

Abstract
T cell exhaustion in cancer is linked to poor clinical outcomes, where evidence suggests T cell metabolic changes precede functional exhaustion. Direct competition between tumor-infiltrating lymphocytes (TIL) and cancer cells for metabolic resources often renders T cells dysfunctional. Environmental stress produces epigenome remodeling events within TIL resulting from loss of the histone methyltransferase EZH2. Here we report an epigenetic mechanism contributing to the development of metabolic exhaustion in TIL. A multi-comics approach revealed a Cdkn2a.Arf-mediated, p53-independent mechanism by which EZH2 inhibition leads to mitochondrial dysfunction and the resultant exhaustion. Reprogramming T cells to express a gain-of-function EZH2 mutant resulted in an enhanced ability of T cells to inhibit tumor growth in vitro and in vivo. Our data suggest that manipulation of T cell EZH2 within the context of cellular therapies may yield lymphocytes that are able to withstand harsh tumor metabolic environments and collateral pharmacologic insults.
Keywords: EZH2, T cell, metabolism, immunotherapy, epigenetics
Introduction
The ability of the immune system to sustain T cell function in a wide range of environments provides the immunosurveillance necessary to eliminate genetically-damaged cells, protecting the host from developing overt malignancies. Cancer cells that escape the initial immune response can form solid tumors, which undergo further immunoediting, driving the generation of an immunosuppressive microenvironment resulting in restricted T cell infiltration and effector function1. Features of the immune-hostile tumor environment include direct repression and recruitment of immunosuppressive cell populations, culminating in failure of the antitumor immune response and disease progression2,3. The continuous T cell activation signaling in sustained antitumor responses promotes expression of inhibitory receptors (PD-1, CTLA-4, Tim-3, and LAG-3) which negatively regulate activation and effector functions, resulting in intrinsically-driven T cell exhaustion programs. Importantly, T cells can still display an exhausted phenotype in the absence of inhibitory molecules4. In the context of solid tumors, one driving force of T cell exhaustion results from inhibitory metabolic parameters encountered by tumor-infiltrating lymphocytes such as glucose deprivation and mitochondrial dysfunction5,6. Recent discoveries have highlighted various metabolic pathways as key intrinsic mechanisms through which T cells regulate their fate7,8. Nutrient availability is a primary driver of metabolic programming, and is a necessary signal for maintenance of self-tolerance and protecting the host from tissue damage9,10. Loss of T cell metabolic regulatory signals can be illustrated by the strong correlation between metabolic syndrome (obesity, hyperglycemia, dyslipidemia, and hypertension) and autoimmune diseases11. T cells, like cancer cells, primarily utilize aerobic glycolysis for their energy needs. However, mitochondria are critical organelles for maintaining the integrity of effector T cells and the formation of memory T cells12. Tumor-infiltrating lymphocytes (TILs) show a progressive loss of mitochondrial function and a reduction in glucose uptake (metabolic exhaustion) which is largely independent of checkpoint blockade or regulatory cell suppression13.
Recent studies have begun to highlight the metabolic underpinnings of T cell function and raised the possibility of metabolic manipulation aimed at vastly improving cancer immunotherapy14,15. The dynamic interplay between epigenetics and metabolic pathways has been revealed as a primary mechanism cells use to sense and respond to environmental pressures16. One such metabolic and epigenetic circuit is observed when T cells experience glucose deprivation and downregulate the epigenetic modifier, EZH2. During T cell activation EZH2 is rapidly upregulated and tumor-induced glucose deprivation suppresses EZH2 function resulting in an overtly immunosuppressive environment17. EZH2 is a histone methyltransferase which functions as the catalytic subunit of the polycomb repressive complex-2 (PRC2). The PCR2 complex represses genes through methylation events (di- and trimethylation) of Lysine-27 on histone 318. The tri-methylation state, H3K27me3, is associated with long-term transcriptional repression. Importantly, mis-regulation of EZH2 in cancer has led to the development of inhibitors, which have been proposed for combination with immunotherapies19,20. However, the role of EZH2 in T cell function must be a therapeutic consideration, as the hostile metabolic environment of solid tumors innately drives EZH2 suppression within T cells, which would only be furthered by systemic EZH2 inhibitor therapy. EZH2high CD8+ T cells are associated with improved T cell function, providing clear rationale for mitigating anti-EZH2 effects, whether from anti-cancer therapies or those intrinsic to the metabolic environment T-cells face within solid tumors17,21,22. Here, we seek to characterize the relationship between the loss of EZH2 and metabolic-stress-induced exhaustion in T cells.
Materials and Methods
Mouse models
The C57BL/6 (stock #000664), CD45.1:B6.SJL-Ptprca Pepcb/BoyJ (stock # 002014), Arf-KO:B6.129X1-Cdkn2atm1Cjs/KaiJ (stock# 029676), Rag1-KO: B6.129S7-Rag1tm1Mom/J (stock#) and the major histocompatibility complex (MHC) class I-restricted OVA specific TCR OT-I: C57BL/6-Tg(TcraTcrb)1100Mjb/J mice were purchased from The Jackson laboratory. The Lck-EZH2Y641F transgenic mice were provided as a gift by Dr. Keith Humphries (BC Cancer Agency) and screened by PCR prior to experimentation. The animal experiments described in this study were reviewed and approved by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee (IACUC). Mice were bred pathogen free standards and transferred to open top cages for experimental procedures. For tumor experiments 8–15 week old mice were randomized to match age and gender across experimental groups. No association of sex or weight with response was observed or expected.
Tumor challenge
Mice were injected in the subcutaneous flank space with 1×106 tumor cells suspended in 100μl of PBS. Tumor growth was monitored daily with caliper measurements of tumor length and width. Survival was plotted on Kaplan-Meier curves, the number of days post-tumor engraftment where the tumor was less than 1000 mm3 (unless noted differently), with no ulceration. Tumor challenge studies were performed in all cases at least twice. For in vivo EZH2i, starting on day 5, mice were injected (orally) with vehicle (0.5% w/v methyl cellulose and 0.1% Tween-80) or 250mg/kg EPZ6438 twice daily for 5 consecutive days.
Primary cell culture
Naïve lymphocytes (CD8+, CD4+, or CD4+CD25+) were isolated from single cell suspension of murine splenocytes using magnetic selection (Miltenyi). Purified lymphocytes were then activated using of 5 μg/mL plate bound antiCD3e (Biolegend), 2 μg/mL soluble CD28 (Biolegend) and 50U/mL IL-2 (Peprotech) for indicated times. Lymphocytes were cultured in RPMI (Life Technologies) with 10% fetal bovine serum, 55 μM 2-mercaptoethanol, 2 mM glutamine, penicillin, and streptomycin at 37°C and 5% CO2.
Cell lines
The B16F10 mouse cell line was purchased from ATCC. The B16SIY cell line was a kind gift from Thomas Gajewski (University of Chicago). Tumor cell lines were cultured in DMEM supplemented with 10% FBS, penicillin and streptomycin at 37°C and 5% CO2. All tumor cell lines were screened for contaminating pathogens (Ectromelia, EDIM, LCMV, MAV1, MAV2, MHV, MPV, MVM, Mycoplasma pulmonis, Polyoma, PVM, REO3, Sendai, TMEV) by IDEXX laboratories and do not contain any pathogens or mycoplasma.
Generation of MC38.SIINFEKL-
MC38 cells were engineered to express DsRed fused in frame with 3 repeated sequences encoding the model antigen SIINFEKL followed by an AAY lilnker. The construct was generated by digesting a gBlock (IDT) encoding 3X-SIINFEKL-AAY with flanking XhoI/BamHI cut sites. 100 ng of the gBlock and 1 ug of pRetro-dsRed-monomer-N1 (Takara: 632465) was digested with XhoI and BamHI and gel purified (QIAGEN). Purified digested gBlock and pRetro vector were ligated together and transformed into Stbl3 (Fisher) cells. The insert region of the pRetro-SIINFEKL-dsRed vector was sequenced to confirm that the insert was in-frame with dsRed without intervening stop codons. To generate the MC38.SIINFEKL.dsRed cell line, Phoenix (ATCC) cells were transfected with the pRetro-SIINFEKL-dsRed vector using Lipofectamine 3000 (Fisher) following manufacturer’s protocol. 48 hours after transfection, supernatant was collected, filtered through a 0.45 μm filter, and added to tumor cells with polybrene at 10 μg/mL. After expansion cells were sorted based on dsRed expression. The cells were sorted 3 more times for the top 5% dsRed positive to ensure no dsRed cells remained.
