score and CYT score. The seven overlap genes were shown. (B) Representative mutated protein regions that are related to immune response in cancer. The mutations in the EGF_CA domain of LTBP4 gene are shown. The up-panel shows the mutations in the LTBP4 and the domain region is marked by green and mutations are shown as red balls. The immune score, MHC score and CYT score distributions of the samples with domain mutations, other region mutations and wild-type (WT) are shown in the bottom-panel. ***, p<0.01, Wilcox’s rank sum test. (C) The overlap of protein regions whose mutations were correlated with immune response scores and immune cell infiltration. Square color reflects the number of overlapped protein regions. ‘No cross’ indicates p<0.05. (D) The overlapped seven genes mutation associated with distinct immune cell infiltration. (E) The distribution of immune cell infiltration for patients with in-domain mutations, outside-domain mutations and wild type of LTBP4 gene. Left for CD8+ T cells and right for neutrophils.
Somatic Mutations Associated with Immune Cell Infiltration
Our above analyses identified candidate protein regions that are potentially correlated with immune response. However, we are still lack of knowledge about the potential mechanism. An emerging role in treatment response has been attributed to immune cell infiltration in human tumors (7). Thus, we next explored to what extent the mutations in these regions were correlated with immune cell infiltration. We first identified the protein regions whose mutations were significantly correlated with the abundance of six tumor-infiltrating immune cells (TIIC) subsets (B cells, CD4 T cells, CD8 T cells, macrophages, neutrophils, and dendritic cells) (50). We also identified the proteins that were associated with B cell receptor (BCR) and TCR diversity, by CPK (CDR3s per kb of reads) that evaluates the clonotype diversity (51). We found that both MHC and CYT scores showed significant association with the expansion of tumor-infiltrating B cells after correction for tumor purity (Figure S5A). In addition, MHC score was weakly associated with TCR diversity, potentially due to clonal expansion of tumor antigen-specific T cell populations. CYT score on the other hand was strongly correlated with CPK (Figure S5B). We found that the protein regions that were correlated with immune score, MHC score and cytolytic activity were significantly overlapped with those correlated with TIIC abundance (Figure 4C). Especially, there were a higher number of protein regions correlated with CD8 T cells, neutrophils, and dendritic cells.
Specifically, we found that seven genes, whose mutations correlated with all three types of immune-related scores, were associated with at least one type of immune cell abundance (Figure 4D). For example, patients with LTBP4 mutations exhibited significantly higher abundance of CD8 T cells and neutrophils (Figure 4E). CD8+ T cells are crucial mediators of anti-tumor immune response and the targets of checkpoint blockade (52), while the role of neutrophils is complicated as they can exert either tumor enhancing or tumor clearance effect (53). Moreover, we found that not only the genomic variants in LTBP4 were correlated with immune cell abundance, but patients with copy number alterations also exhibited significantly different abundance (Figure S5C). These results suggest that the genomic alterations might contribute to immunotherapy response through affecting the immune cell infiltration in cancer.
Critical Genes Enriched for Neoantigens in Cancer
Increasing studies have demonstrated that mutation-derived neoantigens form a major ‘active ingredient’ of successful cancer immunotherapies (3). Next, we predicted the neoantigens that were derived from mutations of TCGA project based on NetMHC (33). There was a limited proportion of neoantigens resulting from mutations in protein coding genes and approximately 1.2% of all mutations in immune-related genes could potentially generate neoantigens (Figure 5A). This is consistent with a recent study showing that neoantigens arose from mutations in only about 60% of the genes (54), while the majority of neoantigens might originate from alternative or noncoding sources. We found that the immunologically-relevant genes (from ImmPort) tended to generate significantly more neoantigens (such as PIK3CA, MUC4, EGFR) compared with coding genes in general (Figure 5A, p=1.18e-4, Fisher’s exact test). Approximately 25% of the mutations in PIK3CA and HRAS could generate neoantigens (Figure 5B). This result is consistent with the observation that RAS plays a role in immunotherapy (55).
Figure-5. Neoantigen enriched in immunologically relevant genes.

