

SUMMARY
The 26S proteasome is specialized for regulated protein degradation and formed by a dynamic regulatory particle (RP) that caps a hollow cylindrical core particle (CP) where substrates are proteolyzed. Its diverse substrates unify as proteasome targets by ubiquitination. We used cryogenic electron microscopy (cryo-EM) to study how human 26S proteasome interacts with M1-linked hexaubiquitin (M1-Ub6) unanchored to a substrate and E3 ubiquitin ligase E6AP/UBE3A. Proteasome structures are available with model substrates extending through the RP ATPase ring and substrate-conjugated K63-linked ubiquitin chains present at inhibited deubiquitinating enzyme hRpn11 and the nearby ATPase hRpt4/hRpt5 coiled coil. In this study, we find M1-Ub6 at the hRpn11 site despite the absence of conjugated substrate, indicating that ubiquitin binding at this location does not require substrate interaction with the RP. Moreover, unanchored M1-Ub6 binds to this hRpn11 site of the proteasome with the CP gating residues in both the closed and opened conformational states.
Keywords: proteasome, ubiquitin, deubiquitinating enzyme, Rpn11, cryo-EM, E6AP
Graphical Abstract
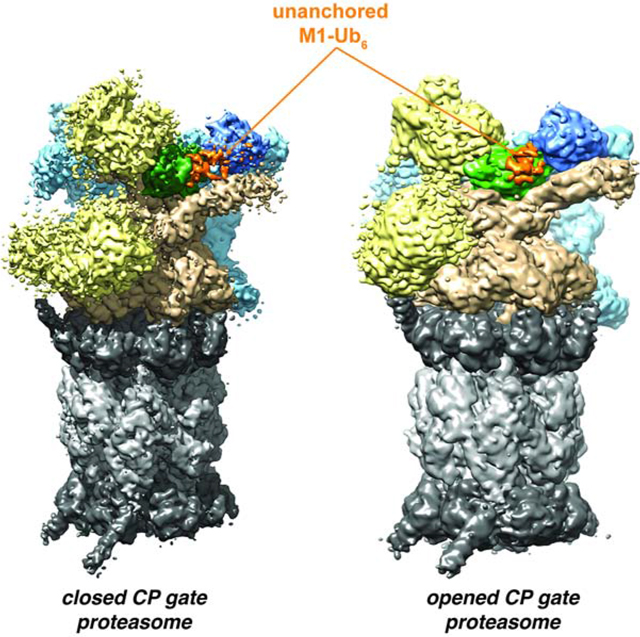
eTOC Blurb
Chen et al. used cryo-EM to study proteasome interaction with unanchored M1-Ub6. Ubiquitin bound hRpn11 of proteasomes in an SA-like conformation with CP gating residues opened and an SD-like conformation with closed gating residues. A second ubiquitin extends along the hRpt4/hRpt5 coiled coil of SA-like proteasome.
INTRODUCTION
The 26S proteasome is a highly dynamic 2.5 MDa molecular machine that is finely evolved for processing and degrading ubiquitinated proteins. It is formed from a cylindrical core particle (CP) that features a hollow interior where substrates are proteolyzed and a regulatory particle (RP) that binds either end of the CP (Bard et al., 2018; Collins and Goldberg, 2017; Ehlinger and Walters, 2013; Finley et al., 2016; Greene et al., 2019a; Wehmer and Sakata, 2016). The RP includes ubiquitin receptors and processing enzymes as well as a hexameric ATPase ring that docks into inter-subunit pockets of the CP α-ring.
Structural studies using cryogenic electron microscopy (cryo-EM) have identified seven conformational states for the proteasome including substrate-free ground state conformations (s1, s2 for yeast and SA, SB for human) as well as proposed substrate-processing conformers (s3, s4, s6 for yeast, and SC, SD(1,2,3) for human) (Bard et al., 2018; Beck et al., 2012; Chen et al., 2016a; Ding et al., 2017; Eisele et al., 2018; Greene et al., 2019a; Huang et al., 2016; Lander et al., 2012; Luan et al., 2016; Matyskiela et al., 2013; Schweitzer et al., 2016; Sledz et al., 2013; Unverdorben et al., 2014; Wehmer et al., 2017; Worden et al., 2017; Zhu et al., 2018). Two recent studies captured model substrates conjugated to K63-linked ubiquitin chains interacting with yeast (de la Pena et al., 2018) or human (Dong et al., 2019) proteasome by inhibiting Rpn11 or ATP hydrolysis respectively. Substrate density threaded through the center of the RP ATPase ring was visible in four distinct s4-like states (1D*, 5D, 5T and 4D) for yeast proteasome (de la Pena et al., 2018) and seven conformational states (EA1, EA2, EB, EC1, EC2, ED1, and ED2) for human proteasome (Dong et al., 2019). These structures define distinct configurations adopted by the ATPase ring during substrate interaction and translocation into the CP.
Despite this progress, the peripheral regions of the 26S proteasome where the substrate receptors are located remain only vaguely defined. Three major ubiquitin receptors (Rpn1, Rpn10, and Rpn13) have been identified (Husnjak et al., 2008; Schreiner et al., 2008; Shi et al., 2016; Young et al., 1998), along with structures that define their unique ubiquitin-binding mechanisms (Husnjak et al., 2008; Lu et al., 2020; Lu et al., 2017; Schreiner et al., 2008; Shi et al., 2016; VanderLinden et al., 2017; Wang et al., 2005; Zhang et al., 2009). Rpn1 and Rpn13 also contribute two of the three proteasome deubiquitinating enzymes (DUB), respectively Usp14/Ubp6 (Lee et al., 2016; Leggett et al., 2002; Shi et al., 2016) and Uch37/UCHL5 (Chen and Walters, 2015; Gandhi et al., 2006; Hamazaki et al., 2006; Qiu et al., 2006; Sahtoe et al., 2015; VanderLinden et al., 2015; Yao et al., 2006). The third deubiquitinating enzyme, Rpn11, is seated over the ATPase channel during substrate engagement (Lander et al., 2012; Lasker et al., 2012; Matyskiela et al., 2013; Unverdorben et al., 2014), from where it liberates substrates from ubiquitin chains by hydrolysis at the substrate ligation site (Yao and Cohen, 2002). The hRpn10 N-terminal von Willebrand factor type A (VWA) domain is also proximal to hRpn11 and well-resolved by cryo-EM (Beck et al., 2012; Lander et al., 2012; Lasker et al., 2012); however, the hRpn10 portion beyond the VWA is not visible in proteasome structures, most likely due to the intrinsic dynamics of its ubiquitin-binding UIM region (Wang et al., 2005). Beyond the UIMs, hRpn10 binds E3 ubiquitin ligase E6AP/UBE3A (Avagliano Trezza et al., 2019; Buel et al., 2020; Kuhnle et al., 2018) at 12 nM affinity through a dedicated RAZUL (Rpn10 AZUL-binding domain) that itself is also intrinsically disordered, but forms an intermolecular 4-helix bundle with the E6AP AZUL (Amino-terminal Zinc-binding domain of Ubiquitin E3a Ligase) (Buel et al., 2020).
Ubiquitin was captured bound to Rpn11 in proteasome samples with K63-linked ubiquitin chains anchored to substrates (de la Pena et al., 2018; Dong et al., 2019). Rpn11-bound ubiquitin was not observed when the ubiquitin chain was removed from substrate (Dong et al., 2019). Ubiquitin has only been observed in this state at Rpn11 when its activity was inhibited by a Zn chelator and the substrate was therefore still attached and threaded through the ATPase ring (de la Pena et al., 2018).
Here we use cryo-EM to study binding of unanchored ubiquitin chains and E6AP to the 26S proteasome. Consistent with previous studies (Beck et al., 2012; Chen et al., 2016a; da Fonseca et al., 2012; de la Pena et al., 2018; Dong et al., 2019; Eisele et al., 2018; Huang et al., 2016; Lander et al., 2012; Lasker et al., 2012; Schweitzer et al., 2016; Wehmer et al., 2017; Zhu et al., 2018), hRpn13 and the C-terminal portion of hRpn10 (including the UIMs and RAZUL) are not defined in our reconstructions and neither is RAZUL-binding E6AP. We are able to observe two conformational states of human 26S proteasome in complex with non-cleavable M1-linked hexaubiquitin (Ub6) however. More specifically, despite the absence of conjugated substrate, we find ubiquitin at hRpn11 in both the ground (SA) state and substrate-processing (SD) state of the proteasome.