IFN-γ ELISPOT Assay
Mice were injected in the subcutaneous flank space with 1×106 tumor cells suspended in 100μl of PBS. Spleens were harvested 7 days after injection for analysis. The enzyme-linked Immunospot assay (ELISPOT) was conducted with the BD mouse IFN-γ kit according to the manufacturer’s protocol. Splenocytes were plated at 106 cells/well and stimulated overnight with, SIINFEKL peptide (160 nM), or PMA (50 ng/ml) and ionomycin (0.5 μM). IFN-γ spots were detected using biotinylated antibody and avidin-peroxidase and developed using AEC substrate (Sigma-Aldrich).
In vitro killing assay
T cell killing assays were carried out using pre-activated CD8+ T cells from wt (control) or OT-1 (tumor specific) mice. Activated T cells were washed and labeled with Cell Trace violet to enable their subsequent discrimination from target cells. Target cells (MC38SIINFEKL) were plated for 16 hours prior to culturing with T cells. The co-cultures were conducted at different target: effector (T:E) ratios for 10 hours. Target cell viability was then determined by the percentage of Annexin V- and PI- (propidium iodide) cells.
Adoptive transfers
For in vivo survival experiments, 2×106 activated CD8+ T cells/mouse were injected i.v. into CD45.1+ C57BL/6 mice. Cells were recovered two days later from the peripheral blood, spleen, and lymph nodes then analyzed by flow cytometry. For adoptive cellular treatment experiments, 4×106 CD8+ OT-I+ or CD8+ OT-I+ Lck-EZH2+ pre-activated T cells/mouse were injected intravenously into tumor-bearing (MC8SIINFEKL) mice and measured for tumor volume growth.
Western blotting and quantitative PCR
For immunoblot analysis, harvested cells were lysed on ice for 30 minutes with radioimmunoprecipitation assay buffer (RIPA) (10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl). Lysates were then cleared by centrifugation and protein concentrations were determined by bicinchoninic acid (BCA) protein assay (Thermo). Electrophoresis was performed using bis-Tris gels 4–12% (Invitrogen) and then samples were transferred to polyvinylidene difluoride (PVDF; Millipore) membranes. Detection was performed using Western Lightning Plus ECL enhanced chemiluminescent substrate (Perkin-Elmer) according to the manufacturer’s instructions. See supplemental methods for the information of antibodies used.
For QPCR, RNA was extracted from activated lymphocytes using an RNAeasy purification kit (Qiagen) and complementary DNA was synthesized (Bio-Rad). Target Primer sets below used were used with Sybr-Green (Bio-Rad) to measure relative transcript levels. Ubiquitin was used as a house keeping gene. ΔΔCT was used to calculate relative fold change.
FACS and TIL sort
Mitochondrial Dyes-
Mitochondrial mass was analyzed using Mitotraker™ Green FM (Thermo) at 100nM according to manufacturer protocol. Mitochondrial membrane potential was analyzed using tetramethylrhodamine methyl ester (TMRM) (Thermo) at 50nM according to manufacturer protocol. Mitochondrial superoxide was analyzed using MitoSOX™ at 1μM according to manufacturer protocol.
Cell cycle was analyzed by fixing cells in 70% ethanol overnight and staining with PI/RNase Staining Buffer (BD Biosciences). FACS data were analyzed with Flow Jo /Dean-Jett Fox (DJF) model (BD Biosciences).
TIL purification-
Tumor dissociation was performed using mechanical separation and was resuspended in FACs Buffer (PBS with 1% FBS) at 1×106 cells/100ul and blocked with FcBlocker (BD Biosciences). The following antibodies were used to define immune populations: anti-CD3 (Biolegend), anti-CD45, anti-CD19, anti-CD8, anti-CD4, anti-CD11b, anti-Gr1, and anti-NK1.1. DAPI was used as a viability stain. For cell sorting tumors, dead cells were depleted prior to staining using dead cell removal magnetic beads (Miltenyi). FACSAria was used for cell sorting.
EM imaging
T cells were harvested and then fixed for 2 h at room temperature with 2.5% glutaraldehyde and 0.05% malachite green in 0.1 M sodium cacodylate buffer, pH 6.8. Samples were post-fixed for 30 min with 0.5% osmium tetroxide and 0.8% potassium ferricyanide in 0.1 M sodium cacodylate, for 1 h in 1% tannic acid, and for 1 h in 1% uranyl acetate at room temperature. Specimens were dehydrated with a graded ethanol series and embedded in resin. Thin sections were cut with an RMC MT-7000 ultramicrotome (Ventana) stained with 1% uranyl acetate and lead citrate before viewing at 200 keV on a FEI Tecnai F20 Transmission Electron Microscope. Digital images were acquired with an AMT digital camera system23.
Metabolic phenotyping
Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were measured using the Seahorse XFe bioanalyzer. 2 ×105 T cells per well (≥8 wells per sample) were spun onto Cell-Tak(Corning) coated seahorse 96 well plates and preincubated at 37°C for ~20 min in the absence of CO2. OCR and ECAR were measured in XF media (non-buffered RPMI 1640 containing 10 mM glucose, 2mM L-glutamine, and 1 mM sodium pyruvate) under basal conditions and in response to 2 μM oligomycin, 2 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP), 10mM 2-Deoxy-D-glucose, and 500 nM rotenone + 500 nM antimycin A.
ATP measurements-
Relative ATP levels were determined using Cell titer Glo™ (Promega) according to manufacturer’s protocol. 1×105 activated T cells were used per 96 well to determine relative luminescence unit for each condition.
Cell death experiments
Activated CD8+ lymphocytes were plated in fresh media at 0.5×106 cells/mL in 96 well plates. Cells were treated with glucose or glutamine withdrawal, H2O2 (Sigma), 2-DeoxyGlucose (Sigma), Staurosporine (Seleckchem) or vehicle control at indicated doses for 24 hours. Cell viability was determined by staining with Annexin V-FITC and DAPI staining. For withdrawal of glucose and glutamine cell were cultured in dialyzed FBS.
Histone mass spectrometry and PTM identification
Histone mass spectrometry was carried out as described previously24. Histones were purified by acid extraction and resolved on a 4–12% gradient SDS-PAGE gel. Histone bands were visualized by Coomasie staining. Histones were excised from the gel, destained, and treated with 20 μL/band of 30% D6-acetic anhydride in 50 mM ammonium bicarbonate, which chemically adds isotopically heavy acetylation on unmodified and monomethylated lysines. Histones were then digested in-gel with 125 ng/band sequencing-grade trypsin at 37°C for overnight. Acidified tryptic peptides were separated using a 2.5 μm Waters XSelect CSH resin on a 150 mm × 0.075 mm column using a nanoAcquity UPLC system (Waters, Milford, MA). Peptides were separated with a 60-minute chromatography gradient, with a 40-minute linear separation gradient from 97% buffer A [0.1% formic acid in water (v/v)], 3% buffer B [0.1% formic acid (v/v), 99.9% acetonitrile (v/v)], to 80% of buffer A, 20% buffer B. Eluted peptides were ionized by electrospray (2150 V) and analyzed on an Orbitrap Fusion Lumos mass spectrometer (Thermo) using data-dependent acquisition. Spectral count data was exported in tabular format and analyzed using R. Additional details regarding histone mass spectrometry and PTM identification are available in the supplementary materials.
FASP bHPLC TMT mass spectrometry
Purified proteins were reduced, alkylated, and digested using filter-aided sample preparation25. Tryptic peptides were separated into 36 fractions on a 100 × 1.0 mm Acquity BEH C18 column (Waters) using an UltiMate 3000 UHPLC system (Thermo) with a 40 min gradient from 99:1 to 60:40 buffer A:B ratio under basic pH conditions, and then consolidated into 12 super-fractions. Each super-fraction was then further separated by reverse phase Jupiter Proteo resin (Phenomenex) on an in-line 200 × 0.075 mm column using a nanoAcquity UPLC system (Waters). Peptides were eluted using a 60 min gradient from 97:3 to 67:33 buffer A:B ratio. Eluted peptides were ionized by electrospray (2.15 kV) followed by mass spectrometric analysis on an Orbitrap Fusion Tribrid mass spectrometer (Thermo) using multi-notch MS3 parameters. Scaffold Q+S (Proteome Software) was used to verify MS/MS based peptide and protein identifications (protein identifications were accepted if they could be established with less than 1.0% false discovery and contained at least 2 identified peptides; protein probabilities were assigned by the Protein Prophet algorithm and to perform reporter ion-based statistical analysis26. Additional details regarding TMT mass spectrometry are available in the supplementary materials.