(A) The left panel shows the proportion of neoantigens in immune gene compared to all coding genes (p=1.18e-4, Fisher’s exact test). The right panel shows the distribution of binding affinity of HLAs in wild-type and mutated peptides (p<2.2e-16, Wilcox’s rank sum test). (B) Number of neoantigens in immune-related genes. The barplots show the number of neoantigens generated from each immune-related gene. The red dot line shows the number of neoantigens/number of total mutations in each gene. (C) Distribution of the mutations in PIK3CA that generate neoantigens. (D) Distribution of the mutations in EGFR that generate neoantigens. (E) The left and right panels show the 3D structure of PIK3CA and EGFR, red dots represent the mutations in the same cluster. The middle panels show the proportion of functional mutations, the green bars for mutations encoding neoantigens and grey bars for all screened mutations. **p<0.05 for Fisher’s exact test.
Next, we focused on mutations in PIK3CA and EGFR that give rise to neoantigens. Based on the viability-based functional screen in two cell lines (Ba/F3 and MCF10A) (21), we found that the mutations that encode neoantigens were likely to be activating mutations in both cell lines (Figure 5C–5D). For example, we identified 46 missense mutations that could generate neoantigens in PIK3CA. Among these mutations, 41 mutations were in the functional dataset. We found that 40 mutations were activating mutations and one was inactivating mutations. Moreover, we identified a mutation cluster (including R38C/H, E81K, R88Q, R93W, K111E and R115L) in PIK3CA (Figure 5E). For EGFR, we identified 17 mutations that could generate neoantigens. In total, 14 mutations were screen for function and 11 mutations were activating, two were neutral and 1 inconclusive in two cell lines (Figure 5D). We also found seven mutations formed a cluster in 3D (including R108K, R252/P/C/H and A189V/D, Figure 5E). We hypothesize that functional mutations which cause a conformational change in the protein may be more likely to be recognized by MHC molecules for immune pruning, but if these mutations subsequently suppress inflammatory signaling it may allow for proliferation of tumor cells with activating mutations.
Prioritization of Candidate Immune Responders Based on Immunome Network
Our above results indicate widespread genomic and transcriptome perturbations in the cancer immunome network. An integrated landscape of these perturbations offers a basis for further advancing our understanding of the immune regulation in cancer. Thus, we proposed a Network-based Integrative model to Prioritize Potential immune respondER genes (NIPPER) (Figure 6A). Briefly, we first identified the genes whose mutations or expression were significantly correlated with immune scsores, MHC scores and CYT scores. These genes were then mapped to an immune-related protein-protein interaction (PPI) network, which consisted of 6,060 interactions among 1,202 immune-related genes. Next, the mutational effects were propagated in the PPI network. Finally, the genes with driver probability greater than expected were identified as immune response-related signature genes. This process was repeated for all cancer types. Here, the genes positively/negatively correlated with immune-related scores were analyzed, separately.
Figure-6. Network-based immune responder candidate identification.

(A) Flowchart to identify potential personalized immune responder genes and candidate drugs for each patient. For each cancer type, expression or mutations of genes that are correlated with immune response scores are mapped to an immune gene-gene interaction network. The immune response correlation is propagated via network links and new responder genes are identified based on random walk method. Drug perturbed gene signatures (n=6,100) are integrated for identification of the drugs that enriched the candidate immune responder genes (FDR<0.01) in specific patients. (B) Top ranked immune response score-related genes across cancer types. Genes are ranked by the number of cancer types that show correlation between gene expression and immune response scores. (C) Top-ranked 39 genes are identified and the corresponding pathways are shown. The subnetworks formed by these 39 genes are shown in the up-panel with node colors indicating whether they are identified by the CRISPR-Cas9. (D) The expression of PDIA3 (linked more known essential immune genes) is correlated with patients’ survival in GBM. (E) ROC curve for the classification of patient response to immunotherapy based on candidate genes. (F) Kaplan-Meier survival curves for patients classified by the average expression of candidate genes.