RESULTS
Addition of unanchored M1-Ub6 and E6AP to 26S proteasome yields two conformational states with extra density
We incubated human 26S proteasome with 10-fold molar excess non-cleavable M1-Ub6 and 5-fold molar excess E6AP (Figure 1A) for cryo-EM studies. To inhibit chain hydrolysis by proteasome DUBs, Gly76 of the first five ubiquitin moieties was substituted with valine. Sample conditions were screened in the presence of 1.5 mM ATP-γ-S or ATP as well as other published conditions, such as inclusion of NP40 (Chen et al., 2016a; Huang et al., 2016). Best results were found with the more slowly hydrolyzed ATP-γ-S, consistent with previous studies that demonstrate ATP-γ-S to compete with ATP for the proteasome ATPases and to increase capture of the 26S proteasome in multiple states (Sledz et al., 2013; Unverdorben et al., 2014; Zhu et al., 2018). Cryo-EM data was collected with a K2 Summit direct electron detector on a Titan Krios microscope, with a characteristic micrograph displayed in Figure S1A. A total of 1,050,369 particles were picked and extracted from 5,971 motion-corrected micrographs. Three rounds of reference-free 2D classification was done with manual inspection and triaging of particles not reflective of proteasome to yield 593,942 sorted particles (Figure 1B and S1B). The single-particle images of singly (RP-CP) or doubly (2RP-CP) capped proteasomes were separated in the following 3D classification, while incompletely capped proteasomes were discarded (Figure S1B). The single-particle images of 2RP-CP were converted to singly capped proteasome (RP-CP) by performing two-fold symmetry expansion in RELION (Scheres, 2012) (Figure S1B). The combined RP-CP single-particle reconstruction was refined to 4.1 Å resolution based on a Fourier Shell Correlation (FSC) value of 0.143 (Figure 1C, black, S1C and S1D). A local resolution map illustrates that only the CP achieves this resolution whereas the RP subcomplex is defined at lower resolution (Figure S1E), consistent with the previously reported multiple conformational states of the RP in the 26S proteasome (Chen et al., 2016a; de la Pena et al., 2018; Ding et al., 2017; Dong et al., 2019; Eisele et al., 2018; Huang et al., 2016; Luan et al., 2016; Matyskiela et al., 2013; Schweitzer et al., 2016; Unverdorben et al., 2014; Wehmer et al., 2017; Worden et al., 2017; Zhu et al., 2018). To better define the RP conformational states, we conducted a masked classification approach, as applied in previous studies (Bai et al., 2015; Scheres, 2016) with 3D classification focusing on the RP subcomplex with the CP subtracted (Figure S1B).
Figure 1. Two conformational states with extra density are resolved for 26S proteasome incubated with unanchored M1-Ub6 and E6AP.

(A) SDS-PAGE gel for human 26S proteasome, E6AP, non-cleavable M1-Ub6, and the mixture of 26S proteasome:E6AP:non-cleavable M1-Ub6 at 1:5:10 molar ratio.
(B) Representative reference-free 2D class averages computed by using RELION (Scheres, 2012). Scale bar, 500 Å.
(C) Fourier Shell Correlation (FSC) plot for the global reconstruction map (black) or final refined maps for the SA-like (blue) or SD-like (green) state. FSC value of 0.143 is indicated by a dashed line with an insert table listing resolution value (Å) for clarity.
(D) Side-by-side comparison of cryo-EM density maps of the SA-like state generated by ATP-γ-S-bound human 26S proteasome mixed with non-cleavable M1-Ub6 and E6AP (right, blue) and SA state of free human 26S proteasome (left, grey, EMD: 8666). Both cryo-EM density maps are overlaid with a ribbon diagram of the human 26S proteasome SA state structure (PDB: 5VFS). The extra density of SA-like proteasome is circled (orange). In (D) and (E), RP and CP subcomplexes are colored in grey and black respectively.
(E) Side-by-side comparison of cryo-EM density maps of the SD-like state generated by ATP-γ-S-bound human 26S proteasome mixed with non-cleavable M1-Ub6 and E6AP (right, green) and SD state of free human 26S proteasome (left, grey, EMD: 8337). The cryo-EM density maps are overlaid with a ribbon diagram of the human 26S proteasome SD state structure (PDB: 5T0J). The extra density of SD-like proteasome is circled (orange).
(F, G) Cross section planes of cryo-EM density maps illustrating the CP α-ring including gating residues (grey scale) and C-terminal domains of ATPase subunits (rainbow colors) with HbYX motifs interdigitated into the CP inter-subunit pockets for the SA-like (F) or SD-like (G) state. The CP gate is closed in the SA-like proteasome (F) and opened in the SD-like proteasome (G).
See also Figure S1.
The 26S proteasome has been well studied by cryo-EM and we therefore focused our analyses on single-particle images from classes that showed different density compared to these previous studies. After 3D auto-refinement and polishing (Figure S1B), two conformational states were found to contain extra density not present in available structures for free human 26S proteasome (Chen et al., 2016a; Zhu et al., 2018). These states were separately refined to 4.8 Å and 5.7 Å resolution (Figure 1C, blue and green, and S1F–S1K).
The multiple states of the 26S proteasome differ by the quantity of ATPase C-terminal hydrophobic-tyrosine-X (HbYX) motifs that are inserted into the CP α-ring, which modulates the status of the CP gating residues and thus substrate accessibility into the proteasome catalytic center (Smith et al., 2007). Best superposition to available 26S proteasome structures was found to the SA and SD states previously reported for human proteasome (Chen et al., 2016a; Zhu et al., 2018); we thus name the two states SA-like and SD-like respectively (Figure 1D and 1E). The correlation of map densities was calculated by using UCSF Chimera (Pettersen et al., 2004) for the SA and SA-like states as well as the SD and SD-like states to be 0.92 and 0.81 respectively. We therefore used the previously determined structures to guide our analyses. For the SA-like state, two ATPase HbYX motifs are inserted into the CP α-ring (Figure 1F), as found for the SA state and identified to be from hRpt3 and hRpt5 insertion into the CP α1–α2 and α5–α6 pockets respectively. The CP gate is correspondingly closed in the SA-like state, as indicated by the packed density of the N-terminal CP α-subunit amino acids (Figure 1F). ATP binds Walker A motifs located next to a short linker between the small and large AAA+ (ATPase Associated with various cellular Activities) subdomains of each ATPase; nucleotide densities were observed at all hRpt subunits of the SA-like state (Figure S1L). All of these attributes are consistent with the previously reported SA state for human proteasome and s1 state for yeast proteasome (Chen et al., 2016a; Dong et al., 2019; Eisele et al., 2018; Wehmer et al., 2017; Zhu et al., 2018).
The SD state is distinguished by the insertion of five ATPase C-terminal tails, specifically hRpt1, hRpt2, hRpt3, hRpt5, and hRpt6, into CP α–pockets (Chen et al., 2016a; Eisele et al., 2018; Wehmer et al., 2017; Zhu et al., 2018), as was observed for the SD-like state (Figure 1G). The CP gate is correspondingly open, illustrated by absence of density at the central CP α-ring channel (Figure 1G). The nucleotide-binding sites were found to be partially occupied (Figure S1M), with poorer fitting compared to the SA-like state, most likely due to SD state heterogeneity, as noted previously in a study that was able to further sub-classify this state by recording 175,250 particles for SD state (Zhu et al., 2018); such sub-classification was not possible with our 52,593 particles for the SD-like state.
The SA-like and SD-like states that we observe differ from the free 26S proteasome SA and SD states respectively by extra density proximal to hRpn11 (Figure S1B), as discussed below.
hRpn11 of an SA-like proteasome binds unanchored M1-ubiquitin chains
The structure of human 26S proteasome bound to K63-ubiquitinated Saccharomyces cerevisiae Cdk inhibitor Sic1 with an N-terminal Pro-Pro-Pro-Ser motif (Sic1PY) was recently reported, demonstrating Sic1PY to thread through the ATPase ring (Dong et al., 2019). In this study, two states similar to SA were reported; namely EA1 and EA2 for which ubiquitin is visible near the N-terminal hRpt4/hRpt5 coiled coil domain or at hRpn11 respectively. Our SA-like state matches well to the EA2 state with a density map correlation of 0.95, despite the absence of a conjugated substrate and interaction with the ATPase ring (Figure 2A). The extra density corresponding to ubiquitin in the EA2 state (Figure 2A, left panel, ribbon diagram shown in orange) is similarly observed for the SA-like state (Figure 2A, right panel, circled in orange). We thus refined the atomic model for the SA-like state based on the EA2 structure, with a correlation coefficient between the model calculated map and experimentally derived map of 0.74. Moreover, the global root mean square deviation (r.m.s.d.) of the SA-like structure to the EA2 structure (PDB ID: 6MSD) is 0.714 Å.
Figure 2. Unanchored M1-Ub6 binds near hRpn11 and extends along the hRpt4/5 coiled coil of the 26S proteasome with gating residues in the closed conformation.

(A) Side-by-side comparison of cryo-EM density maps of identical orientation for the SA-like state generated by ATP-γ-S-bound human 26S proteasome mixed with non-cleavable M1-Ub6 and E6AP (blue, right) and EA2 state (EMD: 9217) of human 26S proteasome with K63-ubiquitinated Sic1PY (grey, left). The EA2 density map is transparent and overlaid with a ribbon diagram of the fitted structure (PDB: 6MSD) highlighting ubiquitin in orange. An 8 Å low-pass filter was applied to all displayed density maps.
(B) Refined cryo-EM density map for the SA-like state with segmentation and coloring for the CP α-ring (dark grey), CP β-ring (light grey), RP ATPase ring (beige), hRpn1 and hRpn2 (yellow), hRpn10 VWA (blue), hRpn11 (green), and remaining lid components (light blue). The extra density corresponding to ubiquitin is colored orange.
(C, D) An enlarged view of the hRpn11 region for the SA-like density map colored as in (B) with a threshold of 0.009 (C) or 0.006 (D) and overlaying a ribbon diagram of the crystal structure (PDB: 5U4P) of ubiquitin (orange)-bound yeast Rpn11 (green)/Rpn8 (not shown). For visualization of the crystal structure, the cryo-EM density corresponding to Rpn11 and ubiquitin are rendered transparent. In (D), an 8 Å low-pass filter was applied to the density map and the extra density extending from the hRpn11-bound ubiquitin is colored in pink with a ribbon diagram for ubiquitin (PDB: 1D3Z) overlaid.
See also Figure S2.