RNA sequencing
RNA sequencing was carried out as before27. For total RNA-seq, the Sequence library was prepared from 500 ng of total RNA using Illumina’s (San Diego, CA) TruSeq RNA Sample Preparation Kit v2 following the manufacturer’s protocol. cDNA Libraries were validated on the Arkansas Children’s Research Institute (ACRI) Genomics core Fragment Analyzer for fragment size peak of approximately 260 bp and were quantified by a Qubit fluorometer. Equal amounts of each library were pooled for sequencing on the NextSeq 500 platform using a high output flow cell to generate approximately 25 million 75-base reads per sample. cDNA libraries were constructed using Illumina’s TruSeq stranded mRNA sample preparation kit according to the manufactures protocol. All sequencing was conducted by the Center for Translational Pediatric Research Genomics Core Lab at Arkansas Children’s Research Institute (Little Rock, AR). For each comparison, edgeR’s t-tests relative to a threshold (glmTreat(), lfc=1) method correcting for batch effects was used to identify differentially expressed genes between experimental groups. Genes with a fold-change (FC) > 2 and multiple test corrected (FDR) p-values < 0.05 were selected for further comparisons between treatments and analyzed by Ingenuity Pathway Analysis (IPA) for biological involvement. Additional details regarding RNA sequencing analysis are available in the supplementary materials.
ChIP and ChIP-seq
ChIP-qPCR and ChIP-Seq were carried out as before28. For H3K27me3 and H3K36me3 ChIP-Seq, 40 million of cells were cross-linked with 1% formaldehyde for 10 minutes, followed by addition of glycine to stop crosslinking. After washing, cell lysis and sonication, the chromatin samples were incubated with antibody-conjugated Dynabeads (Invitrogen) at 4 degrees Celsius. Beads bound with chromatin were then subject to extensive washing and elution. Eluted chromatin was de-crosslinked overnight at 65 degrees, followed by protein digestion with proteinase K and DNA purification with Qiagen PCR purification kit. The obtained ChIP DNA samples were submitted to the UNC-Chapel Hill High-Throughput Sequencing Facility (HTSF) for preparation of multiplexed libraries and deep sequencing with an Illumina High-Seq platform according to the manufacturer’s instructions. Tag counts between DMSO controls and EZH2i treated samples were then determined across the entire mouse genome using Bedtools multicov and log2 fold change and percent change (i.e. decrease or increase) was calculated29. To compare DMSO controls to EZH2i treated samples, Deeptools bamCoverage tool was used to calculate tag coverage for the following mouse genes: cdkn2a, Igf2bp3, Dab2ip, Gzma, Upp1, Tgm2, Fbxo2, and Kit30. Additional details regarding ChIP sequencing analysis are available in the supplementary materials.
Quantification and Statistical Analysis
Please refer to the appropriate methods section for proteomics and sequencing data set statistical methods. Otherwise, comparisons for two groups were calculated using unpaired two-tailed student’s t-tests, comparisons for more than two groups were calculated using one-way ANOVA followed by Bonferroni’s multiple comparison tests. Comparisons over time were calculated using two-way ANOVA followed by Bonferroni’s multiple comparison tests. For Kaplan-Meier plots of survival log-rank test was used to determine P values. Data was analyzed using GraphPad Prism 7, R Studio, and Microsoft Excel.
Results
EZH2 inhibition induces T cell exhaustion and dysfunction
To establish the repression of EZH2 (H3K27me3) during tumor infiltration we purified TILs from an immune suppressive murine model of melanoma (B16F10)31. TILs from B16F10 tumors have been demonstrated to be exhausted and here we show infiltrating CD4+ and CD8+ T cells displayed a reduction in H3K27me3 compared to lymphocytes from tumor draining lymph nodes and in vitro activated T cells (Figure 1A). To model acute inhibition of EZH2 in activated CD8+ T cells, we used highly specific and effective small molecule inhibitors (EZH2i) (Figure 1B). We believe this approach, in contrast to genetic deletion, more accurately models the loss of T cell EZH2 activity, which occurs within the tumor environment. Further, this approach also reveals potential unintended consequences impacting T cell function which could occur with systemic EZH2i anti-cancer therapies. In vitro inhibition of EZH2 in primary CD8+ T cells lead to a minor effect on their in vitro proliferative capacity (Figure 1C).
Figure 1: Systems approach uncovers drivers of T cell exhaustion resulting from EZH2-inhibtion.

(A) Western blot analysis of TDLN and B16F10 melanoma TIL populations. FACS Cell sorting was used to purify CD4+ and CD8+ lymphocyte populations and in vitro activated (CD3e/CD28) CD8+ T cells were used as a positive control. (B) Western blot analysis of in vitro pre-activated, primary CD8+ T cells treated with EZH2i (EPZ6438 2.5μM) for 24 or 48 hours. In all experiments, CD8+ T cells were purified prior to activation. (C) The proliferation of pre-activated CD8+ T cells ±EZH2i was determined using trypan blue exclusion, n=5 and error bars represent SEM. (D) Subset of genes involved in T cell effector function and exhaustion from RNAseq and proteomic analysis of EZH2i-treated pre-activated CD8+ T cells. The RNAseq heat map was generated using logCPM values (Z-score) and asterisks signify an adjusted P value <0.05 (Holm-Sidak). The protein heatmap was generated from normalized tandem mass tag intensities (Z-score) and asterisks signify an adjusted P value <0.05 (Holm-Sidak). (E) Subset of exhaustion associated transcription factors from RNAseq dataset. (F) C57BL/6 (immune competent) or Rag1−/− (immune compromised) mice were injected subcutaneously with 1×106 B16SIY cells. Starting on day 5, mice were injected (orally) with vehicle (0.5% w/v methyl cellulose and 0.1% Tween-80) or 250mg/kg EPZ6438 twice daily for 5 consecutive days. Tumor growth curves depict an average tumor volume in each group, n=5–6 and error bars indicate the SEM. Kaplan-Meier survival of recipient mice (tumor size> 500mm3). P value denotes statistical significance by Log Rank Test. (G) Significantly altered gene lists from Ingenuity Pathway Analysis (IPA) of proteomic data. (H) Venn diagram comparing significant genes identified in RNAseq and proteomics data sets.
RNA sequencing and proteomic analysis was performed on in vitro activated cytotoxic T cells (CD8+) treated post-activation (48 hours) with EZH2i for 48 hours as in Figure 1B. RNA sequencing and proteomic datasets, revealed an exhausted phenotype induced by the in vitro inhibition of EZH2 (Figure 1D). A significant increase in inhibitory receptors (Pdcd1, Lag3, Tigit) and loss of memory markers (Cxcr3, Cd62L) was consistent at both transcript and protein levels, indicating EZH2i-effects negatively control T cell function. Further, the exhaustion driving transcription factors Nr4a (Nrfa1, Nrfa2, Nrfa3), Tox and Tox2 were also elevated in EZH2i treated CD8+ T cells (Figure 1E). In an in vitro setting, EZH2i had no effect on the ability of OT-1+ T cells to kill MC38SIINFEKL tumor cells. Target tumor cells were engineered to express the ovalbumin antigen SIINFEKL and were validated using Elispot (Figure S1A, B). This suggests that EZH2i does not simply lead to a difference in the strength of activation.
In vivo, EZH2 inhibition blocks immune control of the highly immunogenic murine model of melanoma, B16F10SIY (Figure 1F). B16F10SIY cells are engineered to express the model antigen, SIY (SIYRYYGL) and have a well-documented growth delay in immune competent recipient mice due to strong immune detection32. B16F10 cells are resistant to EZH2 inhibition and provide an opportunity to assess the effects of EZH2 inhibitors (EZH2i) on T cell antitumor activity33. Treatment with EZH2i increased the growth rate of B16F10SIY tumors in vivo and decreased survival in WT mice. This was not observed in the immune-compromised, RAG1−/− mice, suggesting a role in the repression of TILs. However, this does not inform us on the extent or the capacity in which T cells require EZH2 for their function.
Multi-omics approach uncovers mitochondrial dysfunction as a driver of EZH2-inhibition induced T cell exhaustion
In addition to the functional markers above, we quantified the relative abundance of 11006 protein coding transcripts and 9301 proteins. EZH2i treatment resulted in 62 transcripts differentially expressed (Log2FC>2.5, p value<0.05) (Figure S2A) and 34 protein levels differentially expressed (Log2FC>1.25, p value<0.05) (Figure S2B). As expected, inhibition of the repressive PRC2 complex, generated mostly increases in transcript and protein levels, visualized through volcano plots (Figure S2C, D). To prioritize upstream candidate genes and pathways contributing to the exhaustion phenotype, we used two approaches. First, Ingenuity pathway analysis of differentially expressed proteins predicted the most significant cellular pathways in EZH2i-treated T cells largely correspond to mitochondrial dysfunction (Figure 1G). The genes that define this gene set include: Gzma, Mapk12, Tgm2, mt-Atp6, Cdkn2a and Cox6a1. Interestingly, elevation of Tec kinase signaling was also identified in the analysis and is known for its role in TCR signaling leading to IL-2 induction which can be sign of over-stimulation and exhaustion34. Our second approach to prioritize candidates was based on the understanding that EZH2 primarily regulates genes at the transcript level, and therefore most upstream candidates are likely different at both the transcript and protein levels. We regarded the genes found in the union of this multi-omics approach as possible drivers of exhaustion in EZH2i-treated T cells (Figure 1H). Taken together, these data suggest EZH2i-driven T cell exhaustion is, at least in part, due to mitochondrial dysfunction.