Based on the proposed network-based model, we identified 39 genes as signature genes in 33 cancer types (Figure 6B). High expressions of these genes were likely to be correlated with immune-related scores. These genes were involved in 11 immune-related pathways, and 8 genes (such as STAT1, B2M, TAP1 and TAP2) had been identified as essential genes in immune response. Moreover, we found that these candidate immune response-related genes were clustered together as modules in the PPI network (Figure 6C). Seven of the 8 essential immune genes were linked in a dense module, and STAT1 was clustered together with more novel immune response-related genes. Interestingly, we identified one candidate gene-PDIA3, which linked 7 known essential genes. The expression of this gene was correlated with the patients’ survival in several cancer types, including CESC, LUAD, HNSC and GBM (Figure 6D and Figure S6). These observations suggest that this gene may play critical roles in immunotherapy, which is consistent with the conclusion of one recent study (56).
Next, we validated these genes in a cohort of melanoma that underwent treatment with adoptive T cell transfer (24). We found that the expression of these genes could classify the responder and non-responder patients with an AUC of 0.713 (Figure 6E). This is similar with the results based on PD1 or PDL1 expression. In another cohort of patients with melanoma treated with anti-PD1, we found that the power of NIPPER was higher than PD1 and PDL1 expression-based and similar as TMB-based methods (Figure S7). We also used TIDE to compare our gene signatures with other public signatures in 17 datasets (26). Our NIPPER signatures achieve AUCs>0.6 in most (9/15) datasets (Figure S8). Moreover, the average expression of these genes was significantly associated with improved overall survival (Figure 6F, log-rank p=0.02). Taken together, these results indicate that signaling networks identified by NIPPER are relevant to response to multiple immunotherapy modalities, and may offer insight on critical signaling cascades that can be targeted to improve immunotherapy outcomes.
Mutation-Perturbed HLA Binding and Potential Immune-related Drugs
Next, we evaluated mutations in the genes identified by NIPPER in different cancers. Here, all mutations in prioritized genes across cancer types were analyzed. We found that the mutations in these genes were likely to alter the binding affinity of HLAs (Figure 7A, p<0.001, Wilcox’s rank sum test). Specifically, we identified 135 mutations in 28 genes that perturbed binding of HLAs (Figure 7B and Table S4). These mutations were distributed in various types of cancer. We identified six mutations in PIK3R1 (encoding p85α) that reduced binding of HLAs (Figure 7B) and some mutations clustered in a possibly functional spot (Figure 7C). Consistently, we identified two mutations (N564D and K567E) that increased growth of cancer cell lines (Figure 7D). These results suggest that our network-based method helps identify cancer functional mutations by their change in binding affinity to HLAs.
Figure-7. Candidate HLA-perturbing mutations and immune-response related small molecules.

(A) The distribution of HLA binding affinity for wild-type (yellow) and mutated peptides (blue) for all NIPPER prioritized genes. ***p<0.001, Wilcox’s rank sum test. (B) The number of mutations that alter binding of HLAs. (C) The 3D structure of candidate mutations in PIK3R1. (D) The relative growth rate of wild-type and mutant PIK3R1 cells in two cell line models. (E) Candidate immune-response related drugs and the corresponding p-values in cancer types predicted by network-based method.
Identification of candidate small molecule drugs is critical for the immunotherapy in cancer. Herein we identified candidate drugs that could increase the expression of identified immune gene signatures based on GSEA analysis (Figure 6A). We obtained more than 6,000 small molecules and their perturbation gene signatures from The Connectivity Map (57). We identified 49 small molecules that significantly perturbed the expression of immune gene signatures (p<0.05, Figure 7E and Table S5). Interestingly, we identified propofol as the top one small molecule for increased immune response. Evidence had shown that propofol could possibly induce a favorable immune response in terms of preservation of IL-2/IL-4 and CD4+/CD8+ T cell ratio in the perioperative period for breast cancer (58). In vitro and in vivo studies had also suggested that propofol could independently reduce migration of cancer cells and metastasis (59). Taken together, these results suggest that the network-based integrative model not only can identify immune response-related gene signatures, it also helps prioritize candidate drugs for immunotherapy in cancer.