Segmentation based on the atomic model for the SA-like structure demonstrates that hRpn13 as well as the hRpn10 UIMs and RAZUL were missing from our cryo-EM density maps (Figure 2B), identical to other SA state structures (Chen et al., 2016a; Dong et al., 2019; Huang et al., 2016; Zhu et al., 2018). A ubiquitin molecule could be fit however to the extra density at hRpn11 (Figure 2B). A ubiquitin at this position was observed in a crystal structure of the yeast Rpn11-Rpn8 heterodimer complexed with monoubiquitin (Worden et al., 2017). Superposition of this crystal structure with the region containing extra density for the SA-like state reveals an excellent fit (Figure 2C), whereas the E6AP HECT (Homologous to the E6AP Carboxyl Terminus) domain or E6AP AZUL:hRpn10 RAZUL complex does not fit well to the extra density (Figure S2A and S2B). Moreover, the cryo-EM density supports C-terminal residues R74 and G75 of the hRpn11-bound ubiquitin forming a three-stranded β-sheet with the hRpn11 β-hairpin (Figure 2D), as was observed in crystal structure of the yeast Rpn11:Rpn8:ubiquitin complex (Worden et al., 2017).
Examination of the SA-like density map at lower threshold (a level of 0.006 compared to the value of 0.009 for Figure 2C) reveals additional scattered density beyond the N-terminus of the hRpn11-bound ubiquitin (Figure 2D). This density likely reflects the presence of a more dynamic ubiquitin moiety linked to M1 of the better resolved hRpn11-bound ubiquitin (Figure 2D). In the EA2 state of human 26S proteasome mixed with K63-ubiquitinated Sic1PY (Dong et al., 2019), low resolution density for a second ubiquitin was observed near the hRpt4/hRpt5 coil coiled domain (Figure S2C), although at a different location compared to what we observe for M1-Ub6 (Figures S2D and 2D).
The SA-like and EA2 proteasome differ for the N-terminal region of hRpn1, which is translated towards the CP in the SA-like state (Figure S2E). Moreover, ubiquitin density was observed bound to the hRpn1 ubiquitin-binding T1 site (Shi et al., 2016) for the EA2 state (Dong et al., 2019), but no density corresponding to ubiquitin was observed at hRpn1 T1 in our SA-like state (Figure S2E). This difference could be because hRpn1 has a higher affinity for K63-linked ubiquitin chains compared to M1-linked chains (Shi et al., 2016).
The substrate-processing state of the proteasome binds unanchored M1-ubiquitin chains
Three SD states, SD(1,2,3), are reported for human 26S proteasome bound to ATP-γ-S that feature an open state for the CP α-ring gating residues and fluctuations between these states that affect relative positioning of the ATPase ring, CP, hRpn1, and lid subcomplex (Zhu et al., 2018). Of these states, SD3 superimposes best onto the SD-like state observed in this study with unanchored M1-Ub6 (Figure 3A and Table S1). We thus used the atomic model structure of the SD3 state (PDB ID: 5VFR) to refine the SD-like state. The resulting correlation coefficient between the model calculated map and experimentally derived map was 0.75. Most but not all regions of the refined SD-like state closely resemble the SD3 structure. For example, the region of hRpn10 and hRpn9 that contacts the Rpt4/Rpt5 N-terminal coil coiled domain is similar to the SD3 state and not the SD(1,2) state (Figure S3A). However, the flexible N-terminus of hRpn1 is most similar to the orientation found in the SD2 state (Figure S3B).
Figure 3. Proteasome with CP gating residues in the opened configuration bind M1-Ub6 at hRpn11.

(A) Side-by-side cryo-EM density maps of identical orientation for the SD-like state of ATP-γ-S-bound human 26S proteasome incubated with 10-fold molar excess non-cleavable M1-Ub6 and 5-fold molar excess E6AP (right, green) and the SD3 state of ATP-γ-S-bound human 26S proteasome (left, grey, EMD: 8665). The SD3 density map is transparent and overlaid with a ribbon diagram of the fitted structure (PDB: 5VFR). Extra density that appears in the SD-like state is indicated by an orange circle. A low-pass filter of 8 Å was applied to the density maps in (A), (C), and (D).
(B) Refined cryo-EM density map with segmentation for the SD-like state with a color scheme that follows Figure 2B.
(C, D) Cryo-EM density maps of identical orientation to (A) for the ED1 state (EMD: 9221) of human 26S proteasome with K63-ubiquitinated Sic1PY (C) or the s4 state (EMD: 9045) of yeast 26S proteasome with K63-ubiquitinated substrate (D). In (D), the s4 density map is transparent and overlaid with a ribbon diagram of the fitted structure (PDB: 6EF3) with ubiquitin highlighted in orange.
(E) Enlarged view of the density map for the hRpn11 region of the SD-like proteasome with a threshold of 0.005 and overlaying a ribbon diagram of yeast Rpn11 (green) and ubiquitin (orange) from the deposited structure of yeast 26S proteasome complexed with K63-ubiquitinated substrate (PDB: 6EF3). For visualization, the density corresponding to hRpn11 and ubiquitin are rendered transparent.
Similar to the SA-like proteasome (Figure 2B), hRpn13 and the hRpn10 UIMs and RAZUL were not visible; neither was E6AP, as illustrated by the segmented cryo-EM density map (Figure 3B). We also did not observe any density corresponding to substrate within the ATPase ring (Figure S3C), as expected for unanchored M1-Ub6. Such density was apparent for the ED1 state of human 26S proteasome with K63-ubiquitinated Sic1PY (Figure S3D) (Dong et al., 2019).
The ED states observed for 26S proteasome bound to K63-ubiquitin conjugated to Sic1PY did not reveal any density for ubiquitin (Figure 3C) (Dong et al., 2019); however, a similar study (de la Pena et al., 2018) with yeast 26S proteasome and K63-ubiquitinated substrate performed with a Zn chelator to inhibit Rpn11 activity indicated ubiquitin to be near Rpn11 of a corresponding s4 state (Figure 3D). The SD-like state for 26S proteasome with non-cleavable unanchored M1-Ub6 similarly revealed extra density near hRpn11 (Figure 3A, right panel, and 3B). These comparisons suggest that hRpn11 hydrolyzes ubiquitin chains at this location and that the SD-like configuration was trapped because M1-Ub6 is non-cleavable.
The ubiquitin observed near Rpn11 of the yeast s4 state (Figure 3D) corresponds well with the extra density of the SD-like state (Figure 3A, right panel, and 3B), providing strong evidence for this extra density belonging to ubiquitin. An enlarged view of this region for SD-like proteasome (Figure 3E) illustrates an interaction with ubiquitin that is similar to that observed for SA-like proteasome (Figure 2C). This finding demonstrates that the substrate processing state of human 26S proteasome with the gating residues opened is productive for ubiquitin binding at this hRpn11 location.
E6AP does not influence binding of unanchored M1-Ub6 at the Rpn11 location of the 26S proteasome
NMR experiments indicate that E6AP bound to the hRpn10 RAZUL is tethered but not docked against the 26S proteasome (Buel et al., 2020), allowed by flexibility of the hRpn10 ubiquitin-binding UIM region (Wang et al., 2005; Zhang et al., 2009). To further test whether the extra density observed in the SA-like or SD-like states could originate or be influenced by E6AP, we acquired a cryo-EM dataset of the 26S proteasome with 10-fold molar excess non-cleavable unanchored M1-Ub6 and no E6AP added. 1645 micrographs were obtained for this sample yielding 164,645 particles for refinement, which was performed as described above for the sample that also contained E6AP and illustrated in Figure S4A with parameters listed in Table S2 (dataset 1). Similar to the analyses with E6AP added, two density maps were obtained with extra density compared to free 26S proteasome that resembled the SA-like and SD-like states; these were resolved to 6.75 Å and 6.47 Å (Figure S4B) resolution respectively. The density maps of 26S proteasome and M1-Ub6 with and without E6AP in the SA-like (Figure S4C) or SD-like (Figure S4D) state fit well with correlations of 0.974 and 0.978 respectively. Refined segmentation maps based on Figure 2B and 3B similarly illustrated extra ubiquitin density at hRpn11 for the sample lacking E6AP in the SA-like (Figure S4E) and SD-like (Figure S4F) state respectively.
We also acquired a second set of cryo-EM data to achieve better resolution with 6220 micrographs, which yielded 863,217 particles for refinement as described in Figure S4G and Table S2 (dataset 2). We were unable to combine the two datasets, which were recorded in different facilities with different pixel size, however, the resolution was improved to 5.8 Å for the SA-like state and 5.96 Å for the SD-like state (Figure S4H–S4K). Density map correlations calculated by UCSF Chimera (Pettersen et al., 2004) to the corresponding states with E6AP also present yielded values of 0.942 and 0.925 respectively. M1-Ub6 density was similarly observed at hRpn11 in SA-like (Figure 4A and 4B) and SD-like (Figure 4C and 4D) states. Thus, this density at hRpn11 clearly belongs to ubiquitin.
Figure 4. M1-Ub6 is visible at hRpn11 in SA-like and SD-like proteasome states in the absence of E6AP.

(A, B) Side-by-side comparison of the refined cryo-EM density maps with segmentation for the SA-like state of ATP-γ-S-bound human 26S proteasome mixed 10-fold molar excess non-cleavable M1-Ub6 without (A) or with (B) E6AP.
(C, D) Side-by-side comparison of the refined cryo-EM density maps with segmentation for the SD-like state of ATP-γ-S-bound human 26S proteasome mixed 10-fold molar excess non-cleavable M1-Ub6 without (A) or with (B) E6AP. The color scheme follows that of Figure 2B. The orientation displayed in (A)-(D) is identical for the CP.
See also Figure S4.