Loss of EZH2 function leads to metabolic exhaustion in CD8+ T cells
T cell activation leads to rapid and robust metabolic changes essential to support proliferation and function35. To further understand possible metabolic changes in response to EZH2 inhibition, we measured oxygen consumption rates (OCR) and extracellular acidification rates (ECAR). Metabolic flux analysis of CD8+ T cells treated with EZH2i revealed a defect in basal oxygen consumption rates (OCR) and substantial loss of spare respiratory capacity (OCRMax-OCRBasal), which are key measurements of mitochondrial respiration and oxidative phosphorylation (OxPhos) (Figure 2A). Naïve T cells were used for comparison and to illustrate the drastic loss in oxygen consumption. EZH2 inhibition resulted in an increased dependence on glycolytic metabolism, indicated by the ratio of basal ECAR/OCR (Glycolysis/OxPhos) (Figure 2B). Parallel analysis of cytotoxic (CD8+), helper (CD4+, CD25−) and regulatory (CD4+, CD25+) T cells reveal a similar loss in OCR (Figure 2C, S3A). Consistent with mitochondrial dysfunction, cytotoxic, helper and regulatory T cells all display a loss of mitochondrial membrane potential (Figure 2D) and an increase in mitochondrial derived superoxide species (Figure 2E) when treated with EZH2i. The metabolic phenotype induced by EZH2 inhibition was replicated in CD8+ T cells with an alternative EZH2i, GSK503 (Figure S3C, D). Surprisingly, cellular ATP levels were not affected in EZH2i treated T cells (Figure S3E). This suggests glycolytic metabolism compensates for the loss of mitochondrial respiration and provides the necessary ATP in an in vitro, glucose rich environment. Transmission electron microscopy revealed an abnormal mitochondrial morphology in CD8+ T cells treated with an EZH2i (Figure 2F, S3F). We also observed a loss of mitochondrial mass using MitoTracker FM (Figure 2G).
Figure 2: Loss of EZH2 leads to metabolic exhaustion in CD8+ T cells.
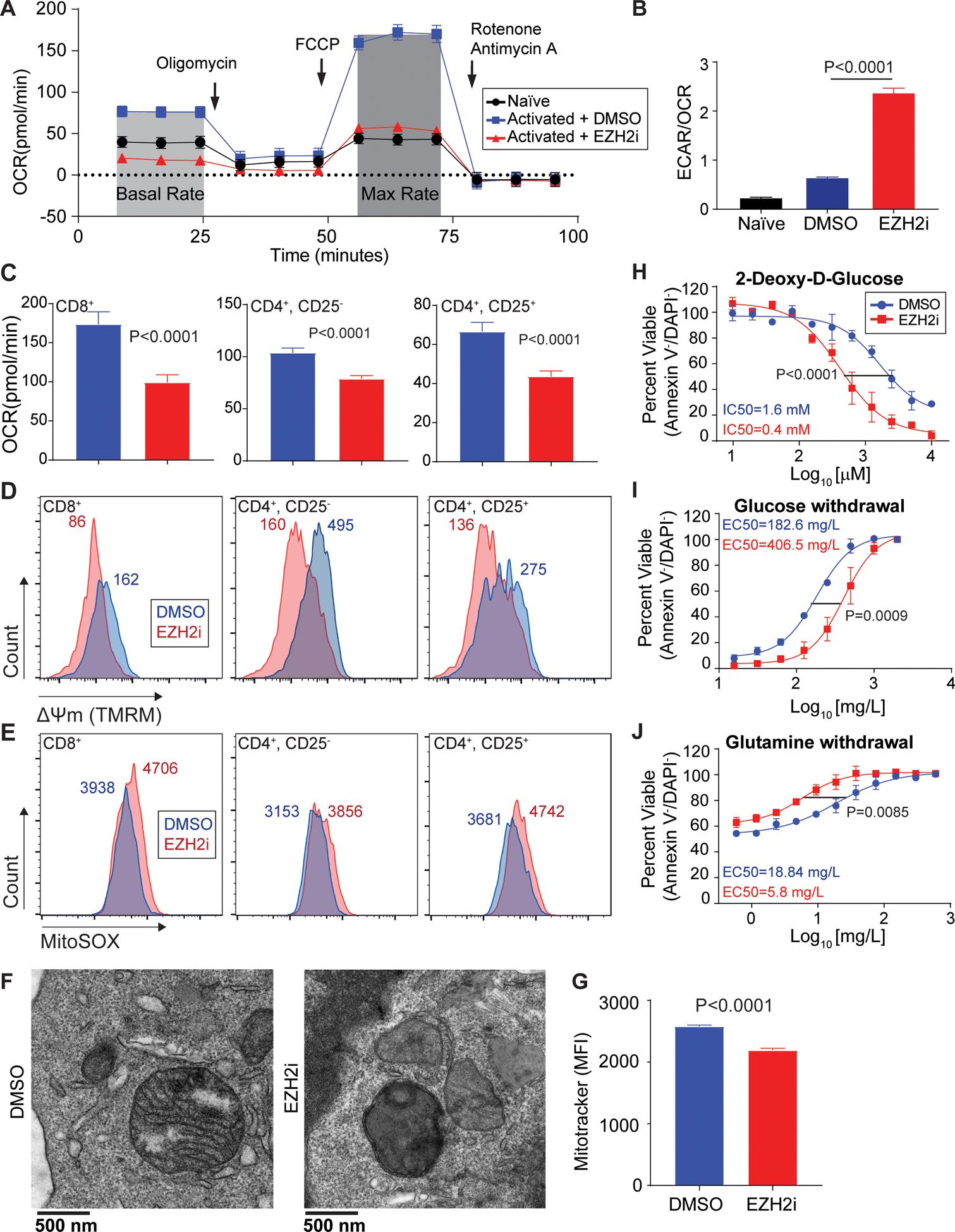
(A) Representative trace of oxygen consumption rate (OCR) in pre-activated CD8+ T cells treated for 48 hours with EZH2i. Arrows indicate injection time of respective inhibitors: Oligomycin (ATP synthase inhibitor), FCCP (Uncoupler), and Rotenone + Antimycin (Complex I & III). Naïve T cells were used for comparison. (B) Glycolytic dependency determined by the ratio of extracellular acidification rates (ECAR) and OCR. Data represent the mean and error bars represent the SEM. P value was determined by unpaired t test. (C-E) Parallel analysis of activated cytotoxic (CD8+), helper (CD4+CD25−), and regulatory (CD4+CD25+) T cells ±EZH2i: (C) OCR, (D) mitochondrial membrane potential (TMRM), and (E) mitochondrial superoxide species (MitoSOX). Histogram median fluorescent intensity (MFI) is indicated. (F) Representative transmission electron microscopy of activated CD8+ T cells ±EZH2i. (G) Relative mitochondrial mass FACS analysis (MitoTracker FM) from T cells ±EZH2i. Data represent the mean (n=3) and error bars represent the SEM. P value was determined by unpaired t test. (H-J) Viability was determined by Annexin-V/DAPI staining in CD8+ T cell cultures treated with: (H) increasing doses of 2-DG, (I) glucose withdrawal, and (J) glutamine withdrawal for 24 hours. Data represent the mean (n=3) and error bars represent SEM. Sum-of-squares F test used for statistical comparison of IC50s.
As the loss of mitochondrial function was clear, we next examined the sensitivity of EZH2-inhibited T cells to metabolic-stress induced cell death. EZH2 inhibition rendered T cells more sensitive to increasing doses of 2-deoxy-D-glucose (glycolysis inhibitor) (Figure 2H) and to the withdrawal of glucose (Figure 2I). Conversely, EZH2 inhibition reduced the sensitivity to the withdrawal of glutamine, a mitochondrial substrate (Figure 2J). EZH2 inhibition also leads to a sensitivity of T cells to oxidative stress (H2O2) (Figure S3G) and pan-kinase inhibition (Staurosporine) (Figure S3H). Thus, EZH2 inhibition causes a dramatic shift in T cell dependency on glycolytic metabolism, rendering them metabolically exhausted and sensitive to further insult including glucose withdrawal.
