

Abstract
Purpose
Mutations in the zona pellucida glycoprotein genes have been reported to be associated with empty follicle syndrome (EFS) and abnormal zona pellucida (ZP). In this study, we performed genetic analysis in the patients with female infertility due to abnormal zona pellucida and empty follicle syndrome to identify the disease-causing gene mutations in these patients.
Methods
We characterized three patients from two independent families who had suffered from empty follicle syndrome or abnormal zona pellucida. Whole exome sequencing and Sanger sequencing were used to identify the mutations in the families. Western blot was used to check the expression of wild type and mutant disease genes.
Results
We identified two novel mutations in these patients, including a novel compound heterozygous mutation (c.507delC, p. His170fs; c.239 G>A, p. Cys80Tyr and c.241 T>C, p. Tyr81His) in ZP1 gene and a compound mutation in ZP2 gene (c.860_861delTG, p.Val287fs and c.1924 C>T, p.Arg642Ter). Expression of the mutant ZP1 protein (p. Cys80Tyr and p. Tyr81His) is significantly decreased compared with the wild-type ZP1. Other three mutations produce truncated proteins.
Conclusions
Our findings expand the mutational spectrum of ZP1 and ZP2 genes associated with EFS and abnormal oocytes and provide new support for the genetic diagnosis of female infertility.
Electronic supplementary material
The online version of this article (10.1007/s10815-020-01926-z) contains supplementary material, which is available to authorized users.
Keywords: ZP1, ZP2, Female infertility, Empty follicle syndrome
Introduction
The zona pellucida (ZP) is an extracellular glycoprotein matrix universally surrounding mammalian eggs, which is essential to mediate sperm binding, prevent postfertilization polyspermy, and provide physical barrier for protecting the developing embryo [1]. The ZP matrix of human is composed of four glycoproteins of the ZP family, ZP1, ZP2, ZP3, and ZP4. It has been reported that in humans, ZP can bind to capacitated spermatozoa and induce acrosomal exocytosis [2–4]. In mouse, ZP1 can covalently cross-link ZP filaments by homodimers held together [5]. Zp1 knockout mice have looser and fragile egg coat [6]. Zp2-null mice develop a thin zona matrix in early follicles and the number of antral stage follicles in ovaries is significantly decreased [7].
Empty follicle syndrome (EFS) is a disease in which no oocytes are obtained in ART (assisted reproduction technology) cycles although follicle development and steroidogenesis levels appear to be normal [8]. Many studies have reported a wide range of EFS prevalence, from 0.045 to 7% of patients undergoing ovum pickup (OPU), based on different inclusion criteria [9]. EFS can be classified into genuine and false types. False EFS is mainly caused by a failure to retrieve oocytes with low HCG (human chorionic gonadotropin, < 40 IU/L) due to an error in the administration or the bioavailability of HCG. Genuine EFS is termed as retrieve no oocytes despite optimal HCG levels on the day of oocyte retrieval, which is caused by dysfunctional folliculogenesis, ovarian aging, or genetic defects. There are three known disease-causing genes associated with EFS, the luteinizing hormone/chorionic gonadotropin receptor (LHCGR), the zona pellucida glycoprotein 1 (ZP1), and the zona pellucida glycoprotein (ZP3) [10–12].
The phenotypes in ZP-related female infertile patients are diverse and can be caused by different mutations in different disease genes. Many mutations in ZP1 gene were associated with EFS, including compound heterozygous mutations (c.170_174del and c.1169_1176del), compound heterozygous mutations of c.181C>T and c.1169_1176delTTTTCCCA [12, 13]. However, two mutations (c.1430 + 1G > T and c.1775-8T > C) in a compound heterozygous state or the homozygous frameshift mutation c.1169_1176delTTTTCCCA in ZP1 produced ZP-free oocytes [14, 15]. Two homozygous mutations (c.1695-2A > G and c.1691_1694dup, respectively) in ZP2 resulted in a thin ZP and in vitro fertilization failure [16].