It is worth noting that the high correlations between the samples with and without E6AP could indicate either that E6AP does not affect the visible portions of the 26S proteasome, consistent with the flexibility of the hRpn10 protein beyond the VWA domain (Buel et al., 2020; Wang et al., 2005; Zhang et al., 2009). However, it could also indicate that E6AP was not retained at the proteasome during cryo-plunging/sample preparation. Future experiments are needed to determine the effect of E6AP at the proteasome.
DISCUSSION
To date the ubiquitin-binding sites contributed by hRpn10 and hRpn13 have not been resolved in human proteasomes. NMR studies have indicated that these two receptors interact dynamically with ubiquitin chains (Lu et al., 2020; Zhang et al., 2009). Such dynamics no doubt stymie the application of cryo-EM to study these interactions. hRpn1 also binds to ubiquitin chains (Shi et al., 2016) and cryo-EM studies have found it too to be dynamic (Ding et al., 2019; Huang et al., 2016; Schweitzer et al., 2016; Wang et al., 2017; Wehmer et al., 2017; Zhu et al., 2018), but ubiquitin of K63-ubiquitinated Sic1PY was observed at the hRpn1 ubiquitin-binding T1 site (Dong et al., 2019). Our study used M1-linked ubiquitin, for which hRpn1 is a poor receptor (Shi et al., 2016) and we in turn did not observe ubiquitin at hRpn1. By contrast, hRpn13 (Chen et al., 2016b) and hRpn10 (Chen et al., 2019) are able to bind M1-Ub chains. Therefore, it is possible that the interactions occurring at hRpn13 and hRpn10 contributes to the trapping of ubiquitin at hRpn11 but that we cannot observe this contribution due to dynamics.
In the SA-like state, the hRpn11-bound ubiquitin chain extends along the N-terminal hRpt4/hRpt5 coil coiled domain (Figure 2D and S2D). hRpt5 was previously found to cross-link to K48-linked Ub4 (Lam et al., 2002), which could be explained by the interaction observed here. More recent in vivo disuccinimidyl sulfoxide (DSSO) cross-linking mass spectrometry experiments identified interactions between ubiquitin K48 and hRpn10 VWA domain K74, K81 and K103 (Wang et al., 2017). The distance between ubiquitin K48 Cα and hRpn10 K74 or K103 Cα is within 28 Å in the SA-like structure (Figure S2F), which is within the estimated DSSO cross-linking capture distance (Cα–Cα < 35 Å) (Wang et al., 2017). These experiments may therefore capture the ubiquitin bound at hRpn11.
Lid component hRpn6 was found to have high variance, with the N-terminal portion smeared out after averaging in a previous study (Wang et al., 2017). We similarly observe hRpn6 V43-K121 variance for the SD-like state, but better resolution and fitting for the SA-like state (Figure S3E). In the SA-like state, the small AAA+ subdomain of hRpt3 contacts the lid subunit hRpn5 TPR (tetratricopeptide repeat) domain residues V131-Y137 (Figure S3F, top panel). These residues are analogous to yeast Rpn5 V125-F131, which were found to be important for maintaining the proteasome conformational landscape (Matyskiela et al., 2013); in particular, loss of these amino acids increased the population of proteasome in substrate-engaged conformations despite absence of substrate (Greene et al., 2019b). We found this interaction between hRpt3 and the hRpn5 TPR domain to be broken in the SD-like state, with hRpt3 instead contacting a different hRpn5 region (Figure S3F, bottom panel), similar to observations made for yeast proteasome (Matyskiela et al., 2013).
Two independent studies have described details of the 26S proteasome with K63-linked ubiquitin chains conjugated to model substrates (de la Pena et al., 2018; Dong et al., 2019). Less is known of how the 26S proteasome interacts with unanchored ubiquitin chains. This state represents the status following cleavage from a substrate and may also represent regions of the chain that are remote from a substrate. A recent report indicated the presence of ubiquitin density localized at Rpn1 in yeast 26S proteasome mixed with unanchored K48-linked tetraubiquitin (Ding et al., 2019). No ubiquitin density was noted in this previous study at Rpn11. Similarly, addition of 40-fold molar excess ubiquitin-aldehyde to yeast 26S proteasome revealed no extra density at Rpn11 and a cryo-EM reconstruction map highly similar to 26S proteasome alone (Aufderheide et al., 2015). These findings are consistent with the reported fast dissociation rate (koff) of ~2.3 s−1 for unanchored ubiquitin chains after Rpn11 cleavage (Bard et al., 2019). We propose that our structure of M1-linked ubiquitin chains at Rpn11 of proteasome in the SD-like state is representative of the moment when the ubiquitin chain is released from substrate following cleavage by Rpn11. Thus, our findings here obtained by using unanchored M1-ubiquitin chains provide new information and most likely we were able to trap this state because our ubiquitin chains are non-cleavable.
There are several implications to our finding that the proteasome binds M1-linked ubiquitin chains at Rpn11 even in the absence of an attached substrate. It implies that the binding of substrate-proximal ubiquitin chains can be sterically occluded from Rpn11 by ubiquitin chains that are not coupled to substrate degradation. Such a scenario is conceivable for either excessively long chains and/or with excess levels of unanchored ubiquitin chains. Either situation may occur with defective DUB activity. Consistent with this possibility, substrate degradation by yeast 26S proteasome is inhibited by excess M1-linked tetraubiquitin and this inhibition occurs without inducing a conformational change to the proteasome (Bard et al., 2019). The lack of requirement for a proximal substrate to bind at Rpn11 also highlights the importance of DUB activity by UCHL5 and Usp14, which trim ubiquitin chains.
We next considered how other ubiquitin linkage types besides M1 might interact at the Rpn11 site of the proteasome. Of particular interest was ubiquitin K48 because it is uniquely oriented towards the Rpn10 VWA domain and Rpt4/5 coiled coil. In the SA-like proteasome, these two regions surround ubiquitin K48 with the Rpn10 VWA within 16 Å and Rpt4/5 within 20 Å (Figure 5A). These distances become even closer for the SD-like state however the movement of these two structural domains to be closer together causes them to be localized to one side of ubiquitin K48 (Figure 5B). In solution, K48 ubiquitin chains adopt a dominant closed conformational state that buries the Rpn11 binding surface (Cook et al., 1992; Trempe et al., 2010) and a dynamic ensemble of extended structures that can adopt a wide range of inter-ubiquitin orientations but with constraint at the linker region where non-covalent interactions are formed between ligated ubiquitin moieties (Lu et al., 2020). The inter-ubiquitin interactions orient K48-attached ubiquitin in a manner that is well-suited for binding to the SD-like proteasome, allowing the inter-ubiquitin interactions to be maintained without steric clashes with RP subunits (Figure 5C). By contrast, K48-linked ubiquitin is highly restricted in the geometry that it can adopt without forming steric clashes in the SA-like proteasome. Only one member of the extended state conformational ensemble could be modeled without forming steric clashes with Rpt4/5 (Figure 5D), indicating possible loss of the inter-ubiquitin interactions or of conformational freedom. This difference suggests that K48-linked chains bind best to Rpn11 when the gating residues are opened.
Figure 5. Enlarged view of hRpn11-bound ubiquitin for the proteasome SA-like or SD-like state highlighting the impact of ubiquitin addition at K48.