EZH2 inhibition leads to histone epigenetic reprogramming at the Cdkn2a locus
The connection between EZH2 inhibition and mitochondrial dysfunction has not been previously explored. To further prioritize our target genes driving the mitochondrial phenotype we sought to connect the expression analysis directly to histone epigenetic control. First, to determine the histone PTM landscape post EZH2 inhibition in T cells we performed a high-resolution mass spectrometric analysis of acid-extracted histones from T cells after EZH2 inhibition (Figure S4A). We quantified the relative abundance ~30 unique histone modifications (mono-methylation, di-methylation, tri-methylation, phosphorylation, and acetylation) and combinatorial status where possible (Figure 3A, 3B and S4B–D). Relative spectral counting of PTMs on the functionally distinct histone H3.1 (DNA synthesis) and H3.3 (DNA synthesis independent), revealed a global reduction H3K27me2/3 as expected. We did not detect a large-scale remodeling of the histone landscape, most of the PTMs detected were not altered between the groups. However, aside from K27me/me2/me3 changes we detected significant changes in s28ph, k36me3, and k37me/me2 on H3.1. Interestingly, we observed an elevation of H3K36me3 on both H3.1 and H3.3, a histone PTM thought to be involved in gene elongation and alternative splicing36. Time course analysis revealed that T cells have near undetectable levels of H3K36me3, this changed as H3K27me2/3 is lost due to EZH2 inhibition (Figure 3C). The presence of H3K36me3 was confirmed using parallel reaction monitoring (PRM), targeted mass spectrometry (Figure 3D)37. These data indicate that a loss of the repressive mark, H3K27me3 coincides with a certain degree of accumulation of H3K36me3 in EZH2i treated cells.
Figure 3: Loss of H3K27me3 leads to histone epigenetic reprogramming at the Cdkn2a locus.
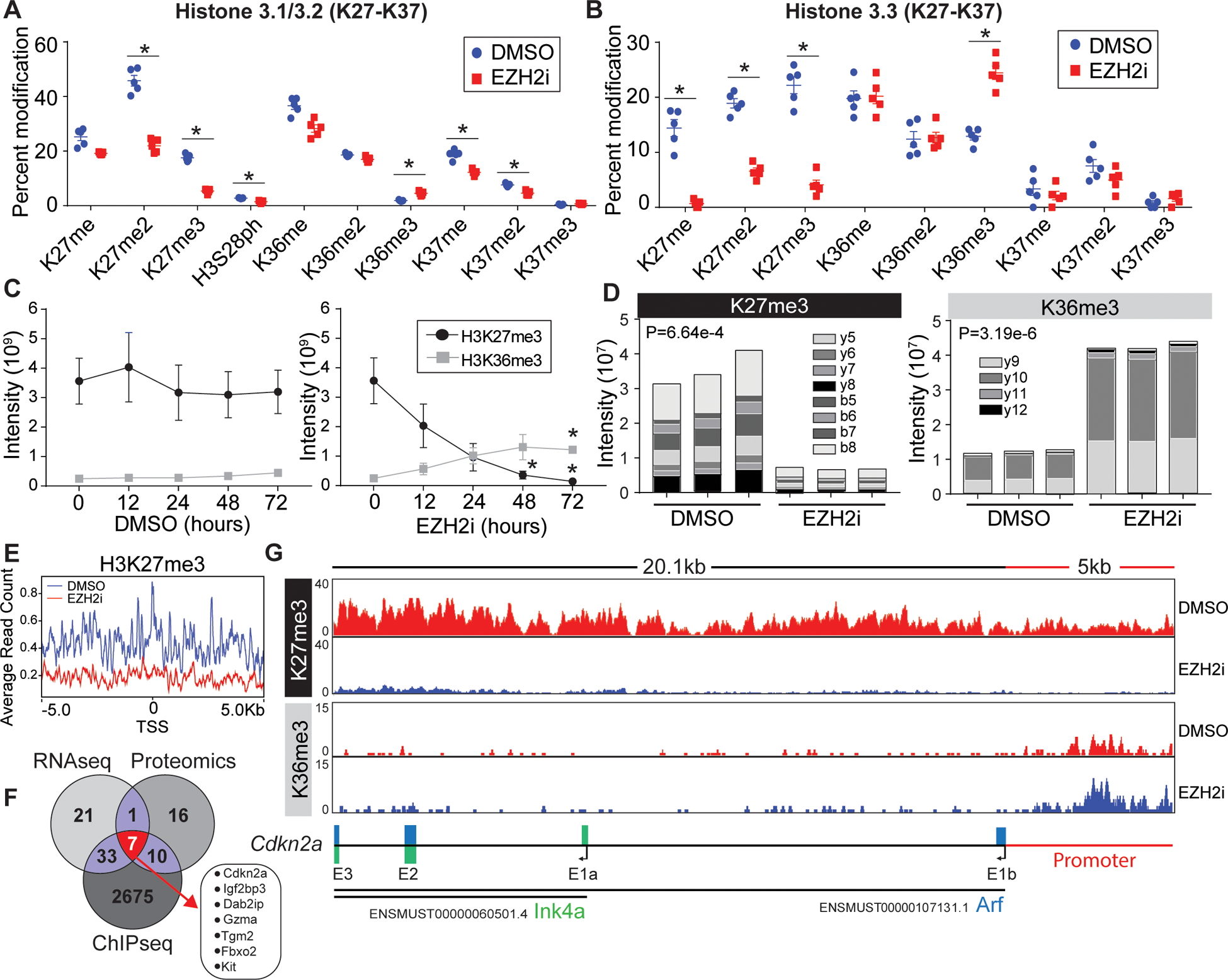
(A, B) Proteomic analysis of acid extracted histones from in vitro activated CD8+ T cells treated post activation with vehicle or EZH2i. Relative abundance of (A) histone H3.1/2 and (B) histone H3.3 post translational modifications are presented as a percent of total peptide spectral counts. Individual replicates (n=5) are plotted as a mean and errors bars represent SEM. Asterisks denote corrected P value <.05 (Holm-Sidak). (C) Proteomic quantification of H3K36me3 and H3K27me3 from time points post EZH2i treatment. Data represent the mean (n=3) and error bars represent SEM. Asterisks signify an adjusted P value <0.05 (2way-ANOVA) (D) Parallel reaction monitoring (PRM) mass spectrometry for H3K36me3 and H3K27me3 from activated T cells 48 hours post EZH2 inhibition. The Specific b and y ion series noted were used to distinguish K36 and K27 trimethylation. Representative data from 3 biological replicates. (E) Average read counts of H3K27me3 relative to the global TSS ±5kb (F) H3K27me3 ChIP-Seq of control and EZH2i treated T cells. 2725 loci with a log2FC<−2.5 were identified, a venn diagram was used to compare genes identified in RNAseq, proteomics, and ChIP-seq data sets. (G) IGV views of H3K27me3 and H3K36me3 read densities at the Cdkn2a locus. Bottom schematic shows the distribution of Arf and Ink4a exons.
In accordance with the histone-proteomics, ChIP-sequencing revealed a genome-wide reduction in H3K27me3 level in the EZH2i treated cells. For instance, in a window of ±5 kb from the TSS, we demonstrated a significant decrease in average read count (ARC) of H3K27me3 (ARC: 0.2), compared to control (0.4) (Figure 3E). While, the EZH2 inhibition doesn’t necessarily impact the occupancy or enrichment of H3K36me3 at the ±5 kb of global TSS site (Figure S5A), we observed enrichment of H3K36me3 at the body of several differentially overexpressed (Log2FC>2.5 genes N=18), including Tgm2, Gzma, Dab2ip or Kit of the most significant candidates from the RNAseq and proteomic data sets (Figure 3F). Interestingly, EZH2i induced an elevation of H3K36me3 at the Cdkn2a promoter (Figure 3G). The Cdkn2a promoter was previously reported to contain bivalent chromatin marks, overlapped at the hypomethylated CpG Island. Induced demethylation resulted in complete loss of H3K27me3 at the gene promoter, leading to a gain in H3K27ac38. Herein, we observed that induced removal of H3K27me3 is concomitant with accumulation of H3K36me3 at the promoter. Increment in gene-expression (Log2FC=4.36) hence can be reasonably correlated to the switch in occupancy from H3K27me3 to the activating H3K36me3, H3K27ac etc. marks at the Cdkn2a promoter in the EZH2 inhibited cells.
The shift in epigenetic regulation at the Ink4a-Arf (Cdkn2a) locus, and release of this gene had significant downstream effects, as evidenced by the gene products presenting as top hits in RNAseq and proteomic data sets. Cdkn2a encodes two tumor suppressors, Ink4a and Arf, which are not expressed in most normal tissues39. Arf (p19Arf) is derived from Cdkn2a through an alternative reading frame product (Arf). Prior studies have revealed Arf exerts potent antiproliferative effects and Cdkn2a is known to be repressed by H3K27me3 in cancer cells40. Background evidence for Cdkn2a regulating cellular responses to stress, along with the high fidelity of this gene through multi-omics analysis implored selection for mechanistic follow up studies. Peptide mapping confirmed Arf as the protein dysregulated at this locus (Figure S6A). Additionally, Arf expression was validated by a series of western blot analysis experiments: time course of EZH2 inhibition (Figure 4A), both CD4+ and CD8+ T cells (Figure 4B), and using an alternative EZH2 inhibitor, CPI-1205 (Figure S6B). Collectively, our genome wide profiling further implicates epigenetic remodeling of the Cdkn2a locus as an event in EZH2 inhibition induced T cell exhaustion.