In this study, we characterize three patients in two unrelated families who diagnosed with primary infertility. No oocytes were retrieved from the patients of family 1 in repeated IVF cycles. The oocytes in the proband of family 2 exhibited a thin ZP.
Materials and methods
Sanger sequencing
This study was approved by the ethics committee on human subject research at Huazhong University of Science and Technology. Written informed consent was obtained from the participants. Two Chinese families with primary infertility were recruited. Total genomic DNA was extracted from whole blood from the patients and the living family members. Sanger sequencing was performed to confirm the ZP1 and ZP2 variants.
Plasmid construction
Human ZP1 and ZP2 gene plasmids were kind gifts from Lei Wang’s lab in Fudan University [14]. Full-length cDNA of ZP1 and ZP2 genes were cloned into pEGFP-C1 vectors separately. Mutations (c.507delC, p. His170fs; c.239 G>A, p. Cys80Tyr, and c.241 T>C, p. Tyr81His) in ZP1 (H170fs; C80Y+Y81H) and mutations (c.860_861delTG, p.Val287fs and c.1924 C>T, p.Arg642Ter) in ZP2 were synthesized by Mut Express II Fast Mutagenesis Kit V2 (Vzyme, China), according to the manufacturer’s protocol.
Cell transfection and western blotting
HEK293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 12% fetal bovine serum (Gibco). The recombinant plasmid was transfected into the cells using Lipofectamine 2000 (Invitrogen, USA). GFP-tagged wild-type (WT) ZP1 expression plasmid, GFP-tagged H170fs mutant ZP1 expression plasmid, GFP-tagged (C80Y+Y81H) mutant ZP1 expression plasmid, GFP-tagged WT ZP2 expression plasmid, GFP-tagged V287fs mutant ZP2 expression plasmid and GFP-tagged R642Ter mutant ZP2 expression plasmid were transfected into HEK293T cells separately. After 36 h, these cells were collected for further analysis. Proteins were extracted using NP40 lysis buffer (100 mM NaCl, 50 mM tris-HCl, and 0.1% NP40 containing 1 mM PMSF). Protein concentration was determined by a BCA assay and electroblotted on PVDF membranes at 200 V for 2 h at 4 °C. Following the transfer, the membrane was blocked with 5% blocking buffer (5% non-fat dried milk and 0.1% Tween-20 in PBS) for 1.5 h at room temperature. The membrane was incubated overnight with first antibody at 4 °C. After incubation, the membrane was washed three times for 10 min with TBST and incubated with a specific secondary antibody at room temperature for 2 h. The membrane was similarly washed three times with TBST, and protein bands on the membrane were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo) with ChemiDoc XRS+.
Results
Clinical characteristics of the affected individuals
The proband of family 1 was a 32-year-old woman with a 10-year history of unexplained primary infertility. She had normal ovarian reserves and regular menstrual cycles. Her chromosomal karyotyping was 46, XX. She underwent ovarian stimulation using the GnRH agonist long protocol. We obtained 10 follicles of diameter more than 14 mm from the patient (May 20, 2019), but no oocytes were retrieved. Estradiol level was 4313 pg/mL when HCG was administered. Oocyte retrieval was performed 37 h after HCG triggering on the left ovary and five cumulus-corona complexes (CCCs) were retrieved but no recognizable oocytes were identified. Through removing the surrounding cumulus cells in hyaluronidase solution (80 IU/mL, SAGE, USA) and repeatedly mechanical aspiration, only a cluster of closely gathered cumulus was found (Fig. 1a–d). The detected estradiol level was 875 pg/mL, and the β-HCG level was 139.9 mIU/mL at the time of oocyte retrieval. Two hours later, oocyte retrieval was performed on the right ovary and no oocyte was obtained. The sister of the proband was a 30-year-old woman who was diagnosed with primary infertility after 3 years of cohabitation with her husband. She had normal ovarian reserves and regular menstrual cycles. Her basal sex hormone level was generally normal. Her chromosomal karyotyping was 46, XX. The patient received two cycles of IVF treatment. In the first attempt (November 15, 2018), a gonadotropin- releasing hormone (GnRH) agonist protocol was performed and had a plasma estradiol level of 1206 pg/mL. The second IVF attempt (March 30, 2019) was carried out with GnRH antagonist protocol and estradiol level reached 2507 pg/mL. No oocytes were obtained in both of the two attempts.