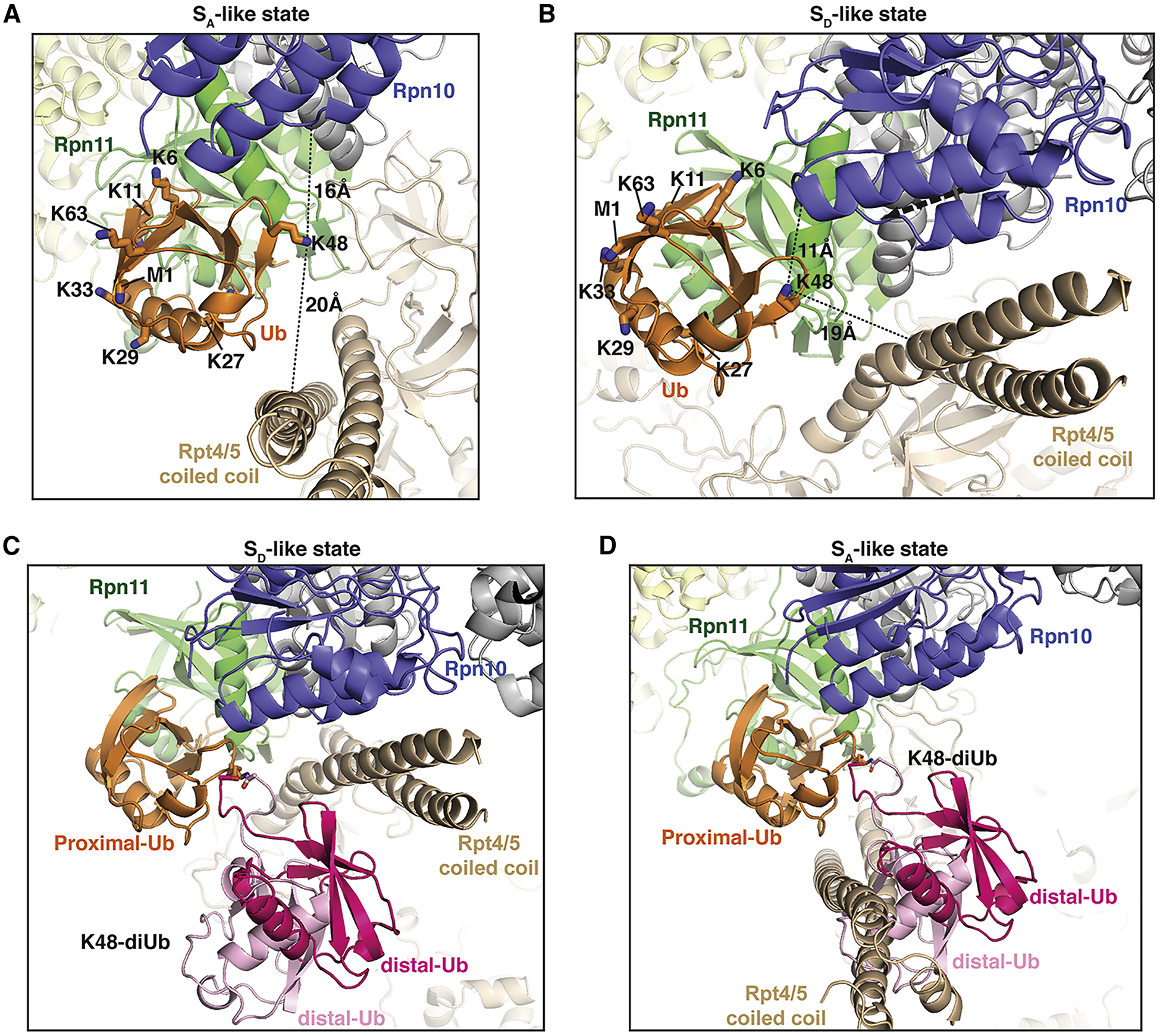
(A-D) Ribbon diagram for an enlarged region of the SA-like (A and D) or SD-like (B and C) proteasome centered on hRpn11-bound ubiquitin (orange) with portions displayed of hRpn11 (green), hRpn10 VWA (blue), the ATPase ring (beige), hRpn2 (yellow), and remaining lid components (grey). In (A) and (B), the heavy atoms of M1 and the ubiquitin lysine residues are displayed and labeled with nitrogen in blue. The shortest distance between K48 Nξ and backbone atoms of hRpn10 VWA or hRpt4/5 coiled coil are displayed and indicated with black dashed lines. In (B) and (C), the crystal structure of ubiquitin-bound yeast Rpn8/Rpn11 (PDB: 5U4P) was aligned to hRpn11 of the SD-like proteasome to generate the displayed models. In (C) and (D), two representative structures are shown following alignment onto Rpn11-bound ubiquitin of the proximal ubiquitin moiety of the K48-linked diubiquitin ensemble (PDB: 6UYI) observed in solution in the extended conformation (Lu et al., 2020). Distal ubiquitin is displayed in either light or dark pink. In (C), no steric clashes occur with distal ubiquitin whereas in (D) the light pink colored distal ubiquitin cannot be accommodated without steric clashes.
See also Figure S5.
These findings further suggest that although K48-linked ubiquitin chains can bind to the Rpn11 site, the ease of proteasome switching between the gate-closed SA-like state and gate-opened SD-like state is likely reduced for this chain type compared to other ubiquitin linkage types that are more remote from RP subunits, such as M1, K11, K29, K33 and K63 (Figure 5A, 5B, S5A, and S5B). This effect may result in tighter coupling between ubiquitin chain hydrolysis by Rpn11 and substrate translocation. More specifically, substrate interaction with the ATPase channel may provide a critical driving force for proteasome conformational switching when ubiquitin chains are linked by K48. By contrast, ubiquitin chains for which conformational switching and in turn Rpn11-catalyzed deubiquitination is facile may more weakly couple these activities with substrate translocation.
Consistent with this model, unanchored ubiquitin chains linked by K11 or K63, but not K48, were readily hydrolyzed by yeast proteasomes with only Rpn11 as an active DUB (Mansour et al., 2015). Moreover, substrates ubiquitinated with M1- or K11-linked ubiquitin chains are reported to not be degraded by the proteasome for an assay that demonstrated robust degradation with K48-linked chains (Martinez-Fonts et al., 2020). Furthermore, in one study, substrates with K63-linked ubiquitin chains were deubiquitinated by the proteasome but not efficiently degraded (Jacobson et al., 2009), albeit there appears to be substrate-dependent effects for this chain type (Martinez-Fonts et al., 2020). Altogether these data support a model in which translocation into the CP for substrates with ubiquitin chains linked by M1, K11, or K63 may not be required for proteasome conformational switching and deubiquitination by Rpn11.
Branched chains, including K11/K48, K29/K48, and K48/K63, have been identified as proteasomal priority signals based on the efficient degradation of their conjugated substrates (Haakonsen and Rape, 2019). These chains may exert an ideal balance of K48 enforced coupling of proteasome conformational switching to substrate translocation and the more rapid ubiquitin chain hydrolysis enabled by K11, K29, and K63 chain types. It is possible that E3 ligases at the proteasome remodel ubiquitin signaling, including by multiubiquitination, to achieve this ideal interaction with the proteasome. E6AP is known to catalyze ubiquitination by K48 linkage (Kim and Huibregtse, 2009; Kim et al., 2007; Ries et al., 2019; Wang and Pickart, 2005) and is recruited to the proteasome by the Rpn10 RAZUL domain (Buel et al., 2020). At the proteasome, E6AP may function coordinately with DUBs to remodel ubiquitin chains with an appropriate distribution of K48-linked ubiquitin for ideal coupling of substrate translocation to proteasome conformational switching and ubiquitin chain hydrolysis. Finally, we do not expect K6-linked ubiquitin chains to be accommodated at Rpn11 because this sidechain is directed towards the Rpn11 contact surface causing unavoidable steric clashes for ubiquitin ligation at this site (Figure 5A, 5B, S5C, and S5D).
Cdc48 has been published to unfold and interact with a ubiquitin moiety from a K48-linked ubiquitin chain to initiate substrate processing (Twomey et al., 2019). The analogous interaction with the proteasome ATPase ring and unanchored M1-ubiquitin chain was not apparent in this study, as we found no evidence of substrate density within the ATPase ring (Figure S3C). Hence, a ubiquitin moiety is not substituting for the model substrates of the previous studies with K63-conjugated ubiquitin chains. Instead, the interaction between Rpn11 and unanchored M1-ubiquitin chains is truly substrate-independent. This study thus illustrates how substrate-independent binding of ubiquitin occurs for this more rigid observable region of the proteasome.
In summary, we find that unanchored M1-ubiquitin chains bind to the proteasome at hRpn11, with the chain extending along the hRpt4/hRpt5 coil coiled domain. This interaction occurs with the CP gating residues in the opened and closed state, with almost equal distribution. In particular, 58% of particles selected with extra density were in the SD-like state and 42% in the SA-like state. In our study, filtered by the presence of extra density compared to free proteasomes, we did not observe density maps that correspond to the SB or SC states, possibly because these two states are less populated for free 26S proteasome even in the presence of ATP-γ-S (Zhu et al., 2018). Future studies are needed to truly understand the impact of E6AP at the proteasome as well as the contribution of the nearby hRpn10 UIMs to binding at hRpn11. Nonetheless, this study demonstrates the site at hRpn11 to be competent for stable interaction with M1-linked ubiquitin chains, which would no doubt facilitate ubiquitin chain hydrolysis by hRpn11.
STAR ★ Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kylie J. Walters (kylie.walters@nih.gov).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The cryo-EM reconstructions generated by this study have been deposited into the Electron Microscopy Data Bank with accession numbers 21691 (SA-like) and 21696 (SD-like) for the sample of human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP; 21699 (SA-like, dataset 1), 21700 (SD-like, dataset 1), 21697 (SA-like, dataset 2), and 21698 (SD-like, dataset 2) for the sample of human 26S proteasome mixed non-cleavable M1-Ub6. The docked coordinates reported in this paper have been deposited into the Protein Data Bank with accession codes PDB: 6WJD (SA-like) and 6WJN (SD-like) for human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP. The raw micrographs and particle data have been deposited in the Electron Microscopy Pilot Image Archive with accession numbers EMPIAR-10401 (sample of human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP), EMPIAR-10402 and EMPIAR-10403 (sample of human 26S proteasome mixed non-cleavable M1-Ub6 dataset 1 and 2). Other data are available from the corresponding author upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Source material was obtained commercially.
METHOD DETAILS
Cryo-EM sample preparation
Commercial human 26S proteasome (Enzo Life Sciences, Inc., BML-PW9310), non-cleavable M1-Ub6 (UBPBio, D4400), and E6AP (UBPBio, K1411) were buffer exchanged separately into 50 mM Tris (pH 7.5), 50 mM NaCl, 1.5 mM ATP-γ-S, 5 mM MgCl2, 2 mM DTT, and 10 μM zinc sulphate by using Zeba™ Micro Spin Desalting Columns (7k MWCO, Thermo Fisher 89883). After buffer exchange, 1 mg/mL proteasome was mixed with 10-fold molar excess M1-Ub6 with or without 5-fold molar excess E6AP.
Cryo-EM grid preparation and imaging
Cryo-EM grids were prepared by applying 2.0 μL of freshly prepared sample on both sides of glow-discharged Quantifoil R1.2/1.3 200 mesh holy carbon copper grids. The grids were blotted for 1.0 s and vitrified by plunging into liquid ethane with a Vitrobot Mark IV (Thermo Fisher Scientific FEI) operated at 18°C and 100% relative humidity. The frozen grids were stored in liquid nitrogen until data collection.