Figure 4: Arf induces metabolic exhaustion independent of p53.

Representative western blot analysis of candidate protein Arf in an (A) EZH2 inhibitor time course and (B) in both pre-activated CD4+ and CD8+ T cells. SV40 transformed MEFs were used as a positive control. (C) Representative western blot analysis of EZH2i treated wt and Arf−/− CD8+ T cells. (D) Oxygen consumption rate (OCR) in activated CD8+ wt or Arf-KO activated T cells treated for 48 hours ±EZH2i. P value was determined by unpaired t test. (E) C57Bl/6 wt or Arf-KO mice were challenged with MC38 tumor cells. Tumor growth curves depict an average tumor volume in each group, n=15–20 and error bars indicate the SEM. Kaplan-Meier survival of recipient mice (tumor size > 1000mm3). P value denotes statistical significance by Log Rank Test.
Arf induces metabolic exhaustion independent of p53
Arf is well known to exert tumor-suppressor activity through inhibition of MDM2, stabilizing p53, which leads to robust cell cycle arrest and cell death41,42. Surprisingly, in this case, Arf had minor effects on T cell survival and cell cycle progression (Figures S6C). Consistent with this finding, in both CD4+ and CD8+ lymphocytes we did not detect a stabilization of p53 upon EZH2-inhibition (Figure 4B). One of the hallmarks of p53 stabilization is the effect on the intrinsic apoptotic machinery (BCL-2 family). For example, p53 is well known to induce the expression of the pro-apoptotic PUMA, which we did not observe (Figure S6D, E). Also, there was no detectable cleavage of the caspase target, PARP (Figure S6E). Interestingly, the transcript levels of p53 dropped in response to EZH2 inhibition (Figure S6F). These data suggest Arf acts independently of p53 in these cells.
It is an intriguing possibility that epigenetic control of Arf directly contributes T cell metabolic exhaustion. Arf has been reported to interact with the mitochondrial protein p32/C1QBP, which was shown to lead to mitochondrial damage43. Additionally, there is an N-terminally truncated Arf protein (smARF) which lacks the residues required for p53 activation and has been reported to localize to mitochondria and trigger mitophagy44,45. To directly assess the contribution of Arf expression to T mitochondrial dysfunction we utilized an Arf knock out (Arf-KO) mouse model, which only disrupts Arf expression at the Cdkn2a locus (Figure 4C)39. Arf deficiency was able to partially restore the loss of OCR induced by EZH2-inhibition in T cells (Figure 4D). ARF plays a role in driving T cell metabolic dysfunction in the tumor environment, though this data suggests that there are additional or compensatory mechanisms at play.
Our data suggest that EZH2 acts as a critical mediator for mitochondrial sufficiency in part through regulation of the Arf locus. To demonstrate the involvement of ARF signaling in T cell dysfunction we challenged Arf deficient (Arf-KO) mice with a murine model colon adenocarcinoma (MC38) (Figure 4E). Constitutive deletion of Arf lead to an enhanced ability to control tumor growth and significantly increased median survival. However, this did not amount to complete protection and is consistent with the partial rescue of the metabolic phenotype.
Exogenous expression of EZH2Y641F improves tumor control
Approaches to enhance the survivability and metabolic sufficiency of tumor specific T cells will be important for advancing this approach46. We assessed whether reprogramming T cells with the gain-of-function EZH2Y641F mutant would enhance their ability to control tumor growth in an ACI mouse model. The EZH2Y641F mutation causes a gain-of-function of EZH2 by altering its substrate specificity. This mutation increases its activity toward the dimethylated substrate and in concert with a WT allele, causing an increase in the trimetylation of H3K2747 (Figure 5A). Tumor-specific T cells were generated from OT-I and OT-I-Lck-EZH2Y641F mice. Activated CD8+ T cells expressing the EZH2 Y641F mutant had slightly elevated rates of oxygen consumption (Figure 5B) and were superior at killing of MC38SIINFEKL tumor cells in vitro (Figure 5C). In a model ACI, tumor (MC38SIINFEKL) bearing mice that received EZH2Y641F positive OT-I T cells were able to control tumor growth significantly better than mice that received either tumor specific (OT-I) and non-specific (wt) control cells (Figure 5D). To determine if EZH2Y641F expression simply enhanced the survival of peripheral T cells in vivo, we adoptively transferred control and EZH2Y641F positive T cells into congenic, CD45.1+ recipient mice and tracked donor (CD45.2+) survival (Figure S7A). We found no significant difference in EZH2Y641F+ T cells within the spleen, lymph nodes or peripheral blood 2 days after transfer. These findings are consistent with the idea that EZH2 plays a role in the protection of TILs from metabolic stress induced dysfunction and that manipulation of EZH2 in ACI therapies would be beneficial.
Figure 5: Exogenous expression of EZH2Y641F in lymphocytes improves tumor killing and adoptive T cell therapy.

(A) Western blot analysis of in vitro activated CD8+ wt and Lck-EZH2Y641F T cells. (B) Oxygen consumption rate of pre-activated CD8+ wt and Lck-EZH2Y641F T cells. P value was determined by unpaired t test. (C) In vitro killing assay was used to determine cytotoxic function. Target cells (MC38 SIINFEKL) were cultured with CellTrace-Violet labeled, pre-activated OT-1 or OT-1- EZH2Y641F CD8+ T cells at a target/effector ratio of 1:0, 1:1, or 1:16. Tumor killing is presented as the percentage of viable (Annexin V−, PI−) tumor cells remaining after 10 hours of co-culture. Data are representative of 2 independent experiments. (D) C57BL/6 mice were inoculated with 5×106 MC8SIINFEKL. After 9 days 4×106 WT, OT-I, or OT-I. EZH2Y641F pre-activated CD8+ T cells were transferred I.V. into recipients and tumor growth was assessed. Tumor growth curves depict an average tumor volume in each group, n=8–10 and error bars indicate the SEM. Asterisk denotes statistical significance P<0.01.
Discussion
Understanding the mechanisms T cells use for protecting metabolic circuits may lead to new strategies and improve the efficacy of cancer immunotherapies. Engineering adoptive T cells, unconstrained by tolerance-based metabolic circuits, may allow for further advancement of cellular therapies against solid tumors. There have been some successes in overcoming inhibitory tumor metabolism, largely through enforcing mitochondrial respiration7,13. For example, the exogenous expression of the transcription factor Pgc1a, which directly controls mitochondrial biogenesis and effectively changes the metabolic circuit. However, recent discoveries have highlighted dynamic metabolic processes as drivers of T cell function. This suggests that genetically or pharmacologically influencing T cell metabolism will negate their ability to dynamically adapt. The goal therefore is to generate T cells that can withstand metabolic stresses, allowing for metabolic plasticity.
The ability of T cells to generate diverse phenotypes is in part due to histone modifications during the extensive chromatin remodeling occurring upon activation48. Genetic deletion studies have recently implicated EZH2 (H3K27me3) in the control T cell effector function and memory precursor formation49,50. However, this approach does not simulate an acute loss of EZH2, which TILs undergo during infiltration. Additionally, genetic deletion of EZH2 can be compensated for by EZH151. Perhaps more importantly, EZH2i therapies are actively being considered for treatment of a variety of advanced solid tumors (eg-NCT02601950, NCT03213665) and in some cases in combination with immune checkpoint inhibition (NCT03525795). These studies are motivated by a recently realized toxicity to T regulatory cells and the ability of H3K27me3 to mediate Th1-type chemokine expression in a tumor cells20,52. However, our work sheds light into the deleterious effects EZH2 inhibition on other T cell subsets and suggests the use of these drugs in combination with immunotherapy may be challenging.
In the present study, we report the identification of EZH2 (H3K27me3) as a protector of T cell metabolic sufficiency. Through a systematic evaluation of H3K27me3 loss, which occurs during tumor infiltration, we have discovered an epigenetic mechanism that contributes to the development of tumor-induced metabolic exhaustion in T cells. Loss of PRC2 repressive activity, through acute inhibition of EZH2, leads to a metabolic insufficiency in T cells. This has been characterized by the loss of mitochondrial respiration, loss of mitochondrial membrane potential, and increased sensitivity to glucose withdrawal. Our data emphasize the coordination of histone epigenetics and T cell metabolic exhaustion. These findings are consistent with recent work defining a role for EZH2 in metabolic reprogramming of cancer cells53,54.