Discussion
Systematically understanding the regulation of immune systems opens more avenues for cancer immunotherapy with a potent clinical efficacy. By integration of pan-cancer omics datasets, we have examined human tumor correlations in the context of immune-related pathways to an extent that is not previously possible. High-throughput gene expression data have been widely used to investigate differentially expressed genes in various types of cancer. Strikingly, we observed extensive immunological gene signatures with widespread perturbation in cancer compared with normal samples. These deregulated immunologically relevant genes are enriched in ‘antigen processing and presentation’, ‘cytokines’ and ‘cytokines receptors’ pathways. Cytokines are critical molecular messengers for cells of the immune system to communicate with one another, which can directly stimulate immune effector cells and enhance tumor cell recognition. Increasing studies have demonstrated that cytokines have broad anti-tumor activity (60). Our analysis revealed that there are widespread perturbations of cytokine pathways in more than 80% of cancer types. A more detailed understanding of cytokine pathway regulation will provide new opportunities for improving cancer immunotherapy.
It is still unclear which factors are key players in regulating the immunome homeostasis. Thus, we systematically analyzed two types of genetic regulation (including CNVs and eQTLs) in immune genes (Figure S9A-C). We found that a number of immunologically relevant genes recurrently dysregulated in various types of cancer. For example, up-regulation of BIRC5 is correlated with CNV amplification (61); and down-regulation of CCL14 is correlated with CNV deletion across cancer types. Moreover, eQTLs analysis revealed a hotspot locus that modulates the expression of DEFB1 (Figure S10A-D), which might function through perturbation of CTCF binding. While a number of immune-related genes show correlation between copy number variations/mutations and gene expression, the relationship is weak and could not fully explain immune-related transcriptome perturbations. Thus, it is likely that other genetic (such as post-transcriptional regulation) or epigenetic variables (such as DNA methylation or histone modification) also contribute to the transcriptome perturbations in cancer.
Prior studies have proposed that the immune score, MHC score and CYT score are correlated with immune response. For example, Rooney et al. quantified the cytolytic activity and identified associated properties across 18 tumor types (25). We found that these three scores that interrogate different aspects of immune signaling are complementary with each other, providing more information than any one signature alone. Further information was gained by utilization of a network-based approach, specifically a random walk with restart network propagation, which considers both the mutations in individual genes as well as the topology of interactions between the corresponding proteins. Integration of the network topology information has been demonstrated to have much higher accuracy in identifying cancer-related genes (62). Thus, we proposed an integrated network model to predict the critical signaling cascades for immunotherapy response. This method identified several well-known immune response-related genes, such as B2M (63), TAP1 (64) and TAPBP (65), as well as several novel genes such as PDIA3. Moreover, we predicted candidate small molecules that may sensitize to immunotherapy based on whether the treatment could induce the expression of these signature immune genes. In particular, propofol was identified as the top-ranked drug candidates for immunotherapy. Propofol is the most commonly used in clinical anesthesia and propofol exhibits a good inhibitory effect on tumor recurrence and metastasis (59). Our current study found that propofol likely induces the expression of immune response-related genes, which may contribute to improved efficacy of immunotherapy.
Taken together, our integrative analysis of the human immunome across cancer types reveals several candidate gene and mutation markers that may be critical for understanding patient responses to immunotherapy. Future studies will need to evaluate the potential molecular functions of our identified signaling cascades by low-throughput experiments.
Supplementary Material
Significance.
This study demonstrates that integration of multi-omics data can help identify critical molecular determinants for effective targeted therapeutics.
Acknowledgments
The authors would like to acknowledge Kevin Drew and Edward M. Marcotte from University of Texas at Austin for helpful discussions. The authors are grateful to contributions from TCGA Research Network Analysis Working Group, and also acknowledge the Biomedical Research Computing Facility at UT Austin and Texas Advanced Computing Center (TACC) for high-performance computing assistance.
Grant Support
This work was supported by National Institutes of Health grant (K22CA214765 to S.Y.), Komen Foundation grant (CCR19609287 to S.Y.), Young Investigator Grant by Breast Cancer Alliance (to N.S.), Liz Tilberis Early Career Award by Ovarian Cancer Research Alliance (Grant# 649968 to N.S.), Alfred P. Sloan Scholar Research Fellowship (FG-2018–10723 to N.S.), Cancer Prevention and Research Institute of Texas grants RR170079 (to B.L.) and RR160093 (to S.G.E.). N.S. is a CPRIT Scholar in Cancer Research with funding from the Cancer Prevention and Research Institute of Texas (CPRIT) New Investigator Grant RR160021. D.J.M. is supported by National Cancer Institute grant K99CA240689. S.A.S. acknowledges support by the NCI (R50RCA211482). A.C. is supported by Department of Defense grant CA191245. C.J.W. is a Scholar of the Leukemia and Lymphoma Society. G.B.M. is supported by a kind gift from the Miriam and Sheldon Adelson Medical Research Foundation, SAC110052 from the Susan G Komen Research Foundation, Breast Cancer Research Foundation, Ovarian Cancer Research Alliance, Prospect Creek Foundation and NCI grant (U01CA217842).