Fig. 1.

Phenotype of the patients’ oocytes. a Normal cumulus-oocyte complexes are shown. b Normal oocyte with surrounding insoluble zona pellucida is shown. c The cumulus-corona complexes of the proband from family 1 are shown. d The cluster of closely gathered cumulus is shown after hyaluronidase dissolution of the proband from family 1. e, f The mature oocytes with thin ZP retrieved from the proband of family 2 are shown. Oocytes show an abnormal ZP with a thin matrix and an enlarged perivitelline space
In family 2, the proband suffered from primary infertility for 4 years. Her husband’s semen examination was normal (Online Resource Table s1). In her first GnRH antagonist protocol cycle (February 21, 2017), five COCs were retrieved. After conventional IVF and denudation, two oocytes were found to be ZP-free, and the others had an abnormal ZP with a thin matrix and an enlarged perivitelline space (Fig. 1e and f). The IVF was canceled because no embryos were available. In the second luteal-phase stimulation attempt (September 9, 2018), seven oocytes were retrieved which also had a thinner ZP matrix and then denuded for ICSI. Three embryos were obtained and cryopreserved, but failed in implantation in subsequent two frozen-thawed embryo transfer (FET) cycles. In her third cycle with GnRH antagonist protocol (June 27, 2019), one oocyte was retrieved and was degenerated after granulosa cell removal. In her fourth luteal-phase stimulation cycle (November 10, 2019), five oocytes, which had an enlarged perivitelline space and thin ZP, were retrieved. Three oocytes were fertilized after ICSI, and three embryos were cryopreserved. The embryos have not been thawed until now.
Identification of mutations in ZP1 and ZP2
Whole exome sequencing was carried out on the two patients (family 1, Fig. 2a), and three heterozygous mutations (c.239 G>A, c.241 T>C and c.507delC) were identified in ZP1. Sanger sequencing of PCR products and T clones which contain exon 2 and exon 3 of ZP1 gene from patients revealed a novel compound heterozygous mutation: c.239 G > A (p. Cys80Tyr) and c.241 T > C (p. Tyr81His) in the same allele of ZP1 gene, and c.507delC (p. His170fs) in another allele of ZP1 gene (Fig. 2b, Online Resource Fig. s1a). The compound heterozygous ZP1 mutation (c.507delC, p. His170fs; c.239 G > A, p. Cys80Tyr and c.241 T > C, p. Tyr81His) was present in both affected females. No mutations in ZP2, ZP3, ZP4 and LHCGR were identified. The heterozygous frameshift mutation c.507delC (p. His170fs) was inherited from the mother of the proband. This mutation was predicted to produce a premature stop codon and probably resulting in the loss of zona pellucida function. Two novel heterozygous missense mutations, c.239 G>A (p. Cys80Tyr) and c.241 T>C (p. Tyr81His), might be inherited from the father of the proband who died years ago and also carried by the unaffected older sister. This compound heterozygous mutation had not previously been reported in public databases, including dbSNP and gnomAD database. Alignment of the ZP1 protein across six species revealed that C80 and Y81 in ZP1 protein are highly conserved (Fig. 2c).
Fig. 2.