Cryo-EM data for human 26S proteasome mixed with 10-fold molar excess non-cleavable M1-Ub6 and 5-fold molar excess E6AP was acquired with a Titan Krios microscope (Thermo Fisher Scientific FEI) operated at 300 kV and equipped with a K2 Summit direct electron detector (Gatan, Table S2). Movies were recorded in the super-resolution mode at a dose rate of 4.58 e−/Å2/s with a total exposure time of 8.0 seconds at 0.2 seconds per frame. Each movie had 40 frames for an accumulated dose of 36.64 e−/Å2. The defocus values ranged from −0.8 to −2.0 μm with an increment step size of 0.2 μm. The pixel size was 1.365 Å. In total, 6,216 movies were collected.
Two cryo-EM datasets were acquired for human 26S proteasome incubated with 10-fold molar excess non-cleavable M1-Ub6 by using Titan Krios microscopes (300 kV) equipped with K2 Summit direct electron detectors (Gatan) in two different facility with different parameters (Table S2). A total of 1,645 movies were collected in the John M. Cowley Center for High Resolution Electron Microscopy at Arizona State University with a pixel size of 1.365 Å whereas 6,220 total movies were collected in the Laboratory of Cell Biology of the Center for Cancer Research of the National Cancer Institute with a pixel size of 1.348 Å; this latter Titan Krios microscope was equipped with a Gatan BioQuantum Image filter. Table S2 lists detailed imaging parameters used for each dataset.
For all datasets, the SerialEM software (Mastronarde, 2005) was employed for automated data collection.
Image processing
All cryo-EM data analyses were done by using RELION 3.0.8 (Zivanov et al., 2018), taking into account defect and gain reference information. The raw movies were gain reference corrected and then aligned to compensate for sample movement and drift by MotionCor2 (Zheng et al., 2017) yielding drift-corrected micrographs with 5 by 5 patches and B-factor of 150. The constant transfer function (CTF) estimation was done by CTFFIND4 (Rohou and Grigorieff, 2015). The micrographs were examined, and those with poor Thon rings were discarded. An initial set of particles were manually picked and 2D class templates generated by 2D classification. Automatic particle picking was run by using the generated 2D class templates in RELION, after which the results were manually inspected to discard contamination and add particles missed by the auto-picking. For the sample with E6AP included, total of 1,050,369 particles were picked and extracted by using a box size of 450 pixels; these were subjected to three rounds of 2D classification to identify and discard false positives or other apparent contamination.
Following 2D classification, 593,942 particles yielded 65 suitable 2D classes with various proteasome orientations that were selected, re-centered and re-extracted. These particles were subjected to one round of unsupervised 3D classification into 6 classes, using a reference map created from a human proteasome model (PDB: 5VFP). One class with 134,217 particles showed single capped proteasome (RP-CP) while another with 30,570 particles showed double capped proteasome (2PR-CP). Auto-refinement was performed with a pixel size of 1.365 Å for each of these two classes. 2RP-CP particles were then converted to pseudo RP-CP particles by performing particle symmetry expansion in RELION. The combined RP-CP and pseudo RP-CP particles were subjected to auto-refinement. To investigate the conformational states of the RP, we subtracted CP signal from the particles and conducted masked 3D classifications with 8 classes focusing on the RP, as described in previous studies (Bai et al., 2015; Scheres, 2016). 3 classes of RP-CP particles (contributing 39.3%) fit well to the free human proteasome SD state map and showed extra density. These classes were combined for further auto-refinement. 2 classes of RP-CP particles (contributing 18.9%) fit well to the free human proteasome SA state map and showed extra density, as did one additional class of pseudo RP-CP particles (contributing 20.5%). These classes were combined for auto-refinement.
After 3D auto-refinement, Bayesian polishing and per-particle CTF refinements were applied for further processing and CTF refinements were iterated until the 3D refinement converged. The final maps were sharpened using a B-factor of −76 Å2 (combined RP-CP and pseudo RP-CP), −104 Å2 (SA-like state), or −133 Å2 (SD-like state). A flowchart of the data processing is displayed in Figure S1B. The two datasets of human 26S proteasome with non-cleavable M1-Ub6 and no E6AP were processed in an identical manner. The numbers of particles used for the final 3D refinement for each sample is listed in Table S2. All resolutions are estimated by applying a soft mask around the protein density and 0.143 FSC value; local resolutions for the density maps were calculated by using a program implemented in RELION (Kucukelbir et al., 2014).
Model building and structure analyses
The atomic models of the EA2 state of human 26S proteasome with K63-ubiquitinated Sic1PY (PDB: 6MSD) or the SD3 state of ATP-γ-S-bound human 26S proteasome (PDB: 5VFR) were fitted as rigid bodies into the SA-like or SD-like state density maps respectively by using UCSF Chimera (Pettersen et al., 2004) (Table S3). The coordinates were further adjusted manually to avoid clashes between fitted rigid bodies by using Coot (Emsley et al., 2010) and structural models further refined by using the Real Space Refine module of the Phenix suite (Adams et al., 2010) (Table S3). All low-pass filter maps were generated by EMAN2 (Tang et al., 2007). Density map correlations were calculated by using UCSF Chimera (Pettersen et al., 2004). Figures were generated by using UCSF Chimera (Pettersen et al., 2004) and Pymol (PyMOL Molecular Graphics System, http://www.pymol.org).
QUANTIFICATION AND STATISTICAL ANALYSIS
All cryo-EM data sets were processed by using RELION 3.0.8 (Zivanov et al., 2018) (Figure S1B, S4A, S4G, and Table S2), with resolution reported based on FSC criterion of 0.143 (Figure 1C, S1C, S1F, S1I, S4B, and S4H). FSC curves were calculated using soft spherical masks and high-resolution noise substitution was used to correct for convolution effects of the masks on the FSC curves (Chen et al., 2013). Refinement statistics for each atomic model are summarized in Table S3. These models were also evaluated based on MolProbity scores and Ramachandran plots.
Supplementary Material
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| ATP-γ-S | Sigma-Aldrich Inc. | A1388 |
| human 26S proteasome | Enzo Life Sciences, Inc. | BML-PW9310 |
| non-cleavable M1-Ub6 | UBPBio | D4400 |
| E6AP/UBE3A | UBPBio | K1411 |
| Zeba™ Micro Spin Desalting Columns (7k MWCO) | Thermo Fisher | 89883 |
| Deposited Data | ||
| SA-like map of human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP | This paper | EMD: 21691 EMPIAR-10401 |
| SA-like structure of human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP | This paper | PDB: 6WJD |
| SD-like map of human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP | This paper | EMD: 21696 EMPIAR-10401 |
| SD-like structure of human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP | This paper | PDB: 6WJN |
| SA-like map of human 26S proteasome mixed non-cleavable M1-Ub6 (dataset 1) | This paper | EMD: 21699 EMPIAR-10403 |
| SD-like map of human 26S proteasome mixed non-cleavable M1-Ub6 (dataset 1) | This paper | EMD: 21700 EMPIAR-10403 |
| SA-like map of human 26S proteasome mixed non-cleavable M1-Ub6 (dataset 2) | This paper | EMD: 21697 EMPIAR-10402 |
| SD-like map of human 26S proteasome mixed non-cleavable M1-Ub6 (dataset 2) | This paper | EMD: 21698 EMPIAR-10402 |
| SA map of human 26S proteasome | (Zhu et al., 2018) | EMD: 8666 |
| SA structure of human 26S proteasome | (Zhu et al., 2018) | PDB: 5VFS |
| SD map of human 26S proteasome | (Chen et al., 2016a) | EMD: 8337 |
| SD structure of human 26S proteasome | (Chen et al., 2016a) | PDB: 5T0J |
| SD1 map of human 26S proteasome | (Zhu et al., 2018) | EMD: 8663 |
| SD1 structure of human 26S proteasome | (Zhu et al., 2018) | PDB: 5VFP |
| SD2 map of human 26S proteasome | (Zhu et al., 2018) | EMD: 8664 |
| SD2 structure of human 26S proteasome | (Zhu et al., 2018) | PDB: 5VFQ |
| SD3 map of human 26S proteasome | (Zhu et al., 2018) | EMD: 8665 |
| SD3 structure of human 26S proteasome | (Zhu et al., 2018) | PDB: 5VFR |
| EA2 map of human 26S proteasome with K63-ubiquitinated Sic1PY | (Dong et al., 2019) | EMD: 9217 |
| EA2 structure of human 26S proteasome with K63-ubiquitinated Sic1PY | (Dong et al., 2019) | PDB: 6MSD |
| ED1 map of human 26S proteasome with K63-ubiquitinated Sic1PY | (Dong et al., 2019) | EMD: 9221 |
| ED1 structure of human 26S proteasome with K63-ubiquitinated Sic1PY | (Dong et al., 2019) | PDB: 6MSJ |
| s4 map (4D) of yeast 26S proteasome with K63-ubiquitinated substrate | (de la Pena et al., 2018) | EMD: 9045 |
| s4 structure (4D) of yeast 26S proteasome with K63-ubiquitinated substrate | (de la Pena et al., 2018) | PDB: 6EF3 |
| crystal structure of ubiquitin-bound yeast Rpn11/Rpn8 | (Worden et al., 2017) | PDB: 5U4P |
| NMR solution structure of ubiquitin | (Cornilescu, 1998) | PDB: 1D3Z |
| crystal structure of E6AP HECT domain | (Huang et al., 1999) | PDB: 1C4Z |
| NMR solution structure of E6AP AZUL/hRpn10 RAZUL | (Buel et al., 2020) | PDB: 6U19 |
| NMR solution structure of K48-linked diubiquitin with hRpn13 and hRpn2 | (Lu et al., 2020) | PDB: 6UYI |
| crystal structure of M1-linked diubiquitin and OPTN | (Li et al., 2018) | PDB: 5WQ4 |
| crystal structure of K11-linked diubiquitin and OTUD2 OUT domain | (Mevissen et al., 2013) | PDB: 4BOS |
| crystal structure of K29-linked diubiquitin | (Kristariyanto et al., 2015) | PDB: 4S22 |
| crystal structure of K33-linked diubiquitin and Trabid NZF-1 | (Michel et al., 2015) | PDB: 5AF6 |
| crystal structure of K63-linked diubiquitin and RNF168 UDM1 | (Takahashi et al., 2018) | PDB: 5XIS |
| crystal structure of K6-linked diubiquitin | (Virdee et al., 2010) | PDB: 2XK5 |
| crystal structure of K6-linked diubiquitin and K6-specific affimer | (Michel et al., 2017) | PDB: 5OHL |
| Software and Algorithms | ||
| SerialEM | (Mastronarde, 2005) | https://bio3d.colorado.edu/SerialEM/ |
| RELION | (Scheres, 2012) | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| MotionCor2 | (Zheng et al., 2017) | https://emcore.ucsf.edu/ucsf-motioncor2 |
| CTFFIND4 | (Rohou and Grigorieff, 2015) | https://grigoriefflab.umassmed.edu/ctffind4 |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| Coot | (Emsley et al., 2010) | https://strucbio.biologie.unikonstanz.de/ccp4wiki/index.php/Coot |
| Phenix suite | (Adams et al., 2010) | https://www.phenix-online.org |
| EMAN2 | (Tang et al., 2007) | https://blake.bcm.edu/emanwiki/EMAN2 |
| Pymol | N/A | http://www.pymol.org |
Highlights.