G.B.M. consults with AstraZeneca, Chrysalis, GSK, ImmunoMET, Ionis, Lilly, PDX Pharmaceuticals, Signalchem Lifesciences, Symphogen, Tarveda, Turbine, Zentalis Pharmaceuticals, receives clinical trials support from AstraZeneca, Lilly, GSK, Genentech, has stock options with Catena Pharmaceuticals, ImmunoMet, SignalChem and Tarveda, sponsored research from Nanostring Center of Excellence and Ionis (Provision of tool compounds), and has licensed technology to Nanostring (DSP patents) and Myriad Genetics (HRD assay). S.A.S. previously advised and has received consulting fees from Neon Therapeutics. S.A.S. reported nonfinancial support from Bristol-Myers Squibb, and equity in Agenus Inc., Agios Pharmaceuticals, BreakBio Corp., Bristol-Myers Squibb, Indiscine and Lumos Pharma, outside the submitted work. C.J.W. is Founder and SAB member of Neon Therapeutics outside the submitted work; in addition, C.J.W. has a patent for PCT/US2011/036665 licensed to Neon Therapeutics, and holds equity in BioNTech, Inc.
Footnotes
Disclosure of Potential Conflicts of Interest
The other authors declare no competing interests.
References
- 1.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 2017;377:2500–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74 [DOI] [PubMed] [Google Scholar]
- 4.Yearley JH, Gibson C, Yu N, Moon C, Murphy E, Juco J, et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin Cancer Res 2017;23:3158–67 [DOI] [PubMed] [Google Scholar]
- 5.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015;527:249–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneider K, Marbaix E, Bouzin C, Hamoir M, Mahy P, Bol V, et al. Immune cell infiltration in head and neck squamous cell carcinoma and patient outcome: a retrospective study. Acta Oncol 2018;57:1165–72 [DOI] [PubMed] [Google Scholar]
- 8.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5 [DOI] [PubMed] [Google Scholar]
- 9.Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016;531:651–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011;12:56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018;173:321–37 e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhattacharya S, Andorf S, Gomes L, Dunn P, Schaefer H, Pontius J, et al. ImmPort: disseminating data to the public for the future of immunology. Immunologic research 2014;58:234–9 [DOI] [PubMed] [Google Scholar]
- 13.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature 2017;548:537–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018;359:801–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH, et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer cell 2017;31:820–32 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 2010;38:e164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2015;24:2125–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ulloa-Montoya F, Louahed J, Dizier B, Gruselle O, Spiessens B, Lehmann FF, et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol 2013;31:2388–95 [DOI] [PubMed] [Google Scholar]
- 19.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999;286:531–7 [DOI] [PubMed] [Google Scholar]
- 20.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016;165:35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng PK, Li J, Jeong KJ, Shao S, Chen H, Tsang YH, et al. Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell 2018;33:450–62 e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nature communications 2017;8:1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu J, Li K, Zhang W, Wan C, Zhang J, Jiang P, et al. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med 2020;12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porta-Pardo E, Godzik A. Mutation Drivers of Immunological Responses to Cancer. Cancer Immunol Res 2016;4:789–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu D, Jaroszewski L, Li Z, Godzik A. AIDA: ab initio domain assembly server. Nucleic acids research 2014;42:W308–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep 2017;18:248–62 [DOI] [PubMed] [Google Scholar]
- 33.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol 2017;199:3360–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J, et al. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 2015;347:1257601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. American journal of human genetics 2008;82:949–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hofree M, Shen JP, Carter H, Gross A, Ideker T. Network-based stratification of tumor mutations. Nature methods 2013;10:1108–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006;313:1929–35 [DOI] [PubMed] [Google Scholar]