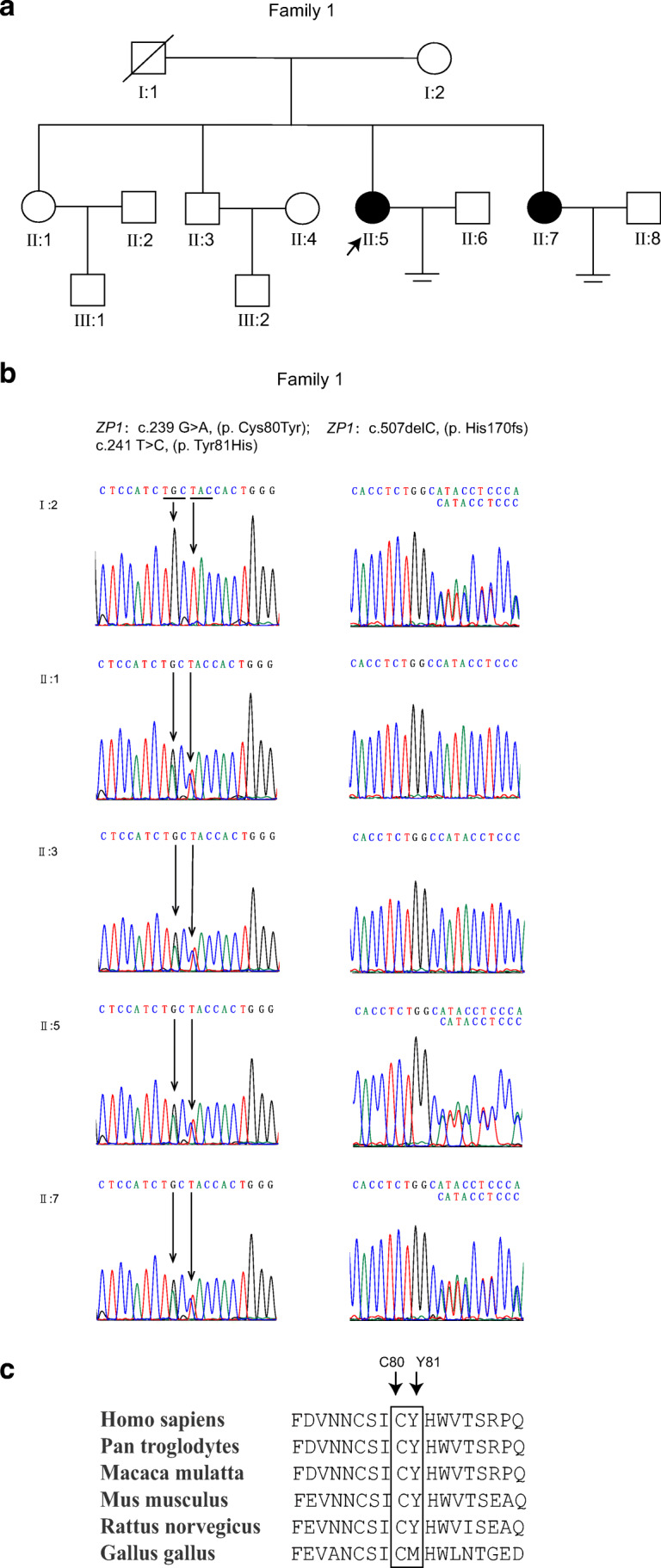
Identification of mutations in ZP1 gene. a Pedigree of the family 1. Squares indicate male family, and circles indicate female. Filled symbols indicate affected individuals; slashes indicate deceased family members and equal signs represent infertility. The arrow indicates the proband of the family. b Chromatograms obtained by direct sequencing of PCR products reveal compound heterozygous mutations of ZP1. c The conservation analysis. Amino acid sequence alingment show that C80 and Y81 in ZP1 protein are highly conserved during evolution.