Cryo-EM reveals M1-Ub6 bound to SA-like proteasome with closed CP gate
M1-Ub6 also binds to SD-like proteasome with opened CP gate
Unanchored M1-Ub6 binds at hRpn11 of SA-like and SD-like proteasome
M1-Ub6 shows second Ub extending along the hRpt4/5 coiled coil in SA-like state
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program through the CCR, NCI, NIH (1 ZIA BC011490 and the CCR FLEX program). Purchase of the Titan Krios in the Eyring Materials Center at Arizona State University was funded by the NSF MRI grant 1531991. The Titan Krios in the Laboratory of Cell Biology of the Center for Cancer Research of the National Cancer Institute is a member of NIH IRP CryoEM (NICE). The EM work performed at the Center for Molecular Microscopy was funded by FNLCR Contract HHSN261200800001E. Z.D. was funded by the Werner H. Kirsten Student Intern Program. This study utilized the computational resources of the High-Performance Computing Biowulf cluster of the NIH (http://hpc.nih.gov).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aufderheide A, Beck F, Stengel F, Hartwig M, Schweitzer A, Pfeifer G.n., Goldberg AL, Sakata E, Baumeister W, and Förster F (2015). Structural characterization of the interaction of Ubp6 with the 26S proteasome. Proc Natl Acad Sci U S A, 8626–8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avagliano Trezza R, Sonzogni M, Bossuyt SNV, Zampeta FI, Punt AM, van den Berg M, Rotaru DC, Koene LMC, Munshi ST, Stedehouder J, et al. (2019). Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat Neurosci 22, 1235–1247. [DOI] [PubMed] [Google Scholar]
- Bai XC, Rajendra E, Yang G, Shi Y, and Scheres SH (2015). Sampling the conformational space of the catalytic subunit of human gamma-secretase. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JAM, Bashore C, Dong KC, and Martin A (2019). The 26S Proteasome Utilizes a Kinetic Gateway to Prioritize Substrate Degradation. Cell 177, 286–298 e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, and Martin A (2018). Structure and Function of the 26S Proteasome. Annual review of biochemistry 87, 697–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck F, Unverdorben P, Bohn S, Schweitzer A, Pfeifer G, Sakata E, Nickell S, Plitzko JM, Villa E, Baumeister W, et al. (2012). Near-atomic resolution structural model of the yeast 26S proteasome. Proc Natl Acad Sci U S A 109, 14870–14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buel GR, Chen X, Chari R, O’Neill MJ, Ebelle DL, Jenkins C, Sridharan V, Tarasov SG, Tarasova NI, Andresson T, et al. (2020). Structure of E3 ligase E6AP with a proteasome-binding site provided by substrate receptor hRpn10. Nat Commun 11, 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, and Henderson R (2013). High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wu J, Lu Y, Ma YB, Lee BH, Yu Z, Ouyang Q, Finley DJ, Kirschner MW, and Mao Y (2016a). Structural basis for dynamic regulation of the human 26S proteasome. Proceedings of the National Academy of Sciences of the United States of America 113, 12991–12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ebelle DL, Wright BJ, Sridharan V, Hooper E, and Walters KJ (2019). Structure of hRpn10 Bound to UBQLN2 UBL Illustrates Basis for Complementarity between Shuttle Factors and Substrates at the Proteasome. Journal of molecular biology 431, 939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Randles L, Shi K, Tarasov SG, Aihara H, and Walters KJ (2016b). Structures of Rpn1 T1:Rad23 and hRpn13:hPLIC2 Reveal Distinct Binding Mechanisms between Substrate Receptors and Shuttle Factors of the Proteasome. Structure 24, 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, and Walters KJ (2015). Structural plasticity allows UCH37 to be primed by RPN13 or locked down by INO80G. Molecular cell 57, 767–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GA, and Goldberg AL (2017). The Logic of the 26S Proteasome. Cell 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook WJ, Jeffrey LC, Carson M, Chen Z, and Pickart CM (1992). Structure of a diubiquitin conjugate and a model for interaction with ubiquitin conjugating enzyme (E2). The Journal of biological chemistry 267, 16467–16471. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Marquardt JL, Ottiger M & Bax A (1998). Validation of Protein Structure from Anisotropic Carbonyl Chemical Shifts in a Dilute Liquid Crystalline Phase. J Am Chem Soc 120, 6836–6837. [Google Scholar]
- da Fonseca PC, He J, and Morris EP (2012). Molecular model of the human 26S proteasome. Mol Cell 46, 54–66. [DOI] [PubMed] [Google Scholar]
- de la Pena AH, Goodall EA, Gates SN, Lander GC, and Martin A (2018). Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Fu Z, Xu C, Wang Y, Wang Y, Li J, Kong L, Chen J, Li N, Zhang R, et al. (2017). High-resolution cryo-EM structure of the proteasome in complex with ADP-AlFx. Cell Res 27, 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Xu C, Sahu I, Wang Y, Fu Z, Huang M, Wong CCL, Glickman MH, and Cong Y (2019). Structural Snapshots of 26S Proteasome Reveal Tetraubiquitin-Induced Conformations. Molecular cell 73, 1150–1161 e1156. [DOI] [PubMed] [Google Scholar]
- Dong Y, Zhang S, Wu Z, Li X, Wang WL, Zhu Y, Stoilova-McPhie S, Lu Y, Finley D, and Mao Y (2019). Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 565, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlinger A, and Walters KJ (2013). Structural insights into proteasome activation by the 19S regulatory particle. Biochemistry 52, 3618–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele MR, Reed RG, Rudack T, Schweitzer A, Beck F, Nagy I, Pfeifer G, Plitzko JM, Baumeister W, Tomko RJ Jr., et al. (2018). Expanded Coverage of the 26S Proteasome Conformational Landscape Reveals Mechanisms of Peptidase Gating. Cell Rep 24, 1301–1315 e1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Chen X, and Walters KJ (2016). Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem Sci 41, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi TK, Zhong J, Mathivanan S, Karthick L, Chandrika KN, Mohan SS, Sharma S, Pinkert S, Nagaraju S, Periaswamy B, et al. (2006). Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38, 285–293. [DOI] [PubMed] [Google Scholar]
- Greene ER, Dong KC, and Martin A (2019a). Understanding the 26S proteasome molecular machine from a structural and conformational dynamics perspective. Curr Opin Struct Biol 61, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene ER, Goodall EA, de la Pena AH, Matyskiela ME, Lander GC, and Martin A (2019b). Specific lid-base contacts in the 26s proteasome control the conformational switching required for substrate degradation. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haakonsen DL, and Rape M (2019). Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends in cell biology 29, 704–716. [DOI] [PubMed] [Google Scholar]
- Hamazaki J, Iemura S, Natsume T, Yashiroda H, Tanaka K, and Murata S (2006). A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J 25, 4524–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, and Pavletich NP (1999). Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2–E3 enzyme cascade. Science 286, 1321–1326. [DOI] [PubMed] [Google Scholar]
- Huang X, Luan B, Wu J, and Shi Y (2016). An atomic structure of the human 26S proteasome. Nature structural & molecular biology 23, 778–785. [DOI] [PubMed] [Google Scholar]
- Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, and Dikic I (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson AD, Zhang NY, Xu P, Han KJ, Noone S, Peng J, and Liu CW (2009). The lysine 48 and lysine 63 ubiquitin conjugates are processed differently by the 26 s proteasome. The Journal of biological chemistry 284, 35485–35494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HC, and Huibregtse JM (2009). Polyubiquitination by HECT E3s and the determinants of chain type specificity. Molecular and cellular biology 29, 3307–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, Gygi SP, and Goldberg AL (2007). Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. The Journal of biological chemistry 282, 17375–17386. [DOI] [PubMed] [Google Scholar]
- Kristariyanto YA, Abdul Rehman SA, Campbell DG, Morrice NA, Johnson C, Toth R, and Kulathu Y (2015). K29-selective ubiquitin binding domain reveals structural basis of specificity and heterotypic nature of K29 polyubiquitin. Molecular Cell 58, 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, and Tagare HD (2014). Quantifying the local resolution of cryo-EM density maps. Nat Methods 11, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnle S, Martinez-Noel G, Leclere F, Hayes SD, Harper JW, and Howley PM (2018). Angelman syndrome-associated point mutations in the Zn(2+)-binding N-terminal (AZUL) domain of UBE3A ubiquitin ligase inhibit binding to the proteasome. The Journal of biological chemistry 293, 18387–18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YA, Lawson TG, Velayutham M, Zweier JL, and Pickart CM (2002). A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature 416, 763–767. [DOI] [PubMed] [Google Scholar]
- Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, and Martin A (2012). Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, and Baumeister W (2012). Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci U S A 109, 1380–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Lu Y, Prado MA, Shi Y, Tian G, Sun S, Elsasser S, Gygi SP, King RW, and Finley D (2016). USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 532, 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, Baker RT, Walz T, Ploegh H, and Finley D (2002). Multiple associated proteins regulate proteasome structure and function. Mol Cell 10, 495–507. [DOI] [PubMed] [Google Scholar]