- 38.Ock CY, Hwang JE, Keam B, Kim SB, Shim JJ, Jang HJ, et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nature communications 2017;8:1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019;380:1103–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity 2018;48:812–30 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res 2012;22:398–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kigel B, Rabinowicz N, Varshavsky A, Kessler O, Neufeld G. Plexin-A4 promotes tumor progression and tumor angiogenesis by enhancement of VEGF and bFGF signaling. Blood 2011;118:4285–96 [DOI] [PubMed] [Google Scholar]
- 44.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nature communications 2015;6:8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Sahni N, Pancsa R, McGrail DJ, Xu J, Hua X, et al. Revealing the Determinants of Widespread Alternative Splicing Perturbation in Cancer. Cell Rep 2017;21:798–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Algarra I, Collado A, Garrido F. Altered MHC class I antigens in tumors. Int J Clin Lab Res 1997;27:95–102 [DOI] [PubMed] [Google Scholar]
- 47.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011;27:1739–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lamar KM, Miller T, Dellefave-Castillo L, McNally EM. Genotype-Specific Interaction of Latent TGFbeta Binding Protein 4 with TGFbeta. PloS one 2016;11:e0150358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 1996;84:443–50 [DOI] [PubMed] [Google Scholar]
- 50.Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, et al. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res 2017;77:e108-e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li B, Li T, Pignon JC, Wang B, Wang J, Shukla SA, et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet 2016;48:725–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ochel A, Cebula M, Riehn M, Hillebrand U, Lipps C, Schirmbeck R, et al. Effective intrahepatic CD8+ T-cell immune responses are induced by low but not high numbers of antigen-expressing hepatocytes. Cellular & molecular immunology 2016;13:805–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang CW, Strong BS, Miller MJ, Unanue ER. Neutrophils influence the level of antigen presentation during the immune response to protein antigens in adjuvants. Journal of immunology 2010;185:2927–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pearson H, Daouda T, Granados DP, Durette C, Bonneil E, Courcelles M, et al. MHC class I-associated peptides derive from selective regions of the human genome. J Clin Invest 2016;126:4690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res 2017;23:3012–24 [DOI] [PubMed] [Google Scholar]
- 56.Zhuang ZM, Wang XT, Zhang L, Tao LY, Lv B. The effect of PDIA3 gene knockout on the mucosal immune function in IBS rats. Int J Clin Exp Med 2015;8:6866–77 [PMC free article] [PubMed] [Google Scholar]
- 57.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017;171:1437–52 e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woo JH, Baik HJ, Kim CH, Chung RK, Kim DY, Lee GY, et al. Effect of Propofol and Desflurane on Immune Cell Populations in Breast Cancer Patients: A Randomized Trial. Journal of Korean medical science 2015;30:1503–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J, Dong W, Wang T, Liu L, Zhan L, Shi Y, et al. Effects of etomidate and propofol on immune function in patients with lung adenocarcinoma. American journal of translational research 2016;8:5748–55 [PMC free article] [PubMed] [Google Scholar]
- 60.Dranoff G Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer 2004;4:11–22 [DOI] [PubMed] [Google Scholar]
- 61.Ghaffari K, Hashemi M, Ebrahimi E, Shirkoohi R. BIRC5 Genomic Copy Number Variation in Early-Onset Breast Cancer. Iranian biomedical journal 2016;20:241–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng F, Jia P, Wang Q, Lin CC, Li WH, Zhao Z. Studying tumorigenesis through network evolution and somatic mutational perturbations in the cancer interactome. Mol Biol Evol 2014;31:2156–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pereira C, Gimenez-Xavier P, Pros E, Pajares MJ, Moro M, Gomez A, et al. Genomic Profiling of Patient-Derived Xenografts for Lung Cancer Identifies B2M Inactivation Impairing Immunorecognition. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23:3203–13 [DOI] [PubMed] [Google Scholar]
- 64.Ling A, Lofgren-Burstrom A, Larsson P, Li X, Wikberg ML, Oberg A, et al. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology 2017;6:e1356143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cui D, Wang J, Zeng Y, Rao L, Chen H, Li W, et al. Generating hESCs with reduced immunogenicity by disrupting TAP1 or TAPBP. Bioscience, biotechnology, and biochemistry 2016;80:1484–91 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.