We performed targeted sequencing of ZP1, ZP2, ZP3, and ZP4 genes in the patient from family 2 (Fig. 3a). According to the Sanger sequencing of PCR products and T clones which contain exon 10 and exon 17 of ZP2 gene from the patient, the proband carried a compound heterozygous mutation in ZP2 gene (c.860_861delTG, p.Val287fs and c.1924 C>T, p.Arg642Ter) and no mutations in ZP1, ZP3, and ZP4 were identified (Fig. 3b, Online Resource Fig. s1b). The frameshift mutation c.860_861delTG (p.Val287fs) was inherited from the father of the proband; the unaffected older sister of the proband also carried this frameshift mutation. This frameshift mutation produced a premature stop codon and was not shown in the dbSNP database. The frequency of the mutation c.1924 C>T (p.Arg642Ter) was 1/223072 (GnomAD_exome), which might be inherited from her mother who died years ago.
Fig. 3.

Identification of mutations in ZP2 gene. a Pedigree of the family 2. b Sanger DNA sequencing reveals mutations in ZP2 gene
Expression of wild-type and mutant ZP1 and ZP2 proteins in cells
To evaluate the effect of the mutations of ZP1 and ZP2 in vitro, HEK293T cells were transfected with the reconstructed vectors. As indicated by western blot analysis, the expression of ZP1 (C80Y+Y81H) was significantly decreased (Fig. 4a and b). All three mutations—ZP1 (p. His170fs), ZP2 (p.Val287fs), ZP2 (p.Arg642Ter)—could generate truncated proteins (Fig. 4a and c).
Fig. 4.

Protein expression of ZP1 and ZP2 in HEK293T cells. a HEK293T cells were seeded in six-well plates and transfected with indicated constructs, and western blotting was performed. NC, negative control. b Relative total protein expression of WT and mutant ZP1 (C80Y+Y81H). Results represent the average ± SEM of three independent experiments (*p < 0.01, n = 3). c Protein expression level of WT and mutant (V287fs, R642Ter) ZP2 in HEK293T cells
Discussion
In this study, we recruited three patients from two independent families with primary infertility. We identified a novel compound heterozygous mutation (c.507delC; c.239 G>A and c.241 T>C) in ZP1 gene from the patients in family 1 who were diagnosed with empty follicle syndrome. A novel compound heterozygous ZP2 mutation (c.860_861delTG and c.1924 C>T) was identified from the patient in family 2 who was diagnosed with primary infertility due to thin zona pellucida.
The compound heterozygous mutation in ZP1 that co-segregate with EFS in family 1 and was absent in public database. A previous study showed that the homozygous frameshift mutation c.507delC (p. His170fs) in ZP1 was carried by a patient from a consanguineous family which no oocytes was obtained in IVF cycle [14]. Those two missense mutations (c.239 G>A, p. Cys80Tyr and c.241 T>C, p. Tyr81His) might be inherited from the father. The older sister who carried these two missense mutations alone had a child. The phenotypic heterogeneity in ZP1-related female infertile patients might be caused by the different mutation site. ZP1 protein has an N-terminal signal sequence (aa 1–26), ZP-N1 domain (aa 36–138), highly conserved trefoil (aa 232–277), and ZP domains (aa 277–553) and a C-terminal transmembrane domain (aa 602–604). Those mutations in ZP1, which produced ZP-free oocytes, were located in the ZP domains. The truncated ZP1 protein might comprise the N-terminal signal sequence, the trefoil domain, and the first half of the ZP domain [14, 15]. Consistent with the former research, the mutation c.507delC was predicted to introduce a premature stop codon, and there would be only the N-terminal signal sequence generated [12, 13]. Those two missense mutations c.239 G>A (p. Cys80Tyr) and c.241 T>C (p. Tyr81His) in the ZP1-N1 domain (aa 36–138) led to decreased expression level of ZP1 protein. Also, a recent report showed that mutation in the ZP1-N1 domain reduces the ability to form disulfide-bonded homodimers, thus hindering human ZP filament cross-linking [17]. Taken together, those might cause failure to interconnect the ZP filaments and insufficient to maintain the oocyte development, thus leading to oocyte degeneration and empty follicles.