- Li F, Xu D, Wang Y, Zhou Z, Liu J, Hu S, Gong Y, Yuan J, and Pan L (2018). Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy 14, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Ebelle DL, Matsuo H, and Walters KJ (2020). An Extended Conformation for K48 Ubiquitin Chains Revealed by the hRpn2:Rpn13:K48-Diubiquitin Structure. Structure 28, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Nowicka U, Sridharan V, Liu F, Randles L, Hymel D, Dyba M, Tarasov SG, Tarasova NI, Zhao XZ, et al. (2017). Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat Commun 8, 15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan B, Huang X, Wu J, Mei Z, Wang Y, Xue X, Yan C, Wang J, Finley DJ, Shi Y, et al. (2016). Structure of an endogenous yeast 26S proteasome reveals two major conformational states. Proceedings of the National Academy of Sciences of the United States of America 113, 2642–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour W, Nakasone MA, von Delbruck M, Yu Z, Krutauz D, Reis N, Kleifeld O, Sommer T, Fushman D, and Glickman MH (2015). Disassembly of Lys11 and mixed linkage polyubiquitin conjugates provides insights into function of proteasomal deubiquitinases Rpn11 and Ubp6. The Journal of biological chemistry 290, 4688–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Fonts K, Davis C, Tomita T, Elsasser S, Nager AR, Shi Y, Finley D, and Matouschek A (2020). The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat Commun 11, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, Lander GC, and Martin A (2013). Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol 20, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, et al. (2013). OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 154, 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MA, Elliott PR, Swatek KN, Simicek M, Pruneda JN, Wagstaff JL, Freund SM, and Komander D (2015). Assembly and specific recognition of K29- and K33-linked polyubiquitin. Molecular Cell 58, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MA, Swatek KN, Hospenthal MK, and Komander D (2017). Ubiquitin Linkage-Specific Affimers Reveal Insights into K6-Linked Ubiquitin Signaling. Molecular Cell 68, 233–246 e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Qiu XB, Ouyang SY, Li CJ, Miao S, Wang L, and Goldberg AL (2006). hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J 25, 5742–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries LK, Sander B, Deol KK, Letzelter MA, Strieter ER, and Lorenz S (2019). Analysis of ubiquitin recognition by the HECT ligase E6AP provides insight into its linkage specificity. The Journal of biological chemistry 294, 6113–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahtoe DD, van Dijk WJ, El Oualid F, Ekkebus R, Ovaa H, and Sixma TK (2015). Mechanism of UCH-L5 activation and inhibition by DEUBAD domains in RPN13 and INO80G. Molecular cell 57, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2016). Processing of Structurally Heterogeneous Cryo-EM Data in RELION. Methods in enzymology 579, 125–157. [DOI] [PubMed] [Google Scholar]
- Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, Elsasser S, Finley D, Dikic I, Walters KJ, and Groll M (2008). Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature 453, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer A, Aufderheide A, Rudack T, Beck F, Pfeifer G, Plitzko JM, Sakata E, Schulten K, Förster F, and Baumeister W (2016). Structure of the human 26S proteasome at a resolution of 3.9 A. Proceedings of the National Academy of Sciences of the United States of America 113, 7816–7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chen X, Elsasser S, Stocks BB, Tian G, Lee BH, Shi Y, Zhang N, de Poot SA, Tuebing F, et al. (2016). Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz P, Unverdorben P, Beck F, Pfeifer G, Schweitzer A, Forster F, and Baumeister W (2013). Structure of the 26S proteasome with ATP-gammaS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proceedings of the National Academy of Sciences of the United States of America 110, 7264–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DM, Chang SC, Park S, Finley D, Cheng Y, and Goldberg AL (2007). Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Molecular cell 27, 731–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi TS, Hirade Y, Toma A, Sato Y, Yamagata A, Goto-Ito S, Tomita A, Nakada S, and Fukai S (2018). Structural insights into two distinct binding modules for Lys63-linked polyubiquitin chains in RNF168. Nat Commun 9, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Trempe JF, Brown NR, Noble ME, and Endicott JA (2010). A new crystal form of Lys48-linked diubiquitin. Acta Crystallogr Sect F Struct Biol Cryst Commun 66, 994–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Ji Z, Wales TE, Bodnar NO, Ficarro SB, Marto JA, Engen JR, and Rapoport TA (2019). Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unverdorben P, Beck F, Sledz P, Schweitzer A, Pfeifer G, Plitzko JM, Baumeister W, and Forster F (2014). Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proc Natl Acad Sci U S A 111, 5544–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLinden RT, Hemmis CW, Schmitt B, Ndoja A, Whitby FG, Robinson H, Cohen RE, Yao T, and Hill CP (2015). Structural basis for the activation and inhibition of the UCH37 deubiquitylase. Molecular cell 57, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLinden RT, Hemmis CW, Yao T, Robinson H, and Hill CP (2017). Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. The Journal of biological chemistry 292, 9493–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virdee S, Ye Y, Nguyen DP, Komander D, and Chin JW (2010). Engineered diubiquitin synthesis reveals Lys29-isopeptide specificity of an OTU deubiquitinase. Nat Chem Biol 6, 750–757. [DOI] [PubMed] [Google Scholar]
- Wang M, and Pickart CM (2005). Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. The EMBO journal 24, 4324–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Young P, and Walters KJ (2005). Structure of S5a bound to monoubiquitin provides a model for polyubiquitin recognition. J Mol Biol 348, 727–739. [DOI] [PubMed] [Google Scholar]
- Wang X, Cimermancic P, Yu C, Schweitzer A, Chopra N, Engel JL, Greenberg C, Huszagh AS, Beck F, Sakata E, et al. (2017). Molecular Details Underlying Dynamic Structures and Regulation of the Human 26S Proteasome. Mol Cell Proteomics 16, 840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehmer M, Rudack T, Beck F, Aufderheide A, Pfeifer G, Plitzko JM, Forster F, Schulten K, Baumeister W, and Sakata E (2017). Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proceedings of the National Academy of Sciences of the United States of America 114, 1305–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehmer M, and Sakata E (2016). Recent advances in the structural biology of the 26S proteasome. Int J Biochem Cell Biol 79, 437–442. [DOI] [PubMed] [Google Scholar]
- Worden EJ, Dong KC, and Martin A (2017). An AAA Motor-Driven Mechanical Switch in Rpn11 Controls Deubiquitination at the 26S Proteasome. Molecular cell 67, 799–811 e798. [DOI] [PubMed] [Google Scholar]
- Yao T, and Cohen RE (2002). A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 419, 403–407. [DOI] [PubMed] [Google Scholar]
- Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, Washburn MP, Conaway RC, Conaway JW, and Cohen RE (2006). Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat Cell Biol 8, 994–1002. [DOI] [PubMed] [Google Scholar]
- Young P, Deveraux Q, Beal RE, Pickart CM, and Rechsteiner M (1998). Characterization of two polyubiquitin binding sites in the 26 S protease subunit 5a. The Journal of biological chemistry 273, 5461–5467. [DOI] [PubMed] [Google Scholar]
- Zhang N, Wang Q, Ehlinger A, Randles L, Lary JW, Kang Y, Haririnia A, Storaska AJ, Cole JL, Fushman D, et al. (2009). Structure of the s5a:k48-linked diubiquitin complex and its interactions with rpn13. Mol Cell 35, 280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Wang WL, Yu D, Ouyang Q, Lu Y, and Mao Y (2018). Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat Commun 9, 1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM reconstructions generated by this study have been deposited into the Electron Microscopy Data Bank with accession numbers 21691 (SA-like) and 21696 (SD-like) for the sample of human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP; 21699 (SA-like, dataset 1), 21700 (SD-like, dataset 1), 21697 (SA-like, dataset 2), and 21698 (SD-like, dataset 2) for the sample of human 26S proteasome mixed non-cleavable M1-Ub6. The docked coordinates reported in this paper have been deposited into the Protein Data Bank with accession codes PDB: 6WJD (SA-like) and 6WJN (SD-like) for human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP. The raw micrographs and particle data have been deposited in the Electron Microscopy Pilot Image Archive with accession numbers EMPIAR-10401 (sample of human 26S proteasome mixed non-cleavable M1-Ub6 and E6AP), EMPIAR-10402 and EMPIAR-10403 (sample of human 26S proteasome mixed non-cleavable M1-Ub6 dataset 1 and 2). Other data are available from the corresponding author upon reasonable request.