We identified the EZH2-inhibition induced mitochondrial dysfunction phenotype using cutting-edge proteomics and gene set enrichment analysis. Using metabolic flux analysis to measure oxygen consumption rates (mitochondrial respiration) and acidification rates (glycolysis) we validated the loss of mitochondrial sufficiency in both CD4+ and CD8+ T cells using multiple specific inhibitors of EZH2. The elevated glycolytic rates induced by EZH2 inhibition is consistent with mitochondrial damage and indicates a strong glycolytic dependence. This metabolic phenotype is consistent with metabolic exhaustion in TILs, which is thought to be a driving force of functional exhaustion55. To mimic the glucose withdrawal present in solid tumors we treated with 2-Deoxy-D-Glucose (glycolysis inhibitor) or altered the concentration of glucose. EZH2 inhibition sensitized T cells to these metabolic stresses a process that we believe occurs during tumor infiltration.
The ability of EZH2 to protect mitochondrial integrity has not been previously explored. Our comparison of proteomics, RNA-sequencing, and ChIP-sequencing approaches greatly narrowed down the H3K27me3-controlled candidate genes involved in driving mitochondrial dysfunction. Surprisingly, we did not find that EZH2 was directly regulating any conical metabolic pathways (fatty acid metabolism, glucose metabolism, electron transport, etc.). However, Cdkn2aArf was a top candidate in all these approaches and known to be repressed by EZH2 (H3K27me3) in cancer cells56. The regulation of the Cdkn2a locus has been previously described in EZH2-KO T cells and was associated prolonged division times leading to a minor reduction in proliferation57. This is likely due to the fact that in activated T cells the expression of Arf does not lead to the canonical stabilization of p53 which is necessary for downstream cell cycle arrest or cell death due to a strong repression of the Trp53 through engagement of TCR signaling58. This observation leads us to propose alternative p53 independent mechanisms. We show that genetic deletion of Arf can rescue the metabolic defect induced during EZH2 inhibition. Thus, we propose that Arf induces mitochondrial dysfunction and therefore the downstream metabolic exhaustion. This p53-independent mechanism has been proposed and is thought to work through an interaction with the mitochondrial protein p32/C1QBP43. p32 is predominantly localized to the mitochondria where it has a functional role in maintaining oxidative phosphorylation59,60. Furthermore, we show that Arf−/− animals have better control of colon adenocarcinoma (MC38) growth.
Therapeutically, this study suggests that enhancing/protecting EZH2 in cytotoxic T cells would be beneficial. One of the most common mutations of EZH2 in cancer cells is Y641F, a gain of function mutation that elevates H3K27me361. We engineered tumor-specific T cells to express the EZH2Y641F mutation and it did, in fact, enhance the ability to control tumor growth in vitro and in adoptive T cell transplant experiments. Additional studies will be necessary to fully explore the potential of utilizing EZH2 gain of function mutants in cellular transplant studies.
Limitations
As outlined above, our results demonstrate that EZH2 through the placement of H3K27me3 contributes to the protection of mitochondrial sufficiency within T cells. However, we did not explore potential non-enzymatic roles for EZH2, which have been proposed to be an important aspect of EZH2 biology62. Understanding the full scope of EZH2 in T cell biology is challenging, especially when paired with conflicting roles of EZH2 within the context of tumor immunology and anti-cancer therapies. There are many important facets to T cells and EZH2 exerts broad regulatory control through modifying the epigenome. While Arf was clearly a top candidate gene in our studies, we did identify other potentially important candidates including Gzma, which has also been implicated in mitochondrial dysfunction63. Nevertheless, our findings provide an innovative perspective on the control of metabolic exhaustion of T cells during tumor infiltration and provides rationale for the clinical development of gain-of-function EZH2 T cell therapies.
Supplementary Material
Statement of significance.
Findings demonstrate that manipulation of T cell EZH2 in cellular therapies may yield cellular products able to withstand solid tumor metabolic-deficient environments.
Acknowledgements
The authors would like to thank the UAMS Biochemistry Department, members of the Tackett laboratory and Drs. J. Opferman, M. Cannon, S. Taverna, K. Raney, T. Chambers and C. O’Brien for their helpful discussion. The authors would also like to thank the UAMS Division of Laboratory Animal Medicine; the UAMS Genetic Models Core; the UAMS Proteomics Core; the UAMS Flow Cytometry Core; the Arkansas Children’s Research Institute Bioinformatics Core.
We acknowledge support from NIH grants F31CA232464 (B. Koss), P20GM121293 (A.J. Tackett), R01CA236209 (A.J. Tackett) and the Scharlau Family Endowed Chair in Cancer Research to A.J. Tackett. This study was additionally supported by the NIH P20GM109005 (NAB).
Footnotes
Conflict of interest statement: The authors have declared that no conflicts of interest exist.
References
- 1.O’Donnell JS; Teng MWL; Smyth MJ Nature Reviews Clinical Oncology. Nature Publishing Group; March 1, 2019, pp 151–167 [DOI] [PubMed] [Google Scholar]
- 2.Jiang Y; Li Y; Zhu B T-cell exhaustion in the tumor microenvironment. Cell Death and Disease 6, e1792 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blank CU; Haanen JB; Ribas A; Schumacher TN The "cancer immunogram" Science 352, 658–660 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Odorizzi PM; Pauken KE; Paley MA; Sharpe A; Wherry EJ Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. The Journal of experimental medicine 212, 1125–1137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKinney EF; Smith KGC Metabolic exhaustion in infection, cancer and autoimmunity. Nature Immunology 19, 213–221 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Buck MD; Sowell RT; Kaech SM; Pearce EL Metabolic Instruction of Immunity. Cell 169, 570–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buck MD; O’sullivan D; Klein RI; Huber TB; Rambold AS; Pearce EL Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming Correspondence. Cell 166, 63–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L; Romero P Metabolic Control of CD8+ T Cell Fate Decisions and Antitumor Immunity. Trends in molecular medicine 24, 30–48 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Wei J; Raynor J; Nguyen TLM; Chi H Frontiers in Immunology. Frontiers Research Foundation; March 9, 2017 [Google Scholar]
- 10.Sukumar M; Roychoudhuri R; Restifo NP Leading Edge Previews Nutrient Competition: A New Axis of Tumor Immunosuppression. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medina G; Vera-Lastra O; Peralta-Amaro AL; Jiménez-Arellano MP; Saavedra MA; Cruz-Domínguez MP; Jara LJ Pharmacological Research. Academic Press; July 1, 2018, pp 277–288 [DOI] [PubMed] [Google Scholar]
- 12.Delgoffe GM; Powell JD Feeding an army: The metabolism of T cells in activation, anergy, and exhaustion. Molecular immunology 68, 492–496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scharping NE; Menk AV; Moreci RS; Whetstone RD; Dadey RE; Watkins SC; Ferris RL; Delgoffe GM The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 701–703 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Kishton RJ; Sukumar M; Restifo NP Cell Metabolism Review Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metabolism 26, 94–109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Bourgeois T; Strauss L; Aksoylar HI; Daneshmandi S; Seth P; Patsoukis N; Boussiotis VA Frontiers in Oncology. Frontiers Media S.A; August 3, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phan AT; Goldrath AW; Glass CK Immunity. Cell Press; May 16, 2017, pp 714–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao E; Maj T; Kryczek I; Li W; Wu K; Zhao L; Wei S; Crespo J; Wan S; Vatan L; Szeliga W; Shao I; Wang Y; Liu Y; Varambally S; Chinnaiyan AM; Welling TH; Marquez V; Kotarski J; Wang H; Wang Z; Zhang Y; Liu R; Wang G; Zou W Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nature immunology 17, 95–103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laugesen A; Højfeldt JW; Helin K Molecular Cell Review Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emran A. Al; Chatterjee A; Rodger EJ; Tiffen JC; Gallagher SJ; Eccles MR; Hersey P Trends in Immunology. Elsevier Ltd; April 1, 2019, pp 328–344 [DOI] [PubMed] [Google Scholar]