The mouse zona pellucida is composed of three glycoproteins—mZP1, mZP2, and mZP3. Zp2 knockout female mice form a thin zona matrix in early follicles consisted of mZP1 and mZP3. However, this zona pellucida is not sustained in preovulatory follicles. Those zona-free oocytes matured and fertilized in vitro can progress to the blastocyst stage, but no live birth has been observed after embryo transplantation [7]. Previous study revealed that ZP2 mediates sperm binding to the zona pellucida, lacking of which oocytes are defective for sperm binding and IVF fail [16, 18]. Consistent with the former research, the mutated ZP2 protein led to the formation of a thin ZP [16]. The frameshift mutation (c.860_861delTG) in ZP2 from family 2 was inherited from the father of the patient, and the unaffected older sister who carried the heterozygous mutation alone had a child. Only the patient who carry the compound heterozygous mutation is infertile.
In conclusion, we have identified two novel compound mutations in ZP1 and ZP2. This finding confirms the important role for ZP in human fertilization, and our study expanded the mutational spectrum, which provides novel evidence for genetic diagnostic of empty follicle syndrome or primary infertility due to abnormal zona pellucida.
Electronic supplementary material
(PDF 509 kb)
Acknowledgments
We would like to thank Sang Qing and Lei Wang from Fudan University for providing the gift of ZP1 and ZP2 plasmids. We also thank all patients for their participation.
Funding information
This work was supported by the National Natural Science Foundation of China (81000079, 81170165, and 81870959 to X.Z.) and supported by the Program for HUST Academic Frontier Youth Team.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Geng Luo and Lixia Zhu contributed equally to this work.
Contributor Information
Lei Jin, Email: leijintongjih@qq.com.
Xianqin Zhang, Email: xqzhang04@hust.edu.cn.
References
- 1.Hughes DC, Barratt CL. Identification of the true human orthologue of the mouse Zp1 gene: evidence for greater complexity in the mammalian zona pellucida? Biochim Biophys Acta. 1999;1447(2–3):303–306. doi: 10.1016/s0167-4781(99)00181-5. [DOI] [PubMed] [Google Scholar]
- 2.Chakravarty S, Kadunganattil S, Bansal P, Sharma RK, Gupta SK. Relevance of glycosylation of human zona pellucida glycoproteins for their binding to capacitated human spermatozoa and subsequent induction of acrosomal exocytosis. Mol Reprod Dev. 2008;75(1):75–88. doi: 10.1002/mrd.20726. [DOI] [PubMed] [Google Scholar]
- 3.Chiu PC, Wong BS, Lee CL, Pang RT, Lee KF, Sumitro SB, et al. Native human zona pellucida glycoproteins: purification and binding properties. Hum Reprod (Oxford, England) 2008;23(6):1385–1393. doi: 10.1093/humrep/den047. [DOI] [PubMed] [Google Scholar]
- 4.Ganguly A, Bukovsky A, Sharma RK, Bansal P, Bhandari B, Gupta SK. In humans, zona pellucida glycoprotein-1 binds to spermatozoa and induces acrosomal exocytosis. Hum Reprod (Oxford, England) 2010;25(7):1643–1656. doi: 10.1093/humrep/deq105. [DOI] [PubMed] [Google Scholar]
- 5.Greve JM, Wassarman PM. Mouse egg extracellular coat is a matrix of interconnected filaments possessing a structural repeat. J Mol Biol. 1985;181(2):253–264. doi: 10.1016/0022-2836(85)90089-0. [DOI] [PubMed] [Google Scholar]