- 20.Goswami S; Apostolou I; Zhang J; Skepner J; Anandhan S; Zhang X; Xiong L; Trojer P; Aparicio A; Subudhi SK; Allison JP; Zhao H; Sharma P Modulation of EZH2 expression in T cells improves efficacy of anti–CTLA-4 therapy. The Journal of Clinical Investigation 128, 3813–3818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He S; Liu Y; Meng L; Sun H; Wang Y; Ji Y; Purushe J; Chen P; Li C; Madzo J; Issa J-P; Soboloff J; Reshef R; Moore B; Gattinoni L; Zhang Y Ezh2 phosphorylation state determines its capacity to maintain CD8+ T memory precursors for antitumor immunity. Nature Communications 8, 2125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karantanos T; Chistofides A; Barhdan K; Li L; Boussiotis VA Regulation of T Cell Differentiation and Function by EZH2. Frontiers in immunology 7, 172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hackstadt T; Scidmore MA; Rockey DD Lipid metabolism in Chlamydia trachomatis-infected cells: Directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proceedings of the National Academy of Sciences of the United States of America 92, 4877–4881 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang SK; Scruggs AM; Donaghy J; Horowitz JC; Zaslona Z; Przybranowski S; White ES; Peters-Golden M Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death & Disease 4, e621–e621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiśniewski JR; Zougman A; Nagaraj N; Mann M Universal sample preparation method for proteome analysis. Nature Methods 6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Nesvizhskii AI; Keller A; Kolker E; Aebersold R A statistical model for identifying proteins by tandem mass spectrometry. Analytical chemistry 75, 4646–4658 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Chiang T; Koss B; Su LJ; Washam CL; Byrum SD; Storey A; Tackett AJ Effect of Sulforaphane and 5-Aza-2’-Deoxycytidine on Melanoma Cell Growth. Medicines 6, 71 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai L; Rothbart SB; Lu R; Xu B; Chen WY; Tripathy A; Rockowitz S; Zheng D; Patel DJ; Allis CD; Strahl BD; Song J; Wang GG An H3K36 Methylation-Engaging Tudor Motif of Polycomb-like Proteins Mediates PRC2 Complex Targeting. Molecular Cell 49, 571–582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinlan AR BEDTools: The Swiss-Army tool for genome feature analysis. Current Protocols in Bioinformatics 2014, 11.12.1–11.12.34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramírez F; Dündar F; Diehl S; Grüning BA; Manke T DeepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Research 42, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leclerc M; Voilin E; Gros G; Corgnac S; De Montpréville V; Validire P; Bismuth G; Mami-Chouaib F Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by Neuropilin-1. Nature Communications 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kline J; Brown IE; Zha Y-Y; Blank C; Strickler J; Wouters H; Zhang L; Gajewski TF Homeostatic proliferation plus regulatory T-cell depletion promotes potent rejection of B16 melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 14, 3156–3167 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Zingg D; Arenas-Ramirez N; Sahin D; Haeusel J; Sommer L; Boyman O; Rosalia RA; Antunes AT The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. CellReports 20, 854–867 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Ferris RL; Lu B; Kane LP Too Much of a Good Thing? Tim-3 and TCR Signaling in T Cell Exhaustion. The Journal of Immunology 193, 1525–1530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michalek RD; Rathmell JC The metabolic life and times of a T-cell. Immunological reviews 236, 190–202 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J; Ahn JH; Wang GG Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cellular and molecular life sciences : CMLS 76, 2899–2916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sowers JL; Mirfattah B; Xu P; Tang H; Park IY; Walker C; Wu P; Laezza F; Sowers LC; Zhang K Quantification of Histone Modifications by Parallel-Reaction Monitoring: A Method Validation. Analytical Chemistry 87, 10006–10014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.King AD; Huang K; Rubbi L; Liu S; Wang CY; Wang Y; Pellegrini M; Fan G Reversible Regulation of Promoter and Enhancer Histone Landscape by DNA Methylation in Mouse Embryonic Stem Cells. Cell Reports 17, 289–302 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamijo T; Zindy F; Roussel MF; Quelle DE; Downing JR; Ashmun RA; Grosveld G; Sherr CJ Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19(ARF). Cell 91, 649–659 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Gil J; Peters G Regulation of the INK4b–ARF–INK4a tumour suppressor locus: all for one or one for all. Nature Reviews Molecular Cell Biology 7, 667–677 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Kamijo T; Weber JD; Zambetti G; Zindy F; Roussel MF; Sherr CJ Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proceedings of the National Academy of Sciences of the United States of America 95, 8292–8297 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y; Xiong Y; Yarbrough WG ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 92, 725–734 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Itahana K; Zhang Y Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer cell 13, 542–553 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grenier K; Kontogiannea M; Fon EA Short mitochondrial ARF triggers Parkin/PINK1-dependent mitophagy. The Journal of biological chemistry 289, 29519–29530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Oosterwijk JG; Li C; Yang X; Opferman JT; Sherr CJ; Prives C Small mitochondrial Arf (smArf) protein corrects p53-independent developmental defects of Arf tumor suppressor-deficient mice. Proceedings of the National Academy of Sciences of the United States of America 114, 7420–7425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu X; Rashida Gnanaprakasam JN; Sherman J; Wang R Frontiers in Oncology. Frontiers Media S.A. 2019 [Google Scholar]
- 47.Berg T; Thoene S; Yap D; Wee T; Schoeler N; Rosten P; Lim E; Bilenky M; Mungall AJ; Oellerich T; Lee S; Lai CK; Umlandt P; Salmi A; Chang H; Yue L; Lai D; Cheng S-WG; Morin RD; Hirst M; Serve H; Marra MA; Morin GB; Gascoyne RD; Aparicio SA; Humphries RK A transgenic mouse model demonstrating the oncogenic role of mutations in the polycomb-group gene EZH2 in lymphomagenesis. Blood 123, 3914–3924 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Roh TY; Cuddapah S; Cui K; Zhao K The genomic landscape of histone modifications in human T cells. Proceedings of the National Academy of Sciences of the United States of America 103, 15782–15787 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobenecker M-W; Park JS; Marcello J; McCabe MT; Gregory R; Knight SD; Rioja I; Bassil AK; Prinjha RK; Tarakhovsky A Signaling function of PRC2 is essential for TCR-driven T cell responses. The Journal of experimental medicine 215, 1101–1113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gray SM; Amezquita RA; Guan T; Kleinstein SH; Kaech SM Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8 + T Cell Terminal Differentiation and Loss of Multipotency. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wassef M; Luscan A; Aflaki S; Zielinski D; Jansen PWTC; Baymaz HI; Battistella A; Kersouani C; Servant N; Wallace MR; Romero P; Kosmider O; Just PA; Hivelin M; Jacques S; Vincent-Salomon A; Vermeulen M; Vidaud M; Pasmant E; Margueron R EZH1/2 function mostly within canonical PRC2 and exhibit proliferation-dependent redundancy that shapes mutational signatures in cancer. Proceedings of the National Academy of Sciences of the United States of America 116, 6075–6080 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagarsheth N; Peng D; Kryczek I; Wu K; Li W; Zhao E; Zhao L; Wei S; Frankel T; Vatan L; Szeliga W; Dou Y; Owens S; Marquez V; Tao K; Huang E; Wang G; Zou W PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Research 76, 275–282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pang B; Zheng XR; Tian J. xia; Gao T. hong; Gu G. yan; Zhang R; Fu YB; Pang Q; Li XG; Liu Q EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1α signaling. Oncotarget 7, 45134–45143 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vantaku V; Putluri V; Bader DA; Maity S; Ma J; Arnold JM; Rajapakshe K; Donepudi SR; von Rundstedt F-C; Devarakonda V; Dubrulle J; Karanam B; McGuire SE; Stossi F; Jain AK; Coarfa C; Cao Q; Sikora AG; Villanueva H; Kavuri SM; Lotan Y; Sreekumar A; Putluri N Epigenetic loss of AOX1 expression via EZH2 leads to metabolic deregulations and promotes bladder cancer progression. Oncogene (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bengsch B; Johnson AL; Kurachi M; Odorizzi PM; Pauken KE; Attanasio J; Stelekati E; McLane LM; Paley MA; Delgoffe GM; Wherry EJ Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 45, 358–373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ito T; Teo YV; Evans SA; Neretti N; Sedivy Correspondence JM Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. CellReports 22, 3480–3492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen G; Subedi K; Chakraborty S; Sharov A; Lu J; Kim J; Mi X; Wersto R; Sung MH; Weng NP Ezh2 regulates activation-induced CD8+ T cell cycle progression via repressing Cdkn2a and Cdkn1c expression. Frontiers in Immunology 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watanabe M; Moon KD; Vacchio MS; Hathcock KS; Hodes RJ Downmodulation of tumor suppressor p53 by T cell receptor signaling is critical for antigen-specific CD4+ T cell responses. Immunity 40, 681–691 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gotoh K; Morisaki T; Setoyama D; Sasaki K; Yagi M; Igami K; Mizuguchi S; Uchiumi T; Fukui Y; Kang D Mitochondrial p32/C1qbp Is a Critical Regulator of Dendritic Cell Metabolism and Maturation. Cell Reports 25, 1800–1815.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Jiang J; Zhang Y; Krainer AR; Xu RM Crystal structure of human p32, a doughnut-shaped acidic mitochondrial matrix protein. Proceedings of the National Academy of Sciences of the United States of America 96, 3572–3577 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wigle TJ; Knutson SK; Jin L; Kuntz KW; Pollock RM; Richon VM; Copeland RA; Scott MP The Y641C mutation of EZH2 alters substrate specificity for histone H3 lysine 27 methylation states. FEBS Letters 585, 3011–3014 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Kim J; Lee Y; Lu X; Song B; Fong KW; Cao Q; Licht JD; Zhao JC; Yu J Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Reports 25, 2808–2820.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martinvalet D; Zhu P; Lieberman J Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22, 355–370 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.