- 6.Rankin T, Talbot P, Lee E, Dean J. Abnormal zonae pellucidae in mice lacking ZP1 result in early embryonic loss. Development (Cambridge, England) 1999;126(17):3847–3855. doi: 10.1242/dev.126.17.3847. [DOI] [PubMed] [Google Scholar]
- 7.Rankin TL, O'Brien M, Lee E, Wigglesworth K, Eppig J, Dean J. Defective zonae pellucidae in Zp2-null mice disrupt folliculogenesis, fertility and development. Development (Cambridge, England) 2001;128(7):1119–1126. doi: 10.1242/dev.128.7.1119. [DOI] [PubMed] [Google Scholar]
- 8.Coulam CB, Bustillo M, Schulman JD. Empty follicle syndrome. Fertil Steril. 1986;46(6):1153–1155. doi: 10.1016/s0015-0282(16)49898-5. [DOI] [PubMed] [Google Scholar]
- 9.Kim JH, Jee BC. Empty follicle syndrome. Clin Exp Reprod Med. 2012;39(4):132–137. doi: 10.5653/cerm.2012.39.4.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yariz KO, Walsh T, Uzak A, Spiliopoulos M, Duman D, Onalan G, King MC, Tekin M. Inherited mutation of the luteinizing hormone/choriogonadotropin receptor (LHCGR) in empty follicle syndrome. Fertil Steril. 2011;96(2):e125–e130. doi: 10.1016/j.fertnstert.2011.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen T, Bian Y, Liu X, Zhao S, Wu K, Yan L, Li M, Yang Z, Liu H, Zhao H, Chen ZJ. A recurrent missense mutation in ZP3 causes empty follicle syndrome and female infertility. Am J Hum Genet. 2017;101(3):459–465. doi: 10.1016/j.ajhg.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L, Fang X, Chen Z, Zhang H, Zhang Z, Zhou P, et al. Compound heterozygous ZP1 mutations cause empty follicle syndrome in infertile sisters. Hum Mutat. 2019. 10.1002/humu.23864. [DOI] [PubMed]
- 13.Yuan P, Li R, Li D, Zheng L, Ou S, Zhao H, Zhang Q, Wang W. Novel mutation in the ZP1 gene and clinical implications. J Assist Reprod Genet. 2019;36(4):741–747. doi: 10.1007/s10815-019-01404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Z, Ni C, Wu L, Chen B, Xu Y, Zhang Z, Mu J, Li B, Yan Z, Fu J, Wang W, Zhao L, Dong J, Sun X, Kuang Y, Sang Q, Wang L. Novel mutations in ZP1, ZP2, and ZP3 cause female infertility due to abnormal zona pellucida formation. Hum Genet. 2019;138(4):327–337. doi: 10.1007/s00439-019-01990-1. [DOI] [PubMed] [Google Scholar]
- 15.Huang HL, Lv C, Zhao YC, Li W, He XM, Li P, Sha AG, Tian X, Papasian CJ, Deng HW, Lu GX, Xiao HM. Mutant ZP1 in familial infertility. N Engl J Med. 2014;370(13):1220–1226. doi: 10.1056/NEJMoa1308851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dai C, Hu L, Gong F, Tan Y, Cai S, Zhang S, Dai J, Lu C, Chen J, Chen Y, Lu G, du J, Lin G. ZP2 pathogenic variants cause in vitro fertilization failure and female infertility. Genet Med. 2019;21(2):431–440. doi: 10.1038/s41436-018-0064-y. [DOI] [PubMed] [Google Scholar]
- 17.Nishimura K, Dioguardi E. Molecular basis of egg coat cross-linking sheds light on ZP1-associated female infertility. Nat Commun. 2019;10(1):3086. 10.1038/s41467-019-10931-5. [DOI] [PMC free article] [PubMed]
- 18.Tsubamoto H, Hasegawa A, Nakata Y, Naito S, Yamasaki N, Koyama K. Expression of recombinant human zona pellucida protein 2 and its binding capacity to spermatozoa. Biol Reprod. 1999;61(6):1649–1654. doi: 10.1095/biolreprod61.6.1649. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 509 